

Page 1

INSTRUCTION MANUAL
INTERNATIONAL
EDITION
Page 2

Table of Contents
English
Cervical-Stim
®
Instruction Manual
Device Description ...................................................................................... 1
The Orthofix Approach to Healing ............................................................... 1
PEMF Stimulation ......................................................................................... 2
Prescription Information .............................................................................. 2
Clinical Data Summary ............................................................................... 3
Data Analysis & Results ............................................................................... 5
Effectiveness Results ..................................................................................... 6
Safety .......................................................................................................... 8
Device Life .................................................................................................. 8
Treatment Instructions .................................................................................. 8
Charging/Recharging the Battery ................................................................... 9
Device Operation ....................................................................................... 10
Wearing the Device .................................................................................... 11
Care and Cleaning ...................................................................................... 12
Travel ......................................................................................................... 12
Storage ...................................................................................................... 12
Disposal ...................................................................................................... 12
Visual and Audio Indicators .......................................................................... 13
Equipment Classification and Device Symbol Descriptions ............................ 14
Service ....................................................................................................... 14
Conformance to Standards .......................................................................... 15
Warranty Information ................................................................................. 15
To learn more about Orthofix, please visit our website at www.orthofix.com.
Package Contents:
1- Cervical-Stim®Cervical Fusion System
1- Literature Pack
1- Charger Unit #270215
1- Comfort Collar
THIS DEVICE IS NONSTERILE.
IT DOES NOT REQUIRE STERILIZATION.
0086
Page 3
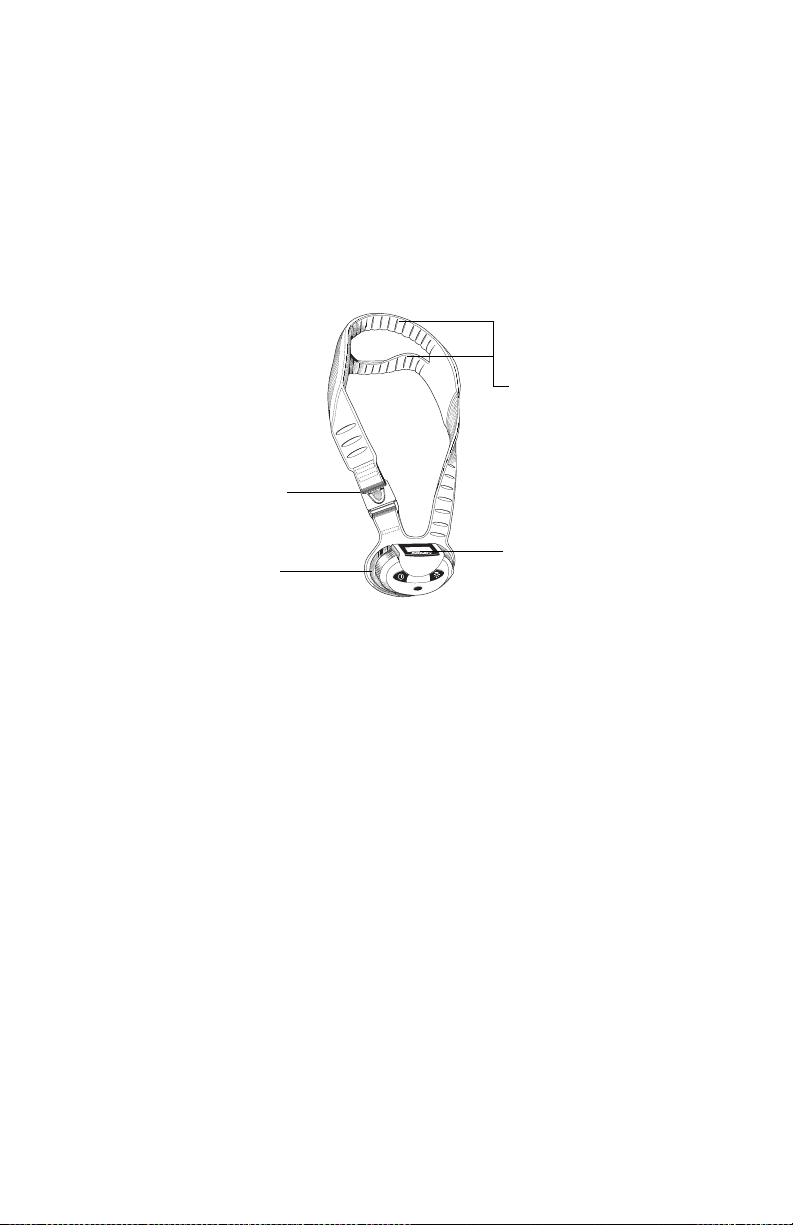
Device Description
The Cervical-Stim®Cervical Fusion System is an external, low-level, Pulsed
Electromagnetic Field (PEMF) device and has been designed with patient comfort
and convenience in mind. It is a single-piece device that is lightweight, flexible
and portable allowing freedom of movement during treatment. A Liquid Crystal
Display (LCD) and audible alarm provide information during treatment such as
operational status, treatment time remaining, battery capacity, etc. See “Visual
and Audio Indicators” for more information.
Cervical-Stim Cervical Fusion System - Model 2505CE
Transducer
Buckle
Joint
Control Unit
The Cervical-Stim is comprised of a control unit and a treatment transducer.
The control unit contains a micro-processor that generates the Cervical-Stim
electrical signal. That signal is converted to a highly uniform, low-energy
magnetic field by the treatment transducer. When the device is centered over
the treatment area, the therapeutic Cervical-Stim PEMF signal is delivered
directly to the fusion site.
The Cervical-Stim is powered by a rechargeable lithium-ion battery pack.
The LCD and audible alarm will alert the patient when the battery is low and
needs to be recharged. See “Charging/Recharging the Battery” for more
information. To ensure that the device is functioning properly, the Cervical-Stim
constantly monitors battery voltage and the electrical signal. If at any time
during treatment, the device stops functioning properly, the LCD will display
an appropriate symbol or error code. See “Visual and Audio Indicators”
for more information.
Control Panel
The Orthofix Approach to Healing
Orthofix is an established worldwide leader in PEMF (pulsed electromagnetic
field) bone growth stimulation technology. Orthofix developed the first
noninvasive, portable PEMF stimulator for cervical fusion with the launch of the
Cervical-Stim Cervical Fusion System in 2005. Orthofix continues to refine and
improve the delivery of pulsed electromagnetic fields for the healing of cervical
spine fusion and salvage of failed cervical spine fusion. Orthofix produces a full
1
Page 4

range of state-of-the-art medical devices for orthopaedics and neurosurgery
that enhance the body's natural healing process, simulate natural function,
protect, restore and heal. Thank you for including Orthofix in your healing
process. To learn more about Orthofix, please visit our website at
www.orthofix.com.
PEMF Stimulation
Pulsed electromagnetic field (PEMF) bone growth stimulation is a safe,
nonsurgical treatment prescribed by a physician to heal nonunion fractures
and promote spinal fusion. Electrical currents have been used to heal bones
since the mid-1800s. However, it wasn’t until the 1950s that scientists made
an important discovery. When human bone is bent or broken, it generates an
electrical field. This low-level electrical field activates the body's own repair
mechanism, which in turn stimulates bone healing.
Orthofix PEMF bone growth stimulators generate a uniform, low-level, pulsed
electromagnetic field similar to the electrical field generated by the body.
The application of PEMF directly to the fusion or fracture site helps activate
and augment the body’s natural healing process to enhance bone fusion.
Prescription Information
Indication
The Cervical-Stim is a noninvasive, pulsed electromagnetic bone growth
stimulator indicated as an adjunct to cervical fusion surgery in patients at
high-risk for non-fusion and nonoperative salvage of failed cervical spine fusion.
English
Contraindications
There are no known contraindications for the Cervical-Stim.
Warnings
• The Cervical-Stim may interfere with the operation of a cardiac pacemaker
or defibrillator. Consultation with the attending cardiologist is recommended.
• The Cervical-Stim should be removed prior to any imaging procedures
(e.g., CT scan, MRI, etc.).
Precautions
• The Cervical-Stim should not be used if there are mental or physical
conditions that may preclude compliance with physician or device
instructions.
• The Cervical-Stim has not been evaluated in treating patients with the
following conditions: osseous or ligamentous spinal trauma, spondylitis,
Paget’s disease, moderate to severe osteoporosis, metastatic cancer, renal
disease, rheumatoid arthritis, uncontrolled diabetes mellitus, patients prone
to vascular migraine headache, seizure, epilepsy, thyroid conditions or
neurological diseases.
• Animal teratological studies performed with this device did not show any
adverse effects in animals. However, the safety of this device for use on
patients who are pregnant or nursing has not been established.
2
Page 5
Adverse Events
Adverse Events Reported at 6 Months by Treatment Group
Adverse Events
Increased Neck Pain
houlder/Arm Pain
S
Re-Injury to Cervical Spine
Adjacent level pathology
Surgical Complications
LBP/Lumbar pathology
Trauma/Injury(not cervical)
Numbness/Tingling
Headache/Migraine
Nonspecific/Unrelated Pain
Nausea
Dizziness/Vertigo
Rash/Discoloration
Rapid/Irregular Heartbeat
Shortness of Breath
Ringing in Ears
Neurologic Symptom/Stroke
Lump in Throat
Diagnosis of Diabetes
Diagnosis of Breast Cancer
Seizure
Death, Unrelated
Tenderness
Screw Broken
Graft Collapse
Carpal Tunnel Syndrome
Choking Sensation
Cardiac Symptoms
Nephrotic Syndrome
Suicide Attempt
Control Group (n=160)
# (%) of
vents
E
0(14.9)
1
10(14.9)
10(14.9)
(4.5)
3
2(3.0)
8(11.9)
2(3.0)
6(8.9)
2(3.0)
2(3.0)
0
2(3.0)
0
0
0
0
1(1.5)
0
0
0
0
0
1(1.5)
1(1.5)
1(1.5)
2(3.0)
1(1.5)
1(1.5)
1(1.5)
1(1.5)
1
# (%)
atients
P
Experiencing
the Event
(5.6)
9
9(5.6)
8(5.0)
(1.9)
3
2(1.3)
8(5.0)
2(1.3)
6(3.8)
2(1.3)
2(1.3)
0
2(1.3)
0
0
0
0
1(0.6)
0
0
0
0
0
1(0.6)
1(0.6)
1(0.6)
2(1.3)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
Cervical-Stim Group (n=163)
of
#*(%) of
Events
6(17.8)
1
16(17.8)
9(10.0)
(8.8)
8
7(7.7)
5(5.5)
5(5.5)
4(4.4)
4(4.4)
3(3.3)
2(2.2)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
0
0
0
0
0
0
0
0
1
# (%)
of
Patients
Experiencing
he Event
t
5(9.2)
1
16(9.8)
9(5.5)
(4.9)
8
5(3.1)
5(3.1)
4(2.5)
4(2.5)
4(2.5)
3(1.8)
2(1.2)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
0
0
0
0
0
0
0
0
TOTAL
1
. % expressed as number of patients experiencing the event / total number of patients in the group
2
. Some patients experienced multiple adverse events
*
There were several adverse events that were more frequently observed in the Cervical-Stim group than in the
control group. Given the types of events, it is unlikely that these adverse events are related to the treatment.
67
2
47
90
2
58
Clinical Data Summary
Study Design
The Cervical-Stim clinical study was a controlled, randomized, parallel
group study of 323 high-risk (smokers, multi-level or both and allograft) adult
subjects with radiographic evidence of compressed cervical nerve roots and
3
Page 6

symptomatic radiculopathy. The purpose of the study was to evaluate the
safety and effectiveness of the PEMF Cervical-Stim device as an adjunct for
high-risk patients who undergo cervical fusion. All subjects underwent
anterior cervical discectomy and fusion using the Smith-Robinson technique
with the Atlantis Plate. Subjects were randomly assigned to either the control
group (standard treatment, n=160) or the treatment group (standard treatment
plus the Cervical-Stim, n=163). Standard treatment was at the physician's
discretion but typically included the standard hospital stay, use of a soft cervical
collar, appropriate medications and physical therapy.
Subjects who met the following inclusion and exclusion criteria were eligible
for participation in the study:
Inclusion Criteria
Adult male or female,18-75 years old with radiographic evidence of
compressed cervical nerve root(s), symptomatic radiculopathy, pain of 5 or
greater on the Visual Analog Scale (VAS) and/or any muscle weakness and
primary cervical spinal fusion performed using the Smith-Robinson technique
with allograft bone and an anterior cervical plate. The fusion procedure must
have been either multi-level (>1 fusion level) or the subject was a smoker
(one pack/day or more) or both; and signed informed consent.
Exclusion Criteria
Traumatic cervical injury, posterior approach or revision fusion, autograft
or bone substitute materials for graft source, history of vascular migraine
headache or prone to uncontrolled seizures or epilepsy (controlled or
uncontrolled) or any neurological diseases or injury; depressed immune
system, regional conditions (Spondylitis, Paget’s disease, rheumatoid arthritis),
infection (systemic or local) within 2 weeks prior to surgery, systemic
conditions (cancer, cardiac arrhythmia, thyroid disease, uncontrolled diabetes
mellitus, renal disease/dysfunction, chronic steroid use or other conditions that
may have affected bone metabolism), cardiac pacemakers, defibrillators, dorsal
column stimulators, hearing aids, cochlear prostheses and cranial stimulators,
subjects who were pregnant, nursing or had planned to become pregnant
within 12 months, subjects that had participated in other clinical studies within
the last 12 months, or had mental or physical conditions which may have
precluded compliance with physician instructions.
English
Evaluation and Follow-Up
Follow-up visits were to have been performed at months 1, 2, 3, 6 and 12
and annually thereafter until the last subject enrolled reached 12 months.
Device Usage
Subjects assigned to the treatment group (Cervical-Stim) were instructed
to wear the device for four hours per day for a minimum of three months
postoperative. Surgeons could, at their discretion, extend the Cervical-Stim
treatment up to six months postoperative.
4
Page 7

Demographic Data
The subjects in this study had a mean age of 46.8 years (range 24 to 73 years).
Of the 323 subjects, 148 (45.8%) were female and 175 (54.2%) were male.
Three hundred one (93.2%) were Caucasian, while 17 (5.3%) were African
American and 5 (1.6%) were Hispanic. One hundred fifty-nine (49.2%) were
nonsmokers and 164 (50.8%) were smokers.
Baseline Demographic Characteristics
ariables
V
Age (years)
Mean
Range
SD
Gender
Female
Male
Race
Caucasian
African-American
Hispanic
Asian
Other
Smoking Status
Nonsmoking
Smoking
1
. P-values of comparison tests between treatment groups using Student’s t-test for numerical
Number of
Subjects
(N = 323)
46.8
24 – 73
9.3
148 (45.8%)
175 (54.2%)
301 (93.2%)
17 ( 5.3%)
5 ( 1.6%)
0
0
159 (49.2%)
164 (50.8%)
Control
(n = 160)
46.7
26 – 72
9.2
75 (46.9%)
85 (53.1%)
150 (93.8%)
7 ( 4.4%)
3 ( 1.9%)
-
-
79 (49.4%)
81 (50.6%)
variables and Pearson x2test for categorical variables.
Cervical-Stim
(n = 163)
46.9
24-73
9.4
73 (44.8%)
90 (55.2%)
151 (92.6%)
10 ( 6.1%)
2 ( 1.2%)
80 (49.1%)
83 (50.9%)
1
P-value
0.846
0.706
0.703
-
-
0.958
Data Analysis and Results
The primary effectiveness endpoint was the increase in frequency of cervical
fusion success by six months postoperatively as assessed by radiographic
evidence. Secondary endpoints were neurological function, VAS pain assessment
and Neck Disability Index. Safety was assessed by the frequency and severity
of adverse events.
Fusion was assessed by radiographs at each visit:
Radiographic fusion was defined as > 50% bony bridging on both the
superior and inferior graft interfaces between adjacent vertebral bodies
AND < 4° angulation (motion) between adjacent fused vertebrae on
flexion/extension lateral films AND absence of radiolucency.
Radiographic non-fusion was defined as < 50% bony bridging at either the
superior or inferior graft interface OR > 4° angulation (motion) between
adjacent fused vertebrae on flexion/extension lateral films OR presence
of radiolucency.
5
Page 8

For purposes of device evaluation, all films were scanned into a central database
and reviewed by two independent, blinded orthopedic surgeons and a blinded,
independent radiologist following completion of the entire study. Films were
viewed and scored using a common protocol. All films at each time point were
evaluated for amount of radiolucency, bony bridging and degree of motion as
evidenced on the flexion/extension cervical spine films. A software program
was used to calculate motion. Results obtained in this fashion were reviewed
and verified by the reviewing orthopedic surgeons. The radiologist’s diagnosis
was considered definitive in the case of a disagreement between the two
orthopedic surgeons.
Effectiveness Results
Of the 323 subjects who were randomized and received surgery, 240 were
evaluable for the effectiveness analysis (Cervical-Stim treatment group, n=122;
control group, n=118). Subjects were deemed unevaluable for the following
reasons: non-existent or non-readable x-rays, subject non-compliance,
protocol violations (inclusion criteria), graft collapse, broken internal hardware,
early study exits due to minor adverse experiences, and one suicide attempt.
The success or failure of these subjects is not known. These unavailable data
could positively or negatively affect the overall success of the study. In order
to assess the impact of the missing data, sensitivity analyses were performed.
These included last observation carried forward and all missing data imputed
as non-fusion. Both of these analyses showed that the results at six months
were still statistically significantly different in favor of the Cervical-Stim group.
In addition, the baseline demographic data from the evaluable population was
compared to the demographic data of the missing subjects. The results of this
analysis indicated there were no significant differences between the evaluable
subjects and the non-evaluable subjects in 14 study variables including key
demographics and clinical parameters.
English
Primary Effectiveness Endpoint
The primary effectiveness endpoint was evidence of radiographic fusion at the
six month time point postoperative. At the six month time point, 102 of the
122 evaluable subjects (84%) in the Cervical-Stim treatment group were
judged to be fused versus 81 of the 118 evaluable subjects (69%) in the control
group (p=0.0065).
Comparison of Radiographic Fusion Outcomes at Six Months
Treatment Group
Control
Cervical-Stim
Number of Subjects
118
122
Number of Subjects
Fused
81
102
Fusion Rate (%)
68.64
83.61
These data show that for patients undergoing cervical fusion surgery, patients
treated adjunctively with the Cervical-Stim experienced an increase in the
frequency of radiographic fusion at six months when compared to the control group.
6
Page 9

An additional analysis was performed to allow for the differences between
the Cervical-Stim treatment group and the control group with respect to
demographic characteristics (gender, age, diagnosis) and risk status (smoking,
multilevel). The overall fusion rate in the Cervical-Stim group remained
statistically significant after adjustment for each of these variables.
Long-term follow-up (12 months) showed no statistical difference between the
two groups with respect to fusion. One hundred sixteen of the 125 evaluable
subjects (92.8%) in the Cervical-Stim treatment group were judged to be fused
at the long-term final endpoint, while 104 of the 120 evaluable subjects
(86.7%) in the control group were judged to be fused.
Overall Radiographic Fusion Fusion Outcomes at 12 Months
Treatment Group
Control
Cervical-Stim
Note: The differences in long-term success rates between treatment groups is not statistically
significant per Pearson x
Number of Subjects
120
125
2
test with the available sample size (x2= 2.5136, p = 0.1129).
Number of Subjects Fused
104
116
Fusion Rate (%)
86.67
92.80
Secondary Effectiveness Endpoints
Secondary endpoints evaluated changes in clinical symptoms. A “clinical success”
with regard to symptoms was defined as no worsening in neurological function,
an improvement in VAS pain assessment and no worsening in Neck Disability
Index. A “clinical failure” with regard to symptoms was defined as failure for
any one of these criteria. There was no statistically significant difference
between the two groups with respect to the percent of subjects considered
a “clinical success” at six months (p=0.8456) or at 12 months (p=0.1129).
7
Page 10

Safety
The adverse events observed in this study are shown in the Adverse Events
Table presented in the Prescription Information section. At six months, the
numbers of subjects who experienced one or more adverse events is similar
in the two groups. A total of 14 severe events were reported in 13 subjects;
nine of the subjects were in the Cervical-Stim treatment group and five
subjects were in the control group. These events included experiences such
as increased pain, shortness of breath, dizziness, unrelated trauma and injury,
unrelated death, surgical complication and adjacent level pathology. For the
nine subjects in the Cervical-Stim treatment group, all severe adverse events
were, in the judgment of the investigators, definitely or probably unrelated to
the device.
Safety data obtained between the six month visit and the final contact with
each subject indicate that 57 adverse events were experienced by a total of
51 subjects between both groups. The number of subjects who experienced
one or more adverse events is similar in the two groups. None of the adverse
events reported between the six month visit and the final contact were severe
and are similar to those reported at six months.
Device Life
The Cervical-Stim can provide up to 270 consecutive (daily) treatments of four
hours each. The overall length of treatment will be determined by the physician
based on the patient and progress toward fusion.
English
Treatment Time
The Cervical-Stim should be worn for four hours per day. At the end of the
daily treatment the device will turn itself off. The device may be turned off
at any time by simply pressing the On/Off button on the control panel.
The Cervical-Stim may be used at any time of day that is convenient for the
patient. It is lightweight and adjustable. And because the Cervical-Stim is
portable, treatment can be received while sitting, walking, reclining, sleeping,
etc. However, since each patient is unique, the overall activity level should be
based on the physician’s instructions.
8
Page 11
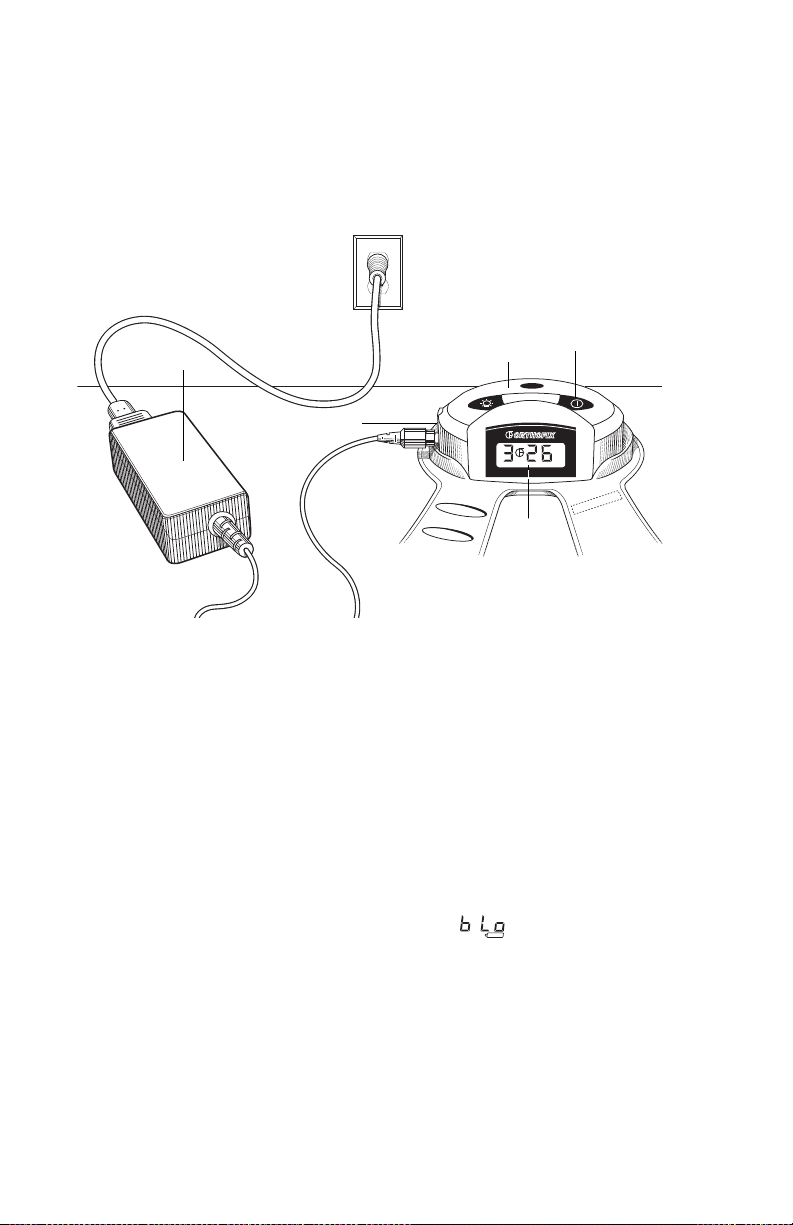
Charging/Recharging the Battery
The Cervical-Stim is powered by a rechargeable lithium-ion battery pack.
A charger unit is provided with the device. Use only the Orthofix charger
to charge the battery.
Note: The Cervical-Stim battery will require charging prior to the first use.
Charger Unit
Charger Port
To charge/recharge the battery, simply plug the barrel connector end of the
charger unit into the charger port located on the control unit. Plug the line
cord securely into the charger unit. Plug the charger into any standard AC wall
outlet. A fully discharged battery will require approximately 4 hours to charge
completely.
The Cervical-Stim battery can be recharged at any time the device is not in use.
It is strongly recommended that the device be recharged after completing daily
treatment.
Note: The Cervical-Stim will not deliver treatment while charging.
When the device is on, the Cervical-Stim LCD will show a battery capacity
symbol. A flashing battery outline, the symbol and an audible beep
indicate a battery low condition and that the battery requires recharging.
See “Visual and Audio Indicators” for more information.
Control Unit
On/Off Button
LCD
9
Page 12
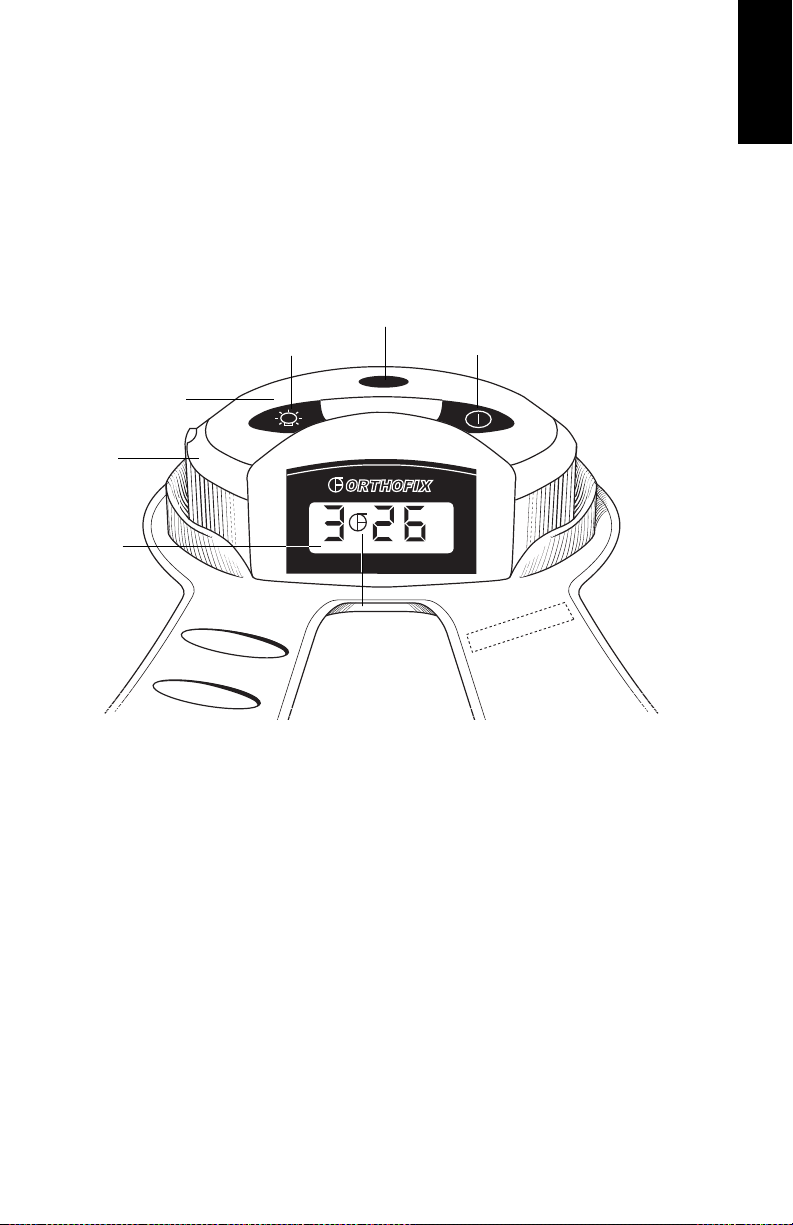
Device Operation
Turning the Device On and Off
The Cervical-Stim is turned on and off by pressing the On/Off button on the
control unit of the device. When the device is on, a sequence of status
messages will display momentarily. The LCD should then show the treatment
time remaining and a flashing Orthofix logo. The flashing logo indicates that
the device is on and functioning normally. (If you do not see this on the display,
contact your provider.) A backlight button is on the control unit. In low light,
press the backlight button for illumination of the LCD.
IR Port
Backlight Button
Control Unit
Charger
Port
LCD
On/Off Button
English
Flashing
Orthofix Logo
Timing of Treatment Sessions
The Cervical-Stim automatically times each treatment session. The timing
begins when the device is turned on using the On/Off button on the control
unit. The LCD shows a countdown of the time remaining in the treatment
session. At the end of daily treatment, the device will turn itself off. To stop
treatment prior to the end of a treatment session, simply press the On/Off
button. To resume treatment, press the On/Off button again. The LCD will
display the remaining treatment time.
Note: For the countdown to function correctly treatment sessions should be
greater than 60 minutes duration.
10
Page 13

Wearing the Device
The Cervical-Stim is intended for the cervical spine and may be worn with or
without a brace. For better comfort, clothing should be worn between the
skin and the Cervical-Stim.
To use the Cervical-Stim, simply slip the device over the head so that it rests
comfortably against the neck and shoulders (see figure below). Or, the device
may be opened at the buckle and placed against the neck and shoulders.
Fasten the buckle like a seatbelt, to secure the unit.
A Comfort Collar is included with Cervical-Stim for improved fit and support.
The Comfort Collar can be inserted or removed from the inside collar of the
device using Velcro Tabs, which attach directly to the material.
Velcro
Tab
Buckle
Joint
11
Page 14

Care and Cleaning
The Cervical-Stim is a sophisticated electronic device and should be handled
with care. Dropping or other mistreatment of the Cervical-Stim may cause
damage to the device.
DO NOT expose the Cervical-Stim to direct sunlight
for long periods of time. The device control unit may be damaged.
DO NOT expose the Cervical-Stim to excessive heat. In warm
climates, the temperature in a car or trunk can exceed 160ºF /
71ºC. Excessive heat can damage the control unit of the device.
DO NOT expose the Cervical-Stim to excessive moisture.
DO NOT dispose of the Cervical-Stim in an incinerator.
DO NOT use solvents to clean the Cervical-Stim.
Clean the device by wiping with a soft, damp cloth.
Travel
Patients should be advised that when traveling by air, it is best to check the
Cervical-Stim with the luggage. If the device is taken on board the airplane,
it should not be worn when passing through passenger screening devices.
The Cervical-Stim could be damaged. The Cervical-Stim user manual should
be taken to quickly and easily identify the device for any security personnel.
English
Storage
The Cervical-Stim should be stored within 14ºF to 113ºF (-10º C to 45º C)
The Cervical-Stim operating temperature range should be within 41ºF to
104ºF (+5º C to 40º C)
Relative Humidity: Up to 95%, non-condensing
Disposal
This device contains lithium batteries. DO NOT dispose of
Cervical-Stim in an incinerator. Dispose of the device properly to
prevent injury.
12
Page 15
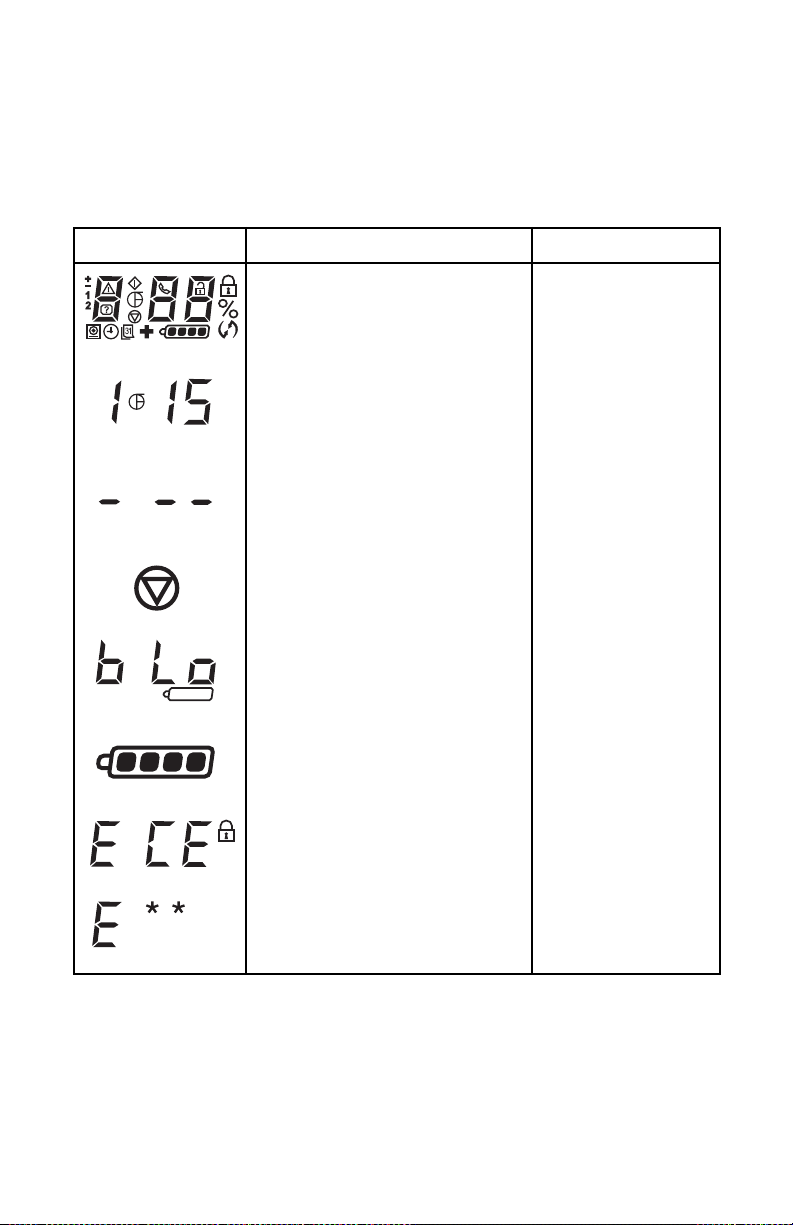
Visual and Audio Indicators
The LCD and audible alarms are designed to provide helpful information
to the user. The chart below shows the various displays and alarms and
their meaning.
Cervical-Stim LCD Visual and Audio Indicators
Symbol /Alarm Description Meaning
all LCD symbols visible and
continuous audible alarm for
approximately 5 seconds
countdown timer displays
remaining treatment time
(hours & minutes)
Orthofix logo flashes
countdown timer
displays three dashes
audible alarm (5 beeps)
steady symbol
for approximately 5 seconds
symbols flash / audible alarm
(approximately 1 beep
per second)
steady symbol indicates
approximate % of charge
symbol filling repeatedly
indicates charge mode
continuous audible alarm
power-on self test
normal treatment
in progress
no treatment time
remaining
treatment complete/
power off
battery low —
recharge required
battery status:
remaining charge
or charging mode
device locked —
call for service
display of any E code
(e.g., E01, E02 . . .)
13
error message —
call for service
Page 16

Equipment Classification and Device Symbol Descriptions
Symbol Meaning
Attention - Refer to Instructions for Use
Type BF Applied Part
On/Off
Backlight Button
Storage Temperature Range
Year of Manufacture for Active Device
Charger Port
Equipment Classifications
• Internally powered equipment
• Type BF applied part
• IEC 529 enclosure rating: IPXO
• Equipment not suitable for use in the presence of a flammable anaesthetic
mixture with air or nitrous oxide
• Mode of operation: intermittent operation
English
The use of accessories other than those specified may result in increased
emissions or decreased immunity of the device.
The battery charger is provided with a 3-wire appliance inlet but considered
double insulated with Class II construction throughout.
For safe usage, follow manufacturer instructions when using the product.
Use of the product in any other manner could have harmful effects and/or void
the warranty.
Note: Inspect the device prior to each use for wear or deterioration.
Do not use if the device does not appear to be in suitable condition.
Service
If you have questions concerning the device or require any assistance, please
contact your local distributor or visit www.orthofix.com for details. There are
no serviceable parts. Notify the manufacturer or local distributor for servicing
needs.
14
Page 17

Conformance to Standards
The Orthofix Cervical-Stim Cervical Fusion System conforms to the following
worldwide series of standards:
UL 60601 - Medical Electrical Equipment, General Requirements for Safety;
(including Electromagnetic Compatibility and Interference)
IEC 60601 - Medical Electrical Equipment, Safety Requirements for Medical
Electrical Systems (including Electromagnetic Compatibility and
Interference)
Warranty Information
Orthofix Inc. warrants the Cervical-Stim®bone growth stimulator to be free
from defects in materials and workmanship for one year from the date of
first use. Provided that all terms and conditions of this Limited Warranty are
complied with, Orthofix Inc. will replace defective components.
This Limited Warranty applies to the product only under normal use and does
not cover any damage or defect caused by accident, misuse, abuse, fire, flood,
and acts of God or by any alteration, tampering, repair or attempted repair by
anyone other than Orthofix Inc. This warranty only applies to the patient for
whom the product is prescribed and is not assignable or transferable.
Defective products covered by this Limited Warranty must be returned to
Orthofix Inc. Attention: Orthofix Returns. You must call your local distributor
to obtain the Return Authorization (RA) number and address prior to returning
the product.
Except as specifically required by applicable law, the foregoing warranty is in
lieu of all other warranties, expressed or implied and Orthofix Inc. specifically
disclaims any and all warranties of merchantability or fitness for a particular
purpose. Under no circumstances shall Orthofix Inc., its authorized
representative, affiliated or subsidiary companies be liable for special,
consequential or incidental damages. The sole remedy with respect to any
defective product shall be limited to replacement.
This Limited Warranty may not be extended or modified except in writing
by Orthofix Inc. No sales person, representative, distributor or doctor is
authorized to make or consent to any extension or modification of the terms
of this Limited Warranty.
For additional information and/or device assistance, contact your local distributor.
15
Page 18

HA N D L EI D I N G
INTERNATIONALE
VERSIE
Page 19

Inhoudsopgave
Nederlands
Cervical-Stim
Beschrijving van het apparaat ...................................................................... 1
De Orthofix-methode voor genezing ............................................................1
PEMF-stimulatie ......................................................................................... 2
Informatie voor voorschrift ......................................................................... 2
Overzicht van klinische gegevens ................................................................ 3
Gegevensanalyse en resultaten .................................................................... 5
Doeltreffendheid - resultaten ...................................................................... 6
Veiligheid ................................................................................................... 8
Gebruiksduur van het apparaat ................................................................... 8
Behandelingsvoorschriften .......................................................................... 8
Laden/herladen van de batterij ..................................................................... 9
Gebruik van het apparaat .......................................................................... 10
Het apparaat dragen .................................................................................. 11
Onderhoud en reiniging ............................................................................. 12
Reizen ...................................................................................................... 12
Opslag ...................................................................................................... 12
Afvoer ...................................................................................................... 12
Visuele en geluidsindicators ....................................................................... 13
Classificatie apparatuur en beschrijving van symbolen op het apparaat ......... 14
Service ..................................................................................................... 14
Naleving van normen ................................................................................ 15
Garantie-informatie ................................................................................... 15
®
handleiding
Ga voor meer informatie over Orthofix naar onze website
op www.orthofix.com.
Inhoud van de verpakking:
1- Cervical-Stim®cervicaal fusiesysteem
1- Literatuur
1- Laadeenheid #270215
I - Comfortkraag
DIT APPARAAT IS NIET STERIEL.
HET BEHOEFT GEEN STERILISATIE.
0086
Page 20
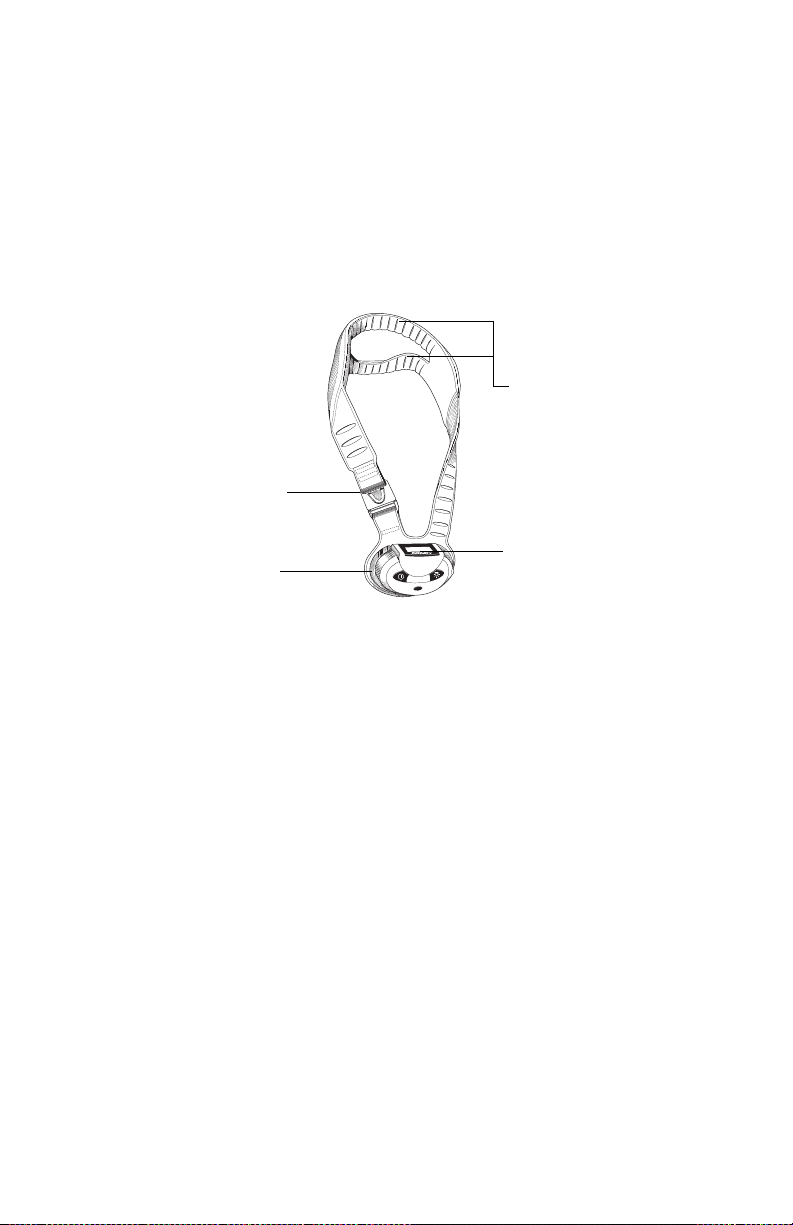
Beschrijving van het apparaat
Het Cervical-Stim®cervicaal fusiesysteem is een apparaat met een extern, laag,
gepulseerd elektromagnetisch veld (PEMF) dat werd ontworpen met het comfort en
gemak van de patiënt in gedachten. Het is een uit één stuk bestaand apparaat dat licht,
flexibel en draagbaar is, waardoor het kan worden verplaatst tijdens de behandeling. Een
Liquid Crystal Display (LCD) en geluidsalarm verschaffen belangrijke feedback tijdens de
behandeling zoals de bedrijfstatus, resterende behandelingstijd, batterijvermogen, etc. Zie
“Visuele en geluidsindicators” voor meer informatie.
Cervical-Stim cervicaal fusiesysteem - model 2505CE
Transductor
Gesp
Gewricht
Regeleenheid
Bedieningspaneel
De Cervical-Stim bestaat uit een regeleenheid en behandelingstransductor. De
regeleenheid bevat een microprocessor die het Cervical-Stim elektrisch signaal
produceert. Dit signaal wordt door de behandelingstransductor omgezet in een zeer
uniform, laag magnetisch veld. Wanneer het apparaat wordt gecentreerd over de te
behandelen plek, wordt het therapeutische Cervical-Stim PEMF-signaal direct aan de
fusieplek geleverd.
De Cervical-Stim wordt gevoed door een oplaadbare lithium-ionbatterij.
De LCD en het geluidsalarm verwittigen de patiënt wanneer de batterij bijna leeg is en
moet worden opgeladen. Zie “Laden/herladen van de batterij” voor meer informatie.
Om te verzekeren dat het apparaat goed werkt, controleert de Cervical-Stim
voortdurend de batterijspanning en het elektrisch signaal. Als het apparaat op een
willekeurig moment tijdens de behandeling niet goed meer werkt, geeft de LCD een
overeenkomstig symbool of foutcode weer. Zie “Visuele en geluidsindicators” voor
meer informatie.
De Orthofix-methode voor genezing
Orthofix is een erkende wereldwijde leider in PEMF (gepulseerde elektromagnetische
veld) botgroeistimulatietechnologie. Orthofix ontwikkelde in 2005 de eerste nietinvasieve, draagbare PEMF-stimulator voor cervicale fusie met de lancering van het
Cervical-Stim cervicaal fusiesysteem. Orthofix blijft de levering van gepulseerde
elektromagnetische velden voor de genezing van cervicale wervelfusie en het redden
van gefaalde cervicale wervelfusie verfijnen en verbeteren. Orthofix produceert een
1
Page 21

breed gamma van ultramoderne medische apparaten voor orthopedie en
neurochirurgie die het natuurlijke genezingsproces van het lichaam verhogen, de
natuurlijke functie simuleren, beschermen, herstellen en genezen. Bedankt dat u
Orthofix gebruikt bij uw genezingsproces. Ga voor meer informatie over Orthofix
naar onze website op www.orthofix.com.
PEMF-stimulatie
Botgroeistimulatie door middel van gepulseerde elektromagnetische velden (PEMF) is
een veilige, niet-chirurgische behandeling die is voorgeschreven door een arts om
fracturen met uitblijvende fusie te genezen en spinale fusie te bevorderen. Elektrische
stroom wordt reeds sinds ongeveer 1850 gebruikt om botten te genezen. Het duurde
echter tot circa 1950 voordat wetenschappers een belangrijke ontdekking deden.
Wanneer menselijk bot wordt gebogen of gebroken, genereert het een elektrisch veld.
Dit lage elektrische veld activeert het eigen herstelmechanisme van het lichaam dat op
zijn beurt de genezing van het bot stimuleert.
Orthofix PEMF botgroeistimulators genereren een uniform, laag, gepulseerd
elektromagnetisch veld dat lijkt op het door het lichaam gegenereerde elektrische veld.
Bij rechtstreekse aanwending van PEMF op de fusie of fractuur wordt het
natuurlijke genezingsproces van het lichaam geactiveerd en verhoogd om de botfusie te
bevorderen.
Informatie voor voorschrift
Indicatie
De Cervical-Stim is een niet-invasieve gepulseerde elektromagnetische
botgroeistimulator die geïndiceerd is als een hulpmiddel voor cervicale fusiechirurgie bij
patiënten met een hoog risico voor uitblijven van de fusie en niet-operatief redden van
gefaalde cervicale wervelfusie.
Nederlands
Contra-indicaties
Er zijn geen bekende contra-indicaties voor de Cervical-Stim.
Waarschuwingen
• De Cervical-Stim kan de werking van een hartpacemaker of defibrillator verstoren.
Het is aanbevolen dat u de behandelende cardioloog raadpleegt.
• De Cervical-Stim moet worden verwijderd vóór beeldvormingsprocedures (bijv. CT
scan, MRI, etc.).
Voorzorgsmaatregelen
• De Cervical-Stim mag niet worden gebruikt als er mentale of lichamelijke condities
zijn die de naleving van de instructies van de arts of het apparaat uitsluiten.
• De Cervical-Stim is niet geëvalueerd bij de behandeling van patiënten met de
volgende aandoeningen: ossaal of ligamentair spinaal trauma, spondylitis, ziekte van
Paget, matige tot ernstige osteoporose, metastatische kanker, nierziekte, reumatoïde
arthritis, ongecontroleerde diabetes mellitus, patiënten die last hebben van vasculaire
hoofdpijn (migraine), convulsie, epilepsie, schildklieraandoeningen of neurologische
ziekten.
• Teratologische studies bij dieren met dit apparaat hebben geen bijwerkingen getoond
bij dieren. De veiligheid van dit apparaat voor gebruik bij patiënten die zwanger zijn
of borstvoeding geven is echter nog niet vastgesteld.
2
Page 22

Bijwerkingen
Bijwerkingen gemeld na 6 maanden door behandelingsgroep
Bijwerkingen
Verergerde nekpijn
ijn in schouder/arm
P
Herbeschadiging van cervicale wervelkolom
Nabijgelegen pathologie
hirurgische complicaties
C
LBP/Lumbale pathologie
Trauma/letsel (niet cervicaal)
evoelloosheid/tintelingen
G
Hoofdpijn/migraine
iet-specifieke/niet verwante pijn
N
Misselijkheid
Duizeligheid/draaierigheid
Uitslag/verkleuring
Snelle/onregelmatige hartslag
Kortademigheid
Oorsuizen
Neurologisch symptoom/beroerte
Dichte keel
Diagnose van diabetes
Diagnose van borstkanker
Convulsie
Overlijden, niet gerelateerd
Gevoeligheid
Schroef gebroken
Falen van graft
Carpaal tunnelsyndroom
Verstikkingsgevoel
Hartsymptomen
Nefrotisch syndroom
Poging tot zelfmoord
Controlegroep (n=160)
aantal (%)
ebeurtenissen
g
0(14,9)
1
10(14,9)
10(14,9)
(4,5)
3
2(3,0)
8(11,9)
(3,0)
2
6(8,9)
2(3,0)
(3,0)
2
0
2(3,0)
0
0
0
0
1(1,5)
0
0
0
0
0
1(1,5)
1(1,5)
1(1,5)
2(3,0)
1(1,5)
1(1,5)
1(1,5)
1(1,5)
aantal (%)
atiënten die de
p
gebeurtenis
ervaren
(5,6)
9
9(5,6)
8(5,0)
(1,9)
3
2(1,3)
8(5,0)
(1,3)
2
6(3,8)
2(1,3)
(1,3)
2
0
2(1,3)
0
0
0
0
1(0,6)
0
0
0
0
0
1(0,6)
1(0,6)
1(0,6)
2(1,3)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
Cervical-Stim groep (n=163)
1
aantal*(%)
gebeurtenissen
16(17,8)
6(17,8)
1
9(10,0)
8(8,8)
(7,7)
7
5(5,5)
5(5,5)
(4,4)
4
4(4,4)
3(3,3)
2(2,2)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
0
0
0
0
0
0
0
0
patiënten die de
aantal (%)
ebeurtenis
g
ervaren
15(9,2)
6(9,8)
1
9(5,5)
8(4,9)
(3,1)
5
5(3,1)
4(2,5)
(2,5)
4
4(2,5)
3(1,8)
2(1,2)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
0
0
0
0
0
0
0
0
1
TOTAAL
1
. % uitgedrukt als aantal patiënten die de gebeurtenis ervaren/totaal aantal patiënten in de groep
2
. Sommige patiënten ervaarden meerdere bijwerkingen
*
Er waren verscheidene bijwerkingen die frequenter werden geobserveerd in de Cervical-Stim groep dan in de
67
2
47
90
2
58
controlegroep. Gezien het soort gebeurtenissen is het onwaarschijnlijk dat deze bijwerkingen in verband staan
met de behandeling.
Overzicht van klinische gegevens
Studieopzet
De Cervical-Stim klinische studie was een gecontroleerde, gerandomiseerde, parallelle
groep studie van 323 volwassen patiënten met hoog risico (rokers, meerniveau of
beide en allotransplantaat) met radiografisch bewijs van samengedrukte cervicale
3
Page 23

zenuwwortels en symptomatische radiculopathie. Het doel van de studie was om de
veiligheid en doeltreffendheid van het PEMF Cervical-Stim apparaat te evalueren als een
hulpmiddel voor patiënten met hoog risico die cervicale fusie ondergaan. Alle patiënten
ondergingen anterieure cervicale discectomie en fusie middels de techniek van SmithRobinson met de Atlantis plaat. De patiënten werden willekeurig toegewezen aan de
controlegroep (standaard behandeling, n=160) of de behandelingsgroep (standaard
behandeling plus de Cervical-Stim, n=163). De standaard behandeling werd bepaald
door de arts maar omvatte gewoonlijk het standaard ziekenhuisverblijf, het gebruik van
een zachte cervicale kraag, de geschikte medicatie en fysiotherapie.
Patiënten die voldeden aan de volgende criteria voor insluiting en uitsluiting kwamen in
aanmerking om deel te nemen aan de studie:
Insluitingscriteria
Volwassen man of vrouw,18-75 jaar oud met radiografisch bewijs van samengedrukte
cervicale zenuwwortel(s), symptomatische radiculopathie, pijn van 5 of hoger op de
Visueel Analoge Schaal (VAS) en/of spierzwakte en primaire cervicale wervelfusie
uitgevoerd met de techniek van Smith-Robinson allotransplantaatbot en een anterieure
cervicale plaat. De fusieprocedure moest ofwel multiniveau (>1 fusieniveau) zijn of de
patiënt was een roker (één pakje/dag of meer) of beide; en een informatie- en
toestemmingsformulier moest ondertekend zijn.
Uitsluitingscriteria
Traumatisch cervicaal letsel, posterieure methode of corrigerende fusie,
autotransplantaat of botvervangingsmateriaal voor transplantaatbron, anamnese van
vasculaire hoofdpijn (migraine) of last hebbend van ongecontroleerde convulsies of
epilepsie (gecontroleerd of ongecontroleerd) of neurologische ziekten of letsel;
verminderd immuunsysteem, plaatselijke aandoeningen (Spondylitis, ziekte van Paget,
reumatoïde arthritis), infectie (systemisch of plaatselijk) 2 weken vóór chirurgie,
systemische aandoeningen (kanker, hartaritmieën, schildklierziekte, ongecontroleerde
diabetes mellitus, nierziekte/stoornis, chronisch gebruik van steroïden of andere
aandoeningen die het botmetabolisme kunnen hebben beïnvloed), hartpacemakers,
defibrillators, dorsale kolomstimulators, gehoorapparaten, cochleaire prothesen en
schedelstimulators, patiënten die zwanger waren, borstvoeding gaven of van plan waren
om zwanger te raken binnen 12 maanden, patiënten die hadden deelgenomen aan
andere klinische studies in de laatste 12 maanden of mentale of lichamelijke
aandoeningen hadden die naleving van de aanwijzingen van de arts uitsloten.
Nederlands
Evaluatie en nazorg
Nazorgbezoeken werden uitgevoerd na maand 1, 2, 3, 6 en 12 en jaarlijks hierna tot de
laatst ingeschreven patiënt 12 maanden bereikte.
Gebruik van apparaat
Patiënten die werden toegewezen aan de behandelingsgroep (Cervical-Stim) werden
geïnstrueerd om het apparaat vier uur per dag te dragen gedurende ten minste drie
maanden na de operatie. Chirurgen konden naar eigen oordeel de Cervical-Stim
behandeling tot zes maand verlengen na de operatie.
4
Page 24

Demografische gegevens
De patiënten in deze studie hadden een gemiddelde leeftijd van 46,8 jaar (tussen 24 en
73 jaar). Van de 323 patiënten waren er 148 (45,8%) vrouwen en 175 (54,2%)
mannen. Driehonderdéén (93,2%) waren blanken, terwijl 17 (5,3%) Afro-Amerikanen
en 5 (1,6%) Hispanics waren. Honderdnegenenvijftig (49,2%) waren niet-rokers en
164 (50,8%) rokers.
Baseline demografische karakteristieken
ariabelen
V
Aantal patiënten
N = 323)
(
Controle
n = 160)
(
Cervical-Stim
n = 163)
(
-waarde
P
1
Leeftijd (jaar)
Gemiddelde
ereik
B
SD
Geslacht
Vrouw
Man
Ras
Blank
Afro-Amerikaan
Hispanic
Aziaat
Andere
Rookstatus
Niet-roker
Roker
1
. P-waarden van vergelijkende testen tussen behandelingsgroepen met gebruik van de t-test van
46,8
4 – 73
2
9,3
148 (45,8%)
175 (54,2%)
301 (93,2%)
17 (5,3%)
5 (1,6%)
0
0
159 (49,2%)
164 (50,8%)
46,7
6 – 72
2
9,2
75 (46,9%)
85 (53,1%)
150 (93,8%)
7 (4,4%)
3 (1,9%)
79 (49,4%)
81 (50,6%)
-
-
46,9
4-73
2
9,4
73 (44,8%)
90 (55,2%)
151 (92,6%)
10 (6,1%)
2 (1,2%)
80 (49,1%)
83 (50,9%)
0,846
0,706
0,703
-
-
0,958
de student voor numerieke variabelen en Pearson x2test voor categorie variabelen.
Gegevensanalyse en resultaten
Het primaire eindpunt voor doeltreffendheid was de verhoging in de frequentie van
geslaagde cervicale fusies met zes maanden na de operatie zoals beoordeeld door
radiografisch bewijs. Secundaire eindpunten waren neurologische functie, VAS
pijnbeoordeling en Neck Disability Index. De veiligheid werd beoordeeld door de
frequentie en ernst van de bijwerkingen.
De fusie werd beoordeeld via röntgenfoto's bij elk bezoek:
Radiografische fusie werd omschreven als >
superieure en inferieure interfaces van het transplantaat tussen nabijgelegen
wervellichamen EN <
4° hoekvorming (beweging) tussen nabijgelegen
gefuseerde wervels op flexie/extensie laterale films EN afwezigheid van
radiolucentie.
50% benige overbrugging op zowel de
Radiografische gefaalde fusie werd omschreven als < 50% benige overbrugging op
ofwel de superieure of inferieure interface van het transplantaat OF> 4°
hoekvorming (beweging) tussen nabijgelegen gefuseerde wervels op
flexie/extensie laterale films OF aanwezigheid van radiolucentie.
5
Page 25

Voor evaluatie van het apparaat werden alle films gescand in een centrale database en
bekeken door twee onafhankelijke, geblindeerde orthopedische chirurgen en een
geblindeerde, onafhankelijke radioloog na het voltooien van de studie. De films werden
bekeken en een score toegekend met behulp van een gebruikelijk protocol. Alle films
werden op elk tijdpunt geëvalueerd voor de hoeveelheid radiolucentie, benige
overbrugging en mate van beweging zoals aangetoond op de flexie/extensiefilms van de
cervicale wervelkolom. Een softwareprogramma werd gebruikt om de beweging te
berekenen. De zodanig verkregen resultaten werden bekeken en gecontroleerd door
de orthopedische chirurgen. De diagnose van de radioloog werd als doorslaggevend
beschouwd in het geval dat er geen akkoord was tussen de twee orthopedische
chirurgen.
Doeltreffendheid - resultaten
Van de 323 patiënten die werden gerandomiseerd en een operatie ondergingen, waren
240 evalueerbaar voor de doeltreffendheidanalyse (Cervical-Stim behandelingsgroep,
n=122; controlegroep, n=118). Patiënten werden als niet evalueerbaar beschouwd
om de volgende redenen: niet bestaande of niet leesbare röntgenfoto's, niet-naleving
door patiënt, schendingen van het protocol (insluitingscriteria), falen van transplantaat,
gebroken interne bevestigingsmiddelen, vroegtijdige beëindiging van studie wegens
lichte bijwerkingen en één poging tot zelfmoord. Het succes of falen van deze
patiënten is niet bekend. Deze niet beschikbare gegevens kunnen het algemene succes
van de studie positief of negatief beïnvloeden. Om de invloed van ontbrekende
gegevens te beoordelen werden gevoeligheidsanalyses uitgevoerd. Deze omvatten
laatste overgebrachte observaties en alle ontbrekende gegevens die als gefaalde fusie
werden toegeschreven. Beide analyses toonden aan dat de resultaten na zes maanden
nog steeds statistisch wezenlijk verschillend waren in het voordeel van de Cervical-Stim
groep. Bovendien werden de baseline demografische gegevens van de evalueerbare
populatie vergeleken met de demografische gegevens van de ontbrekende patiënten.
De resultaten van deze analyse gaven aan dat er geen wezenlijke verschillen waren
tussen de evalueerbare patiënten en de niet-evalueerbare patiënten in 14
studievariabelen waaronder belangrijke demografische gegevens en klinische
parameters.
Nederlands
Primair eindpunt voor doeltreffendheid
Het primaire eindpunt voor doeltreffendheid was bewijs van radiografische fusie
zes maanden na de operatie. Zes maanden na de operatie werden 102 van de 122
evalueerbare patiënten (84%) in de Cervical-Stim behandelingsgroep als gefuseerd
beoordeeld tegenover 81 van de 118 evalueerbare patiënten (69%) in de controlegroep
(p=0,0065).
Vergelijking van radiografische fusieresultaten na zes maanden
Behandelingsgroep
Controle
Cervical-Stim
Aantal patiënten
118
122
Aantal gefuseerde
patiënten
81
102
Fusiepercentage (%)
68,64
83,61
Deze gegevens tonen aan dat, voor patiënten die cervicale fusiechirurgie ondergingen, de
patiënten die bijkomend met de Cervical-Stim werden behandeld een stijging in de
frequentie van radiografische fusie na zes maanden ervoeren wanneer vergeleken met de
controlegroep.
6
Page 26

Een bijkomende analyse werd uitgevoerd om de verschillen tussen de Cervical-Stim
behandelingsgroep en de controlegroep wat betreft demografische karakteristieken
(geslacht, leeftijd, diagnose) en risicostatus (roken, meerniveau) in aanmerking te nemen.
Het algemene fusiepercentage in de Cervical-Stim groep bleef statistisch significant na
aanpassing voor elk van deze variabelen.
Nazorg op lange termijn (12 maanden) toonde geen statistisch verschil tussen de twee
groepen wat betreft fusie. Honderd en zestien van de 125 evalueerbare patiënten
(92,8%) in de Cervical-Stim behandelingsgroep werden als gefuseerd beoordeeld op het
laatste eindpunt op lange termijn, terwijl 104 van de 120 evalueerbare patiënten (86,7%)
in de controlegroep als gefuseerd werden beoordeeld.
Algemene radiografische fusieresultaten na 12 maanden
Behandelingsgroep
Controle
Cervical-Stim
NB: Het verschil in de succespercentages op lange termijn tussen behandelingsgroepen is niet
statistisch significant volgens de Pearson x
2
= 2,5136, p = 0,1129).
(x
Aantal patiënten
120
125
Aantal gefuseerde
patiënten
104
116
2
test met de beschikbare testgrootte
Fusiepercentage (%)
86,67
92,80
Secundaire eindpunten voor doeltreffendheid
Secundaire eindpunten evalueerden wijzigingen in klinische symptomen. Een “klinisch
succes” met betrekking tot de symptomen werd omschreven als geen verslechtering
van de neurologische functie, een verbetering in VAS pijnbeoordeling en geen
verslechtering in Neck Disability Index. Een “klinische mislukking” met betrekking tot
de symptomen werd omschreven als een mislukking voor één van deze criteria. Er was
geen statistisch wezenlijk verschil tussen de twee groepen wat betreft het percentage
patiënten dat als een “klinisch succes” werd beschouwd na zes maanden (p= 0,8456) of
na 12 maanden (p=0,1129).
7
Page 27

Veiligheid
De in deze studie geobserveerde bijwerkingen zijn weergegeven in de tabel met
bijwerkingen in het deel Informatie in voorschrift. Na zes maanden is het aantal
patiënten dat één of meer bijwerkingen ervoer gelijk in de twee groepen. Een totaal
van 14 ernstige gebeurtenissen werd gemeld bij 13 patiënten; negen van de patiënten
waren in de Cervical-Stim behandelingsgroep en vijf patiënten in de controlegroep.
Deze gebeurtenissen waren ervaringen zoals ergere pijn, kortademigheid, duizeligheid,
niet verwant trauma en letsel, niet verwant overlijden, chirurgische complicatie en
nabijgelegen pathologie. Voor de negen patiënten in de Cervical-Stim
behandelingsgroep waren alle ernstige bijwerkingen naar het oordeel van de
onderzoekers beslist of waarschijnlijk niet gerelateerd aan het apparaat.
Veiligheidsgegevens verkregen tussen het bezoek na zes maanden en het laatste contact
met elke patiënt geven aan dat 57 bijwerkingen werden ervaren door totaal 51 patiënten
in beide groepen. Het aantal patiënten dat één of meer bijwerkingen ervoer is gelijk in
de twee groepen. Geen van de bijwerkingen die werden gemeld tussen het bezoek na
zes maanden en het laatste contact waren ernstig en zijn gelijk aan diegene gemeld na
zes maanden.
Gebruiksduur van het apparaat
De Cervical-Stim kan maximaal 270 opeenvolgende (dagelijkse) behandelingen verschaffen
van elk vier uur. De totale duur van de behandeling wordt bepaald door de arts op basis
van de patiënt en de voortgang tot fusie.
Behandelingstijd
De Cervical-Stim moet dagelijks vier uur worden gedragen. Aan het einde van de
dagelijkse behandeling wordt het apparaat automatisch uitgeschakeld. Het apparaat kan
op elk moment worden uitgeschakeld door eenvoudig te drukken op de Aan/uit-knop
op het bedieningspaneel.
Nederlands
De Cervical-Stim kan op elk willekeurig moment van de dag dat het meest geschikt is
voor de patiënt worden gebruikt. Het apparaat is licht en regelbaar. En aangezien de
Cervical-Stim draagbaar is, kan men behandeling krijgen terwijl men zit, wandelt, rust,
slaapt, etc. Elke patiënt is echter uniek en het algemene activiteitsniveau dient
gebaseerd te zijn op de instructies van de arts.
8
Page 28
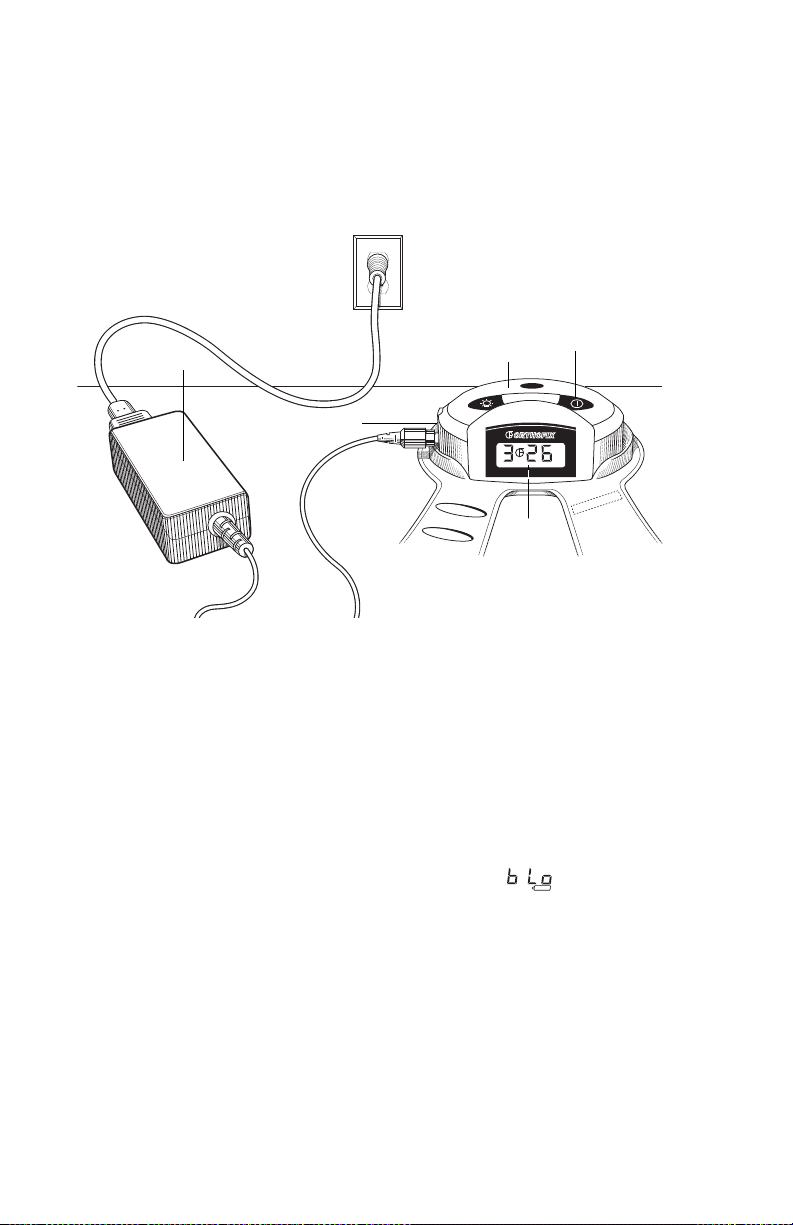
Laden/opnieuw laden van de batterij
De Cervical-Stim wordt gevoed door een oplaadbare lithium-ionbatterij. Er is is bij het
apparaat een laadeenheid geleverd. Gebruik uitsluitend de Orthofix-lader om de batterij
op te laden.
Opmerking: De Cervical-Stim batterij moet voor het eerste gebruik worden opgeladen.
Laadeenheid
Regeleenheid
Aan/uit-knop
Laadapparaat
Poort
LCD
Om de batterij te laden/opnieuw te laden steekt u het busuiteinde van de laadeenheid
in de laadpoort op de regeleenheid. Steek de stekker van het netsnoer stevig in de
laadeenheid. Steek de stekker van de lader in een standaard wandcontactdoos. Voor
een volledig lege batterij duurt het ongeveer 12 uur voordat deze volledig geladen is.
De Cervical-Stim batterij kan op elk willekeurig moment dat het apparaat niet wordt
gebruikt worden geladen. Het wordt sterk aanbevolen om het apparaat op te laden na
het voltooien van de dagelijkse behandeling.
Opmerking: de Cervical-Stim levert geen behandeling tijdens het laden.
Wanneer het apparaat aan staat, toont de Cervical-Stim LCD een symbool met het
batterijvermogen. Een knipperende batterij, het symbool en een hoorbare
pieptoon geven een bijna lege batterij aan die moet worden opgeladen. Zie “Visuele en
geluidsindicators” voor meer informatie.
9
Page 29
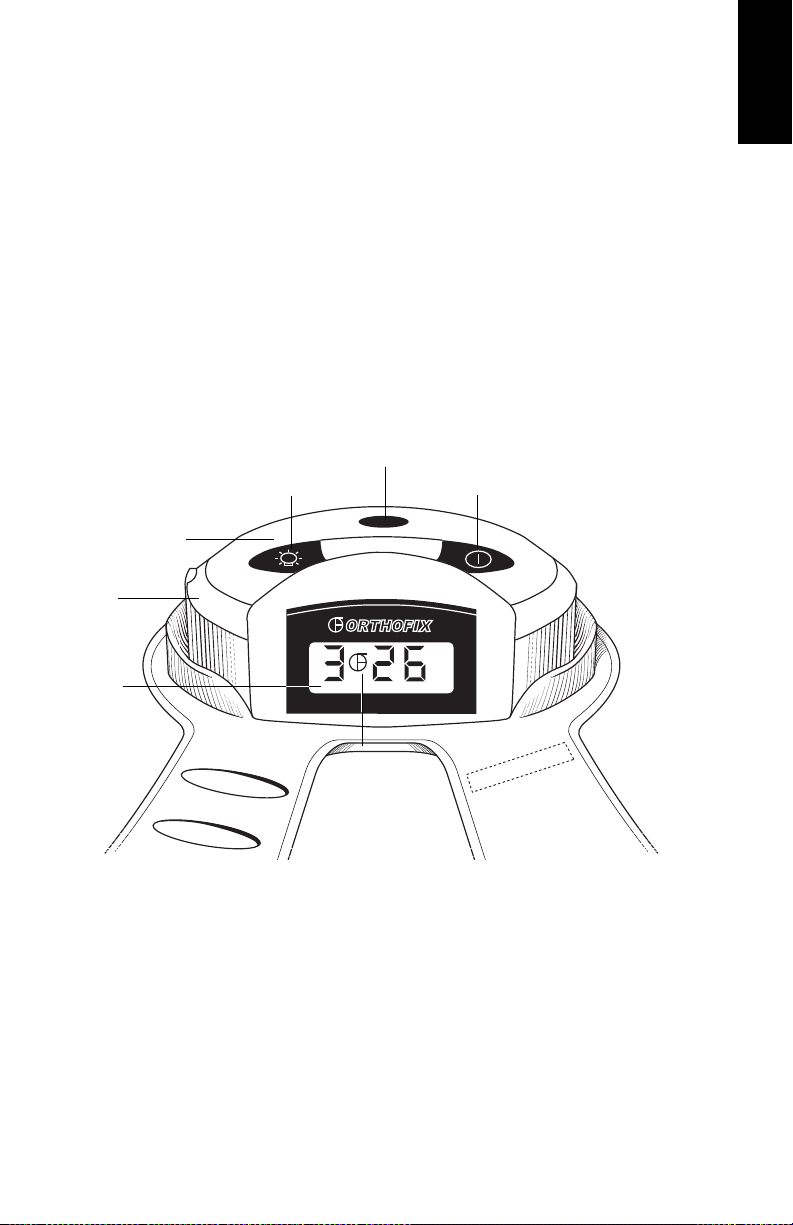
Gebruik van het apparaat
In/uitschakelen van het apparaat
De Cervical-Stim wordt in- en uitgeschakeld door de aan/uit-knop in te drukken op de
regeleenheid van het apparaat. Wanneer het apparaat aan staat, wordt kort een reeks
statusberichten weergegeven. De LCD hoort vervolgens de resterende behandelingstijd
en een knipperend Orthofix-logo weer te geven. Het knipperende logo geeft aan dat
het apparaat aan staat en normaal werkt. (Als u dit niet op het scherm ziet, dient u
contact op te nemen met uw leverancier.) Er bevindt zich een achterverlichtingsknop op
de regeleenheid. Bij weinig licht drukt u op de achterverlichtingsknop voor verlichting
van het LCD.
Om de behandeling te stoppen voor het eind van de dagelijkse behandelingssessie drukt
u eenvoudig op de aan/uit-knop. Om de behandeling te hervatten drukt u opnieuw op
de aan/uit-knop. Het LCD geeft de resterende behandelingstijd weer.
Opmerking: de behandelingssessies moeten langer dan 60 minuten duren voor een
goede werking van de aftelfunctie.
IR-poort
Nederlands
Achterverlichtingsknop
Regeleenheid
Laadapparaat
Poort
LCD
Knipperend
Orthofix-logo
Aan/uit-knop
Timing van behandelingssessies
De Cervical-Stim berekent automatisch elke behandelingssessie. De timing begint
wanneer het apparaat wordt ingeschakeld met de aan/uit-knop op de regeleenheid. Het
LCD toont hoe de resterende tijd in de behandelingssessie wordt afgeteld. Aan het
einde van de dagelijkse behandeling wordt het apparaat automatisch uitgeschakeld. Om
de behandeling te stoppen voor het eind van een behandelingssessie drukt u eenvoudig
op de aan/uit-knop. Om de behandeling te hervatten drukt u opnieuw op de aan/uitknop. Het LCD geeft de resterende behandelingstijd weer.
Opmerking: de behandelingssessies moeten langer dan 60 minuten duren voor een
goede werking van de aftelfunctie.
10
Page 30

Het apparaat dragen
De Cervical-Stim is bedoeld voor de cervicale wervelkolom en kan met of zonder een
beugel worden gedragen. Voor meer comfort moet men kleding dragen tussen de huid
en de Cervical-Stim.
Om de Cervical-Stim te gebruiken schuift u eenvoudig het apparaat over uw hoofd
zodat het comfortabel tegen de hals en schouders rust (zie afbeelding hieronder). Of
het apparaat kan worden geopend bij de gesp en tegen de hals en schouders worden
geplaatst. Maak de gesp vast zoals een autogordel om de eenheid vast te zetten.
De Cervical-Stim wordt geleverd met een comfortkraag voor een betere pasvorm en
betere ondersteuning. De comfortkraag kan in de binnenkraag van het hulpmiddel
worden ingebracht of daaruit worden verwijderd met behulp van de klittenbandflappen
die zich rechtstreeks aan het materiaal vasthechten
Velcro
Gesp
Gewricht
11
Page 31

Onderhoud en reiniging
De Cervical-Stim is een geavanceerd elektronisch apparaat en moet met zorg worden
behandeld. Indien u de Cervical-Stim laat vallen of anderszins verkeerd behandelt, kan
dit schade aan het apparaat veroorzaken.
Stel de Cervical-Stim NIET gedurende lange periodes bloot aan direct
zonlicht. De regeleenheid van het apparaat kan beschadigd worden.
Stel de Cervical-Stim NIET bloot aan overmatige hitte. In warme klimaten
kan de temperatuur in een auto of koffer boven de 71ºC stijgen.
Overmatige hitte kan de regeleenheid van het apparaat beschadigen.
Stel de Cervical-Stim NIET bloot aan overmatig vocht.
Werp de Cervical-Stim NIET weg in een verbrandingsoven.
Gebruik GEEN oplosmiddelen om de Cervical-Stim te reinigen.
Reinig het apparaat door het af te nemen met een zachte, vochtige doek.
Reizen
Patiënten dienen ervan op de hoogte te zijn dat zij de Cervical-Stim het best inchecken
bij de bagage wanneer zij per vliegtuig reizen. Als het apparaat aan boord van het
vliegtuig wordt genomen, mag het niet worden gedragen wanneer men door de
screeningapparaten voor passagiers gaat. De Cervical-Stim kan hierdoor worden
beschadigd. De handleiding van de Cervical-Stim moet worden meegenomen om snel
en eenvoudig het apparaat te identificeren voor veiligheidspersoneel.
Nederlands
Opslag
De Cervical-Stim moet worden opgeborgen bij een temperatuur van -10º C tot 45º C
De bedrijfstemperatuur van de Cervical-Stim moet zich tussen +5º C en 40º C bevinden
Relatieve vochtigheidsgraad: maximaal 95%, niet-condenserend
Afvoer
De Cervical-Stim bevat lithiumbatterijen; deze mogen niet worden afgevoerd in een
verbrandingsoven. Voer het apparaat op de gepaste wijze af.
12
Page 32

Visuele en geluidsindicators
Het LCD en geluidsalarmen zijn bedoeld om nuttige informatie aan de gebruiker te
verschaffen. Het schema hieronder toont de verschillende schermen en alarmen en
hun betekenis.
Visuele en geluidsindicators op het Cervical-Stim LCD
Symbool/Alarm Beschrijving Betekenis
alle LCD-symbolen zichtbaar en
constant geluidsalarm gedurende
ongeveer 5 seconden
aftelfunctie toont resterende
behandelingstijd
(uren en minuten)
Orthofix-logo knippert
aftelfunctie geeft drie streepjes
weer geluidsalarm (5 pieptonen)
gelijkblijvend symbool gedurende
ongeveer 5 seconden
symbolen knipperen/geluidsalarm
(ongeveer 1 pieptoon per
seconde)
gelijkblijvend symbool geeft
laadpercentage bij benadering aan
symbool dat herhaaldelijk wordt
gevuld geeft laadmodus aan
constant geluidsalarm
zelftest bij opstarten
normale behandeling
in uitvoering
geen resterende
behandelingstijd
behandeling voltooid/
uitschakelen
batterij bijna leeg —
opladen vereist
batterijstatus:
resterend vermogen
of laadmodus
apparaat vergrendeld
— bel voor service
weergave van een E code
(foutcode)
(bijv., E01, E02 . . .)
13
foutbericht — bel
voor service
Page 33

Classificatie apparatuur en beschrijving van symbolen op het apparaat
Symbool Betekenis
Attentie - Raadpleeg gebruiksaanwijzing
Toegepast onderdeel type BF
Aan/uit
Achterverlichtingsknop
Opslagtemperatuurbereik
Jaar van productie voor actief apparaat
Laadpoort
Classificatie van de apparatuur
• Intern gevoede apparatuur
• Toegepast onderdeel type BF
• IEC 529 behuizing klasse: IPXO
• Apparatuur niet geschikt voor gebruik in de aanwezigheid van een ontvlambaar
narcotisch mengsel met lucht of lachgas
• Bedrijfsmodus: intermitterend bedrijf
Nederlands
Het gebruik van andere accessoires dan de vermelde kan resulteren in verhoogde
emissie of verlaagde immuniteit van het apparaat.
De batterijlader is geleverd met een driedraads toestel maar wordt als dubbel
geïsoleerd beschouwd met Klasse II constructie over het hele apparaat.
Volg de instructies van de fabrikant voor een veilig gebruik van het product.
Een andere aanwending van het product kan leiden tot schadelijke effecten en/of de
garantie ongeldig maken.
Opmerking: Inspecteer het apparaat voor elk gebruik op slijtage of achteruitgang.
Niet gebruiken als het apparaat niet in geschikte conditie lijkt te zijn.
Service
Als u vragen hebt over het apparaat of hulp vereist, kunt u contact opnemen met uw
plaatselijk distributeur of een bezoek brengen aan onze website www.orthofix.com
voor details. Er zijn geen onderdelen die door de gebruiker kunnen worden
gerepareerd. Stel de fabrikant of plaatselijk distributeur op de hoogte wanneer u
service nodig hebt.
14
Page 34

Naleving van normen
Het Orthofix Cervical-Stim cervicaal fusiesysteem voldoet aan de volgende
wereldwijde normen:
UL 60601 - Medische elektrische apparatuur, algemene veiligheidsvereisten;
(waaronder elektromagnetische compatibiliteit en interferentie)
IEC 60601 - Medische elektrische apparatuur, veiligheidsvereisten voor medische
elektrische systemen (waaronder elektromagnetische compatibiliteit
en interferentie)
Garantie-informatie
Orthofix Inc. garandeert dat de Cervical-Stim®botgroeistimulator vrij is van defecten in
materiaal en vakmanschap gedurende één jaar na de datum van eerste gebruik.
Orthofix Inc. zal alle defecte onderdelen vervangen mits aan alle voorwaarden van deze
beperkte garantie zijn voldaan.
Deze beperkte garantie geldt alleen voor het product bij normaal gebruik en dekt geen
schade of defecten veroorzaakt door ongelukken, misbruik, verkeerd gebruik, brand,
overstroming en force majeure of door wijziging, sabotage, reparatie of gepoogde
reparatie door iemand anders dan Orthofix Inc. Deze garantie geldt alleen voor de
patiënt voor wie het product is voorgeschreven en kan niet worden afgestaan of
overgedragen. Defecte producten gedekt door deze beperkte garantie moeten worden
geretourneerd aan Orthofix Inc. Attention: Orthofix Returns. U moet uw plaatselijk
distributeur bellen om het autorisatienummer voor terugsturen (RA) en adres te
verkrijgen voordat u het product retourneert.
Behalve zoals specifiek vereist door de toepasselijke wet vervangt de voorafgaande
garantie alle andere garanties, expliciet of impliciet en Orthofix Inc. wijst specifiek alle
garanties van verkoopbaarheid of geschiktheid voor en bepaald doel af. In geen geval is
Orthofix Inc., haar erkende vertegenwoordiger, gelieerde ondernemingen of
dochterondernemingen aansprakelijk voor speciale, incidentele of gevolgschade. Het
enige verhaal ten opzichte van een defect product is beperkt tot vervanging.
Deze beperkte garantie mag niet worden verlengd of gewijzigd behalve schriftelijk door
Orthofix Inc. Verkopers, vertegenwoordigers, distributeurs of artsen zijn niet
gemachtigd om een verlenging of wijziging van de voorwaarden van deze beperkte
garantie uit te voeren of hiermee akkoord te gaan.
Neem contact op met uw plaatselijk distributeur voor nadere informatie en/of hulp voor
het apparaat.
15
Page 35

MA N U E L D ´U T I L IS AT I O N
ÉDITION
INTERNATIONALE
Page 36

Table des matières
Français
Manuel d´utilisation Cervical-Stim
Description du dispositif ................................................................................................ 1
Comment Orthofix envisage la guérison ........................................................................1
Stimulation par CEMP ................................................................................................... 2
Information concernant la prescription ..........................................................................2
Résumé des données cliniques ..................................................................................... 3
Analyse des données et résultats ................................................................................... 5
Résultats en matière d´efficacité ................................................................................... 6
Sécurité .......................................................................................................................... 8
Durée de vie du dispositif .............................................................................................. 8
Instructions pour le traitement ...................................................................................... 8
Charge/recharge de la batterie ..................................................................................... 9
Fonctionnement du dispositif ....................................................................................... 10
Port du dispositif .......................................................................................................... 11
Entretien et nettoyage ................................................................................................. 12
Déplacements .............................................................................................................. 12
Rangement ................................................................................................................... 12
Mise au rebut ............................................................................................................... 12
Indicateurs visuels et sonores ...................................................................................... 13
Classement du matériel et description des symboles du dispositif ............................. 14
Réparations .................................................................................................................. 14
Conformité aux normes .............................................................................................. 15
Informations sur la garantie ......................................................................................... 15
®
Pour de plus amples renseignements concernant Orthofix, veuillez consulter notre site
Internet à www.orthofix.com.
Contenu de l´emballage :
1- Système de fusion cervicale Cervical-Stim
1- documentation
1- chargeur, modèle n° 270215
1- Col de Confort
CE DISPOSITIF N´EST PAS STÉRILE.
IL N´EST PAS NÉCESSAIRE DE LE STÉRILISER.
®
0086
Page 37

Description du dispositif
Le système de fusion Cervical-Stim®est un dispositif externe à champs
électromagnétiques pulsés (CEMP) de faible intensité conçu en tenant compte du confort
du patient et de la facilité d´utilisation. Il s´agit d´un dispositif en une seule pièce, léger,
souple et portatif permettant la liberté de mouvements pendant le traitement. Un
affichage à cristaux liquides (LCD) et une alarme sonore assurent un retour d´information
pendant le traitement, notamment l´état opérationnel, la durée de traitement restante, la
charge des piles, etc. Pour de plus amples renseignements, consulter la section
« Indicateurs visuels et sonores ».
Système de fusion cervicale Cervical-Stim - Modèle n° 2505CE
Transducteur
Boucle
Joint
Unité de commande
Tableau de commande
Le dispositif Cervical-Stim se compose d´une unité de commande et d´un transducteur
de traitement. L´unité de commande contient un microprocesseur générant le signal
électrique du Cervical-Stim. Ce signal est converti par le transducteur de traitement en
un champ magnétique de faible intensité mais extrêmement uniforme. Lorsque le
dispositif est centré sur la zone de traitement, le signal CEMP thérapeutique du
dispositif est envoyé directement au site de fusion.
Le dispositif est alimenté par une batterie de piles au lithium-ion rechargeables.
L´affichage LCD et l´alarme sonore avertissent le patient lorsque la batterie est faible
et doit être rechargée. Pour de plus amples renseignements, consulter la section
« Charge/recharge de la batterie ». Pour assurer un bon fonctionnement, le dispositif
surveille en permanence la tension de la batterie et le signal électrique. Si à un moment
quelconque du traitement le dispositif s´arrête de fonctionner correctement, un
symbole ou un code d´erreur approprié apparaît dans l´affichage LCD. Pour de plus
amples renseignements, consulter la section « Indicateurs visuels et sonores ».
Comment Orthofix envisage la guérison
Orthofix est un leader mondial reconnu dans le domaine de la technologie
de stimulation de la croissance des os par CEMP (champ électromagnétique pulsé).
Orthofix a développé le premier stimulateur par CEMP portable non invasif pour la
fusion cervicale en lançant le système de fusion cervicale Cervical-Stim en 2005.
Orthofix continue perfectionner et à améliorer les processus de génération de champs
électromagnétiques pulsés destinés à consolider les spondylodèses du rachis et à
récupérer les spondylodèses non réussies. Orthofix produit une gamme complète
1
Page 38

de dispositifs médicaux de pointe pour les spécialités orthopédiques et
neurochirurgicales qui améliorent le processus de guérison naturelle du corps, stimulent
la fonction naturelle, protègent, restaurent et guérissent. Merci d´inclure Orthofix dans
votre processus de guérison. Pour de plus amples renseignements concernant Orthofix,
veuillez consulter notre site Internet à www.orthofix.com.
Stimulation par CEMP
La stimulation de la croissance osseuse par champs électromagnétiques pulsés (CEMP)
est un traitement sans danger, non chirurgical, prescrit par un médecin pour guérir les
fractures sans consolidation et encourager une spondylodèse. On a utilisé des courants
électriques pour guérir les os depuis le milieu du XIXe siècle. Cependant, ce n´est que
vers 1950 que des chercheurs ont fait une importante découverte. Lorsqu´un os
humain est courbé ou fracturé, il génère un champ électrique. Ce faible champ
électrique active le mécanisme de réparation du corps, ce qui, à son tour, stimule la
guérison de l´os.
Les stimulateurs de croissance osseuse par CEMP d´Orthofix produisent un champ
électromagnétique pulsé, uniforme et faible semblable au champ électrique produit par
le corps. L´application directe de CEMP au site de la fusion ou de la fracture active et
amplifie le processus naturel de guérison du corps pour améliorer la fusion osseuse.
Information concernant la prescription
Indication
Le Cervical-Stim est un stimulateur de croissance osseuse par champ électromagnétique
pulsé non invasif indiqué comme traitement adjuvant des interventions chirurgicales
pour la fusion cervicale chez les patients à haut risque de non-fusion ou une
récupération non-opératoire de fusions de la colonne cervicale ayant échoué.
Français
Contre-indications
Il n´existe aucune contre-indication connue à l´utilisation du Cervical-Stim.
Mises en garde
• Le Cervical-Stim peut interférer avec le fonctionnement d´un stimulateur cardiaque
ou d´un défibrillateur. Il est recommandé de consulter le cardiologue traitant.
• Le Cervical-Stim doit être retiré avant d´effectuer toute procédure d´imagerie (par
ex. tomodensitogramme, IRM, etc.).
Précautions
• Le Cervical-Stim ne doit pas être utilisé lorsque des conditions mentales ou
physiques pourraient mettre en doute l´adhésion aux prescriptions médicales et aux
instructions sur l´utilisation du dispositif.
• Le Cervical-Stim n´a pas été évalué pour le traitement des patients atteints des
affections suivantes : traumatisme du rachis affectant les os ou les ligaments,
spondylite, maladie de Paget, ostéoporose modérée à grave, cancer métastatique,
néphropathie, polyarthrite rhumatoïde, diabète non stabilisé, patients sujets aux
migraines vasculaires, attaques, épilepsie, troubles de la thyroïde ou maladies
neurologiques.
• Des études tératologiques effectuées avec ce dispositif sur des animaux n´ont pas
montré d´effets indésirables. Toutefois, la sécurité de ce dispositif utilisé sur des
patientes enceintes ou qui allaitent n´a pas été établie.
2
Page 39

Événements indésirables :
Événements indésirables signalés après 6 mois au sein du groupe de traitement
roupe témoin (n=160)
Événements indésirables :
Douleur accrue au niveau du cou
ouleurs au niveau de l´épaule/du bras
D
Nouvelle lésion de la colonne cervicale
Pathologie de niveau proximal
omplications chirurgicales
C
Lombalgie/pathologie lombaire
Traumatisme/lésions (non cervicales)
ngourdissements/fourmillements
E
Maux de tête/migraines
ouleurs non spécifiques/sans rapport
D
Nausées
Étourdissements/vertiges
Urticaire/décoloration
Rythme cardiaque rapide/irrégulier
Manque de souffle
Bourdonnement dans les oreilles
Symptômes neurologiques/apoplexie
Grosseur dans la gorge
Diagnostic de diabète
Diagnostic d´un cancer du sein
Attaque
Décès, sans rapport
Sensibilité
Vis cassée
Collapsus de la greffe
Syndrome du canal carpien
Sensation d´étouffement
Symptômes cardiaques
Syndrome néphrotique
Tentative de suicide
G
Nbre (%)
´événements
d
0 (14,9)
1
10 (14,9)
10 (14,9)
(4,5)
3
2(3,0)
8(11,9)
(3,0)
2
6(8,9)
2(3,0)
(3,0)
2
0
2(3,0)
0
0
0
0
1(1,5)
0
0
0
0
0
1(1,5)
1(1,5)
1(1,5)
2(3,0)
1(1,5)
1(1,5)
1(1,5)
1(1,5)
Nbre (%)
atients
p
ressentant
l´événement
(5,6)
9
9(5,6)
8(5,0)
(1,9)
3
2(1,3)
8(5,0)
(1,3)
2
6(3,8)
2(1,3)
(1,3)
2
0
2(1,3)
0
0
0
0
1(0,6)
0
0
0
0
0
1(0,6)
1(0,6)
1(0,6)
2(1,3)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1
de
Groupe Cervical-Stim (n=163)
Nbre*(%)
d´événements
16(17,8)
6(17,8)
1
9(10,0)
8(8,8)
(7,7)
7
5(5,5)
5(5,5)
(4,4)
4
4(4,4)
3(3,3)
2(2,2)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
0
0
0
0
0
0
0
0
Nbre (%)
patients
essentant
r
l´événement
15(9,2)
6(9,8)
1
9(5,5)
8(4,9)
5
5(3,1)
4(2,5)
4
4(2,5)
3(1,8)
2(1,2)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
(3,1)
(2,5)
0
0
0
0
0
0
0
0
1
de
TOTAL
1
. % exprimé sous forme du nombre de patients ayant souffert d´un événement/nombre total de patients dans le groupe
2
. Certains patients ont souffert de plusieurs événements indésirables
*
Plusieurs événements indésirables ont été plus souvent observés chez les patients du groupe Cervical-Stim que chez ceux du groupe
témoin. Étant donné les types d´événements, il est peu probable que ces événements indésirables soient associés au traitement.
67
2
47
90
2
58
Résumé des données cliniques
Conception de l´étude
L´étude clinique Cervical-Stim était une étude contrôlée, randomisée, à groupes
parallèles composée de 323 sujets adultes à risque élevé (fumeurs, multi-niveaux ou
les deux et allogreffes) présentant des preuves radiographiques de racines nerveuses
3
Page 40

cervicales compressées et une radiculopathie symptomatique. Le but de l´étude était
d´évaluer la sécurité et l´efficacité du dispositif en tant que complément pour les
patients à haut risque devant subir une fusion cervicale. Tous les sujets avaient subi une
discectomie et une fusion cervicales antérieures en utilisant la technique de
Smith-Robinson avec plaque Atlantis. Les sujets ont été affectés au hasard soit au
groupe témoin (traitement standard, n=160), soit au groupe de traitement (traitement
standard plus le Cervical-Stim, n=163). Le traitement standard était administré à la
discrétion du médecin mais, en général, comprenait un séjour normal à l´hôpital, le
port d´un collier cervical souple, des médicaments appropriés et une physiothérapie.
Les sujets qui répondaient aux critères d´inclusion et d´exclusion ci-après pouvaient
participer à l´étude :
Critères d´inclusion
Homme ou femme adulte, âgé(e) de 18 à 75 ans présentant les preuves radiographiques
d´une ou de plusieurs racines nerveuses cervicales compressées, d´une radiculopathie
symptomatique, des douleurs de niveau 5 ou plus sur l´échelle visuelle analogue (EVA)
et/ou une faiblesse musculaire ainsi qu´une fusion cervicale primaire réalisée en utilisant
la technique de Smith-Robinson avec allogreffe osseuse et une plaque cervicale
antérieure. La procédure de fusion devait être multi-niveaux (>1 niveau de fusion) ou le
sujet devait être un fumeur (un paquet par jour ou plus), ou les deux ; et devait avoir
signé le consentement éclairé.
Critères d´exclusion
Lésions cervicales traumatiques, fusion par approche postérieure ou de révision,
autogreffon ou matériaux de substitution osseux pour la source de la greffe,
antécédents de migraines vasculaires ou sujet à des attaques incontrôlables ou à
l´épilepsie (contrôlée ou non) ou à tout trouble ou lésion neurologique ; système
immunitaire affaibli, conditions régionales (spondylite, maladie de Paget, polyarthrite
rhumatoïde), infection (systémique ou locale) dans les 2 semaines précédant
l´intervention chirurgicale, conditions systémiques (cancer, arythmie cardiaque, trouble
de la thyroïde, diabète non stabilisé, néphropathie/dysfonction rénale, usage chronique
de stéroïdes ou autres conditions pouvant avoir un effet sur le métabolisme osseux),
stimulateurs cardiaques, défibrillateurs, stimulateurs de la colonne dorsale, appareils
acoustiques, implants cochléaires et stimulateurs crâniens, patientes enceintes, qui
allaitaient ou avaient l´intention de devenir enceintes dans les 12 mois, patients ayant
déjà participé à d´autres études au cours des 12 mois précédents ou patients qui
souffraient de troubles physiques ou mentaux qui pouvaient mettre en doute leur
adhésion aux prescriptions du médecin.
Français
Évaluation et suivi
Des visites de suivi ont dû être réalisées aux mois 1, 2, 3, 6 et 12 et tous les ans par la
suite jusqu´à ce que le dernier sujet enregistré parvienne au 12e mois.
Usage du dispositif
Les sujets assignés au groupe de traitement (Cervical-Stim) ont dû porter le dispositif
quatre heures par jour pendant au moins trois mois après l´intervention chirurgicale.
Les chirurgiens pouvaient, à leur discrétion, prolonger le traitement Cervical-Stim
jusqu´à six mois après l´intervention.
4
Page 41

Données démographiques
Les sujets dans cette étude étaient âgés en moyenne de 46,8 ans (de 24 à 73 ans). Sur
les 323 sujets, 148 (45,8 %) étaient des femmes et 175 (54,2 %) étaient des hommes.
Trois cents un (93,2 %) étaient de race blanche, tandis que 17 (5,3 %) étaient AfroAméricains et 5 (1,6 %) étaient hispaniques. Cent cinquante-neuf (49,2 %) étaient non
fumeurs et 164 (50,8 %) étaient fumeurs.
Caractéristiques démographiques de base
ariables
V
Nombre de sujets
(N = 323)
Témoin
(n = 160)
Cervical-Stim
(n = 163)
Valeur-P
1
Âge (années)
Moyenne
Fourchette
Écart type
Sexe
Féminin
Masculin
Race
Blanche
Afro-américaine
Hispanique
Asiatique
Autre
Usage du tabac
Non-fumeur
Fumeur
1
. Valeurs-P des tests de comparaison entre les groupes de traitement utilisant les tests t
46,8
24 – 73
9,3
148 (45,8 %)
175 (54,2 %)
301 (93,2 %)
17 ( 5,3 %)
5 (1,6 %)
0
0
159 (49,2 %)
164 (50,8 %)
46,7
26 – 72
9,2
75 (46,9 %)
85 (53,1 %)
150 (93,8 %)
7 (4,4 %)
3 (1,9 %)
-
-
79 (49,4 %)
81 (50,6 %)
46,9
24-73
9,4
73 (44,8 %)
90 (55,2 %)
151 (92,6 %)
10 (6,1 %)
2 (1,2 %)
80 (49,1 %)
83 (50,9 %)
0,846
0,706
0,703
-
-
0,958
d´étudiants pour les variables numériques et les tests de Pearson x2pour les variables
catégoriques.
Analyses des données et résultats
Le critère d´efficacité primaire était l´augmentation de la fréquence du nombre de
fusions cervicales réussies au bout de six mois suivant l´intervention chirurgicale telle
qu´évaluée par les preuves radiographiques. Les critères d´efficacité secondaires étaient
la fonction neurologique, l´évaluation des douleurs sur l´EVA et l´échelle d´incapacité
cervicale. La sécurité a été évaluée en fonction de la fréquence et la gravité des
événements indésirables.
La fusion a été évaluée à l´aide des radiographies prises lors de chaque visite :
La fusion radiographique a été définie comme étant >
50 % de rapprochement des
os sur les interfaces tant supérieures qu´inférieures entre les corps
vertébraux adjacents ET un angle de <
4 degrés (déplacement) entre les
vertèbres fusionnés adjacents sur les films latéraux de flexion/extension ET
une absence de radiotransparence.
Le manque de fusion radiographique était défini comme étant < 50 % de
rapprochement des os sur l´interface soit supérieure soit inférieure de la
greffe OU un angle > 4 degrés (déplacement) entre les vertèbres fusionnées
adjacentes sur les films latéraux de flexion/extension OU une présence de
radiotransparence.
5
Page 42

Aux fins de l´évaluation de ce dispositif, tous les films ont été scannés dans une base de
données centrale et examinés par deux chirurgiens orthopédiques indépendants, en
aveugle et un radiologue indépendant en aveugle lorsque l´étude entière fut terminée.
Les films ont été visualisés et notés en utilisant un protocole courant. Chaque film, à
chaque moment temporel, a été mesuré pour évaluer la quantité de radiotransparence,
le rapprochement des os et le degré de mobilité tel que démontré sur les films de
flexion/extension de la colonne cervicale. Un logiciel a été utilisé pour calculer la
mobilité. Les résultats ainsi obtenus ont été examinés et vérifiés par le chirurgiens
orthopédiques chargés de leur évaluation. Le diagnostic du radiologue a été considéré
comme définitif en cas de désaccord entre les deux chirurgiens orthopédiques.
Résultats en matière d´efficacité
Sur les 323 sujets randomisés et ayant subi une intervention chirurgicale, 240 étaient
évaluables lors de l´analyse d´efficacité (groupe de traitement Cervical-Stim, n=122 ;
groupe témoin, n=118). Les sujets étaient estimés ne pas être évaluables pour les
raisons suivantes : manque de radiographies ou radiographies non lisibles, manque de
conformité de la part du sujet, violations du protocole (critères d´inclusion), collapsus
de la greffe, matériau interne cassé, exclusions précoces de l´étude suite à des
événements indésirables mineurs, et une tentative de suicide. Le succès ou l´échec de
ces sujets n´est pas connu. Ces données manquantes pourraient avoir un effet négatif
ou positif sur le succès de l´ensemble de l´étude. Afin d´évaluer l´impact des données
manquantes, des analyses de sensibilité ont été réalisées. Ces dernières comprenaient la
dernière observation transmise et toutes les données manquantes saisies comme nonfusion. Ces deux analyses ont montré que les résultats à six mois étaient toujours très
différents d´un point de vue statistique en faveur du groupe Cervical-Stim. En outre, les
données démographiques de base de la population évaluable ont été comparées aux
données démographiques des sujets manquants. Les résultats de cette analyse ont
indiqué qu´il n´y avait aucune différence importante entre les sujets évaluables et les
sujets non évaluables pour 14 variables de l´étude y compris les données
démographiques clés et les paramètres cliniques.
Français
Critère d´efficacité primaire
Le critère d´efficacité primaire était la preuve de la fusion radiographique au point
temporel de six mois après l´intervention chirurgicale. Au point temporel des six
mois, 102 des 122 sujets évaluables (84 %) du groupe Cervical-Stim ont été jugés
comme étant fusionnés par rapport à 81 des 118 sujets évaluables (69 %) du groupe
témoin (p=0,0065).
Comparaison des résultats des fusions radiographiques à six mois
Groupe de traitement
Témoin
Cervical-Stim
Nombre de sujets
118
122
Nombre de sujets
fusionnés
81
102
Ces données ont montré que pour les patients ayant subi une fusion cervicale, les patients
traités en même temps avec le Cervical-Stim ont démontré une augmentation de la
fréquence des fusions radiographiques à six mois par rapport au groupe témoin.
Taux de fusion (%)
68,64
83,61
6
Page 43

Une analyse supplémentaire a été réalisée pour tenir compte des différences entre le
groupe de traitement Cervical-Stim et le groupe témoin en ce qui concerne les
caractéristiques démographiques (sexe, âge, diagnostic) et le niveau des risques
(fumeur, multi-niveaux). Le taux de fusion général dans le groupe de traitement
Cervical-Stim est resté important du point de vue statistique après avoir tenu compte de
chacune de ces variables.
Un suivi à long terme (12 mois) n´a montré aucune différence statistique entre les deux
groupes en ce qui concerne la fusion. Cent seize des 125 sujets évaluables (92,8 %) du
groupe de traitement Cervical-Stim ont été jugés comme étant fusionnés au dernier
résultat final à long terme, tandis que 104 des 120 sujets évaluables (86,7 %) du groupe
témoin ont été jugés comme étant fusionnés.
Résultats d´ensemble de la fusion radiographique à 12 mois
Groupe de traitement
Témoin
Cervical-Stim
Remarque : les différences de taux de succès à long terme entre les groupes de traitement ne sont pas
importantes d´un point de vue statistique selon le test de Pearson x
disponible (x
Les critères d´efficacité secondaires
Les critères secondaires évaluaient les changements dans les symptômes cliniques.
Un « succès clinique », en ce qui concerne les symptômes, a été défini comme aucune
aggravation de la fonction neurologique, une amélioration de l´évaluation de la douleur
sur l´EVA et aucune aggravation sur l´échelle d´incapacité cervicale. Un « échec
clinique » en ce qui concerne les symptômes a été défini comme un échec en raison de
l´un de ces critères. Il n´y avait aucune différence importante du point de vue
statistique entre les deux groupes en ce qui concerne le pourcentage de sujets
considérés comme étant « un succès clinique » à six mois (p= 0,8456) ou à
12 mois (p= 0,1129).
Nombre de sujets
120
125
2
= 2,5136 ; p = 0,1129).
Nombre de sujets
fusionnés
104
116
Taux de fusion (%)
86,67
92,80
2
avec la taille d´échantillon
7
Page 44

Sécurité
Les événements indésirables observés au cours de cette étude sont présentés dans le
tableau des événements indésirables dans la section Informations sur la prescription.
À six mois, le nombre de sujets ayant ressenti un ou plusieurs des événements
indésirables est semblable pour les deux groupes. Un total de 14 événements graves
ont été signalés chez 13 sujets ; neuf des sujets étaient dans le groupe de traitement
Cervical-Stim et cinq sujets étaient dans le groupe témoin. Ces événements
comprenaient des manifestations telles que : douleurs accrues, manque de souffle,
étourdissements, traumatismes et lésions sans rapport, décès sans rapport,
complications chirurgicales et pathologies de niveau adjacent. Pour les neuf sujets du
groupe de traitement par Cervical-Stim, aucun des événements indésirables graves
n´était associé, selon les investigateurs, définitivement ou probablement au dispositif.
Les données sur la sécurité obtenues entre la visite de six mois et le dernier contact avec
chaque sujet indiquent que 57 événements indésirables ont été ressentis par un total de
51 sujets dans les deux groupes. Le nombre de sujets ayant ressenti un ou plusieurs
événements indésirables est similaire dans les deux groupes. Aucun des événements
indésirables signalés entre la visite à six mois et le dernier contact n´était grave et ils
étaient semblables à ceux signalés à six mois.
Durée de vie du dispositif
Le Cervical-Stim peut fournir jusqu´à 270 traitements consécutifs (quotidiens) de quatre
heures chacun. La durée d´ensemble du traitement sera déterminée par le médecin en
fonction du patient et de l´état d´avancement de la fusion.
Durée du traitement
Le Cervical-Stim doit être porté quatre heures par jour. À la fin du traitement
quotidien, le dispositif s´arrête de lui-même. Le dispositif peut être éteint à tout
moment en appuyant tout simplement sur le bouton On/Off (Marche/Arrêt) situé sur le
tableau de commande.
Français
Il peut être utilisé à tout moment de la journée qui convient le mieux au patient. Il
est léger et réglable. En raison de la portabilité du Cervical-Stim, le traitement peut être
administré assis, en marchant, allongé, en dormant, etc. Cependant, chaque patient
étant unique, son niveau d´activité général devrait être basé sur les instructions du
médecin.
8
Page 45

Charge/recharge de la batterie
Le dispositif est alimenté par une batterie de piles au lithium-ion rechargeables.
Un chargeur est fourni avec le dispositif. Utiliser uniquement le chargeur Orthofix pour
charger la batterie.
Remarque : la batterie du Cervical-Stim doit être chargée avant la première utilisation.
Bouton On/Off
Charg
Prise du
chargeur
Pour charger/recharger la batterie, il suffit de brancher l´extrémité à connecteur
cylindrique du chargeur sur la prise de chargeur située sur l´unité de commande.
Brancher correctement le cordon d´alimentation sur le chargeur. Brancher le chargeur
sur toute prise murale de secteur (courant alternatif). Pour être complètement
rechargée, une batterie complètement à plat devra rester en charge pendant environ
12 heures.
Unité de commande
Affichage
(Marche/Arrêt)
LCD
La batterie peut être rechargée à tout moment lorsque le dispositif n´est pas utilisé.
Il est fortement recommandé de recharger le dispositif après avoir terminé le
traitement quotidien.
Remarque : le Cervical-Stim ne dispensera pas de traitement lorsqu´il est en charge.
Lorsque le dispositif est en marche, un symbole indiquant la charge de la batterie
apparaît dans l´affichage LCD. Un contour de batterie clignotant, le symbole
et un bip sonore indiquent que la charge de la batterie est faible et que cette dernière
doit être rechargée. Consulter la section « Indicateurs visuels et sonores » pour de plus
amples renseignements.
9
Page 46

Fonctionnement du dispositif
Mise en marche et arrêt
On met le Cervical-Stim en marche ou à l´arrêt en appuyant sur le bouton On/Off
(Marche/Arrêt) de l´unité de commande. Lorsque le dispositif est en marche, une série
de messages d´état s´affiche momentanément. L´affichage LCD doit alors indiquer la
durée restante du traitement et un logo Orthofix clignotant. Le logo clignotant indique
que le dispositif est en marche et fonctionne normalement. (S´adresser au fournisseur si
ceci n´est pas visible dans l´affichage.) L´unité de commande comporte un bouton pour
le rétro-éclairage. Quand l´éclairage est faible, rétroéclairer l´affichage LCD en
appuyant sur le bouton.
Pour interrompre le traitement avant la fin de la séance quotidienne, il suffit d´appuyer
sur le bouton On/Off (Marche/Arrêt). Pour reprendre le traitement, appuyer de
nouveau sur le bouton On/Off (Marche/Arrêt). Le temps de traitement restant apparaît
alors dans l´affichage LCD.
Remarque : pour que le compte à rebours fonctionne convenablement, les séances de
traitement doivent être d´une durée supérieure à 60 minutes.
Port IR
Français
Bouton de rétro-éclairage
Bouton On/Off (Marche/Arrêt)
Unité de
commande
Prise du
chargeur
Affichage
LCD
Logo
Orthofix
clignotant
Minutage des séances de traitement
Le Cervical-Stim chronomètre automatiquement chaque séance de traitement. Le
chronométrage commence lorsque le dispositif est mis en marche en appuyant sur le
bouton On/Off (Marche/Arrêt) sur l´unité de commande. L´affichage LCD fait
apparaître le compte à rebours de la durée restante de la séance. À la fin du traitement
quotidien, le dispositif s´arrête de lui-même. Pour arrêter le traitement avant la fin
d´une séance, il suffit d´appuyer sur le bouton On/Off (Marche/Arrêt). Pour reprendre
le traitement, appuyer de nouveau sur le bouton On/Off (Marche/Arrêt). Le temps de
traitement restant apparaît alors dans l´affichage LCD.
Remarque : pour que le compte à rebours fonctionne convenablement, les séances de
traitement doivent être d´une durée supérieure à 60 minutes.
10
Page 47

Port de l´appareil
Le Cervical-Stim a été conçu pour la colonne cervicale et peut être porté avec ou sans
accessoire de soutien. Pour plus de confort, un vêtement doit être porté entre la peau
et le Cervical-Stim.
Pour utiliser le Cervical-Stim, il suffit de faire passer le dispositif par-dessus la tête de
manière à ce qu´il vienne se poser confortablement contre le cou et les épaules (voir la
figure ci-dessous). On peut aussi défaire la boucle du dispositif et le placer entre le cou
et les épaules. et refermer la boucle tout comme avec une ceinture de sécurité pour
attacher l´appareil.
Le Cervical-Stim est muni d'un col de confort pour un ajustement et un soutien
améliorés. Le col de confort peut être inséré à l'intérieur du col du dispositif ou en être
retiré à l'aide de rabats en Velcro, qui s'accrochent directement au matériau.
Velcro
Boucle
Joint
11
Page 48

Entretien et nettoyage
Le Cervical-Stim est un dispositif électronique sophistiqué et doit être manipulé avec
soin. Il risque d´être endommagé en cas de chute ou de manipulation abusive.
NE PAS EXPOSER le Cervical-Stim directement au rayons du soleil
pendant une durée prolongée. L´unité de commande de l´appareil
pourrait être endommagé.
NE PAS exposer le Cervical-Stim à une chaleur excessive. Par temps
chaud, la température dans une voiture ou dans un coffre peut dépasser
71 °C. Les excès de chaleur risquent d´endommager l´unité de
commande.
NE PAS exposer le Cervical-Stim à une humidité excessive.
NE PAS jeter le Cervical-Stim dans un incinérateur.
NE PAS utiliser de solvants pour nettoyer le Cervical-Stim.
Le nettoyer en l´essuyant avec un chiffon doux et humide.
Déplacements
Les patients doivent savoir que lorsqu´ils voyagent en avion, il est préférable
d´enregistrer le Cervical-Stim avec les bagages. Si le dispositif est amené à bord, ne pas
le porter lors du passage aux appareils de contrôle de sécurité. Le Cervical-Stim risque
d´être endommagé. Le manuel d´utilisation du Cervical-Stim doit être emporté pour
que le personnel de sécurité puisse identifier le dispositif rapidement et facilement.
Français
Rangement
Le Cervical-Stim doit être rangé entre -10 °C et 45 °C
La plage de température opérationnelle du Cervical-Stim se situe entre +5 °C et 40 °C.
Humidité relative : jusqu´à 95 %, sans condensation
Mise au rebut
Le Cervical-Stim comprend des piles au lithium ; ne pas mettre au rebus dans un
incinérateur. Jeter le dispositif conformément à la réglementation.
12
Page 49

Indicateurs visuels et sonores
L´affichage LCD et les alarmes sonores sont destinés à fournir des renseignements
utiles à l´utilisateur. Le tableau ci-dessous indique les divers affichages et alarmes ainsi
que leur signification.
Indicateurs visuels (LCD) et sonores du Cervical-Stim
Symboles/Alarmes Description Signification
tous les symboles visibles dans
l´affichage LCD et les alarmes
sonores durent pendant environ
5 secondes
la minuterie du compte à rebours
affiche la durée de traitement
restante (heures et minutes)
le logo Orthofix clignote
la minuterie du compte à rebours
affiche trois tirets alarme sonore
(5 bips)
symbole fixe pendant environ
5 secondes
symboles clignotant/alarme sonore
(environ 1 bip par seconde)
le symbole fixe indique
approximativement la charge
restante en %
le symbole de remplissage répété
indique le mode de charge
alarme sonore continue
auto-test pendant la
mise sous tension
traitement normal en
cours
aucune durée restante
pour le traitement
traitement terminé/
hors tension
batterie faible —
recharge nécessaire
état de la batterie :
charge restante ou
mode de charge
dispositif verrouillé —
appeler le service
après vente
affichage de tout code E
(par ex., E01, E02 . . .)
13
message d´erreur —
appeler le service
après vente
Page 50

Classement du matériel et description des symboles du dispositif
Symbole Signification
Attention - Consulter le mode d´emploi
Type BF pour la partie appliquée
On/Off (Marche/Arrière)
Bouton de rétro-éclairage
Températures minimum/maximum pour le rangement
Année de fabrication du dispositif actif
Prise du chargeur
Catégories de matériel
• Matériel alimenté intrinsèquement
• Type BF pour la partie appliquée
• Niveau de protection de l´enceinte normalisée CEI 529 : IPXO
• Matériel non agréé pour une utilisation en présence d´anesthésiques inflammables
mélangés à de l´air ou à de l´oxyde nitreux
• Mode de fonctionnement : intermittent
L´emploi d´accessoires autres que ceux qui sont spécifiés peut aboutir à l´augmentation
des émissions ou à la réduction d´immunité du dispositif.
Le chargeur de la batterie est fourni avec un réceptacle de prise à 3 fils mais est
considéré comme étant totalement de fabrication de Classe II avec double isolation.
Pour la sécurité du traitement, suivre les instructions du fabricant pendant l´emploi du
dispositif.
Utiliser le dispositif de toute autre manière risquerait de produire des effets nocifs et/ou
d´annuler la garantie.
Français
Remarque : inspecter le dispositif avant chaque utilisation pour déceler les signes
d´usure ou de détérioration.
Ne pas utiliser le dispositif s´il ne paraît pas être en bon état.
Réparations
Pour toute question concernant le dispositif ou pour obtenir de l´aide, s´adresser au
distributeur local ou consulter notre site Internet à www.orthofix.com pour plus de
détails. Aucune pièce n´est susceptible d´être réparée par l´utilisateur. Contacter le
fabricant ou le distributeur local pour tous besoins de réparation.
14
Page 51

Conformité aux normes
Le système de fusion cervicale Cervical-Stim d´Orthofix est conforme aux normes
mondiales suivantes :
UL 60601 - Appareils médicaux électriques, exigences générales en matière de
sécurité ; (y compris la compatibilité électromagnétique et les parasites)
IEC 60601 - Appareils médicaux électriques, exigences en matière de sécurité pour les
systèmes médicaux électriques (y compris la compatibilité et les parasites)
Informations sur la garantie
Orthofix Inc. garantit que le Cervical-Stim®est dépourvu de vices de matériaux et de
fabrication pendant un an à compter de la date de première utilisation. Sous réserve
que toutes les modalités de cette garantie limitée sont respectées, Orthofix Inc.
remplacera les composants défectueux.
Cette garantie limitée s´applique au dispositif exclusivement en cas d´usage normal et
ne couvre aucun dommage ou défaut résultant : d´accident, de mauvais usage, d´abus,
d´incendie, d´inondation et de catastrophes naturelles ou de toute modification,
falsification, réparation ou tentative de réparation par quiconque autre que Orthofix Inc.
La présente garantie ne s´applique qu´au patient à qui le dispositif est prescrit et n´est
ni cessible ni transférable. Les dispositifs défectueux couverts par la garantie limitée
doivent être retournés à Orthofix Inc., À l´attention de : Orthofix Returns (Retour de
produits Orthofix). Il est indispensable d´appeler le distributeur local pour obtenir le
numéro et l´adresse de l´autorisation de renvoi (RA) avant de renvoyer le produit.
Sauf dans la mesure spécifiquement exigée par la législation, la garantie ci-dessus tient
lieu de toutes autres garanties, explicites ou implicites et, Orthofix Inc. dénie toute
autre garantie de valeur marchande ou d´adaptation à un but ou à un usage particulier.
Orthofix Inc., son distributeur agréé, ses sociétés affiliées ou ses filiales ne pourront en
aucun cas être jugés responsables de dommages indirects. Le recours exclusif relatif à
un produit défectueux est limité au remplacement du produit.
Cette garantie limitée ne peut être ni étendue ni modifiée sauf par écrit et par Orthofix
Inc. Aucun vendeur, représentant, distributeur ou médecin n´est autorisé à pratiquer
ou à consentir une prolongation ou une modification quelconque des conditions de la
garantie limitée.
Pour tous renseignements complémentaires et/ou une assistance concernant le dispositif,
s´adresser au distributeur local.
15
Page 52

GE B R A U CH S A N LE I T U NG
INTERNATIONALE
AUSGABE
Page 53

Inhaltsverzeichnis
Deutsch
Cervical-Stim
Beschreibung des Gerätes ........................................................................... 1
Der Orthofix-Ansatz zur Heilung ................................................................. 1
PEMF-Stimulation ........................................................................................ 2
Verschreibungsinformationen ....................................................................... 2
Zusammenfassung der klinischen Daten ....................................................... 3
Datenanalyse und Ergebnisse ....................................................................... 5
Effektivitätsergebnisse ..................................................................................6
Sicherheit ................................................................................................... 8
Lebensdauer des Gerätes ............................................................................ 8
Behandlungsanweisung ................................................................................ 8
Aufladen des Akkus ...................................................................................... 9
Bedienung des Gerätes .............................................................................. 10
Tragen des Gerätes .................................................................................... 11
Reinigung und Pflege .................................................................................. 12
Reisen ....................................................................................................... 12
Lagerung ................................................................................................... 12
Entsorgung ................................................................................................ 12
Sicht- und Tonanzeigen .............................................................................. 13
Geräteklasse und Erklärung der Symbole am Gerät ..................................... 14
Kundendienst ............................................................................................ 14
Konformität mit Normen ........................................................................... 15
Garantieinformationen ............................................................................... 15
®
Gebrauchsanleitung
Für weitere Informationen über Orthofix besuchen Sie bitte unsere Website
unter www.orthofix.com.
Packungsinhalt:
1- Cervical-Stim®Halswirbelbehandlungssystem
1- Dokumentation
1- Ladegerät Art-Nr. 270215
1- Comfortkraag
DIESES GERÄT IST NICHT STERIL.
ES IST KEINE STERILISATION NOTWENDIG.
0086
Page 54

Beschreibung des Gerätes
Das Cervical-Stim®Halswirbelbehandlungssystem ist ein externes Gerät mit einem
schwachaktiven pulsierenden elektromagnetischen Feld (PEMF), das für den Komfort
und die Annehmlichkeit des Patienten konzipiert ist. Das einteilige Gerät ist leicht,
flexibel und tragbar, sodass die Bewegungsfreiheit während der Behandlung nicht
eingeschränkt ist. Eine Flüssigkristallanzeige (LCD) und ein Alarmton liefern dem
Patienten während der Behandlung wichtige Informationen, wie Betriebsstatus,
restliche Behandlungszeit, Akku-Ladezustand usw. Genauere Informationen sind im
Abschnitt „Sicht- und Tonanzeigen“ enthalten.
Cervical-Stim Halswirbelbehandlungssystem - Modell 2505CE
Transducer
Riemen-
verschluss
Steuerung
Bedienfeld
Der Cervical-Stim enthält eine Steuerung und einen Transducer für die Behandlung. Die
Steuerung enthält den Mikroprozessor, der das elektrische Cervical-Stim-Signal erzeugt.
Das Signal wird durch den Transducer in ein sehr gleichmäßiges, schwaches Magnetfeld
umgewandelt. Wenn das Gerät in der Mitte des zu behandelnden Bereichs angebracht
ist, wird das therapeutische Cervical-Stim PEMF-Signal direkt an den
Behandlungsbereich abgegeben.
Der Cervical-Stim wird durch einen aufladbaren Lithiumion-Akku mit Strom versorgt.
Bei schwachem Akku wird der Patient durch das LCD und den Alarmton darauf
aufmerksam gemacht, dass der Akku aufgeladen werden muss. Genauere
Informationen sind im Abschnitt „Aufladen des Akku“ enthalten. Die Akkuspannung und
das elektrische Signal werden vom Cervical-Stim kontinuierlich überwacht, um die
einwandfreie Funktion des Gerätes zu gewährleisten. Sollte das Gerät während der
Behandlung nicht richtig funktionieren, wird dies auf dem LCD durch ein
entsprechendes Symbol oder eine Fehlermeldung mitgeteilt. Genauere Informationen
sind im Abschnitt „Sicht- und Tonanzeigen“ enthalten.
Der Orthofix-Ansatz zur Heilung
Orthofix ist ein bekanntes und weltweit führendes Unternehmen im Bereich der
PEMF (pulsierendes elektromagnetisches Feld) Stimulationstechnologie für
Knochenwachstum. Orthofix entwickelte den ersten nicht-invasiven, tragbaren
PEMF-Stimulator für die Halswirbelbehandlung, der als Cervical-Stim
Halswirbelbehandlungssystem 2005 auf den Markt gebracht wurde. Orthofix
definiert und verbessert auch weiterhin die Abgabe von pulsierenden
elektromagnetischen Feldern für die Heilung von Halswirbelsäulenverschmelzungen
und Behandlung von fehlgeschlagenen Halswirbelsäulenverschmelzungen. Orthofix
1
Page 55

stellt eine Reihe modernster orthopädischer und neurochirurgischer medizinischer
Geräte her, die die Selbstheilung des Körpers fördern, den natürlichen
Heilungsprozess anregen, schützen, wiederherstellen und heilen. Wir bedanken
uns, dass auch Sie Orthofix in Ihren Heilungsprozess einbinden. Für weitere
Informationen über Orthofix besuchen Sie bitte unsere Website unter
www.orthofix.com.
PEMF-Stimulation
Die Stimulation von Knochenwachstum durch pulsierende elektromagnetische Felder (PEMF) ist
eine sichere, nicht-chirurgische Behandlung, die von einem Arzt verschrieben wird, um
ungleichmäßige Brüche zu heilen und die Wirbelverschmelzung zu fördern. Elektromagnetische
Ströme werden schon seit Mitte des 19. Jahrhunderts für die Knochenheilung eingesetzt. Doch erst
in den 50er Jahren des letzten Jahrhunderts machten Wissenschaftler eine wichtige Entdeckung.
Wenn menschliche Knochen sich biegen oder brechen, erzeugen sie ein elektrisches Feld. Dieses
schwachaktive elektrische Feld aktiviert den Reparaturmechanismus des eigenen Körpers, der
wiederum die Knochenheilung stimuliert.
Orthofix PEMF Knochenwachstumsstimulatoren erzeugen ein gleichmäßiges,
schwachaktives, pulsierendes elektromagnetisches Feld, das dem vom Körper
erzeugten Feld ähnelt. Die Anwendung von PEMF direkt an der Fusions- oder
Bruchstelle hilft bei der Aktivierung und Verbesserung des natürlichen
Heilungsprozesses des Körpers und beschleunigt die Knochenverschmelzung.
Verordnungsinformationen
Verwendungszweck
Der Cervical-Stim ist ein nicht-invasiver Knochenwachstumsstimulator mit einem
pulsierenden elektromagnetischen Feld, der als Zusatzmaßnahme zu operativen
Halswirbelsäuleneingriffen bei Patienten mit einem hohen Risiko von
Nichtverschmelzung und zur nicht-chirurgischen Behandlung von fehlgeschlagenen
Halswirbelsäulenverschmelzungen indiziert ist.
Deutsch
Gegenanzeigen
Es liegen keine bekannten Gegenanzeigen für den Cervical-Stim vor.
Warnhinweise
• Der Cervical-Stim kann die Funktion eines Herzschrittmachers oder Defibrillators
stören. Es wird eine Beratung mit dem zuständigen Kardiologen empfohlen.
• Der Cervical-Stim sollte vor jedem Bildgebungsverfahren entfernt werden
(z. B. CT-Scan, MRI, etc.).
Vorsichtsmaßnahmen
• Der Cervical-Stim sollte nicht verwendet werden, wenn ein mentaler oder
physischer Zustand vorliegt, der die Einhaltung der ärztlichen oder
Gerätanweisungen ausschließt.
• Der Cervical-Stim wurde nicht für die Behandlung von Patienten mit den
folgenden Beschwerden beurteilt: knöchernes oder ligamentäres
Rückenmarkstrauma, Spondylitis, Paget-Krebs, mittlere bis schwere
Osteoporose, Karzinommetastase, Nierenerkrankung, rheumatoide Arthritis,
unkontrollierter Diabetes mellitus, Patienten mit Neigung zu Migräne, Anfällen,
Epilepsie, Schilddrüsenvorfällen oder neurologischen Erkrankungen.
• Teratologische Tierstudien, die mit diesem Gerät durchgeführt wurden, zeigten keine
unerwünschten Wirkungen bei Tieren. Die Sicherheit dieses Geräts für den Einsatz
bei schwangeren oder stillenden Patientinnen wurde jedoch nicht bewiesen.
2
Page 56

Unerwünschte Ereignisse
Von einer Behandlungsgruppe nach 6 Monaten gemeldete unerwünschte Ereignisse
ontrollgruppe (n=160)
Unerwünschte Ereignisse
Verstärkte Nackenschmerzen
chmerz in Schulter/Arm
S
Erneute Verletzung der Halswirbelsäule
Pathologie angrenzender Stärke
hirurgische Komplikationen
C
LBP/Lumbalpathologie
Trauma/Verletzung (nicht zervikal)
aubheit/Prickeln
T
Kopfschmerzen/Migräne
nspezifischer/unverbundener Schmerz
U
Übelkeit
Schwindel/Vertigo
Hautausschlag/Entfärbung
Schneller/unregelmäßiger Herzschlag
Kurzatmigkeit
Klingeln in Ohren
Neurologische Symptome/Schlaganfall
Knoten im Rachen
Diagnose von Diabetes
Diagnose von Brustkrebs
Anfall
Tod, ohne Verbindung
Empfindlichkeit
Schraube kaputt
Transplantatkollaps
Karpaltunnelsyndrom
Erstickungsgefühl
Herzsymptome
Nephrose
Suizidversuch
K
Zahl (%) der
reignisse
E
0(14,9)
1
10(14,9)
10(14,9)
(4,5)
3
2(3,0)
8(11,9)
(3,0)
2
6(8,9)
2(3,0)
(3,0)
2
0
2(3,0)
0
0
0
0
1(1,5)
0
0
0
0
0
1(1,5)
1(1,5)
1(1,5)
2(3,0)
1(1,5)
1(1,5)
1(1,5)
1(1,5)
Zahl (%)
atienten, die
P
das Ereignis
erlebt haben
(5,6)
9
9(5,6)
8(5,0)
(1,9)
3
2(1,3)
8(5,0)
(1,3)
2
6(3,8)
2(1,3)
(1,3)
2
0
2(1,3)
0
0
0
0
1(0,6)
0
0
0
0
0
1(0,6)
1(0,6)
1(0,6)
2(1,3)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1
der
Cervical-Stim-Gruppe (n=163)
Zahl*(%) der
Ereignisse
16(17,8)
6(17,8)
1
9(10,0)
8(8,8)
(7,7)
7
5(5,5)
5(5,5)
(4,4)
4
4(4,4)
3(3,3)
2(2,2)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
1(1,1)
0
0
0
0
0
0
0
0
1
Zahl (%)
Patienten, die
as Ereignis
d
erlebt haben
15(9,2)
6(9,8)
1
9(5,5)
8(4,9)
(3,1)
5
5(3,1)
4(2,5)
(2,5)
4
4(2,5)
3(1,8)
2(1,2)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
1(0,6)
0
0
0
0
0
0
0
0
der
GESAMT
1
. Zahl der Patienten in %, die das Ereignis erlebt haben/Gesamtzahl von Patienten in der Gruppe
2
. Einige Patienten erlebten mehrere unerwünschte Ereignisse
*
Es gab mehrere unerwünschte Ereignisse, die in der Cervical-Stim-Gruppe häufiger auftraten als in der
67
2
47
90
2
58
Kontrollgruppe. Die Art der Ereignisse macht es unwahrscheinlich, dass diese unerwünschten Ereignisse mit der
Behandlung in Zusammenhang stehen.
Zusammenfassung der klinischen Daten
Studienauslegung
Bei der klinischen Cervical-Stim-Studie handelte es sich um eine kontrollierte,
randomisierte Parallelgruppenstudie mit 323 erwachsenen Versuchspersonen mit
hohem Risiko (Raucher, mehrstufig oder beides und Allotransplantat) mit
radiografischem Nachweis von gestauchten Halsnervenwurzeln und symptomatischer
3
Page 57

Radikulopathie. Der Zweck der Studie lag in der Bewertung der Sicherheit und
Effektivität des PEMF Cervical-Stim-Geräts als Zusatzmaßnahme für Risikopatienten, die
eine Halswirbelverschmelzung erhalten. Bei allen Versuchspersonen wurde eine
Halswirbeldiskektomie und -verschmelzung mithilfe der Smith-Robinson-Technik mit der
Atlantisplatte durchgeführt. Die Versuchspersonen wurden entweder in die
Kontrollgruppe (Standardbehandlung, n=160) oder die Behandlungsgruppe
(Standardbehandlung plus Cervical-Stim, n=163) randomisiert. Die Standardbehandlung
unterlag dem Arzt, umfasste aber normalerweise den normalen Krankenhausaufenthalt,
die Verwendung einer weichen Halskrause, entsprechender Medikation und
Physiotherapie.
Versuchspersonen, die die folgenden Einschluss- und Ausschlusskriterien erfüllten,
waren für die Teilnahme an der Studie geeignet:
Einschlusskriterien
Männlicher oder weiblicher Erwachsener,18-75 Jahre alt mit radiografischem Nachweis
von gestauchtem(n) Halsnervenwurzel(n), symptomatischer Radikulopathie,
Schmerzgrad 5 oder höher auf der Visual-Analog-Skala (VAS) und/oder Myoparese und
einer primären Halswirbelverschmelzung, die mithilfe der Smith-Robinson-Technik mit
Allotransplantatknochen und einer anterioren Halswirbelplatte durchgeführt wurde.
Das Verschmelzungsverfahren muss entweder mehrstufig (>1 Verschmelzungsstufe)
gewesen sein oder die Versuchsperson muss ein Raucher sein (ein Päckchen/Tag oder
mehr) oder beides sein und die Einverständniserklärung muss unterschrieben
worden sein.
Ausschlusskriterien
Traumatische Halswirbelverletzung, posteriore Ansatz- oder Revisionsverschmelzung,
Autotransplantat oder Knochenersatzmaterial als Transplantatquelle, Migräneanfälligkeit
oder Neigung zu unkontrollierten Anfällen oder Epilepsie (kontrolliert oder
unkontrolliert) oder jegliche neurologische Erkrankung oder Verletzung; geschwächtes
Immunsystem, regionale Zustände (Spondylitis, Paget-Krebs, rheumatoide Arthritis),
Infektion (systemisch oder lokal) innerhalb von zwei Wochen vor der Operation,
systemische Zustände (Krebs, Herzrhythmusstörung, Schilddrüsenerkrankung,
unkontrollierter Diabetes mellitus, Nierenerkrankung/-fehlfunktion, chronische
Steroidverwendung oder andere Zustände, die den Knochenmetabolismus
beeinträchtigt haben könnten), Herzschrittmacher, Defibrillatoren,
Hintersäulenstimulatoren, Hörhilfen, kochleare Prothesen und Schädelstimulatoren,
schwangere oder stillende Versuchspersonen bzw. Versuchspersonen, die in den
nächsten 12 Monaten schwanger werden möchten, Versuchspersonen, die innerhalb
der letzten 12 Monate an anderen klinischen Studien teilgenommen haben oder
mentale oder physische Zustände aufweisen, die die Einhaltung der ärztlichen
Anweisungen ausschließen könnten.
Deutsch
Bewertung und Nachsorge
Nachsorgeuntersuchungen wurden in den Monaten 1, 2, 3, 6 und 12 durchgeführt und
danach jährlich, bis die letzte eingeschriebene Versuchsperson 12 Monate erreichte.
Gerätverwendung
Versuchspersonen, die der Behandlungsgruppe (Cervical-Stim) zugewiesen wurden,
wurden angewiesen, das Gerät für mindestens drei Monate nach der Operation täglich
vier Stunden zu tragen. Chirurgen konnten, sofern gewünscht, die Cervical-StimBehandlung auf sechs Monate nach der Operation verlängern.
4
Page 58

Demografische Daten
Die Versuchspersonen in dieser Studie hatten ein Durchschnittsalter von 46,8 Jahren
(zwischen 24 und 73 Jahren). Von den 323 Versuchspersonen waren 148 (45,8 %)
weiblich und 175 (54,2 %) männlich. 301 (93,2 %) waren Weiße, während 17 (5,3 %)
Afroamerikaner und 5 (1,6 %) Hispanier waren. 159 (49,2 %) waren Nichtraucher
und 164 (50,8 %) waren Raucher.
Grundlegende demografische Eigenschaften
Kontrolle
n = 160)
(
46,7
6 – 72
2
9,2
75 (46,9 %)
85 (53,1 %)
150 (93,8 %)
7 (4,4 %)
3 (1,9 %)
79 (49,4 %)
81 (50,6 %)
-
-
SD)
Zahl der
ersuchspersonen
V
(N = 323)
46,8
4 – 73
2
9,3
148 (45,8 %)
175 (54,2 %)
301(93,2 %)
17 (5,3 %)
5 (1,6 %)
0
0
159 (49,2 %)
164 (50,8 %)
ariablen
V
Alter (Jahre)
Mittel
ereich
B
Standardabweichung
Geschlecht
Weiblich
Männlich
Rasse
Weißer
Afroamerikaner
Hispanier
Asiaten
Andere
Raucherstatus
Nichtraucher
Raucher
1
. P-Werte von Vergleichstests zwischen Behandlungsgruppen mithilfe von T-Tests mit Studenten
(
für nummerische Variablen und Pearson x2Test für kategorische Variablen.
Cervical-Stim
n = 163)
(
46,9
4-73
2
9,4
73 (44,8 %)
90 (55,2 %)
151 (92,6 %)
10 (6,1 %)
2 (1,2 %)
80 (49,1 %)
83 (50,9 %)
1
-Wert
P
0,846
0,706
0,703
-
-
0,958
Datenanalyse und Ergebnisse
Der primäre Effektivitätsendpunkt war der Anstieg der Häufigkeit von erfolgreichen
Halswirbelverschmelzungserfolgen innerhalb von sechs Monaten nach der Operation,
die durch radiografische Nachweise bewertet wurden. Sekundäre Endpunkte waren
neurologische Funktion, VAS-Schmerzbewertung und Neck Disability Index. Die
Sicherheit wurde nach der Häufigkeit und Schwere von unerwünschten Ereignissen
bewertet.
Die Verschmelzung wurde durch Radiografien bei jedem Besuch bewertet:
Radiografische Verschmelzung wurde definiert als >
der superioren und inferioren Transplantatgrenzfläche zwischen aneinander
grenzenden vertebralen Körpern UND <
zwischen aneinander grenzenden verschmolzenen Wirbeln auf
Flexion/Extension lateraler Filme UND fehlender Strahlendurchlässigkeit.
Radiografische Nichtverschmelzung wurde definiert als < 50 %
Knochenüberbrückung an entweder der superioren oder inferioren
Transplantatgrenzfläche ODER > 4° Abknicken (Beweglichkeit) zwischen
aneinander grenzenden verschmolzenen Wirbeln auf Flexion/Extension
lateraler Filme ODER vorhandener Strahlendurchlässigkeit.
5
50 % Knochenüberbrückung an
4° Abknicken (Beweglichkeit)
Page 59

Zum Zweck der Gerätebewertung wurden alle Filme in eine zentrale Datenbank
gescannt und von zwei unabhängigen, blinden Unfallchirurgen und einem blinden,
unabhängigen Radiologen nach Abschluss der gesamten Studie überprüft. Die Filme
wurden überprüft und nach einem allgemeinen Protokoll bewertet. Alle Filme wurden
stets auf Strahlendurchlässigkeit, Knochenüberbrückung und Grad der Beweglichkeit
bewertet, die durch die Flexion/Extension auf den Halswirbelfilmen nachgewiesen
wurden. Die Berechnung des Beweglichkeit erfolgte durch ein Softwareprogramm.
Die auf diese Art erhaltenen Ergebnisse wurden überprüft und durch die
überprüfenden Unfallchirurgen verifiziert. Die Diagnose des Radiologen wurde als
ausschlaggebend angesehen, wenn die beiden Unfallchirurgen sich nicht einig waren.
Effektivitätsergebnisse
Von den 323 Versuchspersonen, die randomisiert und operiert wurden, konnten
240 für die Bewertung der Effektivitätsanalyse verwendet werden (Cervical-StimBehandlungsgruppe, n=122; Kontrollgruppe, n=118). Versuchspersonen wurden aus
den folgenden Gründen nicht in die Bewertung einbezogen: nicht vorhandene oder
nicht erkennbare Röntgenaufnahmen, fehlendes Einverständnis der Versuchsperson,
Protokollverletzungen (Einschlusskriterien), Transplantatkollaps, kaputte interne
Metallteile, früher Ausstieg aus der Studie aufgrund von leichten unerwünschten
Vorkommnissen und ein Suizidversuch. Der Erfolg oder Misserfolg dieser
Versuchspersonen ist unbekannt. Diese nicht verfügbaren Daten könnten den
Gesamterfolg der Studie positiv oder negativ beeinflussen. Um den Einfluss dieser
fehlenden Daten einzuschätzen, wurden Sensitivitätsanalysen durchgeführt. Darin
enthalten waren der Vortrag der letzten Beobachtung und die Einordnung der
fehlenden Daten als Nichtverschmelzung. Beide Analysen zeigten, dass die Ergebnisse
nach sechs Monaten statistisch gesehen immer noch signifikant positiver für die
Cervical-Stim-Gruppe waren. Außerdem wurden die grundlegenden demografischen
Daten der beurteilten Population mit den demografischen Daten der fehlenden
Versuchspersonen verglichen. Die Ergebnisse dieser Analyse deuteten an, dass es bei
14 Studienvariablen einschließlich grundlegender Demografiewerte und klinischer
Parameter keine signifikanten Unterschiede zwischen den beurteilten und den nicht
beurteilten Versuchspersonen gab.
Deutsch
Primärer Effektivitätsendpunkt
Der primäre Effektivitätsendpunkt war der Nachweis für die radiografische
Verschmelzung zum 6-Monatszeitpunkt nach der Operation. An diesem 6Monatszeitpunkt wurden 102 der 122 beurteilten Versuchspersonen (84 %) in der
Cervical-Stim-Behandlungsgruppe als verschmolzen bewertet, verglichen mit 81 der
118 beurteilten Versuchspersonen (69 %) in der Kontrollgruppe (p=0,0065).
Vergleich der radiografischen Verschmelzungsergebnisse nach sechs Monaten
Behandlungsgruppe
Kontrolle
Cervical-Stim
Zahl der
Versuchspersonen
118
122
Zahl der
Versuchspersonen mit
Verschmelzung
81
102
Verschmelzungsrate
(%)
68,64
83,61
Diese Daten zeigen, dass es bei Patienten, die nach einem chirurgischen
Halswirbelsäuleneingriff zusätzlich mit Cervical-Stim behandelt wurden, im Vergleich zur
Kontrollgruppe, zu einer größeren Häufigkeit von radiografischen Verschmelzungen nach
sechs Monaten kam.
6
Page 60

Zusätzlich wurden Analysen durchgeführt, um die Unterschiede zwischen der CervicalStim-Behandlungsgruppe und der Kontrollgruppe in Bezug auf demografische Faktoren
(Geschlecht, Alter, Diagnose) und Risikostatus (Raucher, mehrstufig) einzubeziehen. Die
Gesamtverschmelzungsrate in der Cervical-Stim-Gruppe blieb nach Anpassung für jede
dieser Variablen statistisch signifikant.
Die Langzeit-Nachsorge (12 Monate) zeigte keine statistischen Unterschiede zwischen
den beiden Gruppen in Bezug auf die Verschmelzung. 116 der 125 beurteilten
Versuchspersonen (92,8 %) in der Cervical-Stim-Behandlungsgruppe wurden am finalen
Langzeitendpunkt als verschmolzen bewertet, während 104 der 120 beurteilten
Versuchspersonen (86,7 %) in der Kontrollgruppe als verschmolzen bewertet wurden.
Allgemeine radiografische Verschmelzungsergebnisse nach 12 Monaten
Behandlungsgruppe
Kontrolle
Cervical-Stim
Hinweis: Die Unterschiede der Langzeiterfolgsraten zwischen den Behandlungsgruppen sind laut
Pearson x
statistisch nicht signifikant.
2
-Test mit der verfügbaren Stichprobengröße (x2= 2,5136, p = 0,1129)
Zahl der
Versuchspersonen
120
125
Zahl der
Versuchspersonen mit
Verschmelzung
104
116
Verschmelzungsrate (%)
86,67
92,80
Sekundäre Effektivitätsendpunkte
Sekundäre Endpunkte bewerteten Änderungen klinischer Symptome. Ein “klinischer
Erfolg” in Bezug auf Symptome wurde definiert als keine Verschlechterung der
neurologischen Funktion, eine Verbesserung der VAS-Schmerzbewertung und keine
Verschlechterung des Neck Disability Index. Ein “klinischer Misserfolg” in Bezug auf
Symptome wurde definiert als Misserfolg bei einem einzigen dieser Kriterien. Es gab
keinen statistisch signifikanten Unterschied zwischen den beiden Gruppen in Bezug auf
den Prozentsatz der Versuchspersonen, die nach sechs Monaten (p=0,8456) oder nach
12 Monaten (p= 0,1129) als “klinischer Erfolg” angesehen wurden.
7
Page 61

Sicherheit
Die unerwünschten Ereignisse, die in dieser Studie beobachtet wurden, sind in der
Tabelle Unerwünschte Ereignisse im Abschnitt Verschreibungsinformationen
zusammengefasst. Nach sechs Monaten ist die Zahl der Versuchspersonen, die ein oder
mehrere unerwünschte Ereignisse erlebt haben, in beiden Gruppen gleich. Insgesamt
wurden bei 13 Versuchspersonen 14 schwere Ereignisse festgestellt; neun
Versuchspersonen befanden sich in der Cervical-Stim-Behandlungsgruppe und fünf in
der Kontrollgruppe. Diese Ereignisse umfassen verstärkten Schmerz, Kurzatmigkeit,
Schwindel, unverbundenes Trauma oder Verletzung, unverbundener Tod, chirurgische
Komplikationen und Pathologie angrenzender Stärke. Bei den neun Versuchspersonen
in der Cervical-Stim-Behandlungsgruppe standen alle schweren unerwünschten
Ereignisse nach Einschätzung der Versuchsleiter definitiv oder wahrscheinlich nicht in
Zusammenhang mit dem Gerät.
Sicherheitsdaten, die zwischen dem Besuch nach sechs Monaten und dem letzten
Kontakt gesammelt wurden, deuten darauf hin, dass 57 unerwünschte Ereignisse bei
insgesamt 51 Versuchspersonen beider Gruppen auftraten. Die Zahl der
Versuchspersonen, bei denen ein oder mehrere unerwünschte Ereignisse auftraten, ist in
beiden Gruppen gleich groß. Keines der zwischen dem Besuch nach sechs Monaten und
dem letzten Kontakt gemeldeten unerwünschten Ereignisse war schwer und vergleichbar
mit denen, die nach sechs Monaten gemeldet wurden.
Lebensdauer des Gerätes
Der Cervical-Stim ist für maximal 270 aufeinander folgende (tägliche) Behandlungen à vier
Stunden ausgelegt. Die Gesamtlänge der Behandlung wird vom Arzt je nach Patient und
Fortschreiten der Verschmelzung bestimmt.
Deutsch
Behandlungszeit
Der Cervical-Stim sollte täglich vier Stunden getragen werden. Am Ende der täglichen
Behandlungszeit schaltet sich das Gerät automatisch aus. Das Gerät kann jederzeit einund ausgeschaltet werden, indem die Ein/Aus-Taste auf dem Bedienfeld gedrückt wird.
Der Cervical-Stim kann zu jeder beliebigen Tageszeit je nach Vorliebe des Patienten
angewandt werden. Das Gerät ist leicht und justierbar. Aufgrund der tragbaren
Ausführung des Cervical-Stim kann die Behandlung im Sitzen, Gehen, Liegen oder im
Schlaf usw. durchgeführt werden. Die Aktivitäten während der Behandlung sind jedoch
gemäß Anweisung des Arztes einzuschränken.
8
Page 62

Aufladen des Akku
Der Cervical-Stim wird durch einen aufladbaren Lithiumion-Akku mit Strom versorgt.
Im Lieferumfang des Gerätes ist ein Ladegerät enthalten. Zum Aufladen des Akku muss
das Orthofix Ladegerät verwendet werden.
Hinweis: Der Cervical-Stim Akku muss vor der erstmaligen Verwendung aufgeladen werden.
Ladegerät
Steuerung
Ein/Aus-Taste
Ladegerätanschluss
LCD
Zum Aufladen des Akku wird einfach der zylinderförmige Stecker des Ladegerätes in
den Ladegerätanschluss an der Steuerung gesteckt. Das Netzkabel fest in das Ladegerät
stecken. Das Ladegerät wird an eine normale Wandsteckdose angeschlossen. Eine
vollständig entladene Batterie benötigt zum vollständigen Aufladen etwa 12 Stunden.
Der Cervical-Stim Akku kann jederzeit aufgeladen werden, wenn das Gerät nicht
verwendet wird. Es wird empfohlen, das Gerät jeden Tag nach der Behandlung
aufzuladen.
Hinweis: Während des Aufladens ist keine Behandlung mit dem Cervical-Stim möglich.
Bei eingeschaltetem Cervical-Stim wird auf dem LCD ein Symbol für die
Batteriekapazität angezeigt. Wenn ein blinkender Batterieumriss in Verbindung mit dem
Symbol erscheint und ein Alarmton ertönt, muss der Akku aufgeladen werden.
Genauere Informationen sind im Abschnitt „Sicht- und Tonanzeigen“ enthalten.
9
Page 63

Bedienung des Gerätes
Ein- und Ausschalten des Gerätes
Zum Ein- und Ausschalten des Cervical-Stim wird die Ein/Aus-Taste an der Steuerung
des Gerätes gedrückt. Beim Einschalten des Gerätes wird kurz eine Sequenz von
Statusmeldungen eingeblendet. Anschließend wird auf dem LCD die verbleibende
Behandlungszeit und ein blinkendes Orthofix Logo angezeigt. Das blinkende Logo zeigt
an, dass das Gerät eingeschaltet ist und fehlerfrei funktioniert. (Wenn dieses Logo nicht
zu sehen ist, melden Sie das bitte Ihrem Lieferanten.) An der Steuerung befindet sich
eine Lichttaste für die Hintergrundbeleuchtung. Diese Taste kann bei schwacher
Beleuchtung gedrückt werden, um die Hintergrundbeleuchtung des LCD einzuschalten.
Um die Behandlung vor dem Ende der verordneten täglichen Behandlungszeit zu
beenden, drücken Sie die Ein/Aus-Taste. Zum Wiederaufnehmen der Behandlung
drücken Sie die Ein/Aus-Taste erneut. Auf dem LCD wird dann die restliche
Behandlungszeit angezeigt.
Hinweis: Die richtige Funktion der Zeitkontrolle ist nur dann gewährleistet, wenn vor
dem Beenden der Behandlung mindestens 60 Minuten verstrichen sind.
IR-Port
Lichttaste
Steuerung
Ladegerätanschluss
Ein/Aus-Taste
Deutsch
LCD
Blinkendes
Orthofix Logo
Zeitsteuerung der Behandlungssitzungen
Der Cervical-Stim steuert automatisch die Zeit für jede Behandlungssitzung. Die
Zeitsteuerung beginnt, wenn das Gerät über die Ein/Aus-Taste auf der Steuerung
eingeschaltet wird. Auf dem LCD wird die jeweils verbleibende Behandlungszeit
angezeigt. Am Ende der täglichen Behandlungszeit schaltet sich das Gerät automatisch
aus. Um die Behandlung vor Ende der Behandlungssitzung zu beenden, drücken Sie
einfach auf die Ein/Aus-Taste. Zum Wiederaufnehmen der Behandlung drücken Sie die
Ein/Aus-Taste erneut. Auf dem LCD wird dann die restliche Behandlungszeit angezeigt.
Hinweis: Die richtige Funktion der Zeitkontrolle ist nur dann gewährleistet, wenn vor
dem Beenden der Behandlung mindestens 60 Minuten verstrichen sind.
10
Page 64

Tragen des Gerätes
Der Cervical-Stim ist für die Halswirbelsäule vorgesehen und kann mit oder ohne Gurt
getragen werden. Zum besseren Komfort sollte zwischen der Haut und dem CervicalStim Kleidung getragen werden.
Um den Cervical-Stim zu verwenden, ziehen Sie das Gerät einfach über den Kopf, so
dass es bequem auf Nacken und Schulter liegt (siehe Abbildung unten). Das Gerät kann
auch am Verschluss geöffnet und um Nacken und Schulter gelegt werden. Schließen Sie
den Riemen wie einen Sicherheitsgurt, um das Gerät zu befestigen.
De Cervical-Stim wordt geleverd met een comfortkraag voor een betere pasvorm en
betere ondersteuning. De comfortkraag kan in de binnenkraag van het hulpmiddel
worden ingebracht of daaruit worden verwijderd met behulp van de klittenbandflappen
die zich rechtstreeks aan het materiaal vasthechten
Velcro
Schnallverschluss
11
Page 65

Reinigung und Pflege
Der Cervical-Stim ist ein kompliziertes elektronisches Gerät und sollte vorsichtig
behandelt werden. Durch Aufprall oder Misshandlung kann der Cervical-Stim
beschädigt werden.
Den Cervical-Stim NICHT längere Zeit direkter Sonneneinwirkung
aussetzen. Die Steuerung des Geräts kann dadurch beschädigt werden.
Den Cervical-Stim KEINEN übermäßigen Temperaturen aussetzen. In
warmen Klimazonen kann die Temperatur in einem Auto oder Kofferraum
auf über 71 ºC ansteigen. Außergewöhnliche Hitze kann die Steuerung des
Geräts beschädigen.
Den Cervical-Stim KEINER übermäßigen Feuchtigkeit aussetzen.
Bei der Entsorgung darf der Cervical-Stim NICHT verbrannt werden.
KEINE Lösungsmittel zum Reinigen des Cervical-Stim verwenden.
Das Gerät mit einem weichen, feuchten Tuch abwischen.
Reisen
Patienten sollten darauf hingewiesen werden, dass sie bei Flugreisen den Cervical-Stim
am Besten mit im Gepäck aufgeben. Wird das Gerät mit an Bord des Flugzeugs
genommen, sollte es nicht getragen werden, wenn der Patient durch die
Passagierkontrolle geht. Der Cervical-Stim könnte dadurch beschädigt werden. Das
Benutzerhandbuch des Cervical-Stim sollte mitgenommen werden, damit das
Sicherheitspersonal das Gerät schnell und einfach identifizieren kann.
Deutsch
Lagerung
Der Cervical-Stim sollte bei Temperaturen zwischen -10 ºC und 45 ºC gelagert werden.
Der Cervical-Stim sollte bei Temperaturen zwischen +5 ºC und 40 ºC betrieben
werden.
Relative Luftfeuchtigkeit: Bis zu 95 %, nicht-kondensierend
Entsorgung
Der Cervical-Stim enthält Lithiumbatterien, die nicht verbrannt werden dürfen. Das
Gerät vorschriftsmäßig entsorgen.
12
Page 66

Sicht- und Tonanzeigen
Das LCD und die Alarmsignale haben den Zweck, dem Benutzer hilfreiche
Informationen zu vermitteln. Die verschiedenen Anzeigen und Alarme sowie deren
Bedeutung sind in der Tabelle unten aufgeführt.
LCD-Symbole und Tonsignale des Cervical-Stim
Symbol/Alarm Beschreibung Bedeutung
Alle LCD-Symbole leuchten und
der Alarm ertönt kontinuierlich für
ca. 5 Sekunden
Auf dem Zähler wird die
verbleibende Behandlungszeit
angezeigt
(Stunden und Minuten)
Orthofix Logo blinkt
Drei Striche auf dem
Behandlungszeitzähler
Alarmton (5 Piepsignale)
Wird ca. 5 Sekunden lang konstant
eingeblendet
Symbol blinkt/Alarm ertönt
(ungefähr 1 Piepsignal pro
Sekunde)
Konstantes Symbol zeigt den
Ladezustand in % (ungefähr)
Ein sich füllendes Symbol zeigt den
Auflademodus an
Kontinuierlicher Alarmton
Selbsttest beim
Einschalten
Normale Behandlung
wird durchgeführt
Behandlungszeit
vollständig abgelaufen
Behandlung fertig/
automatische Abschaltung
Schwacher Akku —
muss aufgeladen
werden
Akkustatus:
Restkapazität oder
Auflademodus
Gerät gesperrt —
Kundendienst anrufen
Fehlercode-Anzeige
(z.B. E01, E02 . . .)
13
Fehlermeldung —
Kundendienst anrufen
Page 67

Geräteklasse und Erklärung der Symbole am Gerät
Symbol Bedeutung
Achtung - Gebrauchsanweisung beachten
Anwendungsteil vom Typ BF
Ein/Aus
Lichttaste für die Hintergrundbeleuchtung
Lagertemperaturbereich
Herstellungsjahr des aktiven Gerätes
Ladegerätanschluss
Geräteklasse
• Gerät mit Eigenspeisung
• Anwendungsteil des Typs BF.
• Schutzart nach IEC 529: IPXO
• Nicht in Gegenwart von brennbaren Anästhetikagemischen
mit Luft oder Stickstoffoxid verwenden
• Betriebsart: Kurzzeitbetrieb
Deutsch
Bei Verwendung anderer Zubehörteile als der spezifizierten Teile besteht die Gefahr
erhöhter Emissionen oder reduzierter Störfestigkeit dieses Geräts.
Das Batterieladegerät weist einen dreiadrigen Eingang auf, ist aber durchgehend in der
Bauweise der Klasse II doppelt isoliert.
Zum sicheren Gebrauch sollte die Gebrauchsanweisung des Herstellers beachtet
werden.
Bei anderweitiger Verwendung des Gerätes besteht die Gefahr schädlicher
Auswirkungen und/oder Nichtigkeit der Garantie.
Hinweis: Das Gerät sollte vor jeder Verwendung auf Abnutzung oder Verschleiß
überprüft werden.
Bei nicht einwandfreiem Zustand darf das Gerät nicht verwendet werden.
Kundendienst
Bei Fragen zum Produkt oder wenn Sie Unterstützung brauchen, wenden Sie sich bitte
an Ihren Vertriebshändler vor Ort oder besuchen Sie unsere Website unter
www.orthofix.com. Das Gerät enthält keine Bauteile, die vom Benutzer gewartet
werden können. Bei Reparaturbedarf muss der Hersteller oder der Vertriebshändler
vor Ort beauftragt werden.
14
Page 68

Konformität mit Normen
Das Orthofix Cervical-Stim Halswirbelbehandlungssystem erfüllt die folgenden weltweit
gültigen Normen:
UL 60601 - Medical Electrical Equipment, General Requirements for Safety;
(einschließlich Electromagnetic Compatibility and Interference)
IEC 60601 - Medical Electrical Equipment, Safety Requirements for Medical Electrical
Systems (einschließlich Electromagnetic Compatibility and Interference)
Garantieinformationen
Orthofix Inc. gewährleistet für ein Jahr ab der erstmaligen Verwendung, dass der
Cervical-Stim
Verarbeitungsfehlern ist. Unter der Voraussetzung, dass alle Bedingungen der
beschränkten Garantie erfüllt sind, verpflichtet sich Orthofix Inc. zum Ersatz der
defekten Komponenten.
Diese beschränkte Garantie gilt nur für das unter normalen Bedingungen verwendete
Produkt und schließt jegliche Schäden oder Defekte aus, die durch Unfall, Missbrauch,
Misshandlung, Brand, Überschwemmung, Naturgewalt oder durch Veränderungen,
Eingriff, Reparatur oder versuchte Reparatur durch dritte unberechtigte Personen
entstehen. Diese Garantie ist nicht übertragbar und gilt nur für den Patienten, dem das
Produkt verschrieben wurde. Defekte Produkte, die unter dieser beschränkten
Garantie geschützt sind, müssen an Orthofix Inc., zu Händen: Orthofix Returns,
zurückgesandt werden. Sie müssen sich an Ihren Vertriebshändler vor Ort wenden, um
vor der Rücksendung des Produkts die Retourennummer (RA) und Adresse zu erhalten.
Außer wenn vom anwendbaren Recht ausdrücklich gefordert, ersetzt die obige
Garantie alle anderen stillschweigenden und ausdrücklichen Gewährleistungen und
Zusagen. Orthofix Inc. lehnt ausdrücklich alle Zusagen hinsichtlich der Marktgängigkeit
oder Eignung für einen bestimmten Zweck ab. Unter keinen Umständen haften
Orthofix Inc., ihre autorisierten Vertreter, Konzernunternehmen oder
Tochtergesellschaften für Sonder-, Folge- oder Nebenschäden. Der ausschließliche
Rechtsbehelf bei einem defekten Produkt ist dessen Ersatz.
Diese beschränkte Garantie darf nur von Orthofix Inc. in schriftlicher Form verlängert
oder modifiziert werden. Kein Verkäufer, Vertreter, Distributor oder Arzt ist berechtigt,
eine Verlängerung oder Änderung der Bedingungen dieser beschränkten Garantie zu
gewähren.
®
Knochenwachstumsstimulator frei von Material- und
Für weitere Informationen und/oder Geräteunterstützung wenden Sie sich bitte an Ihren
Vertriebshändler vor Ort.
15
Page 69

MA N U A L E D I I ST R U Z IO N I
EDIZIONE
INTERNAZIONALE
Page 70

Indice
Italiano
Manuale di istruzioni Cervical-Stim
®
Descrizione del dispositivo ........................................................................ 1
L’approccio Orthofix alla guarigione ............................................................ 1
Stimolazione PEMF ................................................................................... 2
Informazioni per la prescrizione ................................................................. 2
Sintesi dei dati clinici .................................................................................. 3
Analisi dei dati e risultati ............................................................................ 5
Risultati riguardanti l’efficacia ..................................................................... 6
Sicurezza .................................................................................................. 8
Durata del dispositivo ................................................................................ 8
Istruzioni terapeutiche ............................................................................... 8
Carica/ricarica della batteria ........................................................................ 9
Funzionamento del dispositivo .................................................................. 10
Come indossare il dispositivo ................................................................... 11
Manutenzione e pulizia ............................................................................ 12
In viaggio ................................................................................................. 12
Conservazione ......................................................................................... 12
Smaltimento ............................................................................................ 12
Indicatori visivi e acustici ................................................................................ 13
Classificazione dell’apparecchio e descrizione dei simboli ........................... 14
Assistenza ............................................................................................... 14
Conformità alle norme ............................................................................. 15
Informazioni sulla garanzia ........................................................................ 15
Per maggiori informazioni su Orthofix, vi invitiamo a visitare il nostro sito
Internet: www.orthofix.com.
Contenuto della confezione:
1- Sistema di fusione cervicale Cervical-Stim
®
1- Busta documentazione
1- Caricabatteria n. 270215
1- Collare di comfort
IL DISPOSITIVO È FORNITO NON STERILE.
NON OCCORRE STERILIZZARE.
0086
Page 71

Descrizione del dispositivo
Il sistema di fusione cervicale Cervical-Stim®è un dispositivo esterno per l'applicazione di
campo elettromagnetico ad impulsi (PEMF) di bassa intensità progettato tenendo conto in
primo luogo del comfort e della comodità del paziente. Realizzato in un unico pezzo, il
dispositivo è leggero, flessibile e portatile, dando al paziente libertà di movimento durante
il trattamento. Un display a cristalli liquidi (LCD) e un allarme sonoro forniscono
informazioni importanti durante il trattamento, quali ad esempio lo stato di
funzionamento, il tempo di trattamento rimanente, la capacità della batteria, ecc. Si veda
il capitolo “Indicatori visivi e acustici” per ulteriori informazioni.
Sistema per fusione cervicale Cervical-Stim - Modello 2505CE
Trasduttore
Attacco
della fibbia
Centralina di controllo
Pannello di controllo
Il Cervical-Stim è costituito da una centralina di controllo e un trasduttore di
trattamento. La centralina di controllo contiene un microprocessore che genera il
segnale elettrico del Cervical-Stim. Il segnale viene convertito dal trasduttore di
trattamento in campo magnetico altamente uniforme di bassa intensità. Quando il
dispositivo è collocato al centro della zona da trattare, il segnale PEMF ad azione
terapeutica del Cervical-Stim arriva direttamente alla sede desiderata.
Il dispositivo Cervical-Stim è attivato da una batteria ricaricabile a ioni di litio.
Il display e l'allarme acustico avvertono il paziente quando il livello della batteria è basso
ed è necessario ricaricare. Vedere il capitolo “Carica/ricarica della batteria” per
ulteriori informazioni. Per garantire che il dispositivo funzioni correttamente, il CervicalStim controlla continuamente il voltaggio della batteria e il segnale elettrico. Se in
qualunque momento durante il trattamento il dispositivo dovesse smettere di
funzionare correttamente, sul display comparirà un apposito simbolo o codice di errore.
Vedere il capitolo “Indicatori visivi e acustici" per ulteriori informazioni.
L'approccio Orthofix alla guarigione
La Orthofix è riconosciuta come leader mondiale nel campo della tecnologia di
stimolazione della crescita ossea mediante PEMF (Pulsed Electromagnetic Field: campo
elettromagnetico ad impulsi). Orthofix ha messo a punto il primo stimolatore PEMF non
invasivo portatile per la fusione cervicale lanciando il sistema per fusione cervicale
Cervical-Stim nel 2005. Orthofix continua a perfezionare la tecnologia di emissione di
campi elettromagnetici ad impulsi per la guarigione di fusioni spinali e il recupero di
fusioni spinali con esito negativo. Orthofix produce una gamma completa di dispositivi
1
Page 72

medici all'avanguardia per uso in campo ortopedico e neurochirurgico, progettati per
stimolare il processo di guarigione naturale dell'organismo, simulare la funzione naturale,
proteggere, riparare e risanare. Grazie per aver scelto Orthofix come coadiuvante del
vostro processo di guarigione. Per maggiori informazioni su Orthofix, vi invitiamo a
visitare il nostro sito Internet: www.orthofix.com.
Stimolazione PEMF
La stimolazione della crescita ossea mediante l'emissione di campi elettromagnetici ad
impulsi (PEMF) è una tecnica terapeutica sicura, non chirurgica, da applicare su
prescrizione medica per il risanamento di fratture non saldate e per facilitare la fusione
spinale. L'uso di corrente elettrica per la guarigione di lesioni ossee è noto già dalla metà
dell'800. È stato solo negli anni 50, però, che gli studiosi hanno fatto una scoperta
importante. Quando è soggetto a flessione o frattura, l'osso umano produce un campo
elettrico. Il campo elettrico, di bassa intensità, attiva i meccanismi di riparazione propri
dell'organismo, i quali a loro volta stimolano la guarigione.
Gli stimolatori PEMF per la crescita ossea Orthofix generano un campo
elettromagnetico ad impulsi uniforme, di bassa intensità, simile al campo elettrico
generato dall'organismo. L'applicazione di PEMF direttamente sulla sede della fusione o
della frattura attiva e potenzia il processo naturale di guarigione tendente alla
riunificazione dell'osso.
Informazioni per la prescrizione
Indicazioni
Cervical-Stim è uno stimolatore di crescita ossea a impulsi elettromagnetici indicato
come coadiuvante dell'operazione di fusione cervicale in pazienti ad alto rischio di
mancata fusione e per il recupero non chirurgico della fusione cervicale non riuscita.
Italiano
Controindicazioni
Non si conoscono controindicazioni per il Cervical-Stim.
Avvertenze
• Il Cervical-Stim può interferire con il funzionamento di pacemaker cardiaci o
defibrillatori. Si consiglia di consultare il cardiologo curante.
• Il Cervical-Stim deve essere rimosso prima di esami di imaging (ad es. TAC, risonanza
magnetica ecc.).
Precauzioni
• Il Cervical-Stim non deve essere utilizzato in presenza di condizioni mentali o fisiche
che precludano la capacità del paziente di attenersi alle istruzioni del medico e a
quelle relative al dispositivo.
• Il Cervical-Stim non è stato valutato per il trattamento di pazienti affetti dalle seguenti
patologie: trauma a carico delle ossa o dei legamenti spinali, spondilite, morbo di
Paget, osteoporosi di grado medio o grave, cancro metastatico, patologia renale,
artrite reumatoide, diabete mellito non controllato, pazienti soggetti a emicrania
vascolare, attacchi, epilessia, disfunzioni tiroidee o malattie neurologiche.
• Gli studi teratologici condotti sugli animali con questo dispositivo non hanno
evidenziato effetti indesiderati negli animali. Tuttavia, la sicurezza del dispositivo se
usato da pazienti in gravidanza o allattamento non è stata accertata.
2
Page 73

Eventi avversi
Eventi avversi riferiti dopo 6 mesi dal gruppo sottoposto a trattamento
ruppo di controllo (n=160)
G
Eventi avversi
Aumento del dolore al collo
olore alla spalla/al braccio
D
Nuova lesione alle vertebre cervicali
Patologia a livello adiacente
omplicazioni operatorie
C
LBP/patologia lombare
Trauma/lesione (non cervicale)
ntorpidimento/formicolio
I
Cefalea/emicrania
olore non specifico/non inerente
D
Nausea
Capogiri/vertigini
Irritazione/scolorimento cutaneo
Battito cardiaco veloce/irregolare
Fiato corto
Ronzio nelle orecchie
Sintomo neurologico/ictus
Nodo in gola
Diagnosi di diabete
Diagnosi di cancro della mammella
Attacco epilettico
Decesso, non correlato
Dolenzia
Vite rotta
Cedimento dell'innesto
Sindrome del tunnel carpale
Sensazione di soffocamento
Sintomi cardiaci
Sindrome nefrotica
Tentativo di suicidio
TOTALE
1
. % espressa come numero di pazienti in cui si è verificato l'evento/numero totale di pazienti nel gruppo
2
. In alcuni pazienti si sono verificati più eventi avversi
*
Alcuni eventi avversi sono stati osservati più frequentemente nel gruppo trattato con Cervical-Stim che nel gruppo
n. (%) di eventi
0 (14,9)
1
10 (14,9)
10 (14,9)
(4,5)
3
2 (3,0)
8 (11,9)
(3,0)
2
6 (8,9)
2 (3,0)
(3,0)
2
0
2 (3,0)
0
0
0
0
1 (1,5)
0
0
0
0
0
1 (1,5)
1 (1,5)
1 (1,5)
2 (3,0)
1 (1,5)
1 (1,5)
1 (1,5)
1 (1,5)
67
1
n. (%)
azienti in cui si
p
è verificato
l'evento
(5,6)
9
9 (5,6)
8 (5,0)
(1,9)
3
2 (1,3)
8 (5,0)
(1,3)
2
6 (3,8)
2 (1,3)
(1,3)
2
0
2 (1,3)
0
0
0
0
1 (0,6)
0
0
0
0
0
1 (0,6)
1 (0,6)
1 (0,6)
2 (1,3)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
2
47
di
di controllo. Dato il tipo di eventi, è improbabile che siano correlabili al trattamento.
Gruppo Cervical-Stim (n=163)
n. *(%) di eventi
16 (17,8)
16 (17,8)
(10,0)
9
8 (8,8)
7 (7,7)
(5,5)
5
5 (5,5)
4 (4,4)
(4,4)
4
3 (3,3)
2 (2,2)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
0
0
0
0
0
0
0
0
90
1
di
n. (%)
pazienti in cui si
verificato
è
l'evento
15 (9,2)
6 (9,8)
1
9 (5,5)
8 (4,9)
(3,1)
5
5 (3,1)
4 (2,5)
(2,5)
4
4 (2,5)
3 (1,8)
(1,2)
2
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
0
0
0
0
0
0
0
0
2
58
Sintesi dei dati clinici
Disegno dello studio
Lo studio clinico Cervical-Stim consisteva in uno studio controllato, randomizzato, a
gruppi paralleli su 323 soggetti adulti ad alto rischio (fumatori, multi-livello o entrambi,
3
Page 74

e con innesto eterologo) con evidenza radiografica di radici dei nervi cervicali compresse
e radicolopatia sintomatica. Scopo dello studio era valutare la sicurezza e l'efficacia del
dispositivo Cervical-Stim per l'applicazione di PEMF come coadiuvante per pazienti ad
alto rischio sottoposti a fusione cervicale. Tutti i soggetti hanno subito discectomia e
fusione cervicale anteriore con tecnica di Smith-Robinson con piastra Atlantis. I soggetti
sono stati suddivisi con assegnazione casuale al gruppo di controllo (terapia standard,
n=160) o al gruppo di trattamento (terapia standard più Cervical-Stim, n=163). La
terapia standard era a discrezione del medico, ma di norma prevedeva il ricovero
standard, l'uso di un collare cervicale morbido, farmaci appropriati e fisioterapia.
Tra i soggetti candidati per lo studio vi erano pazienti che soddisfacevano i seguenti
criteri di inclusione ed esclusione:
Criteri di inclusione
Adulto di sesso maschile o femminile, tra i 18 e i 75 anni di età con evidenza radiografica
di radice/i dei nervi cervicali compressa/e, radicolopatia sintomatica, dolore di grado 5 o
superiore sulla scala analogica visiva (Visual Analog Scale - VAS) e/o debolezza
muscolare e fusione cervicale primaria eseguita con tecnica di Smith-Robinson con
innesto osseo eterologo e piastra cervicale anteriore. Il soggetto deve: o aver subito un
intervento di fusione multi-livello (livello di fusione >1) o essere un fumatore (un
pacchetto al giorno o più) o entrambe le cose; e aver firmato il consenso informato.
Criteri di esclusione
Lesione cervicale traumatica, fusione con approccio posteriore o di correzione, innesto
omologo o con materiale osseo sostitutivo, anamnesi di emicrania vascolare o soggetto
ad attacchi incontrollati o epilessia (controllata o non controllata) o patologie o lesioni
neurologiche; sistema immunitario depresso, patologie regionali (spondilite, malattia di
Paget, artrite reumatoide), infezione (sistemica o locale) entro le 2 settimane precedenti
l'intervento, patologie sistemiche (cancro, aritmia cardiaca, disfunzione tiroidea, diabete
mellito non controllato, nefropatia/disfunzione renale, uso cronico di steroidi o altre
condizioni che possono aver compromesso il metabolismo osseo), pacemaker cardiaco,
defibrillatore, stimolatore della colonna dorsale, apparecchio acustico, protesi cocleare
e stimolatore cranico, pazienti in gravidanza, in allattamento o che intendevano restare
incinte entro 12 mesi, soggetti che avevano partecipato ad altri studi clinici negli ultimi
12 mesi, o soffrivano di condizioni fisiche o mentali che ne precludevano la conformità
alle istruzioni dei medici.
Italiano
Valutazione e follow-up
Le visite di follow-up dovevano avvenire nel primo, secondo, terzo, sesto e, dodicesimo
mese e successivamente ogni anno finché l'ultimo soggetto inserito nello studio
raggiungeva i 12 mesi.
Uso del dispositivo
Ai soggetti assegnati al gruppo di trattamento (Cervical-Stim) è stato chiesto di
indossare il dispositivo per quattro ore al giorno per un minimo di tre mesi dopo
l'intervento. I chirurghi potevano, a loro discrezione, prolungare il trattamento con
Cervical-Stim fino a sei mesi dopo l'intervento.
4
Page 75

Dati demografici
L'età media dei pazienti ammessi nello studio era di 46,8 anni (intervallo da 24 a 73
anni). Dei 323 soggetti, 148 (45,8%) erano di sesso femminile e 175 (54,2%) di sesso
maschile. Trecentouno (93,2%) erano di razza caucasica, mentre 17 (5,3%) erano
afro-americani e 5 (1,6%) ispanici. Centocinquantanove (49,2%) erano non fumatori e
164 (50,8%) erano fumatori.
Caratteristiche demografiche di partenza
umero di soggetti
ariabili
V
Età (anni)
edia
M
Intervallo
SD
esso
S
Femminile
Maschile
azza
R
Caucasica
Afro-americana
Ispanica
Asiatica
Altro
Fumatore/non fumatore
Non fumatore
Fumatore
1
. Valori P dei test di comparazione tra i gruppi di trattamento con il t-test di Student per le
N
(n = 323)
148 (45,8%)
75 (54,2%)
1
301 (93,2%)
17 (5,3%)
5 (1,6%)
159 (49,2%)
164 (50,8%)
46,8
24 – 73
,3
9
0
0
variabili numeriche e il test di Pearson x2per le variabili categoriche.
ontrollo
C
(n = 160)
46,7
26 – 72
,2
9
75 (46,9%)
5 (53,1%)
8
150 (93,8%)
7 (4,4%)
3 (1,9%)
79 (49,4%)
81 (50,6%)
ervical-Stim
C
(n = 163)
46,9
24-73
,4
9
73 (44,8%)
0 (55,2%)
9
151 (92,6%)
10 (6,1%)
-
-
2 (1,2%)
-
-
80 (49,1%)
83 (50,9%)
Valore P
0,846
0,706
0,703
0,958
1
Analisi dei dati e risultati
Il punto finale principale per l'efficacia era l'aumento della frequenza di riuscita della
fusione cervicale entro sei mesi dall'intervento, documentata dall'evidenza radiografica. I
punti finali secondari erano la funzione neurologica, la valutazione del dolore sulla scala
VAS e l'indice di disabilità del collo. La sicurezza è stata valutata in base alla frequenza e
gravità degli eventi avversi.
La fusione è stata valutata mediante radiografia ad ogni visita:
Radiograficamente, la fusione era definita come riunificazione ossea >
sull'interfaccia superiore e inferiore dell'innesto tra corpi vertebrali adiacenti E
angolazione (movimento) <
4° tra vertebre fuse adiacenti sulle lastre laterali di
flessione/estensione E assenza di radiolucenza.
La mancata fusione in radiografie era definita come riunificazione ossea < 50%
sull'interfaccia superiore o inferiore dell'innesto O angolazione (movimento) > 4° tra
vertebre fuse adiacenti sulle lastre laterali di flessione/estensione O presenza di
radiolucenza.
5
50%
Page 76

Ai fini della valutazione del dispositivo, tutte le lastre sono state scansionate e inserite in
una banca dati centrale ed esaminate in cieco da due chirurghi ortopedici indipendenti e
da un radiologo indipendente al temine dell'intero studio. Le lastre sono state
esaminate e classificate secondo un comune protocollo. Tutte le lastre in ogni punto del
tempo sono state valutate per quantità di radiolucenza, riunificazione ossea e grado di
movimento secondo quanto evidenziato dalle lastre delle vertebre cervicali in
flessione/estensione. Per il calcolo del movimento è stato utilizzato un software. I
risultati ottenuti in questo modo sono stati esaminati e verificati dai chirurghi ortopedici
incaricati dell'esame. La diagnosi del radiologo è stata considerata determinante in caso
di disaccordo tra i due chirurghi ortopedici.
Risultati riguardanti l'efficacia
Dei 323 soggetti randomizzati e sottoposti a intervento, 240 risultavano valutabili per
l'analisi di efficacia (gruppo di trattamento Cervical-Stim, n=122; gruppo di controllo,
n=118). Gli altri soggetti sono stati considerati non valutabili per i seguenti motivi:
radiografie inesistenti o non leggibili, non conformità del soggetto, violazioni del
protocollo (criteri di inclusione), cedimento dell'innesto, rottura della protesi interna,
abbandono precoce dello studio per eventi avversi di modesta entità, e un tentativo di
suicidio. L'esito positivo o negativo in questi soggetti non è noto. I dati non disponibili
potrebbero influire positivamente o negativamente sul successo complessivo dello
studio. Per valutare l'impatto dei dati mancanti sono state eseguite analisi di sensibilità.
Le analisi erano basate sull'ultima osservazione riportata a nuovo e sul calcolo eseguito
considerando tutti i dati non disponibili come mancate fusioni. Entrambe le analisi
hanno dimostrato che i risultati a sei mesi indicavano comunque una differenza
statisticamente significativa a favore del gruppo trattato con Cervical-Stim. Inoltre i dati
demografici di partenza della popolazione valutabile sono stati raffrontati a quelli dei
soggetti non valutabili. I risultati di questa analisi non hanno evidenziato differenze
significative tra i soggetti valutabili e quelli non valutabili in 14 variabili di studio, i dati
demografici principali e i parametri clinici.
Italiano
Punto finale primario per l'efficacia
Il punto finale primario per l'efficacia era l'evidenza radiografica di fusione a sei mesi
dopo l'intervento. A sei mesi dopo l'intervento, in 102 dei 122 soggetti valutabili (84%)
nel gruppo trattato con il Cervical-Stim la fusione era giudicata riuscita, a fronte di 81 dei
118 soggetti valutabili (69%) nel gruppo di controllo (p=0,0065).
Confronto degli esiti radiografici di fusione a sei mesi
Gruppo di
trattamento
Controllo
Cervical-Stim
I dati dimostrano che di tutti i pazienti sottoposti a intervento di fusione cervicale, la
frequenza della fusione radiografica a sei mesi è stata maggiore in quelli che hanno
ricevuto la terapia coadiuvante con Cervical-Stim rispetto al gruppo di controllo.
Numero di soggetti
118
122
Numero di soggetti
sottoposti a fusione
81
102
Percentuale
di fusione (%)
68,64
83,61
6
Page 77

È stata eseguita un'ulteriore analisi per rilevare le differenze tra il gruppo di trattamento
con Cervical-Stim e il gruppo di controllo attribuibili alle caratteristiche demografiche
(sesso, età, diagnosi) e la condizione di rischio (fumatore, multilivello). L'incidenza
complessiva di fusione nel gruppo Cervical-Stim è rimasta statisticamente significativa
dopo la correzione in base a ognuna di queste variabili.
Il follow-up di lungo periodo (12 mesi) non ha evidenziato differenze statisticamente
significative tra i due gruppi relativamente alla fusione. In centosedici dei 125 soggetti
valutabili (92,8%) nel gruppo di trattamento con Cervical-Stim la fusione è stata
giudicata riuscita al punto finale di lungo periodo, mentre per i 120 soggetti valutabili del
gruppo di controllo, la fusione è stata giudicata riuscita in 104 casi (86,7%).
Confronto degli esiti radiografici di fusione a 12 mesi
Gruppo di trattamento
Controllo
Cervical-Stim
N.B.: Le differenze nelle percentuali di successo a lungo termine dei due gruppi di trattamento
non sono statisticamente significative per il test di Pearson x2con il campione disponibile
(x2= 2,5136, p = 0,1129).
Numero di soggetti
120
125
Numero di soggetti
sottoposti a fusione
104
116
Percentuale di fusione
(%)
86,67
92,80
Punti finali secondari per l'efficacia
I punti finali secondari valutavano le variazioni dei sintomi clinici. Il “successo clinico”
relativamente ai sintomi era definito come non peggioramento della funzione
neurologica, miglioramento della valutazione del dolore sulla scala VAS, e non
peggioramento dell'indice di disabilità del collo. L'“insuccesso clinico” relativamente ai
sintomi era definito come mancato soddisfacimento anche di uno solo di questi criteri.
Non sono state riscontrate differenze statisticamente significative tra i due gruppi per
quanto riguarda la percentuale di soggetti considerati “successi clinici” a sei mesi
(p=0,8456) o a 12 mesi (p=0,1129).
7
Page 78

Sicurezza
Gli eventi avversi osservati nello studio sono riportati nella Tabella degli eventi avversi
presentata nella sezione dedicata alle informazioni per la prescrizione. A sei mesi, il
numero di soggetti in cui si sono verificati uno o più eventi avversi è analogo nei due
gruppi. È stato riferito un totale di 14 eventi gravi in 13 soggetti, di cui nove
appartenenti al gruppo Cervical-Stim e cinque al gruppo di controllo. Tali eventi erano
costituiti da esperienze quali aumento del dolore, fiato corto, capogiri, traumi o lesioni
non correlate, decesso non correlato, complicazioni chirurgiche e patologia a livello
adiacente. Per i nove soggetti nel gruppo di trattamento con Cervical-Stim tutti gli
eventi avversi gravi erano, a giudizio dei ricercatori, sicuramente o probabilmente non
correlati con il dispositivo.
I dati di sicurezza raccolti tra la visita del sesto mese e il contatto finale con ciascun
soggetto indicano che 57 eventi avversi si sono verificati in un totale di 51 soggetti di
entrambi i gruppi. Il numero di soggetti in cui si sono verificati uno o più eventi avversi è
analogo nei due gruppi. Nessuno degli eventi avversi riferiti tra la visita del sesto mese e
il contatto finale era grave; gli eventi erano simili a quelli riferiti a sei mesi.
Durata del dispositivo
Il Cervical-Stim può erogare fino a 270 trattamenti consecutivi (giornalieri) di quattro ore
ciascuno. La durata complessiva del trattamento dovrà essere definita dal medico in base
al paziente e all'avanzamento della fusione.
Tempo di trattamento
Il Cervical-Stim deve essere indossato ogni giorno per quattro ore. Al termine del
trattamento giornaliero il dispositivo si spegne automaticamente. Il dispositivo si può
spegnere in qualunque momento premendo il pulsante On/Off posto sul pannello di
controllo.
Italiano
Il Cervical-Stim può essere utilizzato a qualunque ora del giorno che sia la più comoda
per il paziente. Il dispositivo è leggero e regolabile. Inoltre, dato che il Cervical-Stim è
portatile, il paziente può sottoporsi al trattamento in posizione seduta, sdraiata, durante
il sonno, durante una camminata ecc. Comunque, poiché ogni paziente è un caso a sé, il
livello di attività complessivo dovrà essere stabilito in base alle istruzioni del medico.
8
Page 79

Carica/ricarica della batteria
Il dispositivo Cervical-Stim è attivato da una batteria ricaricabile a ioni di litio. Il
dispositivo è provvisto di caricabatterie in dotazione. Utilizzare esclusivamente il
caricabatterie Orthofix per la carica del dispositivo.
N.B.: la batteria del Cervical-Stim deve essere caricata prima del primo utilizzo.
Centralina di
Caricabatterie
Presa del
caricatore
Per caricare/ricaricare la batteria è sufficiente inserire l'estremità cilindrica del
connettore nella presa del caricatore situata sulla centralina di controllo. Collegare
saldamente il cavo nel caricatore. Inserire la spina del caricabatterie in una comune
presa di corrente c.a. Quando la batteria è del tutto scarica possono essere necessarie
fino a 12 ore per la ricarica completa.
La batteria del Cervical-Stim si può ricaricare in qualunque momento quando il
dispositivo non è in uso. Si consiglia vivamente di ricaricare il dispositivo dopo aver
terminato il trattamento giornaliero.
N.B.: il Cervical-Stim non eroga il trattamento durante la carica.
Quando il dispositivo è acceso, sul display del Cervical-Stim appare un simbolo indicante
la capacità della batteria. Un profilo di batteria che appare a intermittenza, il simbolo
e un segnale acustico indicano che il livello di carica è basso e che la batteria
deve essere ricaricata. Vedere il capitolo “Indicatori visivi e acustici” per ulteriori
informazioni.
controllo
Pulsante On/Off
LCD
9
Page 80

Funzionamento del dispositivo
Accensione e spegnimento
Il Cervical-Stim si accende e si spegne premendo il pulsante On/Off posto sulla
centralina di controllo del dispositivo. Quando l'apparecchio è acceso il display
visualizza brevemente una serie di messaggi di stato. Successivamente dovrebbe
apparire il tempo di trattamento residuo e il logo Orthofix a intermittenza, indicante
che il dispositivo è acceso e funziona regolarmente. (Se sul display non appare il logo a
intermittenza, rivolgersi al fornitore del dispositivo). La centralina di controllo presenta
un pulsante per la retroilluminazione. In condizioni di scarsa luce premere il pulsante per
illuminare il display.
Per interrompere il trattamento prima del termine della sessione giornaliera, è
sufficiente premere il pulsante On/Off. Per riprendere il trattamento, premere
nuovamente il pulsante On/Off. Il display visualizzerà il tempo di trattamento residuo.
N.B.: perché il contaminuti funzioni correttamente, le sedute di trattamento devono
avere una durata superiore a 60 minuti.
Presa IR
Pulsante di retroilluminazione
Centralina di
controllo
Presa del
caricatore
Pulsante On/Off
Italiano
Display
Logo Orthofix
intermittente
Programmazione delle sedute terapeutiche
Il Cervical-Stim programma automaticamente il tempo di ogni sessione di trattamento.
Il calcolo del tempo trascorso inizia quando il dispositivo viene acceso con il pulsante
On/Off posto sulla centralina. Il display visualizza il tempo residuo di trattamento per la
sessione in corso. Al termine del trattamento giornaliero il dispositivo si spegne
automaticamente. Per interrompere il trattamento prima del termine della sessione, è
sufficiente premere il pulsante On/Off. Per riprendere il trattamento, premere
nuovamente il pulsante On/Off. Il display visualizzerà il tempo di trattamento residuo.
N.B.: perché il contaminuti funzioni correttamente, le sedute di trattamento devono
avere una durata superiore a 60 minuti.
10
Page 81

Come indossare il dispositivo
Il Cervical-Stim è concepito per le vertebre cervicali e può essere indossato con o senza
apparato di sostegno. Indossare qualche indumento tra la pelle e il Cervical-Stim per
maggiore comodità.
Per l'uso del Cervical-Stim, basta passare il dispositivo sopra la testa in modo che sia
appoggiato comodamente sul collo e sulle spalle (vedere figura di seguito). In alternativa
è possibile aprire la fibbia e appoggiare il dispositivo sul collo e sulle spalle. Allacciare la
fibbia come in una cintura di sicurezza perché l'apparecchio sia ben fissato.
Un collare di comfort è incluso con il Cervical-Stim per un migliore adattamento e
supporto. Il collare di comfort può essere inserito o rimosso dal collare interno del
dispositivo usando le striscette di velcro che si fissano direttamente al materiale.
Velcro
Attacco
della fibbia
11
Page 82

Manutenzione e pulizia
Il Cervical-Stim è un dispositivo elettronico tecnologicamente avanzato e deve essere
maneggiato con la dovuta cura. L'apparecchio può riportare danni in caso di caduta o
se maneggiato in modo improprio.
NON esporre il Cervical-Stim ai raggi diretti del sole per periodi
prolungati. Ciò può danneggiare la centralina di controllo.
NON esporre il Cervical-Stim a calore eccessivo. Nei climi caldi la
temperatura all'interno di un'auto o di un baule può arrivare a più di 71 ºC.
Il calore eccessivo può danneggiare la centralina di controllo del dispositivo.
NON esporre il Cervical-Stim a umidità eccessiva.
NON smaltire l'apparecchio in un inceneritore.
NON utilizzare solventi per la pulizia del Cervical-Stim.
Per pulire l'apparecchio servirsi di un panno morbido inumidito.
In viaggio
Il paziente deve essere informato che nei viaggi in aereo è preferibile affidare il
Cervical-Stim al check-in insieme al bagaglio. Se il dispositivo viene portato in cabina,
non deve essere indossato durante il passaggio attraverso gli apparati di controllo dei
passeggeri. Il Cervical-Stim potrebbe risultarne danneggiato. È consigliabile portare
con sé il manuale del Cervical-Stim per consentire al personale di sorveglianza di
identificare il dispositivo in modo semplice e veloce.
Italiano
Conservazione
Il Cervical-Stim deve essere conservato a una temperatura non inferiore a -10º C e non
superiore a 45º C
L’intervallo della temperatura di funzionamento deve essere compreso tra +5º C e 40º C
Tasso di umidità: max. 95%, senza condensa
Smaltimento
Il Cervical-Stim contiene batterie al litio: non smaltire il dispositivo in un inceneritore.
Smaltire il dispositivo in modo corretto.
12
Page 83

Indicatori visivi e acustici
Il display e i segnali acustici hanno la funzione di fornire informazioni utili all'utilizzatore.
La tabella di seguito illustra i vari simboli e segnali, e il loro significato.
Indicatori visivi del display e segnali acustici del Cervical-Stim
Simbolo/segnale Descrizione Significato
tutti i simboli sul display e i segnali
acustici sono attivi in modo
continuo per circa 5 secondi
il contaminuti segna il tempo di
trattamento residuo
(ore e minuti)
il logo Orthofix appare a
intermittenza
sul contaminuti appaiono tre
lineette
segnale acustico (5 bip)
il simbolo appare fisso per circa 5
secondi
simboli a intermittenza / segnale
acustico (circa 1 bip al secondo)
il simbolo fisso indica il livello
approssimativo di carica (in %)
il simbolo indicante un
riempimento ripetuto rappresenta
la modalità “in carica”
segnale acustico continuo
test autodiagnostico di
dispositivo acceso
trattamento
regolarmente in corso
termine del tempo di
trattamento residuo
trattamento terminato/
apparecchio spento
livello batteria basso
— necessaria ricarica
stato batteria: carica
residua o modalità "in
carica"
dispositivo bloccato —
chiamare assistenza
appare un codice E
(ad es. E01, E02 . . .)
13
messaggio di errore
— chiamare
assistenza
Page 84

Classificazione dell'apparecchio e descrizione dei simboli
Simbolo Significato
Attenzione - Consultare le istruzioni per l'uso
Pezzo applicato di tipo BF
Acceso/spento
Pulsante di retroilluminazione
Temperatura min. e max. di conservazione
Anno di produzione per il dispositivo attivo
Presa per il caricabatterie
Classificazioni dell'apparecchio
• Apparecchio con alimentazione interna
• Pezzo applicato di tipo BF
• Classificazione del grado di protezione secondo la norma IEC 529: IPXO
• Apparecchio non adatto all'uso in presenza di miscela anestetica infiammabile
con aria o ossido di azoto.
• Modalità di funzionamento: a intermittenza
L'uso di accessori diversi da quelli specificati può dar luogo a un aumento delle emissioni
o a una riduzione dell'immunità del dispositivo.
Il caricabatterie è dotato di presa a tre fili ma è considerato interamente in doppio
isolamento con struttura di Classe II.
Per una sicurezza ottimale seguire le istruzioni del produttore durante l'uso.
L'uso del prodotto secondo altre modalità può avere effetti nocivi e/o annullare la
garanzia.
Italiano
N.B.: controllare il dispositivo prima di ogni uso per escludere segni di usura o
deterioramento.
Non utilizzare il dispositivo se appare in condizioni non adatte all'uso.
Assistenza
In caso di domande sul dispositivo o per richiedere assistenza si prega di rivolgersi al
rivenditore di zona o visitare il sito Internet www.orthofix.com per i dettagli. Il
dispositivo non presenta pezzi riparabili dall'utilizzatore. Per le vostre esigenze di
assistenza, vi preghiamo di rivolgervi al produttore o al rivenditore di zona.
14
Page 85

Conformità alle norme
Il sistema per fusione cervicale Cervical-Stim Orthofix è conforme alle seguenti serie di
norme internazionali:
UL 60601 - Apparecchiature mediche elettriche, Requisiti generali di sicurezza;
(compresa Compatibilità e Interferenza elettromagnetica)
IEC 60601 - Apparecchiature mediche elettriche, Requisiti generali di sicurezza;
(compresa Compatibilità e Interferenza elettromagnetica)
Informazioni sulla garanzia
La Orthofix Inc. garantisce che lo stimolatore di crescita ossea Cervical-Stim®è esente
da difetti relativi a materiali o fabbricazione; tale garanzia avrà durata di un anno dalla
data del primo utilizzo. A condizione che siano stati rispettati i termini della presente
garanzia limitata, la Orthofix Inc. si impegna a sostituire gli eventuali componenti
difettosi.
La presente garanzia limitata si riferisce al prodotto utilizzato in condizioni normali ed
esclude eventuali danni o difetti causati da incidenti, uso improprio, uso eccessivo,
incendio, allagamento e disastri naturali, nonché modifiche, manipolazioni, riparazioni o
tentate riparazioni da parte di chiunque altro che non sia la Orthofix Inc. La presente
garanzia si riferisce solo al paziente al quale il prodotto è stato prescritto e non è
cedibile o trasferibile. I prodotti difettosi a cui si applica la presente garanzia limitata
devono essere resi alla Orthofix Inc. all'attenzione di: Orthofix Returns. È necessario
rivolgersi al rivenditore locale per ottenere un numero di Autorizzazione Resi (RA) e
l'indirizzo prima di restituire il prodotto.
Salvo quanto specificamente previsto dalle leggi vigenti, la presente garanzia è da
intendersi come sostitutiva di ogni altra garanzia, espressa o sottintesa, e la Orthofix Inc.
specificamente respinge ogni e qualsiasi garanzia di commerciabilità o idoneità a un
particolare scopo. La Orthofix Inc. e le società sue rappresentanti, consociate o
controllate non saranno in alcun caso responsabili di eventuali danni speciali, indiretti o
incidentali. L'unico provvedimento in caso di prodotti danneggiati consisterà nella
sostituzione del prodotto.
La presente garanzia limitata può essere ampliata o modificata solo per iscritto dalla
Orthofix Inc. Venditori, rappresentanti, distributori o medici non sono autorizzati ad
apportare o autorizzare ampliamenti o modifiche dei termini della presente garanzia
limitata.
Per ulteriori informazioni e/o assistenza per il dispositivo, rivolgersi al rivenditore di zona.
15
Page 86

MA N U A L D E I N ST R U C CI O N E S
EDICIÓN
INTERNACIONAL
Page 87

Índice
Español
Manual de instrucciones de
Cervical-Stim
Descripción del dispositivo .......................................................................... 1
El enfoque de Orthofix para la consolidación ............................................... 1
Estimulación mediante CEMP ..................................................................... 2
Información de prescripción ........................................................................ 2
Resumen de datos clínicos .......................................................................... 3
Análisis de los datos y resultados ................................................................. 5
Resultados sobre la efectividad .................................................................... 6
Seguridad ................................................................................................... 8
Vida útil del dispositivo ............................................................................... 8
Instrucciones de tratamiento ....................................................................... 8
Cómo cargar/recargar la batería ................................................................... 9
Funcionamiento del dispositivo .................................................................. 10
Cómo usar el dispositivo ............................................................................... 11
Cuidado y limpieza .................................................................................... 12
Viajes ....................................................................................................... 12
Almacenamiento ....................................................................................... 12
Eliminación ............................................................................................... 12
Indicadores visuales y de audio .................................................................. 13
Clasificación del equipo y descripciones de los símbolos del dispositivo ....... 14
Servicio técnico ......................................................................................... 14
Conformidad con las normas ..................................................................... 15
Información de la garantía ......................................................................... 15
®
Para obtener más información acerca de Orthofix, visite nuestro sitio web en
www.orthofix.com.
Contenido del paquete:
1- Sistema de fusión cervical Cervical-Stim
®
1- Paquete de material impreso
1- Unidad de carga n.º 270215
1- Cuello acolchado
ESTE DISPOSITIVO NO ESTÁ ESTERILIZADO.
NO REQUIERE ESTERILIZACIÓN.
0086
Page 88

Descripción del dispositivo
El sistema de fusión cervical Cervical-Stim®es un dispositivo externo de Campo
electromagnético pulsado (CEMP) y ha sido diseñado teniendo en cuenta su
comodidad y conveniencia. Es un dispositivo de una sola pieza, que es ligero, flexible
y portátil, lo que proporciona libertad de movimiento durante el tratamiento. Una
pantalla de cristal líquido (LCD) y una alarma audible proporcionan importante
información al paciente durante el tratamiento, como el estado operativo, el tiempo
de tratamiento restante, la capacidad de la batería, etc. Para obtener más
información, consulte “Indicadores visuales y de audio”.
Sistema de fusión cervical Cervical-Stim – Modelo 2505CE
Transductor
Unión con
hebilla
Panel de control
Unidad de control
Cervical-Stim incluye una unidad de control y un transductor de tratamiento. La
unidad de control contiene un microprocesador que genera la señal eléctrica de
Cervical-Stim. El transductor de tratamiento convierte dicha señal en un campo
magnético de baja energía altamente uniforme. Cuando el dispositivo está centrado
sobre el área de tratamiento, la señal CEMP terapéutica de Cervical-Stim es
aplicada directamente en el lugar de la fusión.
Cervical-Stim funciona con un paquete de baterías de ion-litio recargables.
La LCD y la alarma audible advertirán al paciente cuando la batería esté baja y deba
ser recargada. Para obtener más información, consulte “Cómo cargar/recargar la
batería”. A fin de asegurar que el dispositivo esté funcionando correctamente,
Cervical-Stim monitorea constantemente la tensión de la batería y la señal eléctrica.
Si en algún momento durante el tratamiento el dispositivo deja de funcionar
correctamente, la LCD mostrará el símbolo o código de error correspondiente.
Para obtener más información, consulte “Indicadores visuales y de audio”.
El enfoque de Orthofix para la consolidación
Orthofix es una empresa líder establecida a nivel mundial en la tecnología de
estimulación del crecimiento óseo mediante campos electromagnéticos pulsados
(CEMP). Orthofix desarrolló el primer estimulador portátil no invasivo mediante
CEMP para la fusión cervical con el lanzamiento del sistema de fusión cervical
Cervical-Stim ocurrido en 2005. Orthofix sigue perfeccionando y mejorando la
aplicación de campos electromagnéticos pulsados para la consolidación de la fusión
cervical y el rescate de la fusión cervical fallida. Orthofix produce una gama
completa de dispositivos médicos de última generación para ortopedia y
neurocirugía, que mejoran el proceso de consolidación natural del cuerpo,
1
Page 89

estimulan la función natural, protegen, restauran y consolidan. Gracias por incluir a
Orthofix en su proceso de consolidación. Para obtener más información acerca de
Orthofix, visite nuestro sitio web en www.orthofix.com.
Estimulación mediante CEMP
La estimulación del crecimiento óseo mediante campos electromagnéticos pulsados
(CEMP) es un tratamiento seguro y no quirúrgico prescrito por un médico para
sanar las fracturas que no consolidan y favorecer la fusión espinal. Las corrientes
eléctricas se han usado para la consolidación ósea desde mediados del siglo XIX.
No obstante, no fue sino hasta la década de 1950 que los científicos hicieron un
importante descubrimiento. Cuando un hueso humano se tuerce o fractura, genera
un campo eléctrico. Este campo eléctrico de bajo nivel activa el mecanismo de
reparación propio del cuerpo, que a su vez estimula la consolidación ósea.
Los estimuladores del crecimiento óseo mediante CEMP de Orthofix generan un
campo electromagnético pulsado de bajo nivel y uniforme similar al campo
eléctrico generado por el cuerpo. La aplicación del CEMP directamente en el lugar
de la fusión o fractura ayuda a activar y aumentar el proceso de consolidación
natural del cuerpo, a fin de mejorar la fusión ósea.
Información de prescripción
Indicación
Cervical-Stim es un estimulador no invasivo del crecimiento óseo mediante campo
electromagnético pulsado indicado como complemento a la cirugía de fusión
cervical en pacientes de alto riesgo de falla de fusión y como tratamiento de
rescate no quirúrgico de la fusión cervical fallida.
Español
Contraindicaciones
No se conocen contraindicaciones para el uso de Cervical-Stim.
Advertencias
• Cervical-Stim puede interferir con el funcionamiento de un marcapasos o un
desfibrilador. Se recomienda que consulte con el cardiólogo que atiende al paciente.
• El Cervical-Stim debe quitarse antes de someterse a cualquier procedimiento de
diagnóstico por imágenes (p.ej., tomografía computarizada, resonancia
magnética, etc.).
Precauciones
• El Cervical-Stim no debe usarse si existen condiciones mentales o físicas que pudieran
impedir el cumplimiento de las instrucciones del médico y del dispositivo.
• El tratamiento con Cervical-Stim no se ha evaluado en pacientes con las
siguientes afecciones: traumatismo óseo o ligamentoso, espondilitis, enfermedad
de Paget, osteoporosis moderada a grave, cáncer metastásico, enfermedad
renal, artritis reumatoidea, diabetes mellitus no controlada, pacientes propensos
a cefaleas vasculares migrañosas, convulsiones, epilepsia, afecciones de la
tiroides o enfermedades neurológicas.
• Estudios teratológicos en animales realizados con este dispositivo no mostraron
ningún efecto adverso en animales. Sin embargo, no se ha establecido la seguridad
del uso de este dispositivo en pacientes que están embarazadas o amamantando.
2
Page 90

Eventos adversos
Eventos adversos reportados a los 6 meses por el grupo del tratamiento
rupo de control (n = 160)
Eventos adversos
Más dolor en el cuello
olor en el hombro/brazo
D
Nueva lesión en la región cervical
Patología a nivel adyacente
Complicaciones quirúrgicas
LBP/Patología lumbar
Trauma/Lesión (no cervical)
Entumecimiento/Hormigueo
Dolor de cabeza/Migraña
Dolor no específico/no relacionado
Náuseas
Mareos/Vértigo
Erupción cutánea/Decoloración
Latidos rápidos/irregulares del corazón
Respiración entrecortada
Zumbido en los oídos
Síntoma neurológico/Apoplejía
Nódulo en la garganta
Diagnóstico de diabetes
Diagnóstico de cáncer del seno
Convulsión
Muerte, no relacionada
Sensibilidad
Rotura de tornillo
Fallo del injerto
Síndrome del túnel carpiano
Sensación de ahogo
Síntomas cardíacos
Síndrome nefrótico
Intento de suicidio
G
N.º (%) de
ventos
e
0 (14,9)
1
10 (14,9)
10 (14,9)
(4,5)
3
2 (3,0)
8 (11,9)
2 (3,0)
6 (8,9)
2 (3,0)
2 (3,0)
0
2 (3,0)
0
0
0
0
1 (1,5)
0
0
0
0
0
1 (1,5)
1 (1,5)
1 (1,5)
2 (3,0)
1 (1,5)
1 (1,5)
1 (1,5)
1 (1,5)
1
de
N.º (%)
acientes que
p
experimentaron
el evento
(5,6)
9
9 (5,6)
8 (5,0)
(1,9)
3
2 (1,3)
8 (5,0)
2 (1,3)
6 (3,8)
2 (1,3)
2 (1,3)
0
2 (1,3)
0
0
0
0
1 (0,6)
0
0
0
0
0
1 (0,6)
1 (0,6)
1 (0,6)
2 (1,3)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
Grupo Cervical-Stim (n = 163)
N.º*(%) de
eventos
6 (17,8)
1
16 (17,8)
9 (10,0)
(8,8)
8
7 (7,7)
5 (5,5)
5 (5,5)
4 (4,4)
4 (4,4)
3 (3,3)
2 (2,2)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
1 (1,1)
0
0
0
0
0
0
0
0
1
N.º (%)
de
pacientes que
experimentaron
l evento
e
5 (9,2)
1
16 (9,8)
9 (5,5)
(4,9)
8
5 (3,1)
5 (3,1)
4 (2,5)
4 (2,5)
4 (2,5)
3 (1,8)
2 (1,2)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
1 (0,6)
0
0
0
0
0
0
0
0
TOTAL
1
. % expresado como número de pacientes que experimentaron el evento/número total de pacientes en el grupo
2
. Algunos pacientes experimentaron varios eventos adversos
*
Hubo varios eventos adversos que se observaron con más frecuencia en el grupo Cervical-Stim que en el grupo de
control. Por los tipos de eventos dados, es improbable que estos eventos adversos estén relacionados con el
tratamiento.
67
2
47
90
2
58
Resumen de datos clínicos
Diseño del estudio
El estudio clínico de Cervical-Stim fue un estudio de grupo, controlado, paralelo y
aleatorizado de 323 sujetos adultos de alto riesgo (fumadores y/o con fusión de
varios niveles y aloinjerto) con evidencia radiográfica de raíces de nervios cervicales
comprimidas y radiculopatía sintomática. El propósito del estudio fue evaluar la
3
Page 91

seguridad y efectividad del dispositivo de CEMP Cervical-Stim como complemento
para pacientes de alto riesgo sometidos a una fusión cervical. Todos los sujetos
habían sido sometidos a disectomía y fusión cervical anterior usando la técnica
Smith-Robinson con la placa Atlantis. Los sujetos fueron asignados aleatoriamente
al grupo de control (tratamiento estándar, n = 160) o al grupo del tratamiento
(tratamiento estándar más el Cervical-Stim, n = 163). El tratamiento estándar fue
determinado a discreción del médico pero incluyó generalmente la estadía en el
hospital, el uso de un collarín cervical suave, los medicamentos apropiados y la
terapia física.
Los sujetos que cumplieron los siguientes criterios de inclusión y exclusión fueron
elegibles para participar en el estudio:
Criterios de inclusión
Sujeto adulto ya sea hombre o mujer, de18-75 años de edad con evidencia
radiográfica de raíces cervicales comprimidas, radiculopatía sintomática, dolor de
grado 5 o superior en la Escala análoga visual (Visual Analog Scale, VAS) y/o
cualquier debilidad muscular y fusión espinal cervical primaria efectuada mediante
la técnica Smith-Robinson con hueso de aloinjerto y una placa cervical anterior. El
procedimiento de la fusión debió haber sido de varios niveles (nivel de fusión> 1)
y/o el sujeto era un fumador (un paquete diario o más); y firmó el consentimiento
informado.
Criterios de exclusión
Lesión cervical traumática, fusión por vía posterior o de revisión, aloinjerto o
materiales de sustitución ósea como fuente del injerto, historial de cefalea vascular
migrañosa o propensión a convulsiones incontroladas o epilepsia (controlada o no
controlada) o alguna enfermedad o lesión neurológica; depresión del sistema
inmunológico, afecciones regionales (espondilitis, enfermedad de Paget, artritis
reumatoidea), infección (sistémica o local) durante las 2 semanas previas a la
cirugía, trastornos sistémicos (cáncer, arritmia cardíaca, enfermedad de la tiroides,
diabetes mellitus incontrolada, enfermedad/disfunción renal, uso crónico de
esteroides u otras enfermedades que pudieran haber afectado el metabolismo
óseo), marcapasos cardíacos, desfibriladores, estimuladores de la columna dorsal,
prótesis auditivas, prótesis cocleares y estimuladores craneales, personas
embarazadas, amamantando o que hubieran planeado quedar embarazadas en un
plazo de 12 meses, sujetos que hubieran participado en otros estudios clínicos en
los últimos 12 meses, o que tuvieran trastornos mentales o físicos que pudieran
haber impedido el cumplimiento de las instrucciones del médico.
Español
Evaluación y seguimiento
Las consultas de seguimiento debían efectuarse a los meses 1, 2, 3, 6 y 12, y a
partir de entonces, una vez al año hasta que el último sujeto participante en el
estudio cumpliera 12 meses de tratamiento.
Uso del dispositivo
A los sujetos asignados al grupo de tratamiento (Cervical-Stim) se les dio
instrucciones para que usaran el dispositivo cuatro horas diarias durante un mínimo
de tres meses en el período postoperatorio. Los cirujanos pudieron, a su
discreción, ampliar el tratamiento de Cervical-Stim hasta los seis meses en el
período postoperatorio.
4
Page 92

Datos demográficos
Los sujetos de este estudio tenían una edad media de 46,8 años (rango de 24 a 73
años). De los 323 sujetos, 148 (45,8%) eran mujeres y 175 (54,2%) eran
hombres. Trescientos uno (93,2%) eran blancos, mientras 17 (5,3%) eran
afro-americanos y 5 (1,6%) eran hispanos. Ciento cincuentinueve (49,2%) eran
no fumadores y 164 (50,8%) eran fumadores.
Características demográficas de base
úmero
ariables
V
Edad (años)
edia
M
Rango
Desviación estándar
énero
G
Femenino
Masculino
aza
R
Blanca
Afro-americana
Hispana
Asiática
Otra
Estado de consumo de tabaco
No fumador
Fumador
1
. Valores-P de las pruebas comparativas entre los grupos de tratamiento usando la prueba de la
N
e sujetos
d
N = 323)
(
46,8
4 – 73
2
9,3
48 (45,8%)
1
175 (54,2%)
301 (93,2%)
17 (5,3%)
5 (1,6%)
0
0
159 (49,2%)
164 (50,8%)
T de Student para las variables numéricas y la prueba de la x2de Pearson para las variables
categóricas.
ontrol
C
n = 160)
(
46,7
6 – 72
2
9,2
5 (46,9%)
7
85 (53,1%)
150 (93,8%)
7 (4,4%)
3 (1,9%)
79 (49,4%)
81 (50,6%)
ervical-Stim
C
n = 163)
(
46,9
4-73
2
9,4
3 (44,8%)
7
90 (55,2%)
151 (92,6%)
10 (6,1%)
-
-
2 (1,2%)
-
-
80 (49,1%)
83 (50,9%)
alor-P
v
0,846
0
0,703
0,958
1
,706
Análisis de los datos y resultados
El criterio de valoración primario de la efectividad fue el incremento de la
frecuencia del éxito de la fusión cervical a los seis meses del período
postoperatorio determinado por evidencia radiográfica. Los criterios de valoración
secundarios fueron la función neurológica, la evaluación del dolor según la escala VAS
y el Índice de incapacidad del cuello. La seguridad fue evaluada por la frecuencia y
gravedad de los eventos adversos.
La fusión fue evaluada mediante radiografías efectuadas en cada consulta:
La fusión radiográfica fue definida como >
interfaces del injerto, la superior y la inferior, entre los cuerpos vertebrales
adyacentes Y <
4° de angulación (movimiento) entre las vértebras adyacentes
con fusión observada en las radiografías laterales de flexión/extensión Y
ausencia de radiolucencia.
La no fusión radiográfica fue definida como < 50% de puente óseo en la interfaz
superior o inferior del injerto O > 4° de angulación (movimiento) entre las
vértebras adyacentes con fusión observada en las radiografías laterales de
flexión/extensión O presencia de radiolucencia.
50% de puente óseo en ambas
5
Page 93

Con fines de evaluación del dispositivo, todas las radiografías se escanearon y se
incluyeron en una base de datos central, y las revisaron dos cirujanos ortopédicos
independientes y un radiólogo independiente "a ciegas" al final de todo el estudio.
Las radiografías se revisaron y calificaron usando un protocolo común. En cada
punto temporal, se evaluaron todas las radiografías en cuanto a la cantidad de
radiolucencia, de puente óseo y el grado de movimiento por evidencias mostradas
en las radiografías de flexión/extensión de la región cervical. Se usó un programa
de software para calcular el movimiento. Los cirujanos ortopédicos revisaron y
verificaron los resultados obtenidos de esta manera. El diagnóstico del radiólogo
se consideró definitivo en el caso de un desacuerdo entre los dos cirujanos
ortopédicos.
Resultados sobre la efectividad
De los 323 sujetos aleatorizados y que recibieron cirugía, 240 fueron evaluables
para el análisis de efectividad (grupo de tratamiento con Cervical-Stim, n = 122;
grupo de control, n = 118). Hubo sujetos que no se consideraron evaluables por
las siguientes razones: radiografías no existentes o no legibles, incumplimiento del
sujeto, infracciones del protocolo (criterios de inclusión), fallo del injerto, equipo
interno roto, existencia de estudio anterior debido a experiencias adversas
menores, y un intento de suicidio. No se conoce el estado de éxito o de fracaso de
estos sujetos. Estos datos con los que no se contó pudieran haber afectado de
manera positiva o negativa el éxito general del estudio. Con el fin de evaluar el
efecto de los datos faltantes, se efectuaron análisis de sensibilidad. Estos análisis
consistieron en que la última observación de estos sujetos se trasladara hasta el
final y que todos los datos faltantes se imputaran a una falla de fusión. Estos dos
análisis mostraron que los resultados hallados a los seis meses seguían siendo
todavía significativamente diferentes y favorables, desde el punto de vista
estadístico, al grupo Cervical-Stim. Además, los datos demográficos de referencia
provenientes de la población evaluable fueron comparados con los datos
demográficos de los sujetos faltantes. Los resultados de este análisis indicaron que
no hubo diferencias significativas entre los sujetos evaluables y los no evaluables en
las variables del estudio, incluidos los parámetros clínicos y demográficos clave.
Español
Criterio de valoración primario de la efectividad
El criterio de valoración primario de la efectividad fue la evidencia de fusión
radiográfica en el punto temporal de seis meses del período postoperatorio. En el
punto temporal de los seis meses, 102 de los 122 sujetos evaluables (84%) en el
grupo de tratamiento con Cervical-Stim habían logrado la fusión en comparación
con 81 de los 118 sujetos evaluables (69%) en el grupo de control (p = 0,0065).
Comparación de los resultados de la fusión radiográfica a los seis meses
Grupo de tratamiento
Control
Cervical-Stim
Número de sujetos
118
122
Número de sujetos que
lograron la fusión
81
102
Tasa de fusión (%)
68,64
83,61
Estos datos muestran que de los pacientes sometidos a la cirugía de fusión cervical,
aquellos tratados de modo complementario con el Cervical-Stim experimentaron a los
seis meses un incremento en la frecuencia de la fusión radiográfica en comparación con
el grupo de control.
6
Page 94

Se realizó un análisis adicional para medir las diferencias entre el grupo de
tratamiento con Cervical-Stim y el grupo de control en relación con las
características demográficas (género, edad, diagnóstico) y el estado de riesgo
(estado de consumo de tabaco, varios niveles). La tasa general de fusión en el grupo
Cervical-Stim siguió siendo significativa desde el punto de vista estadístico tras el
ajuste de cada una de estas variables.
El seguimiento a largo plazo (12 meses) no mostró una diferencia estadística entre
los dos grupos con respecto a la fusión. Ciento dieciséis de los 125 sujetos
evaluables (92,8%) del grupo de tratamiento con Cervical-Stim habían logrado la
fusión según el criterio de valoración final a largo plazo, mientras que 104 de los 120
sujetos evaluables (86,7%) del grupo de control lo habían logrado.
Resultados totales de la fusión radiográfica a los 12 meses
Grupo de tratamiento
Control
Cervical-Stim
Nota: las diferencias en las tasas de éxito a largo plazo entre los grupos de tratamiento no son
significativas desde el punto de vista estadístico según la prueba de la x2de Pearson con el
tamaño de la muestra disponible (x2= 2,5136, p = 0,1129).
Número de sujetos
120
125
Número de sujetos que
lograron la fusión
104
116
Tasa de fusión (%)
86,67
92,80
Criterios secundarios de valoración de la efectividad
Los criterios secundarios de valoración evaluaron los cambios en los síntomas clínicos.
Un “éxito clínico” con respecto a los síntomas se definió como el no empeoramiento de
la función neurológica, una mejoría de la evaluación del dolor según la escala VAS y el no
empeoramiento del Índice de incapacidad del cuello. Un “fracaso clínico” con respecto a
los síntomas se definió como el fracaso de alguno de estos criterios. No hubo una
diferencia significativa desde el punto de vista estadístico entre los dos grupos con
respecto al porcentaje de sujetos considerados a los seis meses como “éxito clínico”
(p = 0,8456) o a los 12 meses (p = 0,1129).
7
Page 95

Seguridad
Los eventos adversos observados en este estudio se muestran en la Tabla de
eventos adversos presentada en la sección Información de prescripción. A los seis
meses, el número de sujetos que experimentaron uno o más eventos adversos es
similar en ambos grupos. Se reportó un total de 14 eventos graves en 13 sujetos;
nueve de ellos formaban parte del grupo de tratamiento con Cervical-Stim y cinco
formaban parte del grupo de control. Estos eventos consistieron en experiencias
tales como mayor dolor, respiración entrecortada, mareos, trauma y lesión no
relacionada, muerte no relacionada, complicación quirúrgica y patología a nivel
adyacente. En los nueve sujetos del grupo de tratamiento con Cervical-Stim, todos
los eventos adversos fueron considerados, a juicio de los investigadores, como
definitivamente o probablemente, no relacionados con el dispositivo.
Los datos de seguridad obtenidos durante el tiempo transcurrido entre la consulta
del sexto mes y el contacto final con cada sujeto indican que un total de 51 sujetos
de ambos grupos experimentaron 57 eventos adversos. El número de sujetos que
experimentaron uno o más eventos adversos es similar en ambos grupos. Ninguno
de los eventos adversos reportados entre la consulta del sexto mes y el contacto
final fue grave y los eventos fueron similares a los reportados a los seis meses.
Vida útil del dispositivo
Cervical-Stim puede proporcionar hasta 270 tratamientos consecutivos (diarios) de
cuatro horas cada uno. La duración total del tratamiento la determinará el médico
basado en el paciente y el progreso de la fusión.
Tiempo de tratamiento
Cervical-Stim debe usarse durante cuatro horas al día. Al final del tratamiento
diario, el dispositivo se apagará solo. El dispositivo se puede apagar en cualquier
momento con solo presionar el botón Encendido/Apagado que se encuentra en el
panel de control.
Español
Cervical-Stim puede usarse en el momento del día que sea más conveniente para el
paciente. Es ligero y ajustable. Dado que Cervical-Stim es portátil, el tratamiento
puede recibirse mientras está sentado, caminando, reclinado, durmiendo, etc. No
obstante, dado que cada paciente tiene características únicas, el nivel de actividad
general debe estar basado en las instrucciones del médico.
8
Page 96

Cómo cargar/recargar la batería
Cervical-Stim funciona con un paquete de baterías de ion-litio recargables. El
dispositivo incluye una unidad de carga. Use únicamente el cargador de Orthofix
para cargar la batería.
Nota: la batería de Cervical-Stim debe recargarse antes de usarla por primera vez.
Botón
Unidad de
Unidad de control
carga
Puerto para
el cargador
Para cargar/recargar la batería, simplemente enchufe el extremo cilíndrico del
conector de la unidad de carga en el puerto para cargador que se encuentra en la
unidad de control. Enchufe el cable de alimentación en la unidad del cargador de
modo que quede bien fijado. Enchufe el cargador en cualquier toma de corriente
CA estándar de pared. Se requerirá aproximadamente 12 horas para cargar
completamente una batería totalmente descargada.
Encendido/Apagado
LCD
La batería de Cervical-Stim se puede recargar en cualquier momento en que no se
esté usando el dispositivo. Se recomienda categóricamente recargar el dispositivo
después de completar el tratamiento diario.
Nota: Cervical-Stim no aplicará el tratamiento mientras se esté cargando la batería.
Cuando el dispositivo esté encendido, la LCD de Cervical-Stim mostrará un
símbolo de capacidad de la batería. La figura de una batería intermitente, el
símbolo y un tono audible indican que la batería está baja y que es
necesario recargarla. Para obtener más información, consulte “Indicadores
visuales y de audio”.
9
Page 97

Funcionamiento del dispositivo
Cómo encender y apagar el dispositivo
Cervical-Stim se enciende y se apaga presionando el botón Encendido/Apagado que se
encuentra en la unidad de control del dispositivo. Cuando el dispositivo está
encendido, aparecerá momentáneamente una secuencia de mensajes de estado.
Luego, la LCD mostrará el tiempo de tratamiento restante y un logotipo intermitente
de Orthofix. El logotipo intermitente indica que el dispositivo está encendido y
funciona normalmente. (Si no ve este logotipo intermitente en la pantalla, comuníquese
con su proveedor). La unidad de control tiene un botón de luz de fondo. Cuando haya
poca luz, presione el botón de luz de fondo para que la LCD se ilumine.
Para interrumpir el tratamiento antes de que finalice la sesión de tratamiento diaria,
simplemente presione el botón Encendido/Apagado. Para reanudar el tratamiento,
presione el botón Encendido/Apagado nuevamente. La LCD mostrará el tiempo de
tratamiento restante.
Nota: para que la cuenta regresiva funcione correctamente, las sesiones de
tratamiento deben durar más de 60 minutos.
Puerto para luz infrarroja
Botón de luz de fondo
Unidad
de control
Puerto para
el cargador
Botón Encendido/Apagado
Español
LCD
Logotipo
intermitente de
Orthofix
Cronometraje de las sesiones de tratamiento
Cervical-Stim cronometra automáticamente cada sesión de tratamiento. El
cronometraje comienza al encender el dispositivo usando el botón Encendido/Apagado
que se encuentra en la unidad de control. La LCD muestra una cuenta regresiva del
tiempo restante de la sesión de tratamiento. Al final del tratamiento diario, el dispositivo
se apagará solo. Para interrumpir el tratamiento antes de que finalice una sesión de
tratamiento, simplemente presione el botón Encendido/Apagado. Para reanudar el
tratamiento, presione el botón Encendido/Apagado nuevamente. La LCD mostrará el
tiempo de tratamiento restante.
Nota: para que la cuenta regresiva funcione correctamente, las sesiones de
tratamiento deben durar más de 60 minutos.
10
Page 98

Cómo usar el dispositivo
Cervical-Stim está diseñado para la región cervical y puede usarse con o sin una
órtesis. Para mayor comodidad, se debe usar ropa entre la piel y el Cervical-Stim.
Para usar el Cervical-Stim, basta con que deslice el dispositivo por la cabeza hasta
que quede cómodamente apoyado sobre el cuello y los hombros (véase la figura
que aparece a continuación). También puede abrir el dispositivo con la hebilla y
colocarlo apoyado sobre el cuello y los hombros. Ajuste la hebilla como si fuera un
cinturón de seguridad, para fijar la unidad.
El Cervical-Stim trae un cuello acolchado para mejorar el ajuste y el apoyo.
Esecuello acolchado se puede colocar o quitar del collarín interior del dispositivo
mediante las lengüetas de Velcro, que lo unen directamente al material.
Velcro
Unión
con
hebilla
11
Page 99

Cuidado y limpieza
Cervical-Stim es un dispositivo electrónico sofisticado y se debe manipular con
cuidado. Dejar caer el Cervical-Stim o tratarlo en forma indebida puede dañar el
dispositivo.
NO exponga el Cervical-Stim a la luz solar directa durante períodos
prolongados. La unidad de control del dispositivo se puede dañar.
NO exponga el Cervical-Stim a calor excesivo. En climas cálidos, la
temperatura dentro de un automóvil o un maletero puede ser mayor
de 160 ºF/71 ºC. El calor excesivo puede dañar la unidad de control
del dispositivo.
NO exponga el Cervical-Stim a humedad excesiva.
NO elimine el Cervical-Stim en un incinerador.
NO use solventes para limpiar el Cervical-Stim.
Limpie el dispositivo con un paño suave y húmedo.
Viajes
Se debe aconsejar a los pacientes que cuando viajen por avión, lo mejor es registrar
el Cervical-Stim con el equipaje. Si el dispositivo se lleva a bordo del avión, no debe
llevarse puesto al pasar por los controles de inspección de seguridad. El CervicalStim se podría dañar. Se debe llevar también el manual de instrucciones del
Cervical-Stim a fin de identificar el dispositivo con rapidez y facilidad ante cualquier
personal de seguridad.
Español
Almacenamiento
Cervical-Stim deberá almacenarse a una temperatura que esté entre 14 ºF a 113 ºF
(-10 ºC a 45 ºC)
El rango de temperaturas de funcionamiento para el Cervical-Stim deberá ser de
41 ºF a 104 ºF (+5 ºC a 40 ºC)
Humedad relativa: hasta 95%, sin condensación
Eliminación
Cervical-Stim contiene baterías de litio, no lo elimine en un incinerador. Deseche el
dispositivo apropiadamente.
12
Page 100

Indicadores visuales y de audio
La LCD y las alarmas audibles están diseñadas para proporcionar información útil al
usuario. El cuadro que figura a continuación muestra los diversos símbolos y las
alarmas, y su significado.
Indicadores visuales de la LCD e indicadores de audio de Cervical-Stim
Símbolo/Alarma Descripción Significado
todos los símbolos de la LCD
visibles y la alarma audible
continua durante
aproximadamente 5 segundos
el cronómetro de cuenta regresiva
muestra el tiempo de tratamiento
restante (horas y minutos) el
logotipo de Orthofix parpadea
el cronómetro de cuenta
regresiva muestra tres rayas
alarma audible (5 tonos)
símbolo fijo durante
aproximadamente 5 segundos
los símbolos parpadean/alarma
audible (aproximadamente 1
tono por segundo)
el símbolo fijo indica el % de
carga aproximado
si el símbolo se llena una y otra
vez, indica modo de carga
alarma audible continua
autoprueba de
encendido
tratamiento normal
en proceso
no hay tiempo de
tratamiento restante
tratamiento finalizado/
apague
batería baja —
recargue
estado de la batería:
carga restante o
modo de carga
dispositivo bloqueado
— llame al servicio
técnico
aparece cualquier código E
(p. ej., E01, E02. . .)
13
mensaje de error —
llame al servicio
técnico
 Loading...
Loading...