

Page 1

Orthofix Inc.
U.S.A. Edition - Physician
P/N 571355-0001 Rev C 3/2009
Printed in U.S.A.
Date of Printing: 3/ 2009
CS-0903(A)-PL-US © Orthofix Inc.
Manufactured by:
Orthofix Inc.
1720 Bray Central Drive
McKinney, Texas 75069
Tel 469-742-2500
800-535-4492
Cervical Fusion System
U.S.A. EDITION
PH Y S I CI A N MA N U A L
Page 2

Table of Contents
Cervical-Stim
®
Physician Manual
Device Description ........ .................................... ................... ....................... 1
Prescription Information ................................................. ............................. 2
Clinical Data Summary .................. .................................... ................... ...... 3
Data Analysis & Results ............ ....................................................... ............ 5
Effectiveness Results .................................................... ................................. 6
Secondary Effectiveness Endpoints ............................................ .................... 7
Safety ................................. ....................................................... .................. 8
Device Life .. ....................................................... ......................................... 8
Treatment Time ........................................................................................... 8
Charging/Recharging the Battery ................................................................... 9
Device Operation ...... .................................... ................... .......................... 10
Wearing the Device ............. ....................................................... ................ 11
Care and Cleaning ............ .................................... ................... ................... 12
Travel ..................................................... .................................................... 12
Storage ... .................................... ................... .................................... ........ 12
Disposal ......................................................... ................... .......................... 12
Service .................. ......................... ................... .................................... ....... 12
Visual and Audio Indicators ..... ..................................................................... 13
Equipment Classification and Symbol Description ......................................... 14
Warranty Information ............................... ................... ............................... 16
To learn more about Orthofix, please visit our website at www.orthofix.com.
Package Contents:
1- Cervical-Stim Cervical Fusion System
1- Literature Pack
1- Charging Unit #270215
THIS DEVICE IS NONSTERILE.
IT DOES NOT REQUIRE STERILIZATION.
Page 3
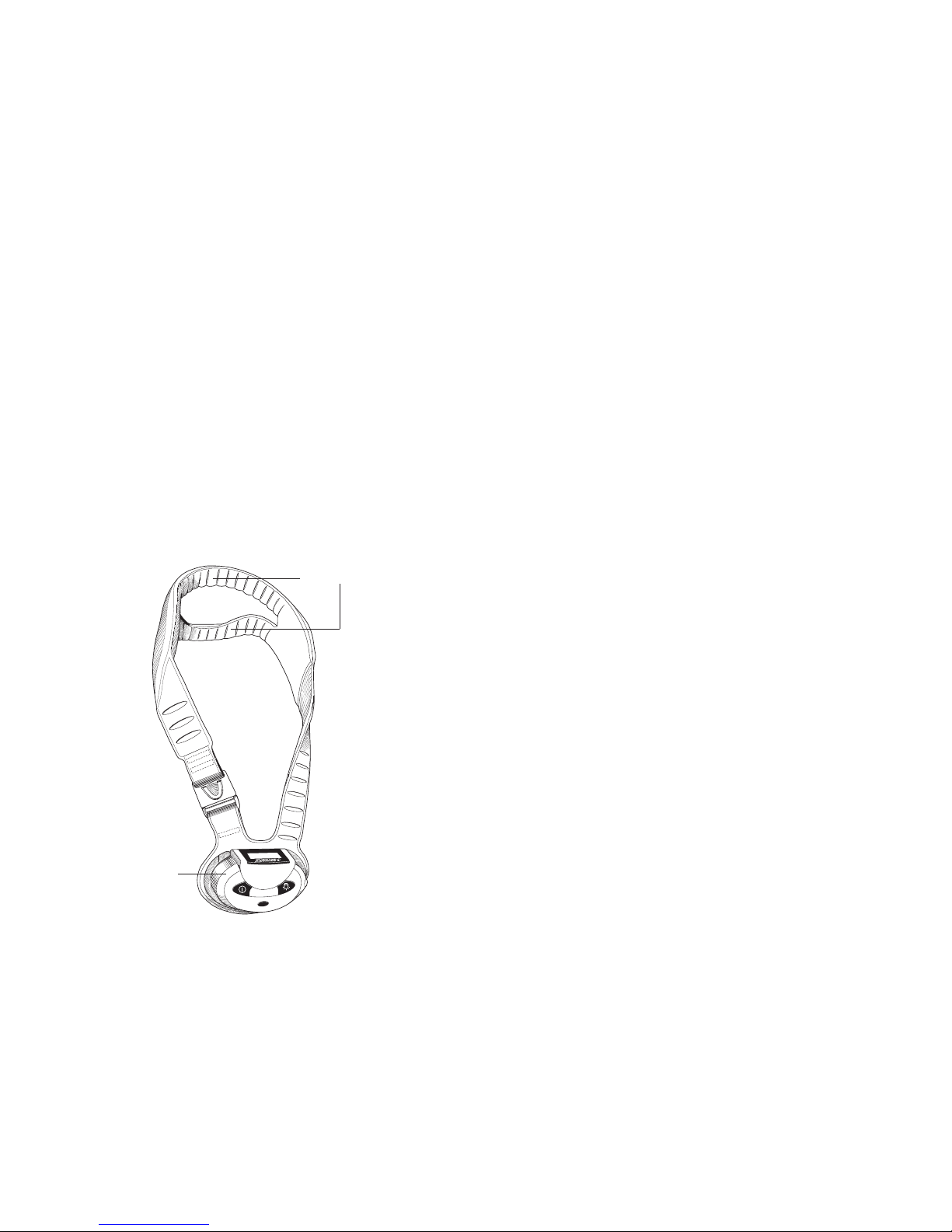
1
Device Description
The Cervical-Stim®Cervical Fusion System is an external, low-level, Pulsed
Electromagnetic Field (PEMF) device. It is a single-piece device that is
lightweight, flexible and portable, allowing freedom of movement during
treatment. A Liquid Crystal Display (LCD) and audible alarm provide
information during treatment (e.g. operational status, treatment time remaining,
battery capacity, etc.). See “Visual and Audio Indicators” for more information.
Cervical-Stim Cervical Fusion System - Model 2505
The Cervical-Stim is comprised of a control unit and a treatment transducer.
The control unit contains a micro-processor that generates the Cervical-Stim
electrical signal. That signal is converted to a highly uniform, low-energy
magnetic field by the treatment transducer. When the device is centered over
the treatment area, the therapeutic PEMF signal is delivered directly to the
fusion site.
The Cervical-Stim is powered by a rechargeable lithium-ion battery pack.
The LCD and audible alarm will alert the patient when the battery is low and
needs to be recharged. See “Charging/Recharging the Battery” for more
information.
2
To ensure that the device is functioning properly, the Cervical-Stim constantly
monitors battery voltage and the electrical signal. If at any time during
treatment the device stops functioning properly, the LCD will display an
appropriate symbol or error code. See “Visual and Audio Indicators” for
more information.
To stop treatment prior to the end of the daily treatment session, simply press
the On/Off button. To resume treatment, press the On/Off button again.
The LCD will display the remaining treatment time.
Prescription Information
Indication
The Cervical-Stim is a noninvasive, pulsed electromagnetic bone growth
stimulator indicated as an adjunct to cervical fusion surgery in patients at
high-risk for non-fusion.
Contraindications
There are no known contraindications for the Cervical-Stim as an adjunct to
cervical spine fusion surgery.
Warnings
• The Cervical-Stim may interfere with the operation of a cardiac pacemaker
or defibrillator. Consultation with the attending cardiologist is recommended.
• The Cervical-Stim should be removed prior to any imaging procedures
(e.g., CT scan, MRI, etc.).
Precautions
• The Cervical-Stim should not be used if there are mental or physical
conditions that may preclude compliance with physician or device
instructions.
• The Cervical-Stim has not been evaluated in treating patients with the
following conditions: osseous or ligamentous spinal trauma, spondylitis,
Paget’s disease, moderate to severe osteoporosis, metastatic cancer, renal
disease, rheumatoid arthritis, uncontrolled diabetes mellitus, patients prone
to vascular migraine headache, seizure, epilepsy, thyroid conditions or
neurological diseases.
• Animal teratological studies performed with this device did not show any
adverse effects in animals. However, the safety of this device for use on
patients who are pregnant or nursing has not been established.
Transducer
Control Unit
Page 4

3 4
symptomatic radiculopathy. The purpose of the study was to evaluate the
safety and effectiveness of the PEMF Cervical-Stim device as an adjunct for
high-risk patients who undergo cervical fusion. All subjects underwent
anterior cervical discectomy and fusion using the Smith-Robinson technique
with the Atlantis Plate. Subjects were randomly assigned to either the control
group (standard treatment, n=160) or the treatment group (standard treatment
plus the Cervical-Stim, n=163). Standard treatment was at the physician's
discretion but typically included the standard hospital stay, use of a soft cervical
collar, appropriate medications and physical therapy.
Subjects who met the following inclusion and exclusion criteria were eligible
for participation in the study:
Inclusion Criteria
Adult male or female, 18-75 years old with radiographic evidence of
compressed cervical nerve root(s), symptomatic radiculopathy, pain of 5 or
greater on the Visual Analog Scale (VAS) and/or any muscle weakness and
primary cervical spinal fusion performed using the Smith-Robinson technique
with allograft bone and an anterior cervical plate. The fusion procedure must
have been either multi-level (>1 fusion level) or the subject was a smoker
(one pack/day or more) or both; and have a signed informed consent form.
Exclusion Criteria
Traumatic cervical injury, posterior approach or revision fusion, autograft
or bone substitute materials for graft source, history of vascular migraine
headache or prone to uncontrolled seizures or epilepsy (controlled or
uncontrolled) or any neurological diseases or injury; depressed immune
system, regional conditions (Spondylitis, Paget’s disease, rheumatoid arthritis),
infection (systemic or local) within 2 weeks prior to surgery, systemic
conditions (cancer, cardiac arrhythmia, thyroid disease, uncontrolled diabetes
mellitus, renal disease/dysfunction, chronic steroid use or other conditions that
may have affected bone metabolism), cardiac pacemakers, defibrillators, dorsal
column stimulators, hearing aids, cochlear prostheses and cranial stimulators,
subjects who were pregnant, nursing or had planned to become pregnant
within 12 months, subjects that had participated in other clinical studies within
the last 12 months, or had mental or physical conditions which may have
precluded compliance with physician instructions.
Evaluation and Follow-Up
Follow-up visits were to have been performed at months 1, 2, 3, 6 and 12
and annually thereafter until the last subject enrolled reached 12 months.
Device Usage
Subjects assigned to the treatment group (Cervical-Stim) were instructed
to wear the device for four hours per day for a minimum of three months
postoperative. Surgeons could, at their discretion, extend the Cervical-Stim
treatment up to six months postoperative.
Clinical Data Summary
Study Design
The Cervical-Stim clinical study was a controlled, randomized, parallel
group study of 323 high-risk (smokers, multi-level or both, and allograft) adult
subjects with radiographic evidence of compressed cervical nerve roots and
Adverse Events Reported at 6 Months by Treatment Group
Increased Neck Pain
S
houlder/Arm Pain
Re-Injury to Cervical Spine
Adjacent Level Pathology
Surgical Complications
LBP/Lumbar Pathology
Trauma/Injury(not cervical)
Numbness/Tingling
Headache/Migraine
Nonspecific/Unrelated Pain
Nausea
Dizziness/Vertigo
Rash/Discoloration
Rapid/Irregular Heartbeat
Shortness of Breath
Ringing in Ears
Neurologic Symptom/Stroke
Lump in Throat
Diagnosis of Diabetes
Diagnosis of Breast Cancer
Seizure
Death, Unrelated
Tenderness
Screw Broken
Graft Collapse
Carpal Tunnel Syndrome
Choking Sensation
Cardiac Symptoms
Nephrotic Syndrome
Suicide Attempt
TOTAL
Adverse Events
# (%) of
E
vents
1
0(14.9)
10(14.9)
10(14.9)
3
(4.5)
2(3.0)
8(11.9)
2(3.0)
6(8.9)
2(3.0)
2(3.0)
0
2(3.0)
0
0
0
0
1(1.5)
0
0
0
0
0
1(1.5)
1(1.5)
1(1.5)
2(3.0)
1(1.5)
1(1.5)
1(1.5)
1(1.5)
67
# (%)
1
of
P
atients
E
xperiencing
the Event
9
(5.6)
9(5.6)
8(5,0)
3
(1.9)
2(1.3)
8(5.0)
2(1.3)
6(3.8)
2(1.3)
2(1.3)
0
2(1.3)
0
0
0
0
1(0.6)
0
0
0
0
0
1(0.6)
1(0.6)
1(0.6)
2(1.3)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
47
2
#*(%) of
E
vents
1
6(17.8)
16(17.8)
9(10.0)
8
(8.8)
7(7.7)
5(5.5)
5(5.5)
4(4.4)
4(4.4)
3(3.3)
2(2.2)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
1(1.1)
0
0
0
0
0
0
0
0
90
# (%)
1
of
P
atients
E
xperiencing
the Event
1
5(9.2)
16(9.8)
9(5.5)
8
(4.9)
5(3.1)
5(3.1)
4(2.5)
4(2.5)
4(2.5)
3(1.8)
2(1.2)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
1(0.6)
0
0
0
0
0
0
0
0
58
2
Control Group (n=160)
Cervical-Stim Group (n=163)
Adverse Events
1
% expressed as number of patients experiencing the event / total number of patients in the group
2
Some patients experienced multiple adverse events
*
There were several adverse events that were more frequently observed in the Cervical-Stim group than in the
control group. Given the types of events, it is unlikely that these adverse events are related to the treatment.
Page 5

For purposes of device evaluation, all films were scanned into a central database
and reviewed by two independent, blinded orthopedic surgeons and a blinded,
independent radiologist following completion of the entire study. Films were
viewed and scored using a common protocol. All films at each time point were
evaluated for amount of radiolucency, bony bridging and degree of motion as
evidenced on the flexion/extension cervical spine films. A software program
was used to calculate motion. Results obtained in this fashion were reviewed
and verified by the reviewing orthopedic surgeons. The radiologist’s diagnosis
was considered definitive in the case of a disagreement between the two
orthopedic surgeons.
Effectiveness Results
Of the 323 subjects who were randomized and received surgery, 240 were
evaluable for the effectiveness analysis (Cervical-Stim treatment group, n=122;
control group, n=118). Subjects were deemed unevaluable for the following
reasons: non-existent or non-readable x-rays, subject non-compliance,
protocol violations (inclusion criteria), graft collapse, broken internal hardware,
early study exits due to minor adverse experiences, and one suicide attempt.
The success or failure of these subjects is not known. These unavailable data
could positively or negatively affect the overall success of the study. In order
to assess the impact of the missing data, sensitivity analyses were performed.
These included last observation carried forward and all missing data imputed
as non-fusion. Both of these analyses showed that the results at six months
were still statistically significantly different in favor of the Cervical-Stim group.
In addition, the baseline demographic data from the evaluable population was
compared to the demographic data of the missing subjects. The results of this
analysis indicated there were no significant differences between the evaluable
subjects and the non-evaluable subjects in 14 study variables including key
demographics and clinical parameters.
Primary Effectiveness Endpoint
The primary effectiveness endpoint was evidence of radiographic fusion at the
six month time point postoperative. At the six month time point, 102 of the
122 evaluable subjects (84%) in the Cervical-Stim treatment group were
judged to be fused versus 81 of the 118 evaluable subjects (69%) in the control
group (p=0.0065).
6
These data show that for patients undergoing cervical fusion surgery, patients
treated adjunctively with the Cervical-Stim experienced an increase in the
frequency of radiographic fusion at six months when compared to the control group.
Treatment Group
Control
Cervical-Stim
Number of Subjects
118
122
Number of Subjects
Fused
81
102
Fusion Rate (%)
68.64
83.61
1
. P-values of comparison tests between treatment groups using Student’s t-test for numerical
variables and Pearson x2test for categorical variables.
Data Analysis and Results
The primary effectiveness endpoint was the increase in frequency of cervical
fusion success by six months postoperatively as assessed by radiographic
evidence. Secondary endpoints were neurological function, VAS pain assessment
and Neck Disability Index. Safety was assessed by the frequency and severity
of adverse events.
Fusion was assessed by radiographs at each visit:
Radiographic fusion was defined as > 50% bony bridging on both the
superior and inferior graft interfaces between adjacent vertebral bodies
AND < 4° angulation (motion) between adjacent fused vertebrae on
flexion/extension lateral films AND absence of radiolucency.
Radiographic non-fusion was defined as < 50% bony bridging at either the
superior or inferior graft interface OR > 4° angulation (motion) between
adjacent fused vertebrae on flexion/extension lateral films OR presence
of radiolucency.
5
V
ariables
Age (years)
Mean
Range
SD
Gender
Female
Male
Race
Caucasian
African-American
Hispanic
Asian
Other
Smoking Status
Nonsmoking
Smoking
Number of
Subjects
(N = 323)
46.8
24 – 73
9.3
148(45.8%)
175 (54.2%)
301(93.2%)
17 ( 5.3%)
5 ( 1.6%)
0
0
159 (49.2%)
164 (50.8%)
Control
(n = 160)
46.7
26 – 72
9.2
75 (46.9%)
85 (53.1%)
150 (93.8%)
7 ( 4.4%)
3 ( 1.9%)
-
-
79 (49.4%)
81 (50.6%)
Cervical-Stim
(n = 163)
46.9
24-73
9.4
73 (44.8%)
90 (55.2%)
151 (92.6%)
10 ( 6.1%)
2 ( 1.2%)
-
-
80 (49.1%)
83 (50.9%)
P-value
1
0.846
0.706
0.703
0.958
Baseline Demographic Characteristics
Comparison of Radiographic Fusion Outcomes at Six Months
Demographic Data
The subjects in this study had a mean age of 46.8 years (range 24 to 73 years).
Of the 323 subjects, 148 (45.8%) were female and 175 (54.2%) were male.
Three hundred one (93.2%) were Caucasian, while 17 (5.3%) were African
American and 5 (1.6%) were Hispanic. One hundred fifty-nine (49.2%) were
nonsmokers and 164 (50.8%) were smokers.
Page 6

8
Safety
The adverse events observed in this study are shown in the Adverse Events
Table presented in the Prescription Information section. At six months, the
numbers of subjects who experienced one or more adverse events is similar
in the two groups. A total of 14 severe events were reported in 13 subjects;
nine of the subjects were in the Cervical-Stim treatment group and five
subjects were in the control group. These events included experiences such
as increased pain, shortness of breath, dizziness, unrelated trauma and injury,
unrelated death, surgical complication and adjacent level pathology. For the
nine subjects in the Cervical-Stim treatment group, all severe adverse events
were, in the judgment of the investigators, definitely or probably unrelated to
the device.
Safety data obtained between the six month visit and the final contact with
each subject indicate that 57 adverse events were experienced by a total of
51 subjects between both groups. The number of subjects who experienced
one or more adverse events is similar in the two groups. None of the adverse
events reported between the six month visit and the final contact were severe
and are similar to those reported at six months.
Device Life
The Cervical-Stim can provide up to 270 consecutive (daily) treatments of four
hours each. The overall length of treatment will be determined by the physician
based on the patient and progress toward fusion.
Treatment Time
The Cervical-Stim should be worn for four hours per day. The device will
automatically turn off when four hours of treatment is reached in a 24-hour
day. The device may be turned off at any time by simply pressing the On/Off
button on the control panel.
The Cervical-Stim may be used at any time of day that is convenient for the
patient. It is lightweight and adjustable. And because the Cervical-Stim is
portable, treatment can be received while sitting, walking, reclining, sleeping,
etc. However, since each patient is unique, the overall activity level should be
based on your instructions.
7
Treatment Group
Control
Cervical-Stim
Number of Subjects
120
125
Number of Subjects Fused
104
116
Fusion Rate (%)
86.67
92.80
Overall Radiographic Fusion Outcomes at 12 Months
Note: The differences in long-term success rates between treatment groups is not statistically
significant per Pearson x
2
test with the available sample size (x2= 2.5136, p = 0.1129).
An additional analysis was performed to allow for the differences between
the Cervical-Stim treatment group and the control group with respect to
demographic characteristics (gender, age, diagnosis) and risk status (smoking,
multilevel). The overall fusion rate in the Cervical-Stim group remained
statistically significant after adjustment for each of these variables.
Long-term follow-up (12 months) showed no statistical difference between the
two groups with respect to fusion. One hundred sixteen of the 125 evaluable
subjects (92.8%) in the Cervical-Stim treatment group were judged to be fused
at the long-term final endpoint, while 104 of the 120 evaluable subjects
(86.7%) in the control group were judged to be fused.
Secondary Effectiveness Endpoints
Secondary endpoints evaluated changes in clinical symptoms. A “clinical success”
with regard to symptoms was defined as no worsening in neurological function,
an improvement in VAS pain assessment and no worsening in Neck Disability
Index. A “clinical failure” with regard to symptoms was defined as failure for
any one of these criteria. There was no statistically significant difference
between the two groups with respect to the percent of subjects considered
a “clinical success” at six months (p= 0.8456) or at 12 months (p= 0.1129).
Page 7

109
Device Operation
Turning the Device On and Off
The Cervical-Stim is turned on and off by pressing the On/Off button on the
control unit of the device. When the device is on, a sequence of status
messages will display momentarily. The LCD should then show the treatment
time remaining and a flashing Orthofix logo. The flashing logo indicates that
the device is on and functioning normally. (If you do not see this on the display,
contact your provider.) A backlight button is on the control unit. In low light,
press the backlight button for illumination of the LCD.
To stop treatment prior to the end of the daily treatment session, simply press
the On/Off button. To resume treatment, press the On/Off button again.
The LCD will display the remaining treatment time.
Timing of Treatment Sessions
The Cervical-Stim automatically times each treatment session. The timing
begins when the device is turned on using the On/Off button on the control
unit. The LCD shows a countdown of the time remaining in the treatment
session. At the end of daily treatment, the device will turn itself off. To stop
treatment prior to the end of a treatment session, simply press the On/Off
button. To resume treatment, press the On/Off button again. The LCD will
display the remaining treatment time.
Note: For the countdown to function correctly, treatment sessions should be
greater than 60 minutes duration.
Control Unit
On/Off Button
Charger
Port
Backlight Button
LCD
Flashing
Orthofix Logo
IR Port
Charging/Recharging the Battery
The Cervical-Stim is powered by a rechargeable lithium-ion battery pack.
A charger unit is provided with the device. Use only the Orthofix charger to
charge the battery.
Note: The Cervical-Stim battery will require charging prior to the first use.
To charge/recharge the battery, simply plug the barrel connector end of the
charger unit into the charger port located on the control unit. Plug the line
cord securely into the charger unit. Plug the charger into any standard AC
wall outlet. A fully discharged battery will require approximately 12 hours
to charge completely.
The Cervical-Stim battery can be recharged at any time the device is not in
use. It is strongly recommended that the device be recharged after completing
daily treatment.
Note: The Cervical-Stim will not deliver treatment while charging.
When the device is on, the Cervical-Stim LCD will show a battery capacity
symbol. A flashing battery outline, the symbol and an audible beep
indicate a battery low condition and that the battery requires recharging.
See “Visual and Audio Indicators” for more information.
Control Unit
Charger Unit
Page 8

1211
Travel
When traveling by air, it is best to check the Cervical-Stim with the luggage.
If the device is taken on board the airplane, it should not be worn when
passing through passenger screening devices. The Cervical-Stim could be
damaged. The Cervical-Stim user manual should be taken to quickly and easily
identify the device for any security personnel.
Storage
The Cervical-Stim should be stored within 14ºF to 113ºF (-10º C to 45º C)
The Cervical-Stim operating temperature range should be within
41ºF to 104ºF (+5º C to 40º C)
Relative Humidity: Up to 95%, non-condensing
Disposal
The Cervical-Stim is for single patient use. Product contains lithium
batteries; do not incinerate. Dispose of device properly to prevent
injury. Please dispose of this product at collection facilities for waste
electrical equipment used in household.
Service
If you have questions concerning the device or require any assistance, please call
469-742-2500 or 800-535-4492. There are no user serviceable parts. Notify the
manufacturer for any servicing needs.
Care and Cleaning
The Cervical-Stim is a sophisticated electronic device and should be handled
with care. Dropping or other mistreatment of the Cervical-Stim may cause
damage to the device.
DO NOT expose the Cervical-Stim to direct sunlight
for long periods of time. The device control unit may be damaged.
DO NOT expose the Cervical-Stim to excessive heat. In warm
climates, the temperature in a car or trunk can exceed 160ºF /
71ºC. Excessive heat can damage the control unit of the device.
DO NOT expose the Cervical-Stim to excessive moisture.
Moisture can damage the electronic components of the device
and the device may stop working.
DO NOT dispose of the Cervical-Stim in an incinerator.
DO NOT use solvents to clean the Cervical-Stim.
Clean the device by wiping with a soft, damp cloth.
Wearing the Device
The Cervical-Stim is intended for the cervical spine and may be worn with or
without a brace. For better comfort, clothing should be worn between the
skin and the Cervical-Stim.
To use the Cervical-Stim, simply slip the device over the head so that it rests
comfortably against the neck and shoulders (see figure below). Or, the device
may be opened at the buckle and placed against the neck and shoulders.
Fasten the buckle like a seatbelt to secure the unit.
Buckle
Joint
Page 9

1413
Visual and Audio Indicators
The LCD and audible alarms are designed to provide helpful information
to the user. The chart below shows the various displays and alarms and
their meaning.
Cervical-Stim LCD Visual and Audio Indicators
The use of accessories other than those specified may result in increased
emissions or decreased immunity of the device.
The battery charger is provided with a 3-wire appliance inlet but considered
double insulated with Class II construction throughout.
For safe usage, follow manufacturer instructions when using the product.
Use of the product in any other manner could have harmful effects and/or void
the warranty.
Note: Inspect the device prior to each use for wear or deterioration.
Do not use if the device does not appear to be in suitable condition.
Symbol /Alarm Description Meaning
all LCD symbols visible and
continuous audible alarm for
approximately 5 seconds
countdown timer displays
remaining treatment time
(hours & minutes)
Orthofix logo flashes
countdown timer
displays three dashes
audible alarm (5 beeps)
steady symbol
for approximately 5 seconds
symbols flash / audible alarm
(approximately 1 beep
per second)
steady symbol indicates
approximate % of charge
symbol filling repeatedly
indicates charge mode
continuous audible alarm
display of any E code
(e.g., E01, E02 . . .)
power-on self test
normal treatment
in Progress
no treatment time
remaining
treatment complete —
power off
battery low —
recharge required
battery status —
remaining charge
or charging mode
device locked —
call for service
error message —
call for service
Equipment Classification and Device Symbol Descriptions
Equipment Classifications
• Internally powered equipment
• Type BF applied part
• IEC 529 enclosure rating: IPXO
• Equipment not suitable for use in the presence of a flammable anaesthetic
mixture with air or nitrous oxide
• Mode of operation: intermittent operation
Symbol Meaning
Attention - Refer to Instructions for Use
Type BF Applied Part
On/Off
Backlight Button
Storage Temperature Range
Year of Manufacture for Active Device
Charger Port
Page 10

1615
Conformance to Standards
The Orthofix Cervical-Stim Cervical Fusion System conforms to the following
worldwide series of standards:
UL 60601 - Medical Electrical Equipment, General Requirements for Safety;
(including Electromagnetic Compatibility and Interference)
IEC 60601 - Medical Electrical Equipment, Safety Requirements for Medical
Electrical Systems (including Electromagnetic Compatibility and
Interference)
Orthofix Compliance Printer Instructions
Patient Compliance Monitoring
The Orthofix Compliance Data Printer is an accessory to the Orthofix line
of Pulsed Electromagnetic Field (PEMF) Bone Growth Stimulators. While
delivering the clinically proven PEMF signal to the treatment site, each Orthofix
Bone Growth Stimulator monitors and tracks patient compliance by recording
the elapsed daily and total patient treatment time. Accumulated compliance
data are retained in a physician-accessed memory within the Bone Growth
Stimulator. This tracking system assists the physician by ensuring that the
patient is compliant with the prescribed treatment time.
Compliance data can be retrieved with the Stimulator alone or in conjunction
with the Orthofix Compliance Printer. When the Compliance Printer is
utilized with the Orthofix stimulator, a hard copy printout of the patient’s
treatment record can be obtained. This is a useful tool in assessing and
documenting compliance to the prescribed daily stimulation.
The Orthofix Compliance Data Printer is a thermal paper printer that allows the
physician to print hard copy patient compliance data. The printer is powered
by connection to standard AC wall outlets (100 – 240 VAC, 50/60 Hz) and
communicates with the Orthofix Bone Growth Stimulators by means of an
infrared interface much like a television remote control.
Please see the Orthofix Compliance Data Monitoring System Guide
For Doctors for complete accessory printer instructions.
Warranty Information
Orthofix Inc. warrants the Cervical-Stim bone growth stimulator to be free
from defects in materials and workmanship for one year from the date of
first use. Provided that all terms and conditions of this Limited Warranty are
complied with, Orthofix Inc. will replace defective components.
This Limited Warranty applies to the product only under normal use and does
not cover any damage or defect caused by accident, misuse, abuse, fire, flood,
and acts of God or by any alteration, tampering, repair or attempted repair by
anyone other than Orthofix Inc. This warranty only applies to the patient for
whom the product is prescribed and is not assignable or transferable.
Defective products covered by this Limited Warranty must be returned to
Orthofix Inc. Attention: Orthofix Returns. You must call a Customer Service
Representative at 1-800-535-4492 or your local distributor to obtain the
Return Authorization (RA) number and address prior to returning the product.
Except as specifically required by applicable law, the foregoing warranty is in
lieu of all other warranties, expressed or implied and Orthofix Inc. specifically
disclaims any and all warranties of merchantability or fitness for a particular
purpose. Under no circumstances shall Orthofix Inc., its authorized
representative, affiliated or subsidiary companies be liable for special,
consequential or incidental damages. The sole remedy with respect to any
defective product shall be limited to replacement.
This Limited Warranty may not be extended or modified except in writing
by Orthofix Inc. No sales person, representative, distributor or doctor is
authorized to make or consent to any extension or modification of the terms
of this Limited Warranty.
For additional information and/or device assistance, contact Orthofix Customer
Service at 800-535-4492.
Page 11

Sistema de fusión cervical
EDICIÓN PARA LOS EE. UU.
Orthofix Inc.
U.S.A. Edition - Physician
P/N 571355-0001 Rev C 3/2009
Printed in U.S.A.
Date of Printing: 3/ 2009
CS-0903(A)-PL-US © Orthofix Inc.
Manufactured by:
Orthofix Inc.
1720 Bray Central Drive
McKinney, Texas 75069
Tel 469-742-2500
800-535-4492
MA N U A L D E L M É DI C O
Page 12

1
Descripción del dispositivo
El sistema de fusión cervical Cervical-Stim®es un dispositivo externo de campo
electromagnético pulsado de bajo nivel. Es un dispositivo de una sola pieza que es
ligero, flexible y portátil, lo que proporciona libertad de movimiento durante el
tratamiento. Una pantalla de cristal líquido (LCD) y una alarma audible proporcionan
información al paciente durante el tratamiento (p.ej., el estado operativo, el tiempo
de tratamiento restante, la capacidad de la batería, etc.). Para obtener más
información, consulte “Indicadores visuales y de audio”.
Sistema de fusión cervical Cervical-Stim – Modelo 2505
Cervical-Stim incluye una unidad de control y un transductor de tratamiento. La
unidad de control contiene un microprocesador que genera la señal eléctrica de
Cervical-Stim. El transductor de tratamiento convierte dicha señal en un campo
magnético de baja energía altamente uniforme. Cuando el dispositivo está centrado
sobre el área de tratamiento, la señal CEMP terapéutica es aplicada directamente en
el lugar de tratamiento.
Cervical-Stim funciona con un paquete de baterías de ion-litio recargables. La LCD y
la alarma audible advertirán al paciente cuando la batería esté baja y deba ser
recargada. Para obtener más información, consulte “Cómo cargar/recargar la
batería”.
Transducer
Control Unit
Índice
Manual del médico para el uso del Cervical-Stim
®
Descripción del dispositivo ............................................................................... 1
Información de prescripción ............................................................................. 2
Resumen de datos clínicos ................................................................................ 3
Análisis de los datos y resultados ...................................................................... 5
Resultados sobre la efectividad ......................................................................... 6
Criterios de valoración secundarios de la efectividad ....................................... 7
Seguridad ........................................................................................................... 8
Vida útil del dispositivo ...................................................................................... 8
Tiempo del tratamiento .................................................................................... 8
Cómo cargar/recargar la batería ....................................................................... 9
Funcionamiento del dispositivo ....................................................................... 10
Cómo usar el dispositivo ................................................................................. 11
Cuidado y limpieza .......................................................................................... 12
Viajes ............................................................................................................... 12
Almacenamiento .............................................................................................. 12
Eliminación ........................................................................................................12
Servicio técnico ............................................................................................... 12
Indicadores visuales y de audio ....................................................................... 13
Clasificación del equipo y descripciones de los símbolos del dispositivo ........ 14
Información de la garantía ............................................................................... 16
Para obtener más información acerca de Orthofix, visite nuestro sitio web
en www.orthofix.com.
Contenido del paquete:
1- Sistema de fusión cervical Cervical-Stim
1- Paquete de material impreso
1- Unidad de carga n.º 270215
ESTE DISPOSITIVO NO ESTÁ ESTERILIZADO.
NO REQUIERE ESTERILIZACIÓN.
Page 13

3
Resumen de datos clínicos
Diseño del estudio
El estudio clínico de Cervical-Stim fue un estudio de grupo, controlado, paralelo y
aleatorizado de 323 sujetos adultos de alto riesgo (fumadores y/o con fusión de
varios niveles y aloinjerto) con evidencia radiográfica de raíces de nervios cervicales
comprimidas y radiculopatía sintomática. El propósito del estudio fue evaluar la
Eventos adversos reportados a los 6 meses por el grupo del tratamiento
Más dolor en el cuello
D
olor en el hombro/brazo
Nueva lesión en la región cervical
Patología a nivel adyacente
Complicaciones quirúrgicas
Dolor en la parte baja de la espalda/Patología lumbar
Trauma/Lesión (no cervical)
Entumecimiento/Hormigueo
Dolor de cabeza/Migraña
Dolor no específico/no relacionado
Náuseas
Mareos/Vértigo
Erupción cutánea/Decoloración
Latidos rápidos/irregulares del corazón
Respiración entrecortada
Zumbido en los oídos
Síntoma neurológico/Apoplejía
Nódulo en la garganta
Diagnóstico de diabetes
Diagnóstico de cáncer del seno
Convulsión
Muerte, no relacionada
Sensibilidad
Rotura de tornillo
Fallo del injerto
Síndrome del túnel carpiano
Sensación de ahogo
Síntomas cardíacos
Síndrome nefrótico
Intento de suicidio
TOTAL
Eventos adversos
# (%) de
e
ventos
1
0 (14.9)
10 (14.9)
10 (14.9)
3
(4.5)
2 (3.0)
8 (11.9)
2 (3.0)
6 (8.9)
2 (3.0)
2 (3.0)
0
2 (3.0)
0
0
0
0
1 (1.5)
0
0
0
0
0
1 (1.5)
1 (1.5)
1 (1.5)
2 (3.0)
1 (1.5)
1 (1.5)
1 (1.5)
1 (1.5)
67
N.º (%)
1
de
p
acientes que
e
xperimentaron
el evento
9
(5.6)
9 (5.6)
8 (5.0)
3
(1.9)
2 (1.3)
8 (5.0)
2 (1.3)
6 (3.8)
2 (1.3)
2 (1.3)
0
2 (1.3)
0
0
0
0
1 (0.6)
0
0
0
0
0
1 (0.6)
1 (0.6)
1 (0.6)
2 (1.3)
1 (0.6)
1 (0.6)
1 (0.6)
1 (0.6)
47
2
N.º *(%) de
e
ventos
1
6 (17.8)
16 (17.8)
9 (10.0)
8
(8.8)
7 (7.7)
5 (5.5)
5 (5.5)
4 (4.4)
4 (4.4)
3 (3.3)
2 (2.2)
1 (1.1)
1 (1.1)
1 (1.1)
1 (1.1)
1 (1.1)
1 (1.1)
1 (1.1)
1 (1.1)
1 (1.1)
1 (1.1)
1 (1.1)
0
0
0
0
0
0
0
0
90
N.º (%)
1
de
p
acientes que
e
xperimentaron
el evento
1
5 (9.2)
16 (9.8)
9 (5.5)
8
(4.9)
5 (3.1)
5 (3.1)
4 (2.5)
4 (2.5)
4 (2.5)
3 (1.8)
2 (1.2)
1 (0.6)
1 (0.6)
1 (0.6)
1 (0.6)
1 (0.6)
1 (0.6)
1 (0.6)
1 (0.6)
1 (0.6)
1 (0.6)
1 (0.6)
0
0
0
0
0
0
0
0
58
2
G
rupo de control (n = 160)
Grupo Cervical-Stim (n = 163)
Eventos adversos
1 % expresado como número de pacientes que experimentaron el evento/número total de pacientes en el grupo
2 Algunos pacientes experimentaron varios eventos adversos
*
Hubo varios eventos adversos que se observaron con más frecuencia en el grupo Cervical-Stim que en el grupo de
control. Por los tipos de eventos dados, es improbable que estos eventos adversos estén relacionados con el
tratamiento.
2
A fin de asegurar que el dispositivo esté funcionando correctamente,
Cervical-Stim monitorea constantemente la tensión de la batería y la señal eléctrica. Si en algún momento durante el tratamiento el dispositivo deja de funcionar correctamente, la LCD mostrará el símbolo o código de error correspondiente. Para obtener más información, consulte “Indicadores visuales y de
audio”.
Para interrumpir el tratamiento antes de que finalice la sesión de tratamiento
diaria, simplemente presione el botón On/Off. Para reanudar el tratamiento, presione el botón On/Off nuevamente. La LCD mostrará el tiempo
de tratamiento restante.
Información de prescripción
Indicación
Cervical-Stim es un estimulador no invasivo del crecimiento óseo
mediante campo electromagnético pulsado indicado como complemento a la cirugía de fusión cervical en pacientes de alto riesgo de falla de
fusión.
Contraindicaciones
No se conocen contraindicaciones para el uso de Cervical-Stim como
complemento de la cirugía de fusión cervical.
Advertencias
• Cervical-Stim puede interferir con el funcionamiento de un marcapasos o un desfibrilador. Se recomienda que consulte con el cardiólogo que atiende al paciente.
• El Cervical-Stim debe quitarse antes de someterse a cualquier procedimiento de diagnóstico por imágenes (p.ej., tomografía computarizada, resonancia magnética, etc.).
Precauciones
• El Cervical-Stim no debe usarse si existen condiciones mentales o
físicas que pudieran impedir el cumplimiento de las instrucciones
del médico y del dispositivo.
• El tratamiento con Cervical-Stim no se ha evaluado en pacientes con
las siguientes afecciones: traumatismo óseo o ligamentoso,
espondilitis, enfermedad de Paget, osteoporosis moderada a grave,
cáncer metastásico, enfermedad renal, artritis reumatoidea, diabetes mellitus no controlada, pacientes propensos a cefaleas vasculares migrañosas, convulsiones, epilepsia, afecciones de la tiroides o
enfermedades neurológicas.
• Estudios teratológicos en animales realizados con este dispositivo
no mostraron ningún efecto adverso en animales. Sin embargo, no
se ha establecido la seguridad del uso de este dispositivo en
pacientes que están embarazadas o amamantando.
Page 14

1
. Valores-P de las pruebas comparativas entre los grupos de tratamiento usando la prueba de la
T de Student para las variables numéricas y la prueba de la x
2
de Pearson para las variables
categóricas.
Análisis de los datos y resultados
El criterio de valoración primario de la efectividad fue el incremento de la frecuencia del éxito de la fusión cervical a los seis meses del período postoperatorio determinado por evidencia radiográfica. Los criterios de valoración secundarios fueron la función neurológica, la evaluación del dolor según la escala VAS y el
Índice de incapacidad del cuello. La seguridad fue evaluada por la frecuencia y
gravedad de los eventos adversos.
La fusión fue evaluada mediante radiografías efectuadas en cada consulta:
La fusión radiográfica fue definida como > 50% de puente óseo en ambas
interfaces del injerto, la superior y la inferior, entre los cuerpos vertebrales adyacentes Y < 4° de angulación (movimiento) entre las vértebras adyacentes con fusión observada en las radiografías laterales de
flexión/extensión Y ausencia de radiolucencia.
La no fusión radiográfica fue definida como < 50% de puente óseo en la
interfaz superior o inferior del injerto O > 4° de angulación
(movimiento) entre las vértebras adyacentes con fusión observada en
las radiografías laterales de flexión/extensión O presencia de radiolucencia.
5
V
ariables
Edad (años)
Media
R
ango
Desviación estándar
Género
Femenino
Masculino
Raza
Blanca
Afro-americana
Hispana
Asiática
Otra
Estado de fumador
No fumador
Fumador
Número
d
e sujetos
(N = 323)
46.8
2
4 – 73
9.3
148 (45.8%)
175 (54.2%)
301 (93.2%)
17 ( 5.3%)
5 (1.6%)
0
0
159 (49.2%)
164 (50.8%)
Control
(
n = 160)
46.7
2
6 – 72
9.2
75 (46.9%)
85 (53.1%)
150 (93.8%)
7 (4.4%)
3 (1.9%)
-
-
79 (49.4%)
81 (50.6%)
Cervical-Stim
(
n = 163)
46.9
2
4-73
9.4
73 (44.8%)
90 (55.2%)
151 (92.6%)
10 ( 6.1%)
2 (1.2%)
-
-
80 (49.1%)
83 (50.9%)
v
alor-P
1
0.846
0.706
0.703
0.958
Características demográficas de base
Datos demográficos
Los sujetos de este estudio tenían una edad media de 46.8 años (rango de 24
a 73 años). De los 323 sujetos, 148 (45.8%) eran mujeres y 175 (54.2%) eran
hombres. 301 (93.2%) eran blancos, mientras que 17 (5.3%) eran afro-americanos y 5 (1.6%) eran hispanos. 159 (49.2%) eran no fumadores y 164
(50.8%) eran fumadores.
4
seguridad y efectividad del dispositivo de CEMP Cervical-Stim como complemento
para pacientes de alto riesgo sometidos a una fusión cervical. Todos los sujetos
fueron sometidos a disectomía y fusión cervical anterior usando la técnica SmithRobinson con la placa Atlantis. Los sujetos fueron asignados aleatoriamente al grupo
de control (tratamiento estándar, n=160) o al grupo del tratamiento (tratamiento
estándar más el Cervical-Stim, n=163). El tratamiento estándar fue determinado a
discreción del médico pero incluyó generalmente la estadía en el hospital, el uso de
un collarín cervical suave, los medicamentos apropiados y la terapia física.
Los sujetos que cumplieron los siguientes criterios de inclusión y exclusión fueron
elegibles para participar en el estudio:
Criterios de inclusión
Sujeto adulto ya sea hombre o mujer, de18-75 años de edad con evidencia radiográfica de
raíces cervicales comprimidas, radiculopatía sintomática, dolor de grado 5 o superior en la
Escala Análoga Visual (VAS) y/o cualquier debilidad muscular y fusión espinal cervical primaria efectuada mediante la técnica Smith-Robinson con hueso de aloinjerto y una placa
cervical anterior. El procedimiento de la fusión debió haber sido de varios niveles (nivel de
fusión > 1) y/o el sujeto era un fumador (un paquete diario o más) y firmó el consentimiento informado.
Criterios de exclusión
Lesión cervical traumática, fusión por vía posterior o de revisión, aloinjerto o materiales de sustitución ósea como fuente del injerto, historial de cefalea vascular
migrañosa o propensión a convulsiones incontroladas o epilepsia (controlada o no
controlada) o alguna enfermedad o lesión neurológica; depresión del sistema
inmunológico, afecciones regionales (espondilitis, enfermedad de Paget, artritis
reumatoidea), infección (sistémica o local) durante las 2 semanas previas a la cirugía,
trastornos sistémicos (cáncer, arritmia cardíaca, enfermedad de la tiroides, diabetes
mellitus incontrolada, enfermedad/disfunción renal, uso crónico de esteroides u
otras enfermedades que pudieran haber afectado el metabolismo óseo), marcapasos cardíacos, desfibriladores, estimuladores de la columna dorsal, prótesis auditivas,
prótesis cocleares y estimuladores craneales, personas embarazadas, amamantando
o que hubieran planeado quedar embarazadas en un plazo de
12 meses, sujetos que hubieran participado en otros estudios clínicos en los últimos
12 meses, o que tuvieran trastornos mentales o físicos que pudieran haber impedido el cumplimiento de las instrucciones del médico.
Evaluación y seguimiento
Las consultas de seguimiento debían efectuarse a los meses 1, 2, 3, 6 y 12, y a partir
de entonces, una vez al año hasta que el último sujeto participante en el estudio
cumpliera 12 meses de tratamiento.
Uso del dispositivo
A los sujetos asignados al grupo de tratamiento (Cervical-Stim) se les dio instrucciones para que usaran el dispositivo cuatro horas diarias durante un mínimo de tres
meses en el período postoperatorio. Los cirujanos pudieron, a su discreción, ampliar
el tratamiento de Cervical-Stim hasta los seis meses en el período postoperatorio.
Page 15

7
Grupo de tratamiento
Control
Cervical-Stim
Número de sujetos
120
125
Número de sujetos que
lograron la fusión
104
116
Tasa de fusión (%)
86.67
92.80
Resultados totales de la fusión radiográfica a los 12 meses
Nota: las diferencias en las tasas de éxito a largo plazo entre los grupos de tratamiento no son sig-
nificativas desde el punto de vista estadístico según la prueba de la x
2
de Pearson con el
tamaño de la muestra disponible (x
2
= 2.5136, p = 0.1129).
Se realizó un análisis adicional para medir las diferencias entre el grupo de
tratamiento con Cervical-Stim y el grupo de control en relación con las características demográficas (género, edad, diagnóstico) y el estado de riesgo (estado
de consumo de tabaco, varios niveles). La tasa general de fusión en el grupo
Cervical-Stim siguió siendo significativa desde el punto de vista estadístico tras el
ajuste de cada una de estas variables.
El seguimiento a largo plazo (12 meses) no mostró una diferencia estadística
entre los dos grupos con respecto a la fusión. 116 de los 125 sujetos evaluables
(92.8%) del grupo de tratamiento con Cervical-Stim habían logrado la fusión
según el criterio de valoración final a largo plazo, mientras que 104 de los
120 sujetos evaluables (86.7%) del grupo de control lo habían logrado.
Criterios de valoración secundarios de la efectividad
Los criterios de valoración secundarios evaluaron los cambios en los síntomas
clínicos. Un “éxito clínico” con respecto a los síntomas se definió como el no
empeoramiento de la función neurológica, una mejoría de la evaluación del
dolor según la escala VAS y el no empeoramiento del Índice de incapacidad del
cuello. Un “fracaso clínico” con respecto a los síntomas se definió como el fracaso de alguno de estos criterios. No hubo una diferencia significativa desde el
punto de vista estadístico entre los dos grupos con respecto al porcentaje de
sujetos considerados como “éxito clínico” a los seis meses (p=0.8456) o a los
12 meses (p= 0.1129).
Con fines de evaluación del dispositivo, todas las radiografías se escanearon y se
incluyeron en una base de datos central, y las revisaron dos cirujanos ortopédicos
independientes y un radiólogo independiente “a ciegas” al final de todo el estudio.
Las radiografías se revisaron y calificaron usando un protocolo común. En cada
punto temporal, se evaluaron todas las radiografías en cuanto a la cantidad de radiolucencia, de puente óseo y el grado de movimiento por evidencias mostradas en
las radiografías de flexión/extensión de la región cervical. Se usó un programa de
software para calcular el movimiento. Los cirujanos ortopédicos revisaron y verificaron los resultados obtenidos de esta manera. El diagnóstico del radiólogo se consideró definitivo en el caso de un desacuerdo entre los dos cirujanos ortopédicos.
Resultados sobre la efectividad
De los 323 sujetos aleatorizados y que recibieron cirugía, 240 fueron evaluables para
el análisis de efectividad (grupo de tratamiento con Cervical-Stim, n=122; grupo de
control, n=118). Hubo sujetos que no se consideraron evaluables por las siguientes
razones: radiografías no existentes o no legibles, incumplimiento del sujeto, infracciones del protocolo (criterios de inclusión), fallo del injerto, equipo interno roto,
existencia de estudio anterior debido a experiencias adversas menores, y un intento
de suicidio. No se conoce el estado de éxito o de fracaso de estos sujetos. Estos
datos con los que no se contó pudieran haber afectado de manera positiva o negativa el éxito general del estudio. Con el fin de evaluar el efecto de los datos faltantes,
se efectuaron análisis de sensibilidad. Estos análisis incluyeron que se trasladara la
última observación de estos sujetos y se imputaran todos los datos faltantes a una
falla de fusión. Estos dos análisis mostraron que los resultados hallados a los seis
meses fueron todavía significativamente diferentes y favorables, desde el punto de
vista estadístico, al grupo Cervical-Stim. Además, los datos demográficos de referencia provenientes de la población evaluable fueron comparados con los datos
demográficos de los sujetos faltantes. Los resultados de este análisis indicaron que
no hubo diferencias significativas entre los sujetos evaluables y los no evaluables en
las 14 variables del estudio, incluidos los parámetros clínicos y demográficos clave.
Criterio de valoración primario de la efectividad
El criterio de valoración primario de la efectividad fue la evidencia de fusión radiográfica en el punto temporal de seis meses del período postoperatorio. En el
punto temporal de los seis meses, 102 de los 122 sujetos evaluables (84%) en el
grupo de tratamiento con Cervical-Stim habían logrado la fusión en comparación
con 81 de los 118 sujetos evaluables (69%) en el grupo de control (p=0.0065).
6
Estos datos muestran que de los pacientes sometidos a la cirugía de fusión cervical,
aquellos tratados de modo complementario con el Cervical-Stim experimentaron un
incremento en la frecuencia de la fusión radiográfica a los seis meses en comparación
con el grupo de control.
Grupo de tratamiento
Control
Cervical-Stim
Número de sujetos
118
122
Número de sujetos
que lograron la fusión
81
102
Tasa de fusión (%)
68.64
83.61
Comparación de los resultados de la fusión radiográfica a los seis meses
Page 16

9
Cómo cargar/recargar la batería
Cervical-Stim funciona con un paquete de baterías de ion-litio recargables.
El dispositivo incluye una unidad de carga. Use únicamente el cargador de
Orthofix para cargar la batería.
Nota: la batería de Cervical-Stim debe recargarse antes de usarla por primera vez.
Para cargar/recargar la batería, simplemente enchufe el extremo cilíndrico del
conector de la unidad de carga en el puerto para cargador que se encuentra en
la unidad de control. Enchufe el cable de alimentación en la unidad del cargador de modo que quede bien fijado. Enchufe el cargador en cualquier toma
de corriente CA estándar de pared. Se requerirá aproximadamente
12 horas para cargar completamente una batería totalmente descargada.
La batería de Cervical-Stim se puede recargar en cualquier momento en que
no se esté usando el dispositivo. Se recomienda categóricamente recargar
el dispositivo después de completar el tratamiento diario.
Nota: Cervical-Stim no aplicará el tratamiento mientras se esté cargando
la batería.
Cuando el dispositivo esté encendido, la LCD de Cervical-Stim mostrará un
símbolo de capacidad de la batería. La figura de una batería intermitente, el
símbolo y un tono audible indican que la batería está baja y que es necesario recargarla. Para obtener más información, consulte “Indicadores visuales
y de audio”.
Unidad de control
Unidad de carga
8
Seguridad
Los eventos adversos observados en este estudio se muestran en la Tabla de
eventos adversos presentada en la sección Información de prescripción. A los
seis meses, el número de sujetos que experimentaron uno o más eventos
adversos es similar en ambos grupos. Se reportó un total de 14 eventos graves
en 13 sujetos; nueve de ellos formaban parte del grupo de tratamiento con
Cervical-Stim y cinco formaban parte del grupo de control. Estos eventos consistieron en experiencias tales como mayor dolor, respiración entrecortada,
mareo, trauma y lesión no relacionada, muerte no relacionada, complicación
quirúrgica y patología a nivel adyacente. En los nueve sujetos del grupo de
tratamiento con Cervical-Stim, todos los eventos adversos fueron considerados,
a juicio de los investigadores, como definitivamente o probablemente, no relacionados con el dispositivo.
Los datos de seguridad obtenidos durante el tiempo transcurrido entre la consulta del sexto mes y el contacto final con cada sujeto indican que un total de 51
sujetos de ambos grupos experimentaron 57 eventos adversos. El número de
sujetos que experimentaron uno o más eventos adversos es similar en ambos
grupos. Ninguno de los eventos adversos reportados entre la consulta del sexto
mes y el contacto final fue grave y los eventos fueron similares a los reportados
a los seis meses.
Vida útil del dispositivo
Cervical-Stim puede proporcionar hasta 270 tratamientos consecutivos (diarios)
de cuatro horas cada uno. La duración total del tratamiento la determinará el
médico basado en el paciente y el progreso de la fusión.
Tiempo del tratamiento
Cervical-Stim debe usarse durante cuatro horas al día. El dispositivo se apagará
automáticamente cuanto hayan transcurrido cuatro horas de tratamiento en un
día (24 horas). Se puede apagar el dispositivo con sólo presionar el botón
On/Off (Encendido/Apagado) que se encuentra en el panel de control.
Cervical-Stim puede usarse en el momento del día que sea más conveniente
para el paciente. Es ligero y ajustable. Dado que Cervical-Stim es portátil, el
paciente puede recibir el tratamiento mientras está sentado, caminando, reclinado, durmiendo, etc. No obstante, dado que cada paciente tiene características únicas, el nivel de actividad general debe estar basado en sus instrucciones.
Page 17

11
Cómo usar el dispositivo
Cervical-Stim está diseñado para la región cervical y puede usarse con o sin
una órtesis. Para mayor comodidad, se debe usar ropa entre la piel y el
Cervical-Stim.
Para usar el Cervical-Stim, basta con que deslice el dispositivo por la cabeza
hasta que quede cómodamente apoyado sobre el cuello y los hombros (véase
la figura que aparece a continuación). También puede abrir el dispositivo con la
hebilla y colocarlo apoyado sobre el cuello y los hombros. Ajuste la hebilla
como si fuera un cinturón de seguridad para fijar la unidad.
Unión
con
hebilla
10
Funcionamiento del dispositivo
Cómo encender y apagar el dispositivo
Cervical-Stim se enciende y se apaga presionando el botón On/Off
(Encendido/Apagado) que se encuentra en la unidad de control del dispositivo.
Cuando el dispositivo está encendido, aparecerá momentáneamente una secuencia
de mensajes de estado. Luego, la LCD mostrará el tiempo de tratamiento restante y
un logotipo intermitente de Orthofix. El logotipo intermitente indica que el dispositivo está encendido y funciona normalmente. (Si no ve este logotipo intermitente en
la pantalla, comuníquese con su proveedor). La unidad de control tiene un botón de
luz de fondo. Cuando haya poca luz, presione el botón de luz de fondo para que la
LCD se ilumine.
Para interrumpir el tratamiento antes de que finalice la sesión de tratamiento diaria,
simplemente presione el botón On/Off. Para reanudar el tratamiento, presione el
botón On/Off nuevamente. La LCD mostrará el tiempo de tratamiento restante.
Cronometraje de las sesiones de tratamiento
Cervical-Stim cronometra automáticamente cada sesión de tratamiento. El
cronometraje comienza al encender el dispositivo usando el botón On/Off que se
encuentra en la unidad de control. La LCD muestra una cuenta regresiva del tiempo
restante de la sesión de tratamiento. Al final del tratamiento diario, el dispositivo se
apagará solo. Para interrumpir el tratamiento antes de que finalice una sesión de
tratamiento, simplemente presione el botón On/Off. Para reanudar el tratamiento,
presione el botón On/Off nuevamente. La LCD mostrará el tiempo de tratamiento
restante.
Nota: para que la cuenta regresiva funcione correctamente, las sesiones de
tratamiento deben durar más de 60 minutos.
Unidad de control
Botón On/Off
Puerto para car-
gador
Botón de luz de
fondo
LCD
Logotipo inter-
mitente de
Orthofix
Puerto para luz infrarroja
Page 18

13
Indicadores visuales y de audio
La LCD y las alarmas audibles están diseñadas para proporcionar información
útil al usuario. El cuadro que figura a continuación muestra los diversos símbolos y las alarmas, y su significado.
Indicadores visuales de la LCD e indicadores de audio de Cervical-Stim
Símbolo/Alarma Descripción Significado
todos los símbolos de la LCD visibles y la alarma audible continua
durante aproximadamente 5
segundos
el cronómetro de cuenta regresiva
muestra el tiempo de tratamiento
restante (horas y minutos)
el logotipo de Orthofix parpadea
el cronómetro de cuenta regresiva
muestra tres rayas
alarma audible (5 tonos)
símbolo fijo durante aproximadamente 5 segundos
los símbolos parpadean/alarma
audible (aproximadamente 1 tono
por segundo)
el símbolo fijo indica el % de
carga aproximado
si el símbolo se llena una y otra
vez, indica modo de carga
alarma audible continua
aparece cualquier código E
(p. ej., E01, E02. . .)
autoprueba de encendido
tratamiento normal en
proceso
no hay tiempo de
tratamiento restante
tratamiento finalizado —
apague
batería baja —
recargue
estado de la batería —
carga restante o modo
de carga
dispositivo bloqueado
—
llame al servicio técnico
mensaje de error
—
llame al servicio técnico
12
Viajes
Cuando se viaje en avión, lo mejor es registrar el Cervical-Stim con el equipaje.
Si el dispositivo se lleva a bordo del avión, no debe llevarse puesto cuando se
pase por los controles de inspección de seguridad. El Cervical-Stim se podría
dañar. Se debe llevar también el manual de instrucciones del Cervical-Stim a fin
de identificar el dispositivo con rapidez y facilidad ante cualquier
personal de seguridad.
Almacenamiento
Cervical-Stim deberá almacenarse a una temperatura que esté entre
14º F a 113º F (-10º C a 45º C).
El rango de temperaturas de funcionamiento para el Cervical-Stim deberá ser
de 41º F a 104º F (+5º C a 40º C)
Humedad relativa: hasta 95%, sin condensación
Disposición
El producto contiene baterías de litio, no incinerar. Disponga del
dispositivo correctamente para prevenir lesiones. Por favor de dar
este producto a los depósitos de equipo electrónico obsoletos de
uso en casa.
Servicio técnico
Si tiene alguna pregunta en relación con el dispositivo o necesita asistencia técnica, sírvase llamar al 469-742-2500 o al 800-535-4492. El dispositivo no tiene
piezas que puedan ser reparadas por el usuario. Notifique al fabricante si necesita algún servicio técnico.
Cuidado y limpieza
Cervical-Stim es un dispositivo electrónico sofisticado y se debe manipular con
cuidado. Dejar caer el Cervical-Stim o tratarlo en forma indebida puede dañar
el dispositivo.
NO exponga el Cervicall-Stim a la luz solar directa durante períodos prolongados. La unidad de control del dispositivo se puede
dañar.
NO exponga el Cervical-Stim a calor excesivo. En climas tórridos,
la temperatura dentro de un automóvil o un maletero puede ser
mayor de 160º F/71º C. El calor excesivo puede dañar la unidad de
control del dispositivo.
NO exponga el Cervical-Stim a humedad excesiva. La humedad
puede dañar los componentes electrónicos del dispositivo y éste
puede dejar de funcionar.
NO deseche el Cervical-Stim en un incinerador.
NO use solventes para limpiar el Cervical-Stim. Limpie el dispositivo
con un paño suave y húmedo.
Page 19

15
Conformidad con las normas
El sistema de fusión cervical Cervical-Stim de Orthofix cumple con la siguiente
serie de normas internacionales:
UL 60601 - Equipo eléctrico del ramo médico, Requisitos generales de seguri-
dad; (incluida la Compatibilidad e interferencia electromagnética)
IEC 60601 - Equipo eléctrico del ramo médico, Requisitos de seguridad para
los sistemas eléctricos del ramo médico (incluida la
Compatibilidad e interferencia electromagnética)
Instrucciones para usar la impresora Orthofix del registro
de cumplimiento
Monitoreo del cumplimiento del tratamiento por parte del paciente
La impresora Orthofix de datos del cumplimiento es un accesorio para la línea
Orthofix de estimuladores del crecimiento óseo por Campo Electromagnético
Pulsado (CEMP). Al mismo tiempo que aplica la señal CEMP clínicamente
probada al lugar del tratamiento, cada Estimulador del crecimiento óseo
Orthofix registra el tiempo transcurrido (diario y total) del tratamiento del
paciente a fin de monitorear y llevar un control del cumplimiento del
tratamiento. Los datos acumulados del cumplimiento se guardan en una
memoria de acceso para el médico que se encuentra dentro del Estimulador
del crecimiento óseo. Este sistema de control le sirve al médico para asegurarle que el paciente está recibiendo el tratamiento necesario para un resultado
exitoso.
Los datos del cumplimiento al tratamiento se pueden recobrar con el
Estimulador solo o en conjunto con la Impresora Orthofix del cumplimiento.
Cuando la impresora del cumplimiento se utiliza con el estimulador Orthofix,
se puede obtener una copia impresa del historial del tratamiento del paciente.
Esto es una herramienta útil para tener acceso y documentar el cumplimiento
por parte del paciente, del tratamiento de estimulación diario indicado.
La impresora Orthofix de los datos de cumplimiento es una impresora de papel
térmico que permite al médico imprimir una copia en papel de los datos del
cumplimiento del paciente. La impresora se alimenta por conexión a una toma
de corriente CA estándar de pared (100 – 240 VCA, 50/60 Hz); y se comunica
con los Estimuladores del crecimiento óseo Orthofix mediante una interfaz
infrarroja muy similar a un control remoto de un televisor.
Sírvase consultar la Guía del sistema de monitoreo de datos de
cumplimiento Orthofix destinado a médicos, para leer las instrucciones
completas de la impresora.
14
Clasificación del equipo y descripciones de los símbolos del dispositivo
Clasificaciones del equipo
• Equipo con alimentación interna
• Pieza aplicada tipo BF
• Clasificación del gabinete según la IEC 529: IPXO
• Equipo no apto para usar en presencia de una mezcla anestésica inflamable
con aire u óxido nitroso
• Modo de funcionamiento: funcionamiento intermitente
El uso de accesorios que no sean los especificados puede provocar un aumento
de las emisiones o una disminución de la inmunidad del dispositivo.
El cargador de la batería incluye una entrada de 3 hilos, pero se considera que
tiene doble aislamiento con construcción Clase II en todo el cargador.
Para un uso seguro, siga las instrucciones del fabricante al usar el producto.
Usar el producto de alguna otra forma puede generar efectos perjudiciales o
anular la garantía.
Nota: antes de cada uso, inspeccione que el dispositivo no haya sufrido des-
gaste ni deterioro. No use el dispositivo si no parece estar en buenas
condiciones.
Símbolo Significado
Atención – Consulte las instrucciones de uso
Pieza aplicada tipo BF
On/Off
Botón de luz de fondo
Rango de temperaturas de almacenamiento
Año de fabricación del dispositivo activo
Puerto para cargador
Page 20

16
Información de la garantía
Orthofix Inc. garantiza que el estimulador del crecimiento óseo Cervical-Stim
no tendrá defectos en los materiales ni en la mano de obra durante un año a
partir de la fecha en que se use por primera vez. Siempre y cuando se cumplan
todos los términos y condiciones de esta Garantía limitada, Orthofix Inc. reemplazará los componentes defectuosos.
Esta Garantía limitada se aplica al producto únicamente en condiciones de uso
normal, y no cubre ningún daño ni defecto provocado por un accidente, uso
indebido, maltrato, incendio, inundación ni casos fortuitos, ni por ninguna
alteración, manipulación, reparación o intento de reparación por parte de algún
tercero que no pertenezca a Orthofix Inc. Esta garantía se aplica únicamente al
paciente al que se le recete el producto, y no se la puede ceder ni transferir.
Los productos defectuosos cubiertos por esta Garantía limitada deben ser
devueltos a Orthofix Inc. Atención: Orthofix Returns. Antes de devolver el
producto, debe llamar al Representante de servicio al cliente al
1-800-535-4492 o a su distribuidor local para obtener el número y dirección
de Autorización de devoluciones (RA).
Excepto que la ley aplicable especifique lo contrario, la garantía precedente
reemplaza a todas las demás garantías, expresas o implícitas, y Orthofix Inc. se
exime específicamente de toda responsabilidad por cualesquiera y todas las
garantías de comerciabilidad o aptitud para un fin en particular. Orthofix Inc.,
sus representantes autorizados, afiliadas o compañías subsidiarias no serán
responsables, bajo ninguna circunstancia, por daños especiales, consecuentes ni
incidentales. El único recurso para subsanar un producto defectuoso será el
reemplazo.
Esta Garantía limitada no podrá ser extendida ni modificada, excepto que
Orthofix Inc. lo haga por escrito. Ningún vendedor, representante, distribuidor
ni médico está autorizado a realizar ni permitir ninguna extensión ni modificación de los términos de esta Garantía Limitada.
Para obtener información adicional y/o asistencia técnica para el dispositivo,
comuníquese con el departamento de Servicio al cliente de Orthofix
al 800-535-4492.
Orthofix Inc.
Edición para los EE.UU. – Paciente
N.º de Ref. 571355-0001 Rev C 3/2009
Impreso en los EE.UU.
Fecha de la impresión: 3/ 2009
CS-0903.A-PL-EO © Orthofix Inc 3/2009
Fabricado por: Orthofix Inc.
1720 Bray Central Drive
McKinney, Texas 75069
Tel. 469-742-2500
800-535-4492
 Loading...
Loading...