

Page 1

GE Healthcare
Instructions 71-7149-00 AN HiTrap ion exchange columns
HiTrap SP HP, 1 ml and 5 ml
HiTrap Q HP, 1 ml and 5 ml
HiTrap™ SP HP and HiTrap Q HP are prepacked, ready to use cation and anion
exchange columns for method scouting, group separations, sample concentration
and sample clean-up of charged biomolecules. HiTrap SP HP and HiTrap Q HP provide
fast, reproducible, and easy separations in a convenient format.
The columns can be operated with a syringe, peristaltic pump or liquid
chromatography system such as ÄKTAdesign
™
or FPLC™ System.
Page 2

Code No. Designation No. supplied
17-1151-01 HiTrap SP HP 5 x 1 ml
17-1152-01 HiTrap SP HP 5 x 5 ml
17-1153-01 HiTrap Q HP 5 x 1 ml
17-1154-01 HiTrap Q HP 5 x 5 ml
Connectorkit
Connectors supplied Usage No. supplied
1/16” male/luer female Connection of syringe to top of
HiTrap column 1
Tubing connector Connection of tubing (e.g. Peristaltic
flangeless/M6 female Pump P1) to bottom of HiTrap column* 1
Tubing connector Connection of tubing (e.g. Peristaltic
flangeless/M6 male Pump P1) to top of HiTrap column** 1
Union 1/16” female/ Connection to original FPLC Sys tem
M6 male through bottom of HiTrap column 1
Union M6 female/ Connection to original FPLC Sys tem
1/16” male through top of HiTrap column 1
Stop plug female, 1/16” Sealing bottom of HiTrap column 2, 5 or 7
* Union 1/16” fe male/M6 male i s also needed .
** Union M6 fema le/1/16” male is als o needed.
Table of contents
1. Description 3
2. Selection of ion exchanger and conditions 5
3. Operation 9
4. Optimization 12
5. Choice of gradient type 13
6. Determination of binding capacity 15
7. Scaling up 17
8. Storage 17
9. Ordering Information 17
p. 2
Page 3

1. Description
Media properties
SP Sepharose™ High Per formance and Q Sepharose High Performance
are strong cation and strong anion exchangers respectively. Both remain
charged and maintain high capacity over broad pH ranges. The functional
groups are coupled to the matrix via chemically stable ether linkages.
Characteristics of HiTrap SP HP and HiTrap Q HP, 1 and 5 ml columns are
listed in Table 1.
Column
HiTrap Q HP and HiTrap SP HP are 1 ml and 5 ml columns made of
polypropylene, which is biocompatible and non-interactive with
biomolecules. The top and bottom frits are manufactured from porous
polyethylene. It is delivered with a stopper on the inlet and a snap-of f end
on the outlet.
The separation can be easily achieved using a syringe together with the
supplied luer adaptor, a peristaltic pump, or in a chromatography system
such as ÄKTA
Note: To prevent leakage it is essential to ensure that the adaptor is tight.
The column cannot be opened or refilled.
™
or FPLC.
p. 3
Page 4

Table 1. HiTrap SP HP and HiTrap Q HP columns characteristics
Column volumes 1 ml or 5 ml
Column dimensions 0.7 × 2.5 cm (1 ml) and 1.6 × 2.5 cm (5 ml)
Total ionic capacity 0.14–0.20 mmol (Cl
0.15–0.20 mmol (H
–
)/ml medium (Q)
+
)/ml medium (SP)
Dynamic binding capacity SP: approx. 55 mg ribonuclease/ml medium
(0.1 M sodium acetate, pH 6.0 at 1 ml/min)
Q: approx. 50 mg HSA/ml medium (20 mM Tris-HCl,
pH 8.2 at 1 ml/min)
Mean particle size 34 μm
Bead structure 6% highly cross-linked spherical agarose
Maximum flow rates HiTrap 1 ml: 4 ml/min, HiTrap 5 ml: 20 ml/min
Recommended f low rates HiTrap 1 ml: 1 ml/min, HiTrap 5 ml: 5 ml/min
Maximum backpressure 0.3 MPa, 3 bar, 42 psi
Chemical stability All commonly used buffers
Charged group SP: – CH
Q: – CH2N+(CH3)
pH stability*
2CH2CH2SO3
3
Sho rt term SP: 3–14, Q : 1–14
Working SP: 4–13, Q: 2–12
Long term SP: 4–13, Q: 2–12
Storage temperature +4° to +30 °C
Storage buffer SP: 20% ethanol, 0.2 M sodium acetate
Q: 20% ethanol
Avoid SP: Oxidizing agents, cationic detergents and
buffers
Q: Oxidizing agents, anionic detergents and
buffers
* Th e ranges given are estimates b ased on our knowledg e and experience.
Pleas e note the follow ing:
pH stability, long term refers to the pH i nterval where the medium is s table over
a long pe riod of time wit hout advers e eff ects on its subsequent chromatographic
performance.
pH stability, short term refers to the pH interval for regeneration, cleaning-in-place and
sanitization procedures.
p. 4
Page 5
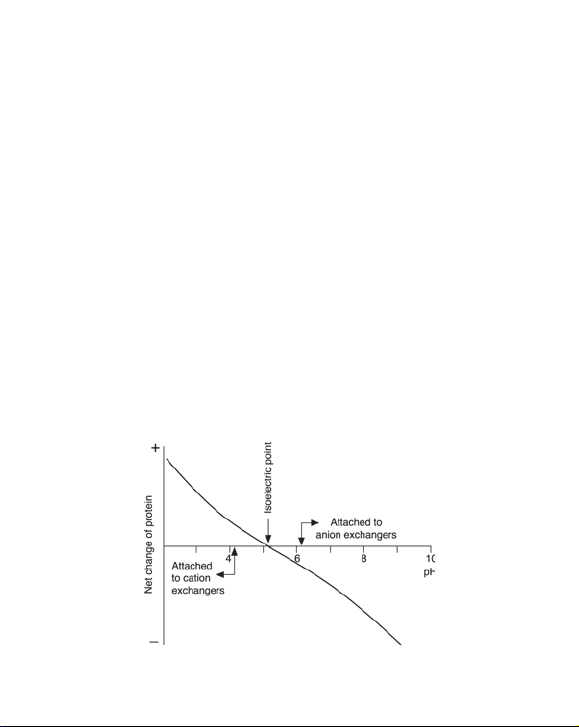
2. Selection of ion exchanger and conditions
Ion exchange chromatography is based on adsorption and reversible
binding of charged sample molecules to oppositely charged groups
attached to an insoluble matrix. The pH value at which a biomolecule carries
no net charge is called the isoelectric point (pI). When exposed to a pH below
its pI, the biomolecule will carry a positive charge and will bind to a cation
exchanger (SP). At pH’s above its pI the protein will carry a negative charge
and will bind to an anion exchanger ( Q). If the sample components are most
stable below their pI’s, a cation exchanger should be used. If they are most
stable above their pI´s, an anion exchanger is used. If stability is high over a
wide pH range on both sides of pI, either type of ion exchanger can be used
(Figure 1).
Selection of buff er pH and ionic strength
Buffer pH and ionic strength are critical for the binding and elution of
material (both target substances and contaminants) in ion exchange
chromatography. Selection of appropriate pH and ionic strength for
the start and elution buffers allows the use of three possible separation
strategies.
Fig 1. The net charge or a protein as a function of pH.
p. 5
Page 6

Strategy 1. Binding and elution of all sample components
Binding is achieved by choosing a start buffer with a low pH for HiTrap SP
HP or a high pH for HiTrap Q HP. The ionic strenght should be kept as low as
possible to allow all components to bind to the ionic exchange (<5 mS/cm).
This results in a concentration of the target substance and a complete
picture of the whole sample. The drawback of this strategy is that the
binding capacity of the ion exchanger for the target substance is dependent
on the amount of contaminant in the sample. Strongly binding contaminants
can also displace bound target protein if a large volume of sample is loaded.
Note: Start conditions are subject to the stability of the sample
components.
Strategy 2. Enrichment of target protein
This is achieved by choosing a start buffer with a pH optimized to allow
maximal binding of target protein, and as high as possible ionic strength
to suppress binding of sample contaminants. This strategy results in a
concentration of the target substances.
Strategy 3. Binding of sample contaminants
This is achieved by choosing a start buffer with a pH and ionic strength
that promotes the binding of some or all contaminating substances but
allows the substance of interest to pass through the column. The drawback
of this approach is that the target substance is not concentrated and the
sample volume applied to the ion exchanger is dependent on the amount of
contaminants in the sample.
Start buff er
The concentration of buf fer required to give effective pH control varies
with the buffer system. A list of suitable buffers and suggested starting
concentrations is shown in Tables 2 and 3, Figs 2 and 3. In the majority of
cases a concentration of at least 10 mM is required to ensure adequate
buffering capacity. The ionic strength of the buf fer should be kept low
(< 5 mS/cm) so as not to interfere with sample binding. Salts also play a
role in stabilizing protein structures in solution and it is important the ionic
strength should not be so low that protein denaturation or precipitation
occurs.
p. 6
Page 7

The buf fering ion should carry the same charge as the ion exchange group
and should have a pKa within 0.5 pH units of the pH used in the separation.
Buffering ions of opposite charge may take par t in the ion exchange process
and cause local disturbances in pH.
Starting pH
Cation exchangers (SP): At least 1 pH unit below the pI of substance to be
bound.
Anion exchangers (Q): At least 1 pH unit above the pI of substance to be
bound.
Table 2. Buffers for cation exchange chromatography.
pH inter val Subst ance Conc . (mM) Counter-ion pKa (25 °C)
1.4–2.4 Mal eic acid 20 Na+ 1.92
2.6–3.6 Methyl malonic acid 20 Na
2.6–3.6 Citric acid 20 Na
3.3–4.3 Lactic acid 50 Na
3.3–4.3 Formic acid 50 Na
3.7–4.7; 5.1–6.1 Succinic acid 50 Na
4.3–5.3 Acetic acid 50 Na
5.2–6.2 Methyl malonic acid 50 Na
5.6–6.6 MES 50 Na
6.7–7.7 Phosphate 50 Na
7.0–8.0 HEPES 50 Na
7.8 – 8 . 8 B I CIN E 50 N a
1
Ref: Handboo k of chemistry a nd physics, 83rd edition, CRC, 2002–2003.
+
or Li+ 3.07
+
3.13
+
3.86
+
or Li+ 3.75
+
4.21; 5.64
+
or Li+ 4.75
+
or Li+ 5.76
+
or Li+ 6.27
+
7.2 0
+
or Li+ 7.5 6
+
8.33
1
p. 7
Page 8
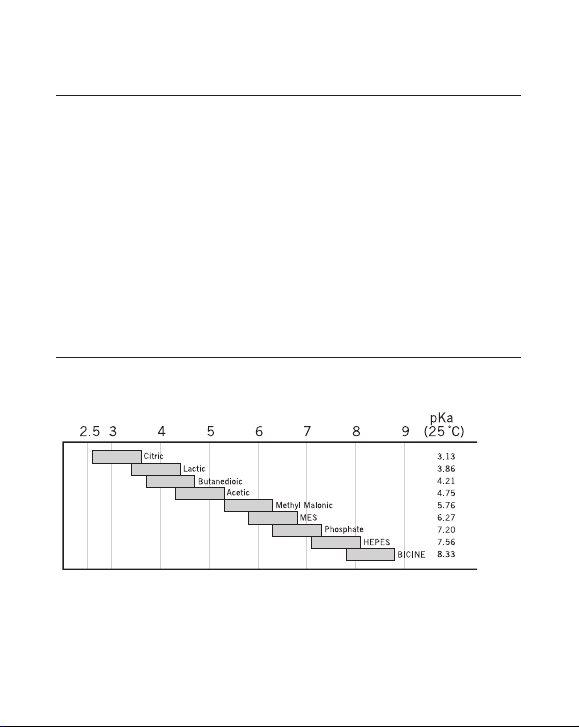
Table 3. Buffers for anion exchange chromatography.
pH inter val Substance Conc. ( mM) Counter-ion pKa (25 °C)
4.3–5.3 N-Methylpiperazine 20 Cl- 4.75
4.8–5.8 Piperazine 20 Cl
5.5–6 .5 L-Histidine 20 Cl
6.0–7.0 bis-Tris 20 Cl
6.2–7.2; 8.6–9.6 bis-Tris propane 20 Cl
7.3–8.3 Triethanolamine 20 Cl
7.6 – 8 .6 Tr i s 20 Cl- 8.07
8.0–9.0 N-Methyldiethanolamine 20 SO
8.0–9.0 N-Methyldiethanolamine 50 Cl
8.4–9.4 Di ethanolamine 20 at pH 8.4 Cl
50 at pH 8.8
8.4–9.4 Propane 1,3-Diamino 20 Cl
9.0–10.0 Ethanolamine 20 Cl
9.2–10.2 Piperazine 20 Cl
10.0–11.0 Propane 1,3-Diamino 20 Cl
10.6–11. 6 Pip eri din e 20 Cl
1
Ref: Handboo k of chemistry a nd physics, 83rd edition, CRC, 2002–2003.
-
or HCOO- 5.33
-
6.04
-
6.48
-
6.65; 9.10
-
or CH3COO- 7.76
2-
8.52
4
-
or CH3COO- 8.52
-
8.88
-
8.88
-
9.50
-
9.73
-
10.55
-
11.12
pH
Fig 2. Recommended buffer substances for cation exchange chromatography.
1
p. 8
Page 9

Fig 3. Recommended buffer substances for anion exchange chromatography.
The columns can be operated with a syringe, peristaltic pump or a
chromatography system.
3. Operation
Buff er preparation
Water and chemicals used for buffer preparation should be of high purity. It
is recommended to filter the buffers by passing them through a
0.22 μm filter immediately before use. See Tables 2 and 3, Figs 2 and 3 for
recommended buffers.
Sample preparation
The sample should be adjusted to the composition of the start buffer.
This can be done by either diluting the sample with start buffer or by
buffer exchange using HiTrap Desalting, HiPrep
column. The sample should be filtered through a 0.22 μm or 0.45 μm filter or
centrifuged immediately before it is applied to the column. See Table 4.
™
26/10 Desalting or PD-10
p. 9
Page 10

p. 10
>5000).
r
>5000).
r
a syringe or pump
to run.
17-5087-01 HiPrep Up to 15–20 ml Prepacked with For desalting and
26/10 15 ml Sephadex G-25 buffer exchange of
Desalting Fine. Requires a protein extracts
pump to run. (M
Table 4. Prepacked columns for desalting and buffer exchange.
Code No Column Loading volume Elution volume Comments Application
17-1408-01 HiTrap 0.1–1.5 ml 1.3–4.0 ml Prepacked with For desalting and buffer
Desalting Sephadex™ G-25 exchange of protein
Superfine. Requires extracts (M
17-0851-01 PD-10 2.5 ml 3.5 ml Prepacked with For desalting and
>5000)
r
>5000).
r
17-0855-01 NICK™ 0.1 ml 0.4 ml Prepacked with For separation of
Sephadex G-25. proteins (M
and nicktranslated
Desalting Sephadex G-25. buffer exchange
Requires only of protein extracts
gravity to run. (M
DNA from radiolabelled
>5000),
r
nucleotides not shorter
than 120 mers, and
similar separations.
17-0853-01 NAP™-5 0.5 ml 1.0 ml Prepacked with For purification of
17-0854 -01 NAP-10 1 .0 ml 1.5 ml Sephadex G-25 proteins (M
17-0852-01 NAP-25 2.5 ml 3.5 ml DNA grade. DNA and oligo -
Requires only nucleotides greater
gravity to run. than 10 bases in length.
Page 11

Purifi cation
1. Fill the syringe or pump tubing with start buffer (low ionic strength).
Remove the stopper and connect the column to the syringe (with the
provided adaptor), “drop to drop” to avoid introducting air into the
column.
2. Remove the snap-off end at the column outlet.
3. Wash out the preservatives with 5 column volumes of start buffer, at
1 ml/min for the 1 ml column and 5 ml/min for the 5 ml column.
4. Wash with 5 column volumes of elution buffer (start buffer with
1 M NaCl).
5. Finally equilibrate with 5–10 column volumes of start buffer.
6. Apply the sample at 1 or 5 ml/min for the 1 ml and 5 ml columns
respectively, using a syringe fit ted to the luer adaptor or by pumping it
onto the column.
7. Wash with at least 5 column volumes of start buffer or until no material
appears in the effluent.
8. Elute with 5–10 column volumes of elution buffer (see section “Choice of
gradient type”).
9. The purif ied fractions can be desalted using a HiTrap Desalting,
HiPrep 26/10 Desalting or a PD-10 columns if necessary.
10. After the completed elution, regenerate the column by washing with
5 column volumes of regeneration buffer (start buffer with 1 M NaCl)
followed by 5–10 column volumes of start buffer. The column is now
ready for a new sample.
For a first experiment the following conditions are recommended:
Flow rate: 1 ml/min using HiTrap 1 ml column
5 ml/min using HiTrap 5 ml column
Start buffer: See Tables 2 and 3
Elution buffer: Start buffer + 1 M NaCl
Gradient volume: 20 ml
p. 11
Page 12

Note: If a P1-pump is used a max flow rate of 1–3 ml/min can be run on
a HiTrap 1 ml column packed with Sepharose High Performance
media.
4. Optimization
If sample composition is unknown, a simple screening test with the aid
of a syringe or pump can be per formed to optimize starting pH and ionic
strength.
1. Set up a series of buf fers with different pH´s, in the range 4–8 (SP) or 5–9
(Q), with 0.5–1 pH unit intervals between each buffer. Make one series
with 1 M NaCl included in the buffers (regeneration buffer) and the other
without NaCl (start buffer).
2. Equilibrate the column, see Purification.
3. Adjust the sample to the chosen start buffer, see Sample preparation.
4. Apply a known constant amount of the sample at 1 or 5 ml/min for the
1 ml and 5 ml columns respectively. Collect eluate.
5. Wash with at least 5 column volumes start buffer or until no material
appears in effluent. Collect eluate.
6. Elute bound material with elution buf fer. 3–5 column volumes is usually
sufficient. Other volumes may be required, depending on the chosen
operational conditions. Collect eluate.
7. Analyze all eluates for example by activity assay and SDS-PAGE and
determine the purity and the amount bound to the column.
8. Perform steps 2–7 for the next buffer pH.
9. Decide which pH should be used for the selected purification strategy.
10. To decide on starting ionic strength conditions, a similar screening is
done, but the buf fer pH is held constant and the ionic strength is varied
in the interval 0–0.5 M, with intervals of 0.05 to 0.1 M salt between each
buffer.
p. 12
Page 13

Further optimization
The recommendations given above will give a sound basis for developing an
efficient purification step. Details of how flow rate, sample loading, particle
size and elution scheme may be optimized to meet the special needs can
be found in the handbook, Ion Exchange Chromatography & Chromatofoucsing, Principles and Methods, Code No. 11-0004-21.
GE Healthcare supplies a wide range of ion exchange chromatography
media for purif ication of biomolecules at all scales. See Ordering information
and visit www.gehealthcare.com/hitrap.
5. Choice of gradient type
1. Stepwise gradients are easy to produce and require minimal equipment.
Eluted peaks are very sharp and elution volumes minimal. However,
care must be exercised in the design of the steps and the interpretation
of results for substances eluted by a sharp change in pH or small
differences in ionic strength. Peaks tend to have sharp fronts and
pronounced tailing since they frequently contain more than one
component.
2. Continuous salt gradients are the most frequently used type of elution.
Many types of gradient forming systems are available. Two buffers of
differing ionic strength, the start and elution buf fer (start buffer
+ 1 M NaCl or higher buffer salt concentration), are mixed together and if
the volume ratio is changed linearly, the ionic strength changes linearly.
Note: Another, but less common, method to desorb bound material is to
increase (SP) or decrease (Q) the pH of the eluent. Continuous pH
gradients are difficult to produce at constant ionic strength, since
simultaneous changes in ionic strength, although small, also occur
(buffering capacities are pH dependent).
p. 13
Page 14

Elution with stepwise ionic strength gradients
Stepwise elution is the sequential use of the same buffer at different ionic
strengths. It is technically simple and fast, and is suitable for syringe
operation. It is often used for sample concentration and sample clean-up.
Stepwise elution gives small peak volumes and the resolution depends on
the difference in elution power between each step.
1. Choose starting conditions as outlined under Optimizing starting
conditions.
2. Equilibrate the column, see Purification.
3. Adjust the sample to the choosen starting pH and ionic strength, see
Sample preparation.
4. Apply the sample at 1 or 5 ml/min for the HiTrap 1 ml or 5 ml column
respectively. Collect eluate.
5. Wash with at least 5 column volumes of start buffer or until no material
appears in effluent. Collect eluate.
6. Elute with the first step ionic strength buffer. The volumes required for
stepwise elution depend on the operating conditions. However, 3–5
column volumes is usually sufficient. Collect eluate.
7. Elute with next ionic strength buffer. Collect eluate.
8. After completed elution, regenerate the column by washing with 5
column volumes of regeneration buffer (start buffer with 1 M NaCl)
followed by 5–10 column volumes of start buffer. The column is now
ready for a new sample.
Elution with continuous ionic strength gradients
Continuous salt gradient elution is the most frequently used type of
elution in ion exchange chromatography. It is very reproducible and leads
to improved resolution, since zone sharpening occurs during elution.
Continuous gradients can be prepared in different ways, depending on
available equipment.
p. 14
Page 15

– A peristaltic pump and a gradient mixer e.g. pump P-1, gradient mixer
GM-1.
– A one pump system, e.g. ÄKTAprime™ plus.
– A two pump system, e.g FPLC or ÄKTA.
1. Choose starting conditions as outlined under Optimizing starting
conditions.
2. Equilibrate the column, see Purification.
3. Adjust the sample to the chosen starting pH and ionic strength, see
Sample preparation.
4. Apply the sample at 1 or 5 ml/min for the HiTrap 1 or 5 ml column
respectively. Collect eluate.
5. Wash with 5–10 column volumes of star t buffer or until no material
appears in effluent.
6. Start the gradient elution. A gradient volume of 10–20 column volumes
and an increase in ionic strength to 0.5 M NaCl is usually sufficient.
7. Regenerate the column by washing with 5 column volumes of start
buffer with 1 M NaCl followed by 5–10 column volumes of star t buffer.
The column is now ready for a new sample.
6. Determination of binding capacity
The amount of sample which can be applied to a column depends on the
capacity of the column and the degree of resolution required. The capacity
is dependent on the sample composition, choosen starting conditions of pH
and ionic strength and the flow rate at which the separation is done. The
influence of flow rate and pH on the capacity for some model proteins are
shown in Figure 4.
Samples were applied until 5% of the start material appeared in the eluent.
The column was then washed with 10 ml start buffer (20 mM Tris-HCl, pH 8.2
or 9.0) before elution with elution buffer (20 mM Tris-HCl, 1.0 M NaCl, pH 8.2
or 9.0).
1. Equilibrate the column, see Purification.
2. Adjust the sample to the chosen starting pH and ionic strength, see
Sample preparation.
3. Determine the concentration of the specific proteins by UV, SDS- PAGE,
ELISA or other appropriate techniques.
p. 15
Page 16

Fig 4. Binding capacity of human IgG, HSA and human transferrin at dif ferent pH´s
on HiTrap Q HP, 1 ml.
4. Apply the sample solution to the column with a pump or a syringe, at a
flow rate equal to the flow rate to be used in the purification method.
Collect fractions and continue sample application until the column is
saturated.
5. Wash the column with 5–10 column volumes start buffer or until no
material appears in the effluent.
6. Elute bound proteins with 3–5 column volumes of elution buffer (start
buffer with 1 M NaCl) and collect eluate.
7. Analyse fractions and eluates from steps 4 and 6 for the specific protein
and determine the breakthrough profile (sample concentration as a
function of the amount of sample applied). The dynamic capacity is the
amount that can be applied without any significant breakthrough. The
total capacity for the specific protein is determined from step 6.
p. 16
Page 17

7. Scaling up
For quick scale-up of purifications (back pressure will increase), two or three
HiTrap ion exchange columns of the same type can be connected in series.
For further scale-up SP Sepharose High Performance and Q Sepharose High
Performance are available in prepacked HiLoad
packs. See Ordering Information.
™
columns or bulk media
8. Storage
HiTrap SP HP: Rinse with water and then wash with 5 column volumes of
20% ethanol, 0.2 M sodium acetate.
HiTrap Q HP: Rinse with water and then with 5 column volumes of 20%
ethanol.
Seal the column with the supplied stoppers. The recommended storage
temperature is +4 to +30 °C.
9. Ordering Information
Produc t No. Supplied Code No.
HiTrap SP HP 5 x 1 ml 17-1151-01
HiTrap SP HP 5 x 5 ml 17-1152-01
HiTrap Q HP 5 x 1 ml 17-1153-01
HiTrap Q HP 5 x 5 ml 17-1154-01
p. 17
Page 18

Related products No. Supplied Code No.
HiTrap IEX Selection Kit 7 x 1 ml 17-6002-33
HiTrap Desalting 5 x 5 ml 17-1408- 01
HiTrap Desalting 100 x 5 ml* 11-0003-29
HiPrep 26/10 Desalting 1 x 53 ml 17-5087-01
HiPrep 26/10 Desalting 4 x 53 ml 17-5087-02
PD-10 Desalting column 30 17-0851-01
HiLoad 16/10 SP Sepharose High Performance 1 x 20 ml 17-1137-01
HiLoad 26/10 SP Sepharose High Performance 1 x 53 ml 17-1138-01
HiLoad 16/10 Q Sepharose High Performance 1 x 20 ml 17-1064-01
HiLoad 26/10 Q Sepharose High Performance 1 x 53 ml 17-1066-01
SP Sepharose High Performance 75 ml 17-1087-01
Q Sepharose High Performance 75 ml 17-1014-01
* Specia l pack size deliv ered on specif ic order.
Accessories No. Supplied Code No.
1/16” male/luer female* 2 18-1112-51
Tubing connector flangeless/M6 female* 2 18-1003-68
Tubing connector flangeless/M6 male* 2 18-1017-98
Union 1/16” female/M6 male* 6 18-1112-57
Union M6 female /1/16” male* 5 18-3858-01
Union luerlock female/M6 female 2 18-1027-12
HiTrap/HiPrep, 1/16” male connector for ÄKTAdesign 8 28-4010-81
Stop plug female, 1/16”
Fingertight stop plug, 1/16”
* One c onnector inc luded in each Hi Trap pac kage.
† Two, five, o r seven stop plu gs female inclu ded in HiTrap pack ages dependi ng on the produc t.
‡
O ne finger tight stop plu g is connected t o the top of each Hi Trap colu mn at delivery .
†
5 11-0004-64
‡
5 11-0003-55
p. 18
Page 19

Realated literature
Ion Exchange Chromatography & Chromatofocusing
Handbook, Principles and Methods 1 11-0004-21
Ion Exchange Columns and Media,
Selection Guide 1 18-1127-31
Convenient Protein Purification,
HiTrap Column Guide 1 18-1129-81
p. 19
Page 20

www.gehealthcare.com/hitrap
www.gehealthcare.com
GE Healthcare Bio-Sciences AB
Björkgatan 30
751 84 Uppsala
Sweden
HiTrap, Sepharose, Sephadex, FPLC, ÄKTA, ÄKTAprime, ÄKTAdesign, Drop Design, NICK, NAP, HiPrep and HiLoad are
trademarks of GE Healthcare companies. GE, imagination at work and GE monogram are trademarks of General Electric
Company.
All goods and services are sold subject to the terms and conditions of sale of the company within GE Healthcare which
supplies them. GE Healthcare reserves the right, subject to any regulatory and contractual approval, if required, to make
changes in specifi cations and features shown herein, or discontinue the product described at any time without notice or
obligation. Contact your local GE Healthcare representative for the most current information.
© 2006 General Electric Company – All rights reserved.
GE Healthcare AB, a General Electric Company.
GE Healthcare Europe GmbH
Munzinger Strasse 5
D-79111 Freiburg
Germany
GE Healthcare UK Ltd
Amersham Place
Little Chalfont
Buckinghamshire, HP7 9NA
UK
GE Healthcare Bio-Sciences Corp
800 Centennial Avenue
P.O. Box 1327
Piscataway, NJ 08855-1327
USA
GE Healthcare Bio-Sciences KK
Sanken Bldg.
3-25-1, Hyakunincho
Shinjuku-ku, Tokyo 169-0073
Japan
71-7149-00 AN 08/2006
Elanders Östervåla 2006
 Loading...
Loading...