

Page 1

Proclaim™ Implantable Pulse Generator
Clinician's Manual
Models 3660, 3661, 3662, 3663, 3665, 3667
Page 2

CAUTION: Federal (USA) law restricts this device to sale by or on the order of a physician.
State of California to cause cancer and birth defects or other reproductive harm. For more information, go to
www.P65Warnings.ca.gov.
™ Indicates a trademark of the Abbott group of companies.
‡ Indicates a third
Bluetooth and Bluetooth logo are registered trademarks of Bluetooth SIG, Inc.
Pat.
©
WARNING: This product can expose you to chemicals including ethylene oxide, which is known to the
http://www.abbott.com/patents
2020 Abbott. All Rights Reserved.
-party trademark, which is property of its respective owner.
Page 3

i
Contents
Prescription and Safety Information ........................................................................... 1
Intended Use ....................................................................................................................... 1
Indications for Use ............................................................................................................... 1
Contraindications ................................................................................................................. 1
MRI Safety Information ......................................................................................................... 1
Warnings ............................................................................................................................. 1
Precautions .......................................................................................................................... 3
Adverse Effects .................................................................................................................... 6
Safety and Effectiveness Studies ........................................................................................... 6
System Overview ....................................................................................................... 6
Product Description ................................................................................................... 7
Package Contents ................................................................................................................ 8
Identifying the IPG ............................................................................................................... 8
Directions for Use ...................................................................................................... 8
Creating an IPG Pocket ........................................................................................................ 9
Connecting a Lead or Extension to the IPG ........................................................................... 9
Implanting the IPG ............................................................................................................. 11
Replacing the IPG .............................................................................................................. 12
Disposing of Explanted Components ................................................................................... 12
Checking the Status of the IPG Battery ..................................................................... 12
Technical Support ................................................................................................... 13
Appendix A: Product Specifications ......................................................................... 13
Storage Specifications ........................................................................................................ 13
Product Materials ............................................................................................................... 13
IPG Specifications .............................................................................................................. 14
Compatibility Guidelines for IPGs with Compatible Headers................................................. 15
Appendix B: System Components and Accessories .................................................. 16
IPGs .................................................................................................................................. 16
Programmers and Controllers ............................................................................................. 16
Leads and Extensions ......................................................................................................... 16
Adapters ............................................................................................................................ 17
Trial System ....................................................................................................................... 17
Appendix C: Battery Longevity Information ............................................................... 18
Energy Factors for Tonic Stimulation Parameters ................................................................ 19
Energy Factors for BurstDR™ Stimulation Parameters ........................................................ 23
Battery Longevity Graphs .................................................................................................... 25
Appendix D: Regulatory Statements ......................................................................... 26
Disposal Guidelines for Battery-Powered Devices ................................................................ 26
Statement of FCC Compliance ............................................................................................ 26
Statement of Compliance With License-Exempt RSS Standard (Canada) ............................. 26
Identification Information for Product Registration ............................................................... 27
Wireless Technology Information ........................................................................................ 27
Radio Transmitter, Cables, Transducers ............................................................................. 28
Quality of Service for Wireless Technology .......................................................................... 28
Appendix E: Symbols and Definitions ....................................................................... 30
Page 4

Page 5

1
Prescription and Safety Information
Read this section to gather important prescription and safety information.
Intended Use
This neurostimulation system is designed to deliver low-intensity electrical impulses to nerve
structures. The system is intended to be used with leads and associated extensions that are
compatible with the system.
Indications for Use
This neurostimulation system is indicated as an aid in the management of chronic, intractable
pain of the trunk and/or limbs, including unilateral or bilateral pain associated with the following:
failed back surgery syndrome and intractable low back and leg pain.
Contraindications
This system is contraindicated for patients who are unable to operate the system or who have
failed to receive effective pain relief during trial stimulation.
MRI Safety Information
Some models of this system are Magnetic Resonance (MR) Conditional, and patients with these
devices may be scanned safely with magnetic resonance imaging (MRI) when the conditions for
safe scanning are met. For more information about MR Conditional neurostimulation components
and systems, including equipment settings, scanning procedures, and a complete listing of
conditionally approved components, refer to the MRI procedures clinician's manual for
neurostimulation systems (available online at
about MR Conditional products, visit the Abbott Medical product information page at
neuromodulation.abbott/MRI-ready.
medical.abbott/manuals). For more information
Warnings
The following warnings apply to this neurostimulation system.
Poor surgical risks. Neurostimulation should not be used on patients who are poor surgical risks
or patients with multiple illnesses or active general infections.
Magnetic resonance imaging (MRI). Some patients may be implanted with the components that
make up a Magnetic Resonance (MR) Conditional system, which allows them to receive an MRI
scan if all the requirements for the implanted components and for scanning are met. A physician
can help determine if a patient is eligible to receive an MRI scan by following the requirements
provided by Abbott Medical. Physicians should also discuss any risks of MRI with patients.
Patients without an MR Conditional neurostimulation system should not be subjected to MRI
because the electromagnetic field generated by an MRI may damage the device electronics and
induce voltage through the lead that could jolt or shock the patient.
Diathermy therapy. Do not use short-wave diathermy, microwave diathermy, or therapeutic
ultrasound diathermy (all now referred to as diathermy) on patients implanted with a
neurostimulation system. Energy from diathermy can be transferred through the implanted system
and cause tissue damage at the location of the implanted electrodes, resulting in severe injury or
death.
Page 6

2
Diathermy is further prohibited because it may also damage the neurostimulation system
components. This damage could result in loss of therapy, requiring additional surgery for system
implantation and replacement. Injury or damage can occur during diathermy treatment whether
the neurostimulation system is turned on or off.
Electrosurgery. To avoid harming the patient or damaging the neurostimulation system, do not
use monopolar electrosurgery devices on patients with implanted neurostimulation systems.
Before using an electrosurgery device, place the device in Surgery Mode using the patient
controller app or clinician programmer app. Confirm the neurostimulation system is functioning
correctly after the procedure.
During implant procedures, if electrosurgery devices must be used, take the following actions:
Use bipolar electrosurgery only.
Complete any electrosurgery procedures before connecting the leads or extensions to the
neurostimulator.
Keep the current paths from the electrosurgery device as far from the neurostimulation
system as possible.
Set the electrosurgery device to the lowest possible energy setting.
Confirm that the neurostimulation system is functioning correctly during the implant
procedure and before closing the neurostimulator pocket.
Implanted cardiac systems. Physicians need to be aware of the risk and possible interaction
between a neurostimulation system and an implanted cardiac system, such as a pacemaker or
defibrillator. Electrical pulses from a neurostimulation system may interact with the sensing
operation of an implanted cardiac system, causing the cardiac system to respond inappropriately.
To minimize or prevent the implanted cardiac system from sensing the output of the
neurostimulation system, (1) maximize the distance between the implanted systems; (2) verify
that the neurostimulation system is not interfering with the functions of the implanted cardiac
system; and (3) avoid programming either device in a unipolar mode (using the device’s can as
an anode) or using neurostimulation system settings that interfere with the function of the
implantable cardiac system.
Other active implanted devices. The neurostimulation system may interfere with the normal
operation of another active implanted device, such as a pacemaker, defibrillator, or another type
of neurostimulator. Conversely, the other active implanted device may interfere with the operation
of the neurostimulation system.
Interference with other devices. Some of this system’s electronic equipment, such as the
programmer and controller, can radiate radiofrequency (RF) energy that may interfere with other
electronic devices, including other active implanted devices. Avoid placing equipment
components directly over other electronic devices. To correct the effect of interference with other
devices, turn off the equipment or increase the distance between the equipment and the device
being affected.
Operation of machines, equipment, and vehicles. Patients using therapy that generates
paresthesia should turn off stimulation before operating motorized vehicles, such as automobiles,
or potentially dangerous machinery and equipment because sudden stimulation changes may
distract them from properly operating it. However, current data shows that most patients using
BurstDR™ stimulation therapy do not experience paresthesia. For patients who do not feel
paresthesia, sudden stimulation changes are less likely to occur and distract them while operating
motorized vehicles, machinery, or equipment.
Page 7

3
Explosive and flammable gases. Do not use a clinician programmer or patient controller in an
environment where explosive or flammable gas fumes or vapors are present. The operation of
these devices could cause them to ignite, causing severe burns, injury, or death.
Keep the device dry. Programmer and controller devices are not waterproof. Keep them dry to
avoid damage. Advise patients to not use their device when engaging in activities that might cause
it to get wet, such as swimming or bathing.
Pediatric use. Safety and effectiveness of neurostimulation for pediatric use have not been
established.
Pregnancy and nursing. Safety and effectiveness of neurostimulation for use during pregnancy
and nursing have not been established.
Device components. The use of components not approved for use by Abbott Medical with this
system may result in damage to the system and increased risk to the patient.
Device modification. Equipment is not serviceable by the customer. To prevent injury or damage
to the system, do not modify the equipment. If needed, return the equipment to Abbott Medical
for service.
Application modification. To prevent unintended stimulation, do not modify the operating system
in any way. Do not use the application if the operating system is compromised (i.e., jailbroken).
Case damage. Do not handle the IPG if the case is pierced or ruptured because severe burns
could result from exposure to battery chemicals.
IPG disposal. Return all explanted IPGs to Abbott Medical for safe disposal. IPGs contain
batteries as well as other potentially hazardous materials. Do not crush, puncture, or burn the IPG
because explosion or fire may result.
Product materials. Neurostimulation systems have materials that come in contact or may come
in contact with tissue. A physician should determine whether or not a patient may have an allergic
reaction to these materials before the system is implanted.
Precautions
The following precautions apply to this neurostimulation system.
General Precautions
Clinician training. Implanting physicians should be experienced in the diagnosis and treatment of
chronic pain syndromes and have undergone surgical and device implantation training.
Patient selection. It is extremely important to select patients appropriately for neurostimulation.
Thorough psychiatric screening should be performed. Patients should not be dependent on drugs
and should be able to operate the neurostimulation system.
Infection. Follow proper infection control procedures. Infections related to system implantation
might require that the device be explanted.
Implantation of two systems. If two systems are implanted, ensure that at least 20 cm (8 in)
separates the implanted IPGs to minimize unintended interaction with other system components.
Implantation of multiple leads. If multiple leads are implanted, leads and extensions should be
routed in close proximity. Nonadjacent leads can possibly create a conduit for stray
electromagnetic energy that could cause the patient unwanted stimulation.
High stimulation outputs. Stimulation at high outputs may cause unpleasant sensations or motor
disturbances, or render the patient incapable of controlling the stimulator. If unpleasant
sensations occur, the device should be turned off immediately.
Page 8

4
Electromagnetic interference (EMI). Some equipment in home, work, medical, and public
environments can generate EMI that is strong enough to interfere with the operation of a
neurostimulation system or damage system components. Patients should avoid getting too close
to these types of EMI sources, which include the following examples: commercial electrical
equipment (such as arc welders and induction furnaces), communication equipment (such as
microwave transmitters and high-power amateur transmitters), high-voltage power lines,
radiofrequency identification (RFID) devices, and some medical procedures (such as therapeutic
radiation and electromagnetic lithotripsy).
Lead movement. Patients should be instructed to avoid bending, twisting, stretching, and lifting
objects over 2 kg (5 lb) six to eight weeks after implantation of a neurostimulation system.
Extension of the upper torso or neck may cause lead movement and alter the stimulation field
(especially with leads in the cervical area), resulting in overstimulation or ineffective stimulation.
Patient training. Instruct patients to use their neurostimulation system only after an authorized
clinician has programmed the device and has trained the patient how to control stimulation and
safely use the system.
Programmer use. Allow only authorized use of the clinician programmer to avoid any
programming changes that may injure a patient.
Sterilization and Storage
Single-use, sterile device. The implanted components of this neurostimulation system are
intended for a single use only. Sterile components in this kit have been sterilized using ethylene
oxide (EtO) gas before shipment and are supplied in sterile packaging to permit direct introduction
into the sterile field. Do not resterilize or reimplant an explanted system for any reason.
Storage environment. Store components and their packaging where they will not come in contact
with liquids of any kind.
Handling and Implementation
Expiration date. An expiration date (or “use-before” date) is printed on the packaging. Do not use
the system if the use-before date has expired.
Handle the device with care. The clinician programmer and patient controller are sensitive
electronic devices that can be damaged by rough handling, such as dropping them on the
ground.
Care and handling of components. Use extreme care when handling system components prior
to implantation. Excessive heat, excessive traction, excessive bending, excessive twisting, or the
use of sharp instruments may damage and cause failure of the components.
Package or component damage. Do not implant a device if the sterile package or components
show signs of damage, if the sterile seal is ruptured, or if contamination is suspected for any
reason. Return any suspect components to Abbott Medical for evaluation.
Exposure to body fluids or saline. Prior to connection, exposure of the metal contacts, such as
those on the connection end of a lead or extension, to body fluids or saline can lead to corrosion.
If such exposure occurs, clean the affected parts with sterile, deionized water or sterile water for
irrigation, and dry them completely prior to lead connection and implantation.
System testing. To ensure correct operation, always test the system during the implant
procedure, before closing the neurostimulator pocket, and before the patient leaves the surgery
suite.
Page 9

5
Hospital and Medical Environments
High-output ultrasonics and lithotripsy. The use of high-output devices, such as an
electrohydraulic lithotriptor, may cause damage to the electronic circuitry of an implanted IPG. If
lithotripsy must be used, do not focus the energy near the IPG.
Ultrasonic scanning equipment. The use of ultrasonic scanning equipment may cause
mechanical damage to an implanted neurostimulation system if used directly over the implanted
system.
External defibrillators. The safety of discharge of an external defibrillator on patients with
implanted neurostimulation systems has not been established.
Therapeutic radiation. Therapeutic radiation may damage the electronic circuitry of an implanted
neurostimulation system, although no testing has been done and no definite information on
radiation effects is available. Sources of therapeutic radiation include therapeutic X rays, cobalt
machines, and linear accelerators. If radiation therapy is required, the area over the implanted
IPG should be shielded with lead. Damage to the system may not be immediately detectable.
Home and Occupational Environments
Security, antitheft, and radiofrequency identification (RFID) devices. Some antitheft devices,
such as those used at entrances or exits of department stores, libraries, and other public places,
and airport security screening devices may affect stimulation. Additionally, RFID devices, which
are often used to read identification badges, as well as some tag deactivation devices, such as
those used at payment counters at stores and loan desks at libraries, may also affect stimulation.
Patients who are implanted with nonadjacent multiple leads and patients who are sensitive to low
stimulation thresholds may experience a momentary increase in their perceived stimulation, which
some patients have described as uncomfortable or jolting. Patients should cautiously approach
such devices and should request help to bypass them. If they must go through a gate or doorway
containing this type of device, patients should turn off their IPG and proceed with caution, being
sure to move through the device quickly.
Scuba diving or hyperbaric chambers. Patients should not dive below 30 m (100 ft) of water or
enter hyperbaric chambers above 4.0 atmospheres absolute (ATA). Pressures below 30 m
(100 ft) of water (or above 4.0 ATA) could damage the neurostimulation system. Before diving or
using a hyperbaric chamber, patients should discuss the effects of high pressure with their
physician.
Wireless use restrictions. In some environments, the use of wireless functions (e.g., Bluetooth®
wireless technology) may be restricted. Such restrictions may apply aboard airplanes, near
explosives, or in hazardous locations. If you are unsure of the policy that applies to the use of this
device, please ask for authorization to use it before turning it on.
Mobile phones. While interference with mobile phones is not anticipated, technology continues to
change and interaction between a neurostimulation system and a mobile phone is possible.
Advise patients to contact their physician if they are concerned about their mobile phone
interacting with their neurostimulation system.
Page 10

6
Adverse Effects
In addition to those risks commonly associated with surgery, the following risks are associated
with using this neurostimulation system:
Unpleasant sensations or motor disturbances, including involuntary movement, caused by
stimulation at high outputs (If either occurs, turn off your IPG immediately.)
Undesirable changes in stimulation, which may be related to cellular changes in tissue
around the electrodes, changes in electrode position, loose electrical connections, or lead
failure
Stimulation in unwanted places (such as radicular stimulation of the chest wall)
Lead migration, causing changes in stimulation or reduced pain relief
Epidural hemorrhage, hematoma, infection, spinal cord compression, or paralysis from
placement of a lead in the epidural space
Cerebrospinal fluid (CSF) leakage
Paralysis, weakness, clumsiness, numbness, or pain below the level of the implant
Persistent pain at the electrode or IPG site
Seroma (mass or swelling) at the IPG site
Allergic or rejection response to implant materials
Implant migration or skin erosion around the implant
Battery failure
Safety and Effectiveness Studies
For information that supports the clinical use of this neurostimulation system, refer to the clinical
summaries manual for spinal cord stimulation (SCS) systems. This neurostimulation system is
similar in technology and intended use to the systems reported in the literature and clinical
studies. Therefore, the literature and clinical studies represent the safety and effectiveness of this
neurostimulation system.
System Overview
This neurostimulation system is designed to deliver electrical stimulation to nerve structures. The
neurostimulation system includes the following main components:
Implantable pulse generator (IPG)
Leads
Clinician programmer
Patient controller
Patient magnet
The IPG delivers electrical pulses through the leads to electrodes near selected nerve fibers in
order to provide therapeutic stimulation. The patient magnet can turn the IPG on and off if the
physician enabled this functionality. Physicians use the clinician programmer to create and
modify programs for a patient. Patients use the patient controller to control their prescribed
programs.
Page 11
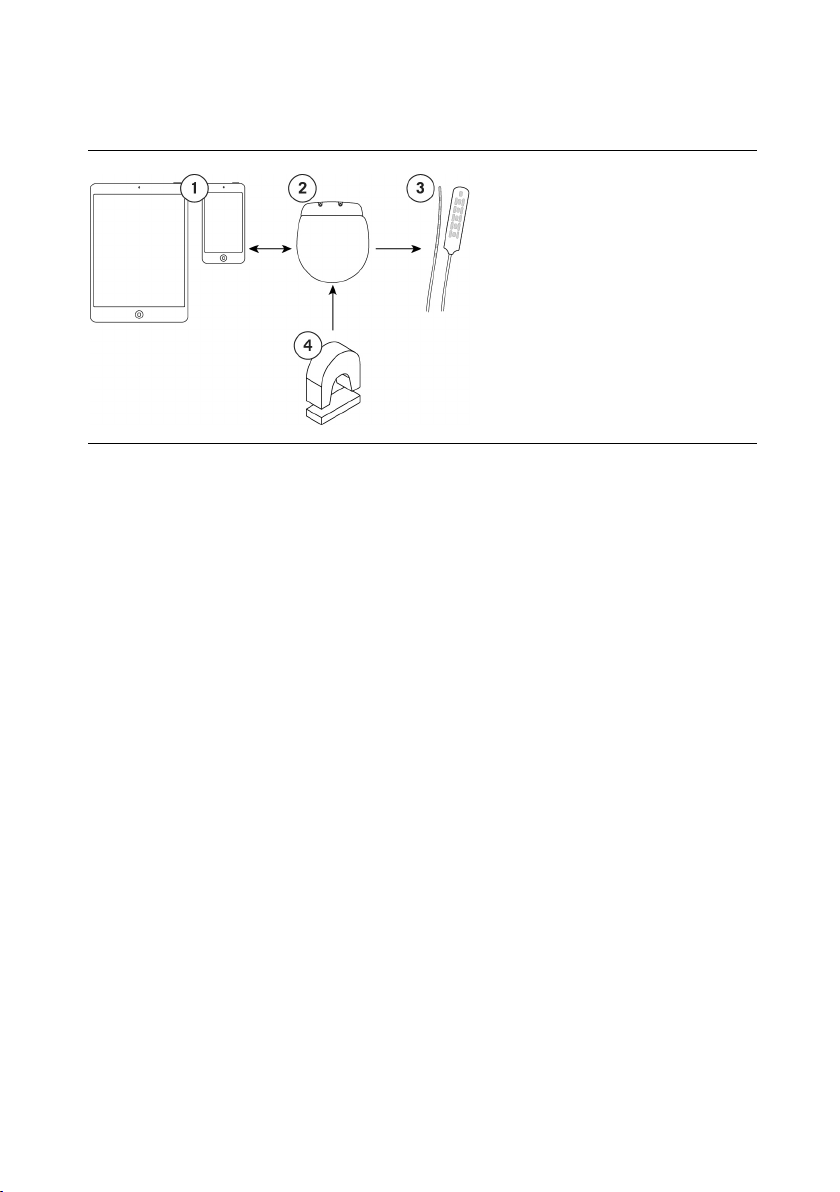
7
The following image shows how the major system components are intended to interact.
Figure 1. Interaction among main system components
1. Clinician programmer or
patient controller
2. IPG
3. Leads
4. Patient magnet
NOTE: This manual provides instructions for implanting the IPG. For instructions for using
other components, see the applicable manuals for those components.
Product Description
This implantable pulse generator (IPG) is an electronic device designed to be connected to one or
more extensions or leads with up to 16 electrodes total. It is powered by a hermetically sealed
battery within a titanium case and uses microelectronic circuitry to generate constant-current
electrical stimulation. The IPG can deliver stimulation with a single program or with multiple
programs. Each program can provide stimulation to a single anatomical area or to multiple areas.
The IPG communicates wirelessly with system programmers and controllers, and IPGs are
available in small and large sizes to accommodate different power needs.
Some models support additional functions:
Upgradeability. Models can receive software upgrades after implantation to provide patients
with additional features as approved by the respective regulatory agencies. To upgrade
features on the IPG, a system programmer is needed.
Compatible header. Models with a compatible header are designed to allow the IPG to
connect to leads or extensions from another manufacturer that meet the compatibility
guidelines (referred to as "IPGs with compatible headers").
For more information about which models provide these additional functions, as well as other IPG
specifications, see the appropriate appendix in this manual.
NOTE: For more information about the neurostimulation system, see the clinician’s
programming manual for this system.
NOTE: In this document, the term "clinician programmer" refers to the St. Jude Medical™
Clinician Programmer device, "patient controller" refers to the St. Jude Medical™ Patient
Controller device, "clinician programmer app" refers to the St. Jude Medical™ Clinician
Programmer software application (app), and "patient controller app" refers to the St. Jude
Medical™ Patient Controller app.
Page 12

8
Package Contents
In addition to the product documentation, the IPG kit contains the following items:
1 IPG (see the appendix in this manual for model numbers)
1 pocket sizer
1 torque wrench (Model 1101)
2 port plugs (Model 1111)
Identifying the IPG
Before implanting the IPG, you can view the model number engraved on the IPG. After
implantation, you can identify the IPG using a radiopaque identification tag that you can view with
standard X-ray procedures. The tag, which is located in the lower left corner of the IPG when the
logo side of the IPG is facing toward you, contains a code in the following format: SJMLN. ‘SJM’
designates Abbott Medical as the manufacturer; ‘LN’ is a letter and a number combination that
identifies the model family (see the following figure).
For the Proclaim™ IPG, the code is SJM A1. To determine the exact model IPG that is implanted,
use the clinician programmer app to communicate with the IPG and view IPG information. See the
clinician’s manual for the clinician programmer for instructions.
Figure 2. Location of the IPG code on a small IPG (left) and large IPG (right)
Directions for Use
Read this section carefully for suggested directions for use related to the IPG. For directions for
use for other system components not covered in this document, see the clinician’s manual for the
appropriate device.
NOTE: Before the surgical procedure, set up communication between the clinician
programmer and the IPG while the IPG is in its sterile packaging to ensure that it is
functional. If the IPG has never established communication with a programmer, you must
first activate the IPG for communication ("wake up" the IPG) by holding a magnet over the
IPG for 10 seconds.
Page 13

9
Creating an IPG Pocket
The following steps outline the suggested procedure to create an IPG pocket:
1. Determine the site for the IPG, ensuring that the lead is long enough to reach the pocket and
provide a strain relief loop.
NOTE: Common sites for IPG implantation are along the midaxillary line, in the upper
buttock along the posterior axillary line (taking care to avoid the belt line), and in the area
over the abdomen just below the lowermost rib. To ensure a flat area is selected, you can
mark a flat area prior to the surgical procedure while the patient is in a sitting position.
CAUTION: Do not place the IPG deeper than 4.0 cm (1.57 in) because the clinician
programmer may not communicate effectively with the IPG.
2. Create the pocket so that the IPG is parallel to the skin surface and no deeper than 4.0 cm
(1.57 in) below the skin surface.
3. Insert and remove the pocket sizer to ensure that the pocket is large enough to
accommodate the IPG, allowing enough extra room for a strain relief loop for each lead or
extension.
Connecting a Lead or Extension to the IPG
The following steps outline the suggested guidelines to connect a lead or extension to the IPG:
WARNING: To avoid harming the patient or damaging the neurostimulation system,
ensure that any electrosurgery procedures are completed before connecting the leads
or extensions to the IPG.
CAUTION: Do not connect a lead or extension with body fluid or saline residue on its
contacts because corrosion can occur and cause failure of the system.
1. If any of the lead or extension contacts came in contact with body fluid or saline, thoroughly
clean the contacts with sterile deionized water or sterile water for irrigation and dry them
completely.
2. To help ensure that the lead or extension can be fully inserted into the IPG header, insert the
torque wrench through the septum on the IPG header, turn the torque wrench clockwise to
tighten the setscrew until the torque wrench clicks, and then loosen the setscrew again by
turning the wrench counterclockwise about 2.5 times.
CAUTION: Use only the torque wrench included in the extension, IPG, or torque
wrench kit. If you need to loosen the setscrew, turn the setscrew (in quarter turns
counterclockwise) just enough to insert or remove the lead or extension from the IPG
header. Retracting the setscrew too far may cause it to come loose and fail to secure
the lead or extension to the IPG.
CAUTION: To avoid sharply bending and damaging the lead or extension when
performing the following step, insert the lead or extension parallel with the header port.
Additionally, try grasping the lead or extension about 5 mm at a time from the opening
of the header port while inserting.
3. Using clean gloves, carefully slide the proximal end of the lead or extension into the IPG
header until it stops. Confirm that the lead or extension is correctly inserted by following
these visual indicators and referring to the corresponding figures that follow:
- For IPGs that connect to Abbott Medical leads or extensions, the first contact band (at
the tip) of the lead or extension extends slightly past the first header contact and is
Page 14

10
Fully inserted
Not fully inserted
Fully inserted
Not fully inserted
visible, the windows between each of the header contacts are clear, and the ninth
contact band of the lead or extension is not visible.
- For IPGs with compatible headers, the windows between each of the header contacts
are clear and none of the contact bands are visible.
Figure 3. Correct versus incorrect insertion of the lead or extension (IPGs with Abbott Medical
leads or extensions)
1. First contact band (tip) is visible past the
first header contact
2. Window between each header contact is
clear
3. Ninth contact band is not visible
4. First contact band (tip) is not visible past
the first header contact
5. Window between each header contact is
partially blocked by contact band
6. Ninth contact band is visible
Figure 4. Correct versus incorrect insertion of the lead or extension (IPGs with compatible
header)
4. Use the clinician programmer app to communicate with the IPG, and test the impedance to
ensure that the lead or extension is fully inserted. See the clinician's manual for the clinician
programmer app for instructions.
5. Insert the torque wrench through the septum and tighten the setscrew, turning it clockwise
until the torque wrench clicks.
NOTE: After removing the torque wrench, check the septum to ensure it has closed. If the
septum did not close, gently reseat the septum flaps.
1. Window between each header contact is
clear
2. Eighth contact band is not visible
3. Window between each header contact is
partially blocked by contact band
4. Eighth contact band is visible
Page 15

11
Figure 5. Tighten the setscrew clockwise
6. If implanting two leads, repeat the previous steps. If implanting a single lead only, insert the
header port plug into the unused port, and use the torque wrench to tighten the setscrew
until the torque wrench clicks.
Figure 6. Insert the port plug
Implanting the IPG
The following steps outline the suggested procedure to implant the IPG:
1. Place the IPG into the IPG pocket with the logo side facing the skin surface and at a depth
not to exceed 4.0 cm (1.57 in).
NOTE: By implanting the IPG with the logo side facing the skin surface, you enhance the
IPG's ability to detect a magnet.
2. Carefully coil any excess lead or extension behind the IPG in loops no smaller than 2.5 cm
(1 in) in diameter to provide strain relief for the lead or extension and IPG connection.
CAUTION: Do not bring the suture needle in contact with an IPG, lead, or extension, or
the component may be damaged.
3. To stabilize the IPG within the pocket, pass suture through the holes at the top of the IPG
header and secure it to connective tissue.
4. Check the entire system by fluoroscopy before closing to ensure proper positioning of the
lead or leads and that it is straight, with no sharp bends or kinks.
Page 16

12
5. Use the clinician programmer app to communicate with the IPG and perform intraoperative
testing to confirm that the system is operational. See the clinician's manual of the clinician
programmer app for instructions.
NOTE: IPG output may not be identical to that of the trial stimulator at the same settings.
6. Ensure that the IPG is away from the pocket incision suture line, close the pocket incision,
and apply the appropriate dressings.
Replacing the IPG
The following steps outline the suggested procedure to replace an IPG:
1. Turn off stimulation or verify that it is turned off.
CAUTION: Exercise care when using sharp instruments or electrocautery around leads
or extensions, or they may be damaged.
2. Open the IPG implant site per normal surgical procedure.
3. Insert the torque wrench through the septum of the IPG header and loosen the setscrew by
turning it counterclockwise.
CAUTION: When performing the following step, do not bend the lead or extension
sharply; or it may be damaged.
4. Gently remove the lead or extension from the IPG header; then clean and dry all
connections, ensuring they are free of fluid and tissue.
5. To complete the IPG replacement procedure, see the following sections: “Connecting a Lead
or Extension to the IPG” (page 9) and “Implanting the IPG” (page 11).
Disposing of Explanted Components
Explanted Abbott Medical components should be returned to Abbott Medical for proper disposal.
To return an explanted component, place it in a container or bag marked with a biohazard label
and coordinate the return with your Abbott Medical representative or Technical Support.
Checking the Status of the IPG Battery
The IPG contains a nonrechargeable battery. The amount of time that the battery will provide
active stimulation depends on the patient’s stimulation settings and daily usage time. To check
the status of the IPG battery, use the clinician programmer app or patient controller app. For more
information about this function, refer to the clinician’s programming manual and the user’s guide
for the patient controller app. For information about estimating longevity of the IPG battery, see
the appropriate appendix in this manual.
NOTE: IPG battery status is available one day after first using the clinician programmer app
to program the IPG.
The following list provides general information about the battery status:
A low-battery warning will appear on the clinician programmer app or patient controller app
when the battery is approaching its end of service.
Stimulation will automatically stop when the battery cannot support stimulation.
Page 17

13
Temperature
Component
Material
Technical Support
For technical questions and support for your product, use the following information:
+1 855 478 5833 (toll-free within North America)
+1 651 756 5833
For additional assistance, call your local Abbott Medical representative.
Appendix A: Product Specifications
NOTE: Not all models are available in all countries. Contact your local representative for
more information.
Storage Specifications
Store the components in this kit according to the following conditions.
Table 1. Storage conditions for components
-20°C–60°C (-4°F–140°F)
Product Materials
The following materials are intended to come into contact with tissue.
Table 2. Product materials for IPG kit
IPG Titanium, silicone rubber
Pocket sizer Polybutylene terephthalate
Port plug Polysulfone
NOTE: These components are not made with natural rubber latex.
Page 18

14
Model
MRI Status
Upgradeable
Features,
Burst
Capable
Compatible
Header
Height
Length
Thickness
Weight
Volume
Power source
Connector
strength
Program
storage capacity
IPG Specifications
The Proclaim™ IPGs have the following physical specifications.
Table 3. IPG specifications
3660 3662
MR
Conditional
Yes No
3661 3663 MR Unsafe Yes Yes
3665 3667 MR Unsafe No No
5.55 cm
(2.19 in)
4.95 cm
(1.95 in)
1.34 cm
(0.53 in)
6.68 cm
(2.63 in)
5.02 cm
(1.98 in)
1.35 cm
(0.53 in)
48.9 g (1.7 oz) 58.3 g (2.1 oz)
30.4 cm3
3
(1.9 in
)
Carbon monofluoride/silver
38.6 cm
(2.4 in
3
3
)
vanadium oxide cell
10 N (Models 3660, 3662, 3665,
3667)
5 N (Models 3661, 3663)
15 programs with 8 stim sets each
Page 19

15
Parameter
Tonic Range
Tonic Steps
Burst Range*
Burst Steps*
range)
200–500 Hz
10 Hz
—
—
Device
Model
The IPG has the following operating parameters.
Table 4. Operating parameters for the IPG
Pulse width 20–1000 µs
Frequency 2–200 Hz 2 Hz — —
500–1200 Hz 20 Hz — —
Burst rate
frequency
Intraburst
frequency
Amplitude 0–25.5 mA 0.1–1.0 mA
NOTE: Columns with * represent operating parameters for BurstDR™ stimulation programs
on IPGs capable of BurstDR stimulation mode.
NOTE: For each tonic program, you have the option to select the amplitude range. For
information on setting the amplitude range, see the clinician's programming manual for this
system.
NOTE: The number of stim sets in use for a tonic program governs the maximum frequency
(1200/number of stim sets).
NOTE: The maximum current depends on the impedance, frequency, and pulse width
settings.
— — 10–60 Hz 10 Hz
— — 250–500 Hz 10 Hz
0–12.75 mA 0.05–0.50 mA
10 µs
(20–500 µs range)
50 µs
(500–1000 µs
50–1000 µs 50 µs
500–1000 Hz 20 Hz
0–12.75 mA 0.05–0.50 mA
Compatibility Guidelines for IPGs with Compatible Headers
IPGs with compatible headers are compatible with the following Medtronic‡ leads and extensions
available before May 5, 2015.
Table 5. Compatible Medtronic leads and extensions
Permanent lead
Extension
3776-45, 3776-60, 3776-75, 3876-45, 3876-60, 3876-75, 3777-45, 3777-60,
3777-75, 3877-45, 3877-60, 3877-75, 3778-45, 3778-60, 3778-75, 3878-45,
3878-60, 3878-75, 39286-30, 39286-65, 39565-30, 39565-65
3708120, 3708140, 3708160, 3708220, 3708240, 3708260, 3708320,
3708340, 3708360, 3876, 3877, 3878
WARNING: The use of Medtronic leads or extensions other than those specified in this table
may increase risk to the patient, including the potential for tissue damage.
Page 20

16
Appendix B: System Components and Accessories
The Proclaim™ neurostimulation system includes the following components.
NOTE: Not all models are available in all countries. Contact your local representative for
more information.
NOTE: The model 3661 and 3663 IPGs are compatible only with the leads and extensions
listed in "Compatibility Guidelines for IPGs with Compatible Headers" (page 15). They are not
compatible with Abbott Medical leads and extensions.
IPGs
3660 Proclaim™ XR 5 implantable pulse generator
3661 Proclaim™ 5 implantable pulse generator
3662 Proclaim™ XR 7 implantable pulse generator
3663 Proclaim™ 7 implantable pulse generator
3665 Proclaim™ 5 implantable pulse generator
3667 Proclaim™ 7 implantable pulse generator
IPG Accessories
1101 Torque wrench
1111 Port plug
Programmers and Controllers
3874 St. Jude Medical™ Clinician Programmer App
3875 St. Jude Medical™ Patient Controller App
Programmer and Controller Accessories
1210 Patient magnet
3884 SCS patient manual and magnet
Leads and Extensions
3100-series percutaneous leads
3200-series paddle leads
3300-series extensions
Lead and Extension Accessories
1100-series stylets
1102 Guide wire for percutaneous leads
1103 Introde-AK™ lead introducer
1105 Lead anchor, butterfly
1106 Lead anchor, long
1109 Strain relief
1112 Tunneling tool, 12 in
1114 Epidural needle, 14 gauge, 4 in (10 cm)
1116 Epidural needle, 14 gauge, 6 in (15 cm)
1120 Tunneling tool, 20 in
Page 21

17
1192 Swift-Lock™ anchor
1194 Cinch™ anchor
1701 SCS accessory kit
1803 Lead and extension insertion tool
Adapters
2311 8-channel adapter, M, 10 cm
2316 8-channel adapter, M, 60 cm
Trial System
3599 St. Jude Medical™ External Pulse Generator
Trial System Accessories
1203 Cleaning cloths
1212 Coin cell batteries
1213 Pouch with adhesive (5)
1214 Pouch without adhesive and belt (5)
1216 EPG header cap
1218 Carrying case
1917 Battery door
3013 Multilead trial cable
3032 External pulse generator, 2-port header
Page 22

18
Appendix C: Battery Longevity Information
The longevity of the IPG battery depends on the following factors:
Programmed settings, such as frequency, pulse width, amplitude, and number of active
electrodes
Program impedance
Hours of stimulation per day
Shelf life of the device between the dates of manufacture and implant
Duration of communication sessions between the IPG and the patient controller or clinician
programmer
To estimate battery longevity manually, perform the following steps. For additional help with
estimating battery longevity, contact Technical Support.
1. Locate the energy factor for the desired stimulation parameters according to the lead
impedance in the tables in one of the following sections:
- For IPGs using tonic stimulation parameters, see "Energy Factors for Tonic Stimulation
Parameters" (page 19).
- For IPGs using BurstDR™ stimulation parameters, see "Energy Factors for BurstDR™
Stimulation Parameters" (page 23).
NOTE: If the desired parameters do not appear in the tables, estimate the energy factor by
choosing a value between the listed energy factors for the closest parameters.
2. For an IPG using multiple areas, determine the energy factor for each area from the previous
step, and add each of these values together.
3. Use the figures in "Battery Longevity Graphs" (page 25) to determine the estimated battery
longevity by finding the energy factor from the previous steps on the curve for the
appropriate model IPG.
Page 23

19
Pulse Width (µs)
Amplitude (mA)
Frequency (Hz)
100
200
300
500
30
1 60
90
30
2 60
90
30
3 60
90
30
4
60
26
35
44
85 90
30
5 60
90
30
23
33
43
81
6 60
90
30
7 60
90
30
8 60
90
30
9 60
90
30
10 60
90
Energy Factors for Tonic Stimulation Parameters
The following tables show energy factors according to various stimulation parameters for tonic
programs.
NOTE: Energy factors are for IPGs that provide 12 hours of daily stimulation. For an IPG that
is providing 24 hours of daily stimulation, double the energy factor shown in the table.
Table 6. Energy factors for various tonic stimulation parameters (350-ohm impedance)
13 14 14 15
18 19 20 23
22 24 26 29
15 17 20 24
21 26 30 40
27 34 41 54
16 19 23 30
24 30 37 51
30 41 51 71
17 22 26 47
34 47 61 122
21 30 38 55
34 51 68 102
46 71 97 148
37 58 78 152
51 81 112 223
29 44 60 92
49 80 112 175
68 115 162 257
31 49 67 125
53 89 125 242
74 129 183 359
33 64 89 139
58 118 169 271
81 173 249 401
41 69 97 182
73 130 186 355
105 190 274 528
Page 24

20
Pulse Width (µs)
Amplitude (mA)
Frequency (Hz)
100
200
300
500
30
1 60
90
30
2
60
21
26
30
40
90
30
3 60
90
30
4 60
90
30
5 60
90
30
6 60
90
30
7 60
90
30
8 60
90
88
156
264
426
30
9 60
90
30
10 60
90
Table 7. Energy factors for various tonic stimulation parameters (500-ohm impedance)
13 14 14 15
18 19 20 23
22 24 26 29
15 17 20 24
27 34 41 54
16 19 23 38
24 30 37 68
30 41 51 97
19 26 33 47
31 44 58 85
41 61 82 122
24 35 47 69
40 62 85 130
54 88 122 190
26 40 54 81
44 71 98 152
61 102 142 274
33 52 72 111
57 96 135 214
80 139 198 317
35 58 94 148
62 107 180 288
43 74 104 190
78 139 200 372
112 203 294 554
53 92 131 210
96 175 254 412
140 258 376 613
Page 25

21
Pulse Width (µs)
Amplitude (mA)
Frequency (Hz)
100
200
300
500
30
1 60
90
30
2
60
21
26
30
40
90
30
3 60
90
30
4 60
90
30
5 60
90
30
6 60
90
30
7 60
90
30
8 60
90
116
211
305
566
30
9 60
90
30
10 60
90
Table 8. Energy factors for various tonic stimulation parameters (700-ohm impedance)
13 14 16 18
18 19 24 28
22 24 31 38
15 17 20 24
27 34 41 54
18 23 28 38
27 37 48 68
36 51 66 97
22 31 40 58
35 53 71 107
47 75 102 156
24 41 55 83
40 73 102 158
54 105 148 232
30 47 63 114
51 85 119 220
71 122 173 325
37 60 84 151
65 112 159 294
92 163 234 436
45 76 108 197
81 144 207 382
58 98 139 220
103 184 265 427
147 269 390 710
62 119 169 271
112 225 326 529
160 330 482 871
Page 26

22
Pulse Width (µs)
Amplitude (mA)
Frequency (Hz)
100
200
300
500
30
1 60
90
30
2
60
24
31
37
51
90
30
3 60
90
30
4 60
90
30
5 60
90
30
6 60
90
30
7 60
90
30
8 60
90
148
269
432
702
30
9 60
90
30
10 60
90
Table 9. Energy factors for various tonic stimulation parameters (1000-ohm impedance)
14 15 16 18
19 21 24 28
24 27 31 38
16 20 23 30
31 41 51 72
20 26 33 47
31 44 58 85
41 61 81 122
24 35 47 69
40 62 85 130
54 88 122 190
30 47 64 98
51 85 119 186
72 122 173 275
37 60 84 131
65 112 160 292
92 163 234 431
49 80 112 195
85 148 211 377
120 215 345 558
58 98 139 243
103 184 293 473
68 119 185 297
124 225 358 580
178 362 529 939
80 153 221 356
147 293 429 699
213 433 635 1126
Page 27

23
Amplitude
(mA)
Intermittent Dosage (On Time/Off Time)
Continuous
Dosage
5 s/15 s
15 s/45 s
30 s/90 s
60 s/180 s
30 s/150 s
30 s/180 s
30 s/360 s
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
Amplitude
(mA)
Intermittent Dosage (On Time/Off Time)
Continuous
Dosage
5 s/15 s
15 s/45 s
30 s/90 s
60 s/180 s
30 s/150 s
30 s/180 s
30 s/360 s
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
225
137
106
93
64
55
29
301
1.8
2.0
Energy Factors for BurstDR™ Stimulation Parameters
The following tables show energy factors according to various stimulation parameters for
BurstDR™ stimulation programs.
NOTE: Energy factors represent IPGs that provide 24 hours of daily stimulation using default
values for the Burst Frequency, Intra-burst Rate, and Pulse Width settings.
NOTE: In neurostimulation therapy, “dose” refers to the delivery of a quantity of energy to
tissue. A difference in “dose” in this context does not imply differences in expected
effectiveness response as it would with a drug. There is no demonstrated difference in safety
or effectiveness among these doses.
Table 10. Energy factors for various BurstDR stimulation parameters (350-ohm impedance)
47 31 25 23 16 13 7 77
71 43 34 28 20 17 10 89
93 53 41 34 24 20 12 104
117 65 49 40 29 25 13 118
146 76 56 46 34 28 16 134
175 106 84 73 50 42 24 240
202 121 95 82 56 48 26 270
230 135 106 91 63 55 29 301
247 151 120 100 70 60 33 330
279 163 133 110 76 65 35 361
Table 11. Energy factors for various BurstDR stimulation parameters (500-ohm impedance)
46 32 25 23 15 13 7 76
71 44 34 29 19 18 9 91
95 56 41 34 24 20 12 105
121 67 49 41 28 25 14 120
151 93 73 64 43 37 21 211
174 108 84 73 50 43 23 240
203 122 96 82 57 48 26 271
264 160 129 111 70 59 32 330
297 196 168 146 97 83 45 512
Page 28

24
Amplitude
(mA)
Intermittent Dosage (On Time/Off Time)
Continuous
Dosage
5 s/15 s
15 s/45 s
30 s/90 s
60 s/180 s
30 s/150 s
30 s/180 s
30 s/360 s
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
Amplitude
(mA)
Intermittent Dosage (On Time/Off Time)
Continuous
Dosage
5 s/15 s
15 s/45 s
30 s/90 s
60 s/180 s
30 s/150 s
30 s/180 s
30 s/360 s
0.2
0.4
71
43
34
29
20
17
10
93
0.6
100
64
51
45
30
26
15
152
0.8
1.0
1.2
1.4
1.6
1.8
2.0
Table 12. Energy factors for various BurstDR stimulation parameters (700-ohm impedance)
49 32 26 23 15 13 8 73
72 43 34 29 20 17 10 90
94 53 41 35 24 21 12 101
125 79 62 54 37 31 17 180
153 95 74 64 43 37 20 208
177 108 85 73 50 42 23 239
209 129 103 90 57 48 27 273
239 159 132 115 80 68 36 417
271 179 149 132 88 75 41 465
299 195 164 146 97 83 44 509
Table 13. Energy factors for various BurstDR stimulation parameters (1000-ohm impedance)
48 32 26 23 15 13 7 77
126 78 62 56 37 31 17 182
151 96 73 64 44 37 20 212
184 127 106 94 62 53 29 333
215 145 119 107 72 61 33 378
241 163 134 120 80 68 37 422
281 208 183 160 106 92 49 556
312 228 197 182 117 100 54 607
Page 29

25
Battery Longevity Graphs
The following figure shows the estimated battery longevity of a newly implanted IPG.
Figure 7. Estimated battery longevity by energy factor for Proclaim™ IPGs (from time of implant)
1. Estimated battery
longevity (years)
2. Energy factor
3. Models 3660, 3661,
and 3665
4. Models 3662, 3663,
and 3667
The following figure shows the estimated longevity of an IPG battery after the low-battery
warning—also called the elective replacement indicator (ERI)—first appears on the clinician
programmer app or patient controller app when the battery is approaching its end of service.
Figure 8. Estimated battery longevity by energy factor for Proclaim IPGs (from time of ERI)
1. Estimated battery
longevity (months)
2. Energy factor
3. Models 3660, 3661, and
3665
4. Models 3662, 3663, and
3667
Page 30

26
Appendix D: Regulatory Statements
This section contains regulatory statements about your product.
Disposal Guidelines for Battery-Powered Devices
This device contains a battery and a label is affixed to the device in accordance with European
Council directives 2002/96/EC and 2006/66/EC. These directives call for separate collection and
disposal of electrical and electronic equipment and batteries. Sorting such waste and removing it
from other forms of waste lessens the contribution of potentially toxic substances into municipal
disposal systems and into the larger ecosystem. Return the device to Abbott Medical at the end of
its operating life.
Statement of FCC Compliance
This equipment has been tested and found to comply with the limits for a Class B digital device,
pursuant to part 15 of the FCC rules. These limits are designed to provide reasonable protection
against harmful interference in a residential installation. This equipment generates, uses, and can
radiate radiofrequency energy and, if not installed and used in accordance with the instructions,
may cause harmful interference to radio communications. However, there is no guarantee that
interference will not occur in a particular installation. If this equipment does cause harmful
interference to radio or television reception, which can be determined by turning the equipment
off and on, the user is encouraged to try to correct the interference by one or more of the following
measures:
Reorient or relocate the receiving antenna.
Increase the separation between the equipment and receiver.
Connect the equipment into an outlet on a circuit different from that to which the receiver is
connected.
Consult the dealer or an experienced radio/TV technician for help.
Operation is subject to the following two conditions:
This device may not cause harmful interference.
This device must accept any interference received, including interference that may cause
undesired operation.
Modifications not expressly approved by the manufacturer could void the user’s authority to
operate the equipment under FCC rules.
Statement of Compliance With License-Exempt RSS Standard (Canada)
This device complies with Industry Canada license-exempt RSS standard(s). Operation is subject
to the following two conditions: (1) this device may not cause interference, and (2) this device
must accept any interference, including interference that may cause undesired operation of the
device.
Page 31

27
Identifier Type
Registration Identifier
Antenna type
Antenna dimensions
Modulation
Magnetic field strength (at 2 m distance)
Electric field strength (at 2 m distance)
Output power (EIRP*)
Range
1–2 m typical
Center frequency
2.44 GHz
Channel
Bandwidth
Data flow
Protocol
Identification Information for Product Registration
This device has a label that contains, among other information, a product identifier in the following
format:
Table 14. Registration identification information
FCC registration number RIASJMRFC
Industry Canada (IC) registration number IC: 8454A-M3660123
Wireless Technology Information
The following table summarizes the technical details of the Bluetooth® Smart wireless technology
as it is implemented in the device.
Table 15. Bluetooth Smart wireless technology information
Embedded patch antenna in header
8.1 mm x 5.1 mm x 4.9 mm
GFSK
16.3 µA/m
6.1 mV/m
1 mW (0 dBm) typical, 10 mW (+10 dBm)
maximum
40 logical channels
2 MHz per channel
Bi-directional
Bluetooth Smart wireless technology
*EIRP = Equivalent isotropically radiated power
Page 32

28
Radio Transmitter, Cables, Transducers
The device contains a radio transmitter/receiver with the following parameters.
Radio transmitter parameters:
Frequency (range): 2.4000 to 2.4835 GHz
Bandwidth (-15 dB): 2.398 to 2.4855 GHz
Channel: 40 logical channels using AFH
Modulation: GFSK
Radiated output power: 10 mW (+10 dBm) maximum
Magnetic field strength (at 2 m distance): 16.3 µA/m
Duty cycle: Variable, but low (<5%)
Semi-duplex capability
The radio receiver in the device is using the same frequency and bandwidth as the transmitter.
Cables and transducers:
Cables and transducers are not used during normal use of the device nor while programming the
device.
Quality of Service for Wireless Technology
Bluetooth® Smart wireless technology enables communication between the generator and the
clinician programmer or patient controller. The quality of the wireless communication link varies
depending on the use environment (operating room, recovery room, and home environment).
After the clinician programmer or patient controller is paired with a generator, the Bluetooth
wireless technology symbol is visible on the clinician programmer or patient controller in the upper
right-hand corner of the screen. When the Bluetooth Smart wireless technology connection is not
active, the symbol appears dimmed.
The quality of service (QoS) should allow wireless data to be transferred at a net rate of
2.5 kB/sec. Each connection interval includes a semi-duplex transmission with a required
acknowledge, a transmission latency in each direction (2x), and a receive-to-transmit mode (RXto-TX) time. Data is resent if not successfully received. Each key press may transmit up to 4 data
packets with up to 20 bytes per packet, depending on the number of packets that need to be
transmitted (i.e., if there is only one packet to transmit, only one packet will be transmitted). If the
interference is high (e.g., the bit error rate exceeds 0.1%), the user may experience what appears
to be a slow connection, difficulty pairing devices, and a need to decrease the distance between
connected devices. For information on how to improve connection issues, please refer to
“Troubleshooting for Wireless and Coexistence Issues” (page 29).
Page 33

29
Wireless Security Measures
The wireless signals are secured through device system design that includes the following:
The generator will encrypt its wireless communication.
Only one patient controller or clinician programmer may communicate with the generator at
the same time.
A unique key for each unit that is checked during each transmission.
Built-in pairing that specifies valid and legitimate pairing among units.
Proprietary authentication in addition to the pairing procedure specified in Bluetooth® Smart
wireless technology, which includes an element of proximity.
A proprietary algorithm that detects and prevents an unauthorized user from attempting to
pair with the generator.
Troubleshooting for Wireless and Coexistence Issues
If you experience issues with the wireless communication between the generator and the clinician
programmer or patient controller, try the following:
Decrease the distance between the devices
Move the devices so they share line of sight
Move the devices away from other devices that may be causing interference
Close the clinician programmer or patient controller application, and turn the clinician
programmer or patient controller off and on
Wait a few minutes and try connecting again
Do not operate other wireless devices (i.e., laptop, tablet, mobile phone, or cordless phone)
at the same time
NOTE: Wireless communications equipment, such as wireless home network devices, mobile
and cordless telephones, and tablets, can affect the device.
Page 34

30
Symbol
Definition
Appendix E: Symbols and Definitions
The following symbols may be used in this document and on some of the products and
packaging:
Table 16. Symbols and definitions
Caution, consult accompanying documents
Consult instructions for use
Follow instructions for use on this website
Magnetic Resonance (MR) Conditional, an item with demonstrated safety in the MR
environment within the defined conditions. At a minimum, address the conditions of
the static magnetic field, the switched gradient magnetic field, and the
radiofrequency fields. Additional conditions, including specific configurations of the
item, may be required.
Magnetic Resonance (MR) Unsafe, an item poses unacceptable risks to the patient,
medical staff, or other persons within an MR environment
Device contains a radio-frequency (RF) transmitter, which may cause RF
interference with other devices near this device.
Single use only
Do not resterilize
Expiration date
Date of manufacture
Manufacturing facility
Temperature limits for storage conditions
Humidity limits
Pressure limits
Do not use if the product sterilization barrier or its packaging is compromised
Page 35

31
Symbol
Definition
Table 16. Symbols and definitions
Catalog number
Manufacturer
Contents quantity
Pulse generator
Accessories
Serial number
Batch code
Unique Device Identification
Prescription use only
Ethylene oxide gas sterilization
Authorized European representative
European conformity, affixed according to the relevant provisions of AIMD directive
90/385/EEC and RE directive 2014/53/EU Annex II. Hereby, Abbott Medical
declares that this device complies with the essential requirements and other relevant
provisions of these directives.
The full text of the European Union RE directive 2014/53/EU declaration of
conformity is available at the following internet address:
www.neuromodulation.abbott/euconformity.
Australian Communications and Media Authority (ACMA) and New Zealand Radio
Spectrum Management (RSM) Regulatory Compliance Mark (RCM)
This equipment is certified for type certification pursuant of Article 38-24 of the
Japan Radio Law
Page 36

Abbott Medical
Abbott Medical
2020-10
*600130718*
6901 Preston Road
Plano, Texas 75024 USA
+1 855 478 5833
+1 651 756 5833
ARTEN600130718 A
The Corporate Village
Da Vincilaan 11 Box F1
1935 Zaventem
Belgium
+32 2 774 68 11
 Loading...
Loading...