

Page 1

Systèmes CPL/SM à trappe d’ions Agilent série 6300
CPL/SMnsouple hautes performances
Page 2

Des niveaux de performances
correspondant à vos applications
et à votre budget
Que vous recherchiez des protéines de faible
abondance, des métabolites de médicaments
dans des matrices complexes ou des résidus
de pesticides dans des produits alimentaires,
il existe un système CPL/SM à trappe d’ions
série 6300 adapté à vos attentes. En outre,
quelle que soit la trappe d’ions série 6300
que vous choisissiez, elle comprendra une
acquisition des données à grande vitesse et
à faible coût pour une compatibilité avec la
chromatographie moderne haute résolution.
La série 6310 rend la SM nhautes
performances économique
La série 6310 est une valeur sûre, dotée de
caractéristiques exceptionnelles de sensibilité, de souplesse et de fiabilité. Elle
associe un changement de polarité rapide
à des capacités d’acquisition dépendantes
des données pour augmenter la quantité de
données que vous pouvez acquérir en une
seule analyse.
La série 6320 offre une sensibilité,
une résolution de masse et une
vitesse de balayage supérieures
La trappe d’ions grande capacité de la série
6320 augmente la sensibilité et les performances à tous les niveaux. La série 6320 a
une vitesse de balayage de 26 000 u/s avec
une résolution supérieure à l’unité, et propose
un mode “ spécial peptides “ pour améliorer
l’identification des protéines de faible
abondance.
Souplesse sans précédent de la CPL/SMnhautes performances
Avec quatre niveaux de performances, une large gamme de choix d’ionisation, un logiciel spécifique de
l’application et une vaste sélection de technologies de CPL, les systèmes CPL/SM à trappe d’ions Agilent
série 6300 s’adaptent à la plupart des types d’analyse. Une technologie éprouvée alliée à la fiabilité
légendaire d’Agilent vous garantit à tout moment les performances dont vous avez besoin.
2
La série 6330 offre une sensibilité
exceptionnelle aux analytes de
faible abondance
Grâce à un nouveau détecteur haute
résolution, la trappe 6330 est probablement
la trappe d’ions la plus sensible disponible
pour les applications réelles. Elle atteint une
sensibilité maximale sans altérer la vitesse
de balayage ni la résolution comme les
trappes d’ions en deux dimensions.
La série 6340 améliore la
caractérisation PTM et
l’identification des protéines
La série 6340 offre les performances exceptionnelles de la série 6330 et permet en outre
des dissociations induites par transfert
d’électrons (ETD). Les ETD constituent une
autre technique de fragmentation fournissant
une image plus complète des séquences
peptidiques et des emplacements des
difications chimiques.
Page 3
3
www.agilent.com/chem/iontrap
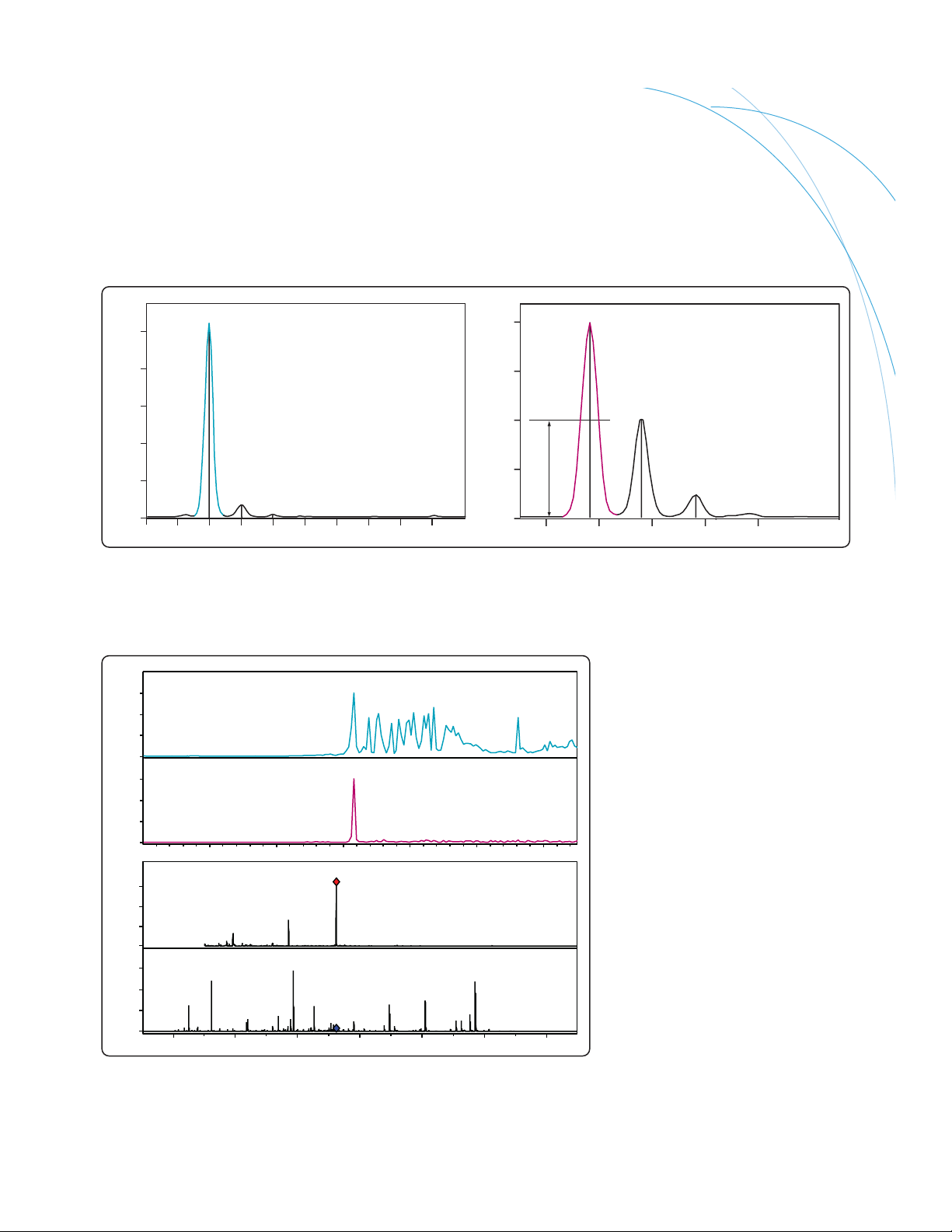
321,9
323,0
0,00
0,25
0,50
0,75
1,00
1,25
x10
6
Intens.
320
321 322 323 324 325 326 327 328 329
m/z
Spectre de SM Pic
à 322 m/z
2121,8
2122,8
2123,8
0
1
2
3
4
2121
2122 2123 2124 2125
m/z
Spectre de SM Pic
à 2 122 m/z
FWHM
R = M/delta M @ 50 %
R = 2 121,8/0,34
Résolution = 6 241
R = M/delta M @ 50 %
R = 322,1/0,27
Résolution = 1 185
x10
6
Intens.
BPC 400-1 200 m/z
1 fmole BSA dans la colonne
0,0
0,5
1,0
1,5
Intens.
0,0
0,5
1,0
1,5
x10
6
1
2 3 4 5
Durée [min]
391,5
518,3
569,9
722,7
Spectre de SM 3,1-3,2 min
249,1
321,1
437,2
535,3
584,3
650,4
704,9
778,4
892,4
1007,5
1167,5
Spectre de SM/SM 722,7 m/z
0
1
2
3
x10
5
Intens.
0
1
2
3
x10
4
200
400
600 800 1000 1200
1400
m/z
x10
6
Fragmento-grammes extraits 722,7 m/z
1 fmole BSA dans la colonne
Une analyse d’une femtomole de digestion de BSA dans une colonne par la trappe 6330 montre
une sensibilité exceptionnelle
Systèmes CPL/SM à trappe d’ions Agilent série 6300
Avec les trappes d’ions tridimensionnelles, vous n’avez pas besoin de sacrifier la vitesse de balayage pour la résolution. Cette analyse de perfluorophosphozines
sur une large gamme de masse 200 - 2 200 m/z, réalisée avec une trappe 6320, démontre une excellente résolution à une vitesse de balayage de 8 100 u/s.
linéaire supérieure. Cette résonance non
linéaire transfère très efficacement de l’énergie
aux ions pour une éjection rapide et précise
depuis la trappe. Vous obtenez ainsi une
association supérieure de gamme de masse, de
résolution de masse et de vitesse de balayage.
Le verrouillage de phase breveté contrôle avec
précision le débit d’éjection des ions, entraînant
une reproductibilité élevée entre balayages et
augmentant la fiabilité de vos résultats.
Une manière plus efficace
d’augmenter la capacité et la
sensibilité de la trappe
La capacité de la trappe a un impact direct
sur la sensibilité. Certains fabricants ont
choisi une géométrie de trappe d’ions en
deux dimensions comme moyen simple
d’augmenter la capacité de la trappe. Toutefois, les trappes d’ions en deux dimensions
compromettent d’autres paramètres de
fonctionnement importants, tels que la
vitesse de balayage et la résolution de
masse. Elles nécessitent également deux
détecteurs pour une sensibilité optimale.
Les détecteurs doubles peuvent diminuer
la fiabilité et augmenter les exigences de
maintenance.
Agilent a augmenté la capacité des trappes
d’ions tridimensionnelles de la série 6300
grâce à un réglage minutieux de la géométrie
du dispositif, aux matériaux, à la fabrication
de précision et aux paramètres de fonctionnement définis. La capacité obtenue est
élevée et offre les avantages de vitesse de
balayage et de résolution inhérents aux
trappes d’ions tridimensionnelles.
Une technologie de trappe d’ions
avancée pour des performances
optimales
Dans les trappes d’ions, la précision de masse,
la résolution de masse et la vitesse de balayage
dépendent considérablement de la vitesse et
du débit d’éjection des ions. La trappe d’ions
multipolaire tridimensionnelle brevetée utilisée
dans la série 6300 crée une résonance non
Page 4
4
Les dissociations induites par
transfert d’électrons améliorent la
couverture des séquences peptidiques et l’emplacement des PTM
La CPL/SM à trappe d’ions Agilent 6340
propose non seulement la fragmentation
CID standard, mais aussi une autre forme
de fragmentation appelée “ dissociations
induites par transfert d’électrons “ (ETD),
capable d’améliorer les analyses protéomiques. Les ETD produisent essentiellement
des ions de séries c et z. Les ETD fournissent
généralement une fragmentation plus régulière sur un peptide que la CID, d’où une
couverture de séquences supérieure. Cette
plus grande couverture de séquences
améliore l’identification des protéines par
une recherche dans une base de données
ou un séquençage de novo.
261,1
331,2
430,3
517,2
673,2
766,3
830,8
1168,0
1243,6
1532,6
1654,6
1904
2002,9
0
200
400
600
200 400 600 800 1000 1200 1400 1600 1800 2000 m/z
z
4
414,1
543,2
680,4
802,3
1033,7
1090,5
1237
1351
1402,7
1661
c
4
[M+3H]
3+
[M+2H]
2+
932,5
IADP
H
DH
T
GF
L
t
E
y
V
ATR
1790
1922
A
RT V
y
Et L F GTHDHE
PDAI
347,4 446,5
684,0
822,3
894,8
943,8
0,0
0,2
0,4
0,6
0,8
1,0
x10
4
~ x 4
559,5
ETD
z
5
z
3
z
2
z
9
z
11
z
13
z
6
z
14
z
15
c
5
c
9
++
c
9
c
6
c
10
c
8
c
11
c
13
++
c
14
++
c
12
c
16
c
14
c
13
c
15
200 400 600 800 1000 1200 1400 1600 1800 2000 m/z
y
4
y
3
y
9++
y
17-H3PO4
3+
[M+2H - H3PO4]
3+
b
15
-H
3
PO
4
3+
b
15
-H
3PO4
++
b
15
++
b
12
++
b
14
++
Phosphopeptide IADPEHDHTGFLtEyVATR
CID
Spectres CID et ETD SM/SM du phosphopeptide IADPEHDHTGFLtEyVATR de la protéine ERK humaine recombinante (ERK 1). Le spectre CID montre une certaine
fragmentation, mais l’ion dominant provient de la perte d’acide phosphorique. Le spectre ETD montre des séries c et z presque complètes. Dans le spectre ETD,
les emplacements des phosphorylations peuvent être identifiés directement à partir du spectre.
En outre, avec les ETD, les modifications
post-traductionnelles, telles que la
phosphorylation ou la glycosylation, restent
généralement fixées à l’acide aminé durant
la fragmentation. Ceci permet de déterminer
plus facilement l’emplacement spécifique
de la modification.
La série 6340 peut acquérir des spectres CID
ou ETD, selon les besoins de votre application.
La série 6340 peut également passer d’une
fragmentation CID à une fragmentation
ETD d’un balayage à un autre, pour l’image
la plus complète possible de l’identité des
protéines et des emplacements précis des
modifications chimiques. Un mode spécial
d’acquisition dépendant des données est
disponible pour déclencher une nouvelle
accumulation et des ETD en cas de pertes
de neutres spécifiées par l’utilisateur.
Davantage de solutions de
fragmentation
La dissociation par excitation rapide est
un nouveau mode de fonctionnement CID.
Il élimine la “ coupure 1/3 “ associée
aux spectres CID traditionnels de trappe
d’ions. Ceci permet de piéger les ions de
produit de masse faible nécessaires pour
les applications telles que l’étiquetage
d’isotopes iTRAQ. Agilent prévoit d’introduire la dissociation par excitation rapide
dans la série 6300 en 2007.
Page 5

5
www.agilent.com/chem/iontrap
Source MALDI avec focalisation
dynamique pulsée pour une
sensibilité et une cohérence
améliorées
Systèmes CPL/SM à trappe d’ions Agilent série 6300
Source
d’ionisation
électrospray fiable
avec nébulisation
orthogonale et gaz de
dessiccation à contrecourant haute capacité
Séparations à nanodébit faciles et
reproductibles
Les puces-CLHP révolutionnaires d’Agilent
intègrent de manière homogène l’enrichissement des échantillons et les colonnes de
séparation d’un système CPL à nanodébit
avec les raccords complexes et le nébuliseur
utilisés en spectrométrie de masse à électronébulisation. La puce obtenue est légèrement
plus grande qu’une lame porte-objet de
microscope, qui fournit une résolution chromatographique exceptionnelle et améliore
la sensibilité SM. Les puces-CLHP sont
beaucoup plus fiables et faciles à utiliser que
les nanocolonnes classiques. Les pucesCLHP sont disponibles pour les applications
à grandes et petites molécules.
L'interface puce-CLHP Cube MS comprend
des mécanismes de manipulation des puces
et les éléments d’une source d’ions à électronébulisation. Elle automatise complètement
la manipulation, le positionnement et les
raccordements des puces pour garantir des
performances maximales avec un effort
minimal.
L'interface puce-CLHP Cube
MS augmente la sensibilité
et la commodité
Les puces-CLHP
fournissent des
performances
chromatographiques
exceptionnelles et
sont faciles à utiliser
Une source
multimodale
peut doubler
le débit
Les choix de sources d’ions
augmentent la polyvalence
de la trappe d’ions
La polyvalence des systèmes de CPL/SM à
trappe d’ions Agilent série 6300 est améliorée par le grand choix possible parmi les
meilleures sources d’ions du secteur. Voici
les sources disponibles :
• L’ionisation par électronébulisation (ESI)
à nanodébit et débit capillaire (microlitre)
standard
• L’ionisation chimique à pression
atmosphérique (APCI)
• Le multimode avec ESI et APCI
simultanées
• La photo-ionisation à pression
atmosphérique (APPI)
• MALDI à pression atmosphérique avec
focalisation dynamique pulsée
Toutes les sources CPL/SM comprennent
la technologie de nébulisation orthogonale
brevetée Agilent qui élimine les réglages
et favorise la propreté du capillaire et de
l’optique ionique. Toutes les sources
comprennent également un système à gaz
de dessiccation à contre-courant haute
capacité qui améliore la sensibilité, la
reproductibilité et la qualité du spectre,
en réduisant les agrégats de solvant et les
ions adduits de phase mobile. La nébulisation
orthogonale et le gaz de dessiccation haute
capacité apportent aux sources d'ions
Agilent une haute tolérance aux composants
non volatils.
Page 6

6
L’acquisition intelligente des données garantit leur qualité
La capacité de rassembler plusieurs niveaux de données SM dans une seule analyse est l’un des nombreux
avantages d’une trappe d’ions. Cependant, le défi ne consiste pas simplement à acquérir des données,
mais également à le faire intelligemment, en acquérant les meilleures de chaque balayage, celles qui sont
les plus instructives. Les systèmes de CPL/SM à trappe d’ions série 6300 offrent un grand choix de fonctionnalités d’acquisition de données intelligentes pour optimiser les données recueillies à partir de chaque
échantillon.
Acquisition dépendante des
données, entièrement automatisée
Tous les instruments de CPL/SM à trappe
d’ions série 6300 peuvent effectuer jusqu’à
5 étapes (SM
5
) de SM/SM dépendante
Fonctionnalité dépendante des données Avantage
Les N précurseurs les plus abondants* Augmente le nombre d’ions précurseurs uniques à partir desquels les données sont acquises.
Particulièrement utile pour les composés coéluants.
Exclusion isotopique Évite l’acquisition de données SM/SM inutiles
à partir des isotopes (
13
C, etc.)
Liste d’inclusion statique Garantit que les données sont acquises à partir des ions cibles, même s’ils sont
peu abondants
Liste d’exclusion statique Élimine l’acquisition de données SM/SM à partir d’ions de solvant et
d’autres composants de bruit de fond connus
Liste préférentielle statique Garantit que les données sont acquises à partir des ions cibles, s’ils sont présents, mais
permet l’acquisition de données à partir d’autres ions en l’absence d’ions cibles
Sélection d’état de charge préférentielle Garantit une sélection des ions peptidiques doublement chargés pour des données CID
SM/SM de meilleure qualité ou des ions peptidiques de charge supérieure pour des
données ETD SM/SM de meilleure qualité
Exclusion active - calcul répété Augmente la quantité de données uniques acquises en empêchant l’acquisition de
données SM/SM à partir du même ion plus du nombre de fois spécifié par l’utilisateur
Exclusion active - temps d’exclusion Supprime des ions de la liste d’exclusion active après un délai spécifié par l’utilisateur pour
garantir une nouvelle acquisition si un ion apparaît dans plusieurs pics chromatographiques
Seuil - absolu* Garantit que les données sont acquises uniquement à partir d’ions
d’abondance spécifiée par l’utilisateur
Seuil - relatif* Garantit que les données sont acquises uniquement à partir d’ions d’abondance spécifiée
par l’utilisateur par rapport à l’ion le plus abondant dans un balayage SM
AutoMS
3
dépendant des pertes de neutres †Améliore l’identification des PTM en effectuant une SM3sur les N ions de produit les plus
abondants montrant les pertes de neutres spécifiées par l’utilisateur à partir de leurs précurseurs
Pseudo-MSn dépendant des pertes Améliore l’identification des PTM en fragmentant les N ions de produit les plus abondants
de neutres
†
correspondant aux pertes de neutres spécifiées par l’utilisateur à partir de leurs précurseurs
Expériences d’étiquetage d’isotopes Fournit une détection et une fragmentation de paires SILE dans des expériences de stables
(SILE)
†
quantification de protéines impliquant des stratégies ICAT, SILAC, 16O/18O, ICPL et
d’autres stratégies d’étiquetage isotopique
ETD dépendante des pertes de neutres
‡
Améliore la détermination de site des PTM en accumulant de nouveau le précurseur et en
effectuant une fragmentation ETD lorsque des pertes de neutres spécifiées par l’utilisateur
sont détectées
* Peut être définie séparément pour une SM >2
†
Disponible uniquement avec les modèles 6330 et 6340
‡
Disponible uniquement avec le modèle 6340
des données, entièrement automatisée. Le
logiciel sophistiqué de la série 6300 comprend
un grand choix de fonctionnalités d’acquisition dépendantes des données statiques
et actives qui vous aident à augmenter
la quantité de données utiles acquises.
Pour plus de simplicité et de commodité,
toutes les données SM/SM acquises à
partir d’une analyse sont stockées dans
un seul fichier.
Page 7

359,2
0,00
0,25
0,50
0,75
1,00
1,25
100 150 200 250 300 350 m/z
O
OH
Balayage complet SM de prednisone
Ion moléculaire à 359 m/z
Balayage complet SM/SM de 359 m/z
Tension fixe de dissociation induite par collision (CID)
313,0
341,1
359,1
Balayage complet SM/SM de 359 m/z
Tension CID (optimisation manuelle)
136,7
146,7
212,8
238,9
266,9
276,9
287,0
295,0
305,0
313,1
323,0
329,0
341,1
359,0
1
2
3
5
x10
Intens.
5
x10
Intens.
4
x10
Intens.
4
x10
Intens.
0,00
0,25
0,50
0,75
1,00
1,25
100 150 200 250 300 350 m/z
100 150 200 250 300 350 m/z
O
O
[M+H]
+
[M+H]
+
[M+H–18]
+
[M+H]
+
[M+H–18]
+
136,8
146,7
160,7
170,7
186,7
196,8
212,8
222,8
236,9
252,9
266,9
276,9
287,0
295,0
305,0
313,0
323,0
0,0
0,5
1,0
1,5
Balayage complet SM/SM de 359 m/z
Montée de tension CID (SmartFrag) et
largeur de fragmentation réglable (AFW)
100 150 200 250 300 350 m/z
[M+H–2(18)]
+
CH
3
CH
3
CH2OH
www.agilent.com/chem/iontrap
7
Mode de résolution maximale
pour des données uniques de
qualité
Utilisant des critères d’acquisition dépendants des données, les trappes d’ions
série 6300 peuvent effectuer un balayage
de résolution maximale sur une fenêtre de
masse étroite autour de chaque ion sélectionné. Les avantages sont nombreux :
données de très haute qualité - les pics
ont souvent une résolution correspondant
à la ligne de base - à partir d’ions uniques
plus nombreux. Vous pouvez utiliser le
mode de résolution maximale pour les
analyses SM et SM/SM.
Mode “ spécial peptides “ pour
une meilleure identification des
protéines
Le mode “ spécial peptides “ utilise le mode
de balayage amélioré de résolution supérieure
durant la SM
1
pour une détermination plus
précise des états de charge +1, +2 et +3
des ions peptidiques. Il utilise le mode de
Systèmes CPL/SM à trappe d’ions Agilent série 6300
La montée de tension CID SmartFrag
automatisée et la largeur de fragmentation
réglable (AFW) fournissent des spectres
SM/SM plus riches et éliminent le besoin
d’optimisation manuelle de la tension CID,
consommatrice de temps
balayage ultra-rapide en SM2et au-delà pour
raccourcir les temps de cycle. Au final, vous
identifiez plus de protéines dans des digestions
protéiques complexes.
Changement de polarité rapide pour
un débit accru
La capacité de rassembler des données d’ionisation
positives et négatives complémentaires dans une
seule analyse vous fait économiser un temps
précieux et des échantillons. Le changement de
polarité rapide de la série 6300 vous permet de
rassembler plus de balayages sur un pic, pour une
meilleure qualité et une plus grande sensibilité des
données.
Des paramètres intelligents fournissent
des résultats précis pour des
non-spécialistes
Pour simplifier le fonctionnement, le logiciel de la
trappe d’ions comprend un mode “ intelligent “. À
partir d’informations simples telles que la masse
cible, la stabilité des composés et la distribution des
ions prévue, et en utilisant une boucle de rétroaction
d’évaluation de données spectrales en temps réel,
le logiciel peut déterminer automatiquement les
paramètres optimaux de la trappe d’ions. Vous avez
toujours un contrôle indépendant total sur les paramètres de sources d’ions pour une compatibilité
complète avec les méthodes de CPL.
De meilleures données SM/SM grâce à
la montée d’énergie de collision
L’obtention d’une fragmentation CID optimale passe
généralement par une optimisation manuelle fastidieuse propre à l’échantillon. La montée d’énergie
de collision unique SmartFrag et la largeur de fragmentation réglable (AFW) garantissent que chaque ion
précurseur reçoit exactement l’énergie dont il a
besoin pour une fragmentation optimale - et de
manière automatique. Ce processus génère un plus
grand nombre d’ions de produit et plus d’informations
structurelles avec moins d’efforts.
Page 8

Les outils logiciels tirent le meilleur parti de vos données
Des performances d’instrument sensibles et reproductibles et des données de haute qualité sont nécessaires, mais pas suffisantes, pour atteindre vos objectifs d’analyse. Il faut utiliser les bons outils logiciels pour
transformer des données de qualité en informations exploitables. Agilent offre un grand choix d’utilitaires
puissants pour vous aider à transformer vos données SM
n
en réponses.
Navigation simplifiée dans les
résultats
Les analyses SMnautomatisées peuvent
générer de très grandes quantités de
données SM multigénérationnelles. Le
logiciel de CPL/SM à trappe d’ions série
6300 simplifie ce processus en stockant
toutes les données d’une analyse dans un
fichier unique. Une arborescence familière
contribue à une navigation simplifiée et
au repérage précis des données dont vous
avez besoin. Des filtres spéciaux vous
aident à extraire rapidement des sousensembles spécifiques de données.
8
Une arborescence vous permet de trouver facilement
les données dont vous avez exactement besoin
Une identification automatisée des composés accélère
les analyses et accroît la productivité
Recherche automatique de
composés
Les fonctions Find Compounds (Trouver les
composés) fournissent de puissants outils
d’extraction et de navigation des données
basées sur les composés, pour les expériences de CPL/SM
n
complexes. Les
fonctions Find Compounds (Trouver les
composés) permettront de
• Repérer toutes les expériences SM
n
uniques
• Générer des entrées de composés et des
chromatogrammes d’ions totaux SM
2
pour chaque ion précurseur SM unique
• Extraire et organiser hiérarchiquement
tous les spectres SM et SM
n
associés par
entrée de composé, et en faire la moyenne
Des fonctions sont également disponibles
pour générer automatiquement une moyenne
de spectres de masse pour tous les pics
chromatographiques intégrés dans des
expériences SM uniquement ou SM
n
manuelles.
Les résultats peuvent être évalués, imprimés
et enregistrés pour un archivage ou un
nouveau traitement.
Page 9

www.agilent.com/chem/iontrap
Systèmes CPL/SM à trappe d’ions Agilent série 6300
9
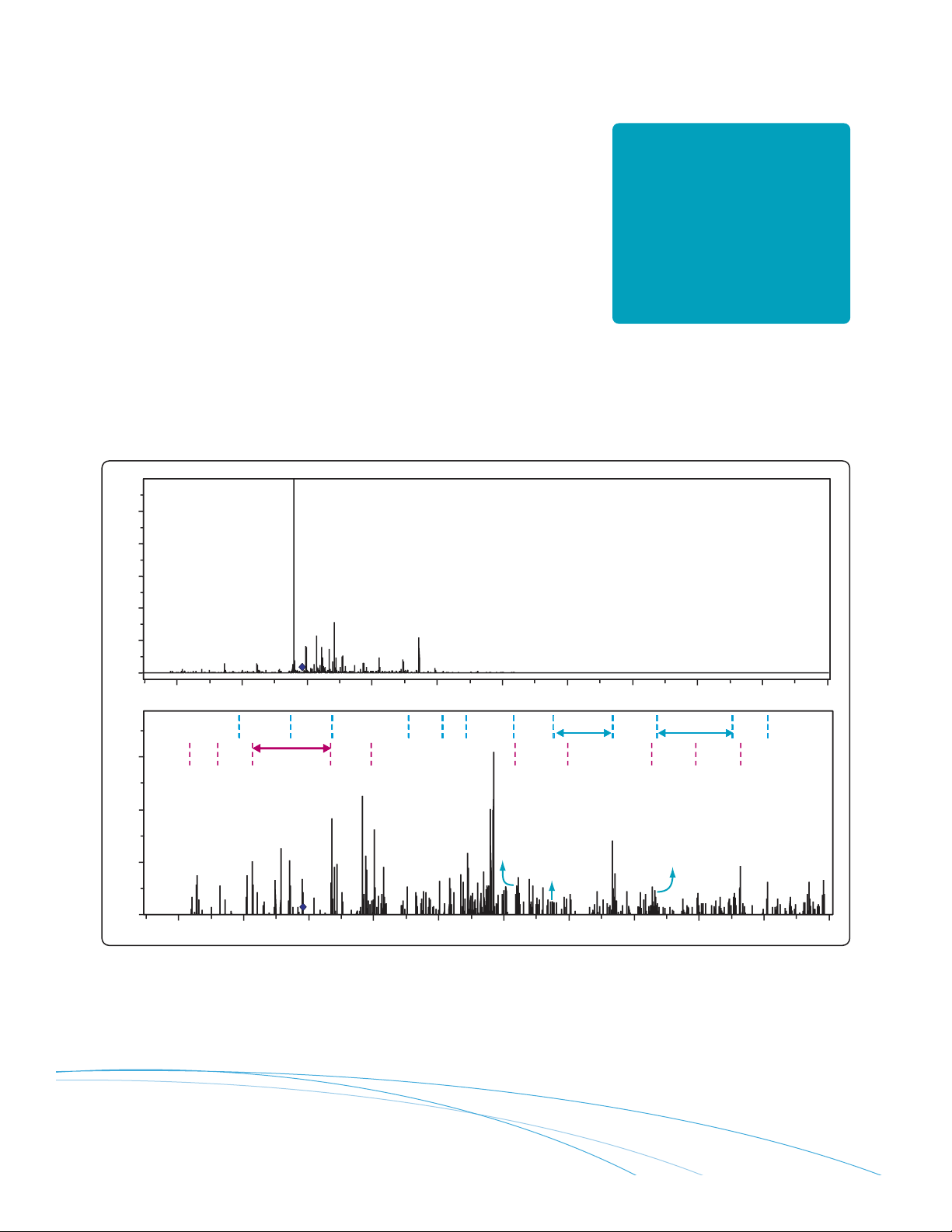
La SM à balayage complet et l’analyse des pertes de neutres postérieure identifient les anthocyanines
O-glycosylées avec de l’arabinose
26
25
24
23
19-22
18
17
16
15
14
13
12
11
10
9
8
4-7
3
2
1
0,0 2,5 5,0 7,5 10,0 12,5 15,0 17,5 20,0 Durée [min]
0
2
4
6
x10
7
Intens.
26
25
24
23
22
21
20
19
18
17
16
15
14
13
12
11
10
9
8
7
6
5
4
3
16,5 17,0 17,5 18,0 18,5 19,0 19,5 20,0 20,5 Durée [min]
0
1
2
3
4
5
x10
7
Intens.
TIC
EICs
Fragmento-grammes
extraits – Détails
TIC & EIC
0
2
4
6
x10
4
Intens.
0
1
2
3
4
5
x10
6
18
19 20 21 22 23 24 25
Durée [min]
Chromatogramme du pic de base
du signal SM
Chromatogramme de l'analyse
des pertes de neutres
(transition 132 u)
Lors de l’analyse d’un foie de rat, le logiciel Dissect a repéré 26 métabolites d’un médicament
antagoniste en moins d’une minute. Les résultats soutiennent la comparaison avec ceux obtenus en
une heure d’identification manuelle fastidieuse. Bien qu’il y ait eu de nombreux composés coéluants,
les spectres de masse reconstruits par le logiciel Dissect étaient relativement purs.
Le programme Dissect automatise
l’identification des composants
mineurs dans des échantillons
complexes
Les composants à l’état de traces dans les
échantillons biologiques sont difficiles à
détecter dans les chromatogrammes d’ions
totaux (TIC) de CPL/SM car des interférences
endogènes produisent d’importants bruits
de fond. L’extraction de signaux à l’état
de traces requiert souvent une analyse de
données consommatrice de temps, et est
compliquée par les élutions successives, la
formation d’ions adduits et les interférences
de matrice.
Le programme Dissect facultatif automatise
le repérage de composants mineurs dans
des données complexes SM uniquement.
Il génère d’abord et intègre des fragmentogrammes extraits (EIC) pour chaque masse,
et décide - selon la symétrie, la largeur et
d’autres facteurs - quels sont les pics chromatographiques valides. Le programme applique
ensuite une logique floue et plusieurs critères
pour éliminer les pointes de bruit et les pics
associés au groupe provenant du même
composé. Enfin, le programme Dissect obtient
des spectres de masse propres pour chaque
composé.
L’analyse des pertes de neutres
identifie les éléments communs
de plusieurs analytes
L’analyse des pertes de neutres est un outil
puissant d’identification des éléments structurels communs de plusieurs analytes. Les
données de balayage complet constituent
un avantage propre à la SM/SM à trappe
d’ions. Elles vous permettent d’effectuer
une analyse des pertes de neutres après
acquisition. Vous n’avez pas besoin de
prévoir les pertes de neutres. Le logiciel de
CPL/SM à trappe d’ions série 6300 comprend
des outils visant à faciliter l’analyse des
pertes de neutres. Des fonctionnalités
associées, telles que la montée d’énergie
de collision, garantissent une fragmentation
optimale de sorte à obtenir de nombreux
ions de produit à analyser.
Page 10

Quantification plus rapide et plus
facile
Le puissant logiciel QuantAnalysis rend la
quantification plus rapide et plus facile. Le
logiciel se compose de trois vues simples
(tableau de travail, chromatogramme/spectre
et courbe d’étalonnage) pouvant être affichées dans une seule fenêtre ou dans des
onglets. Le tableau de travail est constitué
d’une feuille de calcul dans laquelle vous
configurez la quantification et visualisez
les résultats. Vous pouvez le personnaliser
pour n’afficher que les données essentielles,
et enregistrer ces mises en page personnalisées pour les utiliser ultérieurement. Les
trois vues sont liées de manière dynamique,
c’est-à-dire que toute modification apportée
à l’une d’elles est répercutée dans les autres.
Par exemple, la sélection d’une régression
différente pour la courbe d’étalonnage recalcule automatiquement les concentrations.
Extraction du maximum d’informations de chaque échantillon
Le logiciel ACD/SpecManager, disponible
viaAdvanced Chemistry Development, peut
vous aider à extraire le maximum d’informations de vos données SM :
• Algorithme de détection des composants
pour réduire le bruit aléatoire et le bruit
de fond
• ChemSketch, l’outil standard de l’industrie
pour représenter des structures chimiques
• Outils pour simplifier l’ajout et le retrait
de spectres
• Outils pour stocker des chromatogrammes
et des spectres traités, et générer des
rapports
• Outils de corrélation pour faciliter l’interprétation SM en mettant en corrélation
les structures et les spectres SM
• Un lien direct pour un transfert facile des
spectres de masse du logiciel de la trappe
d’ions à ACD/SpecManager
Le logiciel SpecManager fournit également un
support en matière de techniques croisées,
ainsi qu’une plate-forme inter-fournisseurs,
une intégration de données multispectrales
comprenant la RMN, la SM, l’UV-visible
et l’IR.
Éditeur de rapports personnalisé
Un éditeur de rapports personnalisé accélère
et facilite la création de rapports au format
souhaité.
Automatisation utilisateur
L’écriture de scripts Visual Basic offre
des possibilités quasiment illimitées
d’automatisation conçue par l’utilisateur.
Vos méthodes peuvent appeler des scripts
personnalisés pour automatiser l’analyse
des données et la génération de rapports.
Ces scripts peuvent tirer profit d’une large
gamme de commandes propres à la SM.
10
Le tableau de travail (en haut), le chromatogramme/spectre (en bas à gauche) et la courbe
d’étalonnage (en bas à droite) sont liés de manière dynamique. Toute modification d’une vue
se répercute automatiquement dans les autres.
144,8
158,8
170,8
186,9
198,9
212,9
239,0
253,1
271,1
285,1
295,1
313,2
327,2
339,3
379,3
397,3
0
500
1000
1500
2000
2500
3000
Intens.
100
150 200 250 300 350 400 450 500 550
m/z
CH
2
CH
3
CH
3
CH
3
CH
3
HO
H
3
C
H
H
Avec le logiciel ACD/SpecManager, vous pouvez construire des bases de données de corrélations
entre les structures et vos données SM. Faites des recherches dans ces tableaux de corrélations
par rapport aux spectres inconnus.
Page 11

www.agilent.com/chem/iontrap
Le logiciel Spectrum Mill facilite
la protéomique à grand débit
Le logiciel Spectrum Mill vous aide à identifier
des protéines à partir de données SM et
SM/SM par une recherche dans une base
de données ou un séquençage de novo.
Une extraction spectrale intelligente
accélère l’identification des
protéines
Le logiciel Spectrum Mill identifie et exclut
les spectres de bruit et les spectres de
mauvaise qualité avant la recherche dans
la base de données, ce qui accroît considérablement la vitesse de recherche. Cela
réduit également le nombre de faux positifs.
Plusieurs options de recherche
pour l’identification des protéines
augmentent la flexibilité
Le programme de recherche SM/SM comprend le mode identité pour les peptides
non modifiés et le mode variable pour les
modifications.
Le programme de cartographie peptidique
(PMF) réalise un excellent travail d’identification de protéines à partir de spectres SM.
Prise en charge des données ETD
Le logiciel Spectrum Mill prend en charge
les spectres CID standard et les spectres
générés par ETD.
La validation automatique et
manuelle de correspondances
augmente le degré de confiance
Le logiciel Spectrum Mill valide les correspondances rapidement et automatiquement.
Vous pouvez également passer en revue de
manière interactive les correspondances
proposées, en les comparant aux spectres
SM/SM réels. Les spectres non validés
peuvent faire l’objet de nouvelles recherches à l’aide d’autres paramètres ou bases
de données.
Interprétation spectrale de novo
L’algorithme de séquençage de novo génère
une liste classée de séquences peptidiques
potentielles. Il écarte les solutions irréalistes et compense les difficultés spectrales
courantes, telles que le bruit et la fragmentation incomplète.
Informations quantitatives et
qualitatives
Le logiciel Spectrum Mill détermine automatiquement les différences d’abondances
relatives des protéines trouvées. Il prend
également en charge ICAT, iTRAQ et d’autres
technologies d’étiquetage d’isotopes stables.
Accessibilité des données
complexes
Le logiciel Spectrum Mill résume et met en
corrélation les résultats de manière à rendre
les informations accessibles même pour
Systèmes CPL/SM à trappe d’ions Agilent série 6300
11
Le logiciel Spectrum Mill simplifie et accélère l’identification des protéines et le repérage des modifications post-traductionnelles
des utilisateurs de SM non experts. Vous
pouvez comparer d’importants ensembles
de données à travers plusieurs expériences
et résumer les résultats au niveau des
protéines.
Compatibilité avec des données
provenant de plusieurs fournisseurs
Des modules complémentaires sont
disponibles et permettent au logiciel
Spectrum Mill de traiter des formats
de données non-Agilent :
• .RAW (Thermo Finnigan)
• .wiff (SCIEX CPL/SM)
• .pkl (Waters et autres)
MASCAT simplifie les recherches
de protéines Mascot
Le logiciel de la série 6300 inclut le
programme MASCAT à l’intention des
chercheurs qui utilisent habituellement
le programme de recherche de protéines
Mascot. MASCAT concatène les fichiers
au format générique Mascot (*.mgf), fournissant ainsi un moyen de synthétiser les
résultats à partir de plusieurs analyses
CPL/SM/SM en deux dimensions en une
seule recherche de protéines.
Page 12

Spécifications
Précision de masse (Tout)
± 0,2 u dans la gamme de masse standard étalonnée à une résolution normale en mode
balayage complet, avec un étalonnage et des statistiques de cible et d’ions ICC appropriés,
et un équilibre thermique des circuits électroniques et de la source d’ions.
Stabilité de l’axe des masses (Tout)
± 0,2 u de la valeur étalonnée observée sur 8 heures dans une gamme de masse standard
à une résolution normale en mode balayage complet, avec des statistiques de cible et
d’ions ICC appropriées, et un équilibre thermique des circuits électroniques et de la source
d’ions et une température ambiante de 21 °C ± 3 °C (70 °F ± 6 °F)
Sélection de précurseurs monoisotopiques (Tout)
La sélection de précurseurs monoisotopiques est possible sur toute la gamme de masse
standard (50 - 2 200 m/z) à une température ambiante de 21 °C ± 3 °C (70 °F ± 6 °F).
Gamme de masse, résolution de masse et vitesse de balayage
6310 6320, 6330 et 6340
Gamme de masse Résolution Vitesse de balayage Résolution Vitesse de balayage
m/z LMH (u) (u/s) LMH (u) (u/s)
50 - 2 200 ≤ 0,6 13 000 ≤ 0,6 26 000
≤ 0,45 5 500 ≤ 0,35 8 100
≤ 0,35 1 650 ≤ 0,25 800
200 - 4 000 3 - 4 27 000 ≤ 3 27 000
Changement de polarité
Changement de polarité entre balayages avec 1 spectre positif et 1 spectre négatif en
approximativement 1 seconde pour tous les modèles
Sensibilité
Conditions
Colonne à résolution rapide ZORBAX SB-C18 2,1 x 30 mm 3,5 µm
Phase mobile - 25 % d’eau 75 % de méthanol 5 mM d’acétate d’ammonium
Débit - 400 µl/min
Mode - balayage complet SM/SM
SM/SM - transition de l’ion moléculaire protoné (609 m/z) vers la somme des
deux ions de produit les plus abondants
Gamme de balayage des ions de produit - 175 - 650 m/z
Gamme de masse - standard (50 - 2 200 m/z)
Résolution - standard (0,6 u)
6310 6320 6330 6340
Quantité (réserpine - 5 pg 1 pg 250 fg 250 fg
dans la colonne)
Rapport signal sur bruit ≥ 50:1 ≥ 50:1 ≥ 50:1 ≥ 50:1
(balayage complet
SM/SM)
Pour plus d’informations...
Pour en savoir plus :
www.agilent.com/chem/iontrap
Achetez en ligne :
www.agilent.com/chem/store
Trouvez un centre d’assistance
Agilent dans votre pays :
www.agilent.com/chem/contactus
États-Unis et Canada
1-800-227-9770
agilent-inquiries@agilent.com
Europe
info_agilent@agilent.com
Asie-Pacifique
adinquiry_aplsca@agilent.com
Utilisation uniquement en recherche. Ne pas utiliser
dans des méthodes de diagnostic.
Les informations, les descriptions et les spécifications
publiées ici peuvent être modifiées sans préavis.
Agilent Technologies décline toute responsabilité pour
les erreurs éventuelles du présent document ainsi
que pour les dommages fortuits ou consécutifs à la
fourniture, l’utilisation ou la performance de ce dernier.
© Agilent Technologies, Inc. 2006
Imprimé aux Pays-Bas le 11 novembre 2006
5989-5824FR
 Loading...
Loading...