


Cardiac Resynchronization Therapy Efcacy Enhancements (CRTee)
Summary of clinical results
Clinical study
Caution: Federal Law (USA) restricts this device to sale by or on the order of a physician.

The following list includes trademarks or registered trademarks of Medtronic in the United States and
possibly in other countries. All other trademarks are the property of their respective owners.
Brava, Medtronic, Viva

TABLE OF CONTENTS
SUMMARY OF CLINICAL RESULTS ............................................................................................ 2
1 STUDY PURPOSE .................................................................................................................... 2
2 STUDY SCOPE, DESIGN, AND METHODS ............................................................................ 2
3 SUBJECT INCLUSION AND EXCLUSION CRITERIA .......................................................... 3
4 RESULTS ................................................................................................................................. 3
Primary Objectives ...................................................................................................................... 8
Secondary Objective .................................................................................................................... 9
5 ADVERSE EVENTS SUMMARY ........................................................................................... 12
6 DEATH SUMMARY ............................................................................................................... 13
7 CLINICAL STUDY CONCLUSION ....................................................................................... 14
1

IDE Number: G140092
SUMMARY OF CLINICAL RESULTS
1 STUDY PURPOSE
The purpose of the CRTee Study was to demonstrate that the amount of effective CRT pacing
during AF (atrial fibrillation) when CAFRPlus (Conducted AF Response Plus) was applied was
not inferior to the amount of effective CRT pacing during AF when CAFR (Conducted AF
Response) was applied.
Additionally, this clinical study was designed to confirm safety and efficacy of the CRTee
system.
2 STUDY SCOPE, DESIGN, AND METHODS
The study was a prospective, multi-center, randomized, controlled, crossover, global trial.
Sample size called for 75 subjects to be enrolled. Subjects were enrolled into the study prior to
injection of the CRTee software at the baseline visit. Following enrollment, subjects then
completed Phase 1 visit procedures which included an in-office assessment, testing the CRTee
algorithm to verify that it was able to correctly identify effective/ineffective CRT pacing and
randomization. Subjects were randomized into either Group A (CAFR ON first) or Group B
(CAFRPlus ON first).
Randomized subjects completed CareLink transmissions or a save-to media on a weekly basis
for up to 4 weeks until the required amount of AF (at least 2 hours) has been reported. After 4
weeks or when the AF burden has been reported, whichever occurred first, the subject was
brought into the clinic for the Phase 2 visit. At the Phase 2 visit, data was collected from the
device, the in-office assessment was repeated and the subject was crossed over into the opposite
arm. A Holter may have been requested (up to 24 hours) during the Phase 2 visit to better
understand unique algorithm behavior. In the event a Holter was requested, the subject returned
the Holter via mail after 24 hours.
Subjects continued to complete CareLink transmissions or a save-to media on a weekly basis for
up to 4 weeks until the required AF burden (at least 2 hours) had been reported. After 4 weeks or
when the AF burden had been reported, whichever occurred first, the subject was brought into
the clinic for the Phase 3 visit. At the Phase 3 visit, data was collected from the device; the inoffice assessment was repeated. A Holter may have been requested (up to 24 hours) to better
understand unique algorithm behavior. In the event a Holter was requested, the subject returned
the Holter via mail after 24 hours. After the Phase 3 visit was complete, subjects had the option
to have CAFRPlus ON for the remainder of the follow up period, up to 4 months postrandomization, and at that time a final visit/study exit occurred. The CRTee feature set was
removed from the device upon study exit. As a safety feature, after 200 days, the CRTee feature
set was automatically removed from the device to cover cases of subjects lost to follow-up.
2

IDE Number: G140092
3 SUBJECT INCLUSION AND EXCLUSION CRITERIA
Patients who met all inclusion and no exclusion criteria were eligible.
Inclusion Criteria
• Subject was willing to sign and date the study patient Informed Consent form.
• Subject was at least 18 years of age (or older, if required by local law).
• Subject was expected to remain available after enrollment to complete follow-up visits in
both arms of the study
• Subject had a Medtronic Viva or Brava CRT-D device implanted at least 30 days prior to
enrollment.
• Subject had history of AF burden, of at least 6 days of at least 4 hours of AF over any 4
week period within the last 90 days as documented in device diagnostic data OR if
subject had no atrial lead (therefore no device diagnostic data) but clinical evidence of
high AF burden.
• Subject had demonstrated history of being able to complete Left Ventricular Capture
Management (LVCM) documented in device data.
• Subject had a documented % V pacing during AF of less than or equal to 97% within 90
days prior to enrollment or within 10 days after enrollment.
Exclusion Criteria
• Subject had undergone AV node ablation for treatment of AF.
• Subject had complete or 3rd degree AV block.
• Subject had an MI within 30 days.
• Subject had medical conditions that limit study participation (per physician discretion).
• Subject was enrolled in one or more concurrent studies that could confound the study
results as determined by Medtronic.
• Subject had a limited life expectancy for non-cardiac causes that would not allow
completion of the study.
• Subject was pregnant (in the US, all women of child-bearing potential had to undergo a
pregnancy test within seven days prior to CRTee download).
Subject met the exclusion criteria required by local law.
4 RESULTS
The first subject enrollment occurred on 07 October 2014, and enrollment was completed on 08
January 2016. In the 15 months enrollment period, a total of 71 subjects were enrolled and 66
were randomized. Enrollments occurred in 22 investigational centers (13 US, 9 OUS) from 6
countries.
3
IDE Number: G140092
United States
Total OUS
Europe
Patient Accountability
A total of 71 subjects were enrolled at 22 centers, described in Table 1.
Table 1: Enrollment numbers by Geography
Geography Country
Total Total Total 71 (100%) 66 (100%)
Total US Total US 36 (51%) 31 (47%)
UNITED
STATES
Center Enrolled
(n=71)
Cardiac Arrhythmia Services 2 (3%) 1 (2%)
Cardiology Associates, P.A. 1 (1%) 1 (2%)
Carle Physician Group 1 (1%) 0 (0%)
CentraCare Heart & Vascular
Center
Heart Clinics Northwest, P.S. 3 (4%) 2 (3%)
Heartland Cardiology 3 (4%) 1 (2%)
Minneapolis Heart Institute 1 (1%) 1 (2%)
Mount Carmel St Ann's Hospital 10 (14%) 10 (15%)
NC Heart and Vascular Associates 4 (6%) 4 (6%)
North Memorial Heart and Vascular 6 (8%) 6 (9%)
Oklahoma Cardiovascular Research
Group
Park Nicollet Institute 1 (1%) 1 (2%)
1 (1%) 1 (2%)
2 (3%) 2 (3%)
Randomized
(n=66)
Texas Cardiac Arrhythmia
Research Foundation
Total OUS Total OUS 35 (49%) 35 (53%)
Total Europe 26 (37%) 26 (39%)
HUNGARY MH Honvédkorház 8 (11%) 8 (12%)
Azienda Ospedaliera Cardinale
Panico
ITALY
SLOVAKIA VUSCH 3 (4%) 3 (5%)
UNITED
KINGDOM
Ospedale Bolognini 2 (3%) 2 (3%)
Policlinico Universitario Agostino
Gemelli
Hammersmith Hospital, Imperial
College Healthcare NHS Trust
Liverpool Heart and Chest Hospital 1 (1%) 1 (2%)
The Newcastle upon Tyne
Hospitals
4
1 (1%) 1 (2%)
4 (6%) 4 (6%)
3 (4%) 3 (5%)
1 (1%) 1 (2%)
4 (6%) 4 (6%)
IDE Number: G140092
Middle East
Geography Country
Total Middle East 9 (13%) 9 (14%)
SAUDI
ARABIA
SAS Program: V:\CRTee\Reports\Final_Report\Programs\EnrollmentByCenterGeography.sas
Center Enrolled
Prince Sultan Cardiac Center 9 (13%) 9 (14%)
(n=71)
Randomized
(n=66)
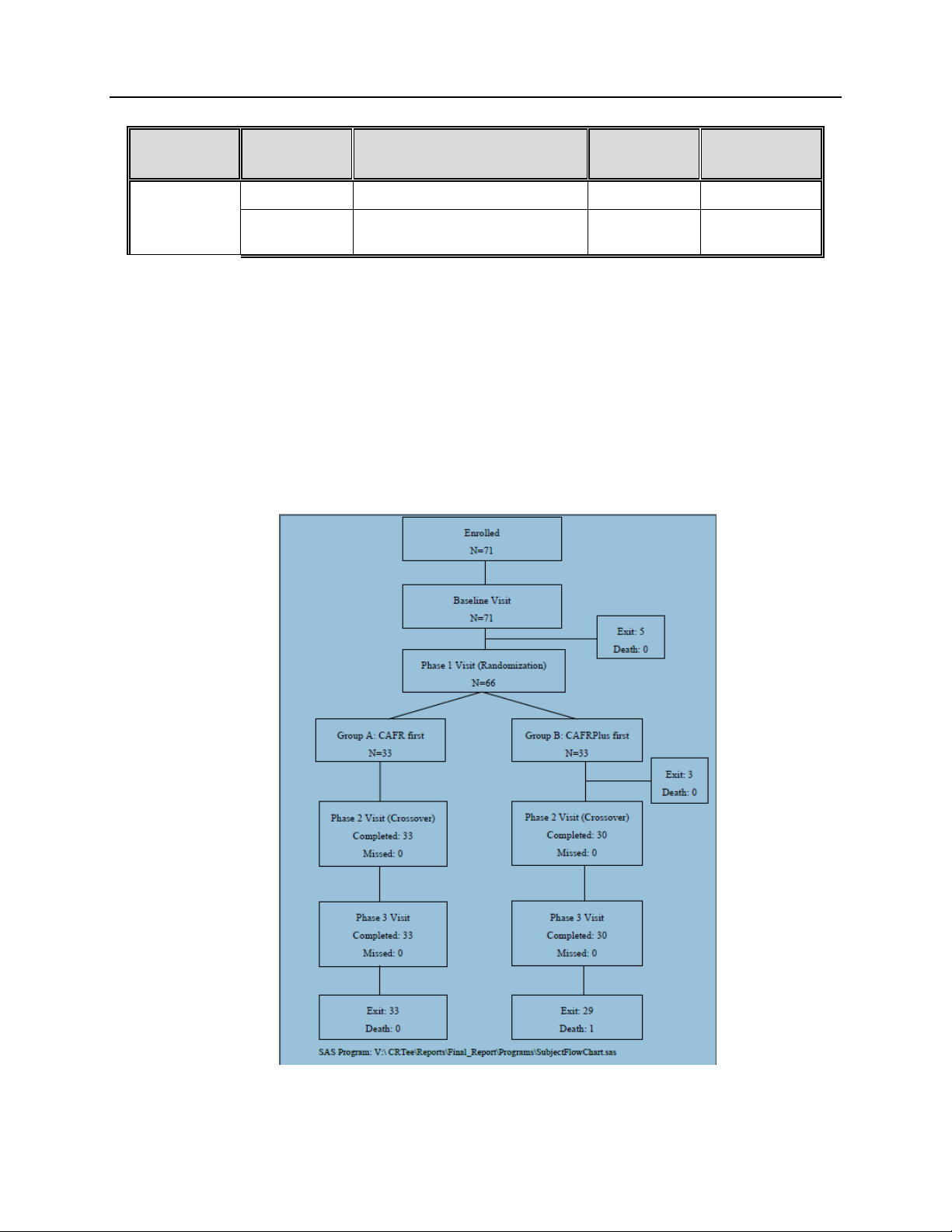
Of the 71 enrolled subjects, 66 were successfully randomized. Figure 1 displays the
accountability of all enrolled subjects. Five subjects did not meet inclusion/exclusion criteria and
were exited from the study prior to being randomized. Sixty-six subjects were randomized 1:1 to
either Group A: CAFR first (33) or Group B: CAFRPlus first (33). After randomization, three
additional subjects were found to not meet inclusion/exclusion criteria and subsequently exited
from the study without undergoing further study procedures. The remaining 63 randomized
subjects went on and completed their study visits. One subject expired after completing phase 3
visit but prior to being exited from the study; the other 62 subjects were all exited from the study
at the time of the final database freeze.
Figure 1: Subject Distribution
5

IDE Number: G140092
Baseline Characteristics
Baseline characteristics for successfully randomized subjects are summarized in Table 2 and
Table 3.
Table 2: Subject Demographics
CAFR first:
Group A
Subject Characteristics
Gender (N,%)
Male 29 (87.9%) 32 (97.0%) 61 (92.4%)
Female 4 (12.1%) 1 (3.0%) 5 (7.6%)
Age (years)
Mean ± Standard Deviation 70.7 ± 11.6 72.4 ± 9.1 71.6 ± 10.4
Median 75.0 75.0 75.0
25th Percentile - 75th Percentile 63 - 77 67 - 79 65 - 79
10th Percentile - 90th Percentile 58 - 82 61 - 83 58 - 83
Minimum – Maximum 31 - 86 49 - 87 31 - 87
Subjects With Measure Available (N,%) 33 (100.0%) 33 (100.0%) 66 (100.0%)
Race (N,%)
Not reportable per local laws or
regulations
Subject/physician chose not to provide
information
(N = 33)
13 (39.4%) 12 (36.4%) 25 (37.9%)
0 (0.0%) 0 (0.0%) 0 (0.0%)
CAFRPlus first:
Group B
(N = 33)
Total
Randomized
(N = 66)
White or Caucasian 19 (57.6%) 20 (60.6%) 39 (59.1%)
Black or African American 1 (3.0%) 1 (3.0%) 2 (3.0%)
Ethnicity (N,%)
Not reportable per local laws or
regulations
Subject/physician chose not to provide
information
Not Hispanic or Latino 22 (66.7%) 24 (72.7%) 46 (69.7%)
Hispanic or Latino 0 (0.0%) 0 (0.0%) 0 (0.0%)
SAS Program: V:\CRTee\Reports\Final_Report\Programs\BaselineDemoSummary.sas
11 (33.3%) 9 (27.3%) 20 (30.3%)
0 (0.0%) 0 (0.0%) 0 (0.0%)
6
IDE Number: G140092
Table 3: Primary Indication for Implant
Primary Indication Type CAFR first:
Group A
(N = 33)
Subject has LVEF less than 35% due to prior
myocardial infarction and is at least 40 days postmyocardial infarction and is in NYHA functional Class
II or III.
Subject has nonischemic dilated cardiomyopathy and
has an LVEF less than or equal to 35% and is in NYHA
functional Class II or III.
Subject is survivor of cardiac arrest due to ventricular
fibrillation or hemodynamically unstable sustained VT
excluding any completely reversible causes.
Subject has hypertrophic cardiomyopathy and has 1 or
more major risk factors for SCD.
Subject has nonischemic heart disease and has an
LVEF of less than or equal to 35% and is in NYHA
functional Class I.
Subject has syncope of undetermined origin with
clinically relevant, hemodynamically significant
sustained VT or ventricular fibrillation induced at
electrophysiological study.
7 (21.2%) 18 (54.5%) 25 (37.9%)
16 (48.5%) 8 (24.2%) 24 (36.4%)
2 (6.1%) 2 (6.1%) 4 (6.1%)
2 (6.1%) 1 (3.0%) 3 (4.5%)
2 (6.1%) 0 (0%) 2 (3.0%)
0 (0%) 2 (6.1%) 2 (3.0%)
CAFRPlus first:
Group B
(N = 33)
Total
Randomized
(N = 66)
Subject has unexplained syncope, significant LV
dysfunction, and nonischemic dilated cardiomyopathy
Pediatric/congenital subject is survivor of cardiac arrest
after evaluation to define the cause of the event and to
exclude any reversible causes.
Subject has LV dysfunction due to prior myocardial
infarction and is at least 40 days post-myocardial
infarction, has an LVEF less than 30%, and is in
NYHA functional Class I.
Subject has documented hemodynamically unstable VT
and/or VT with syncope AND EF equal to or less than
40%.
Subject has structural heart disease and spontaneous
sustained VT, whether hemodynamically stable or
unstable.
SAS Program: V:\CRTee\Reports\Final_Report\Programs\BaselineDemoSummary.sas
2 (6.1%) 0 (0%) 2 (3.0%)
1 (3.0%) 0 (0%) 1 (1.5%)
0 (0%) 1 (3.0%) 1 (1.5%)
0 (0%) 1 (3.0%) 1 (1.5%)
1 (3.0%) 0 (0%) 1 (1.5%)
7

IDE Number: G140092
Follow-up Experience
The follow-up visit compliance for all successfully implanted and randomized subjects is
presented in Table 4.
Table 4: Follow-up Visit Compliance
Visit Expected
Visits
Baseline 66 66 (100%) 66 (100%) 0 (0.0%)
Phase1 66 66 (100%) 64 (97.0%) 0 (0.0%)
Phase2 63 63 (100%) 57 (90.5%) 0 (0.0%)
Phase3 63 63 (100%) 57 (90.5%) 0 (0.0%)
Exit 65 65 (100%) 64 (98.5%) 0 (0.0%)
SAS Program: V:\CRTee\Reports\Final_Report\Programs\VisitSummary.sas
Completed
Visits
In Window
Visits
Missed Visits
Primary Objectives
The primary objective was to demonstrate that the percent effective CRT pacing during AF when
CAFRPlus was applied was not inferior to when CAFR was applied (non-inferiority test).
Null Hypothesis (Ho): μd ≤ -2%
Alternative Hypothesis (Ha): μd > -2%
Where μd was the paired difference in percent effective CRT pacing during AF between when
CAFRPlus was applied and when CAFR was applied, based on subjects’ paired measurements
from the two cross-over follow-up periods, and -2% was the non-inferiority margin selected
based upon clinical judgment.
Results:
The average amount of effective pacing was 80.8% ± 14.3% when CAFR algorithm was ON,
and it increased to 87.8% ± 7.8% when CAFRPlus algorithm was ON (Table 30). The average
increase in effective pacing from CAFR to CAFRPlus was 7.0% ± 8.7%. The difference was
tested using the paired t-test with a non-inferiority margin of -2%, which yielded a one-sided pvalue of <0.0001 (Table 5).
The primary objective of non-inferiority was met.
Table 5: Primary Objective Result - Non-Inferiority Test of CAFRPlus vs. CAFR
Measurement Subjects1 Mean
Difference
95.86% Confidence
Interval2
Non-Inferiority
Margin
Nominal
One-sided
P-value3
Difference in % Effective CRT Pacing
during AF (CAFRPlus - CAFR)
54 7.0 (4.5, 9.5) -2 <0.0001
8

IDE Number: G140092
Measurement Subjects1 Mean
Difference
1
Subjects with a complete dataset of paired CAFR and CAFRPlus measurements (AF >= 2 hours in each arm).
2
Confidence interval coverage probability dictated by the Hwang-Shih-DeCani alpha spending function.
3
P-value from the paired t-test with a non-inferiority margin of -2.
SAS Program: V:\CRTee\Reports\Final_Report\Programs\PrimaryObjective.sas
95.86% Confidence
Interval2
Non-Inferiority
Margin
Secondary Objective
The secondary objective was pre-specified by the study protocol. The secondary objective was to
demonstrate that the percent effective CRT pacing during AF when CAFRPlus was applied was
greater than when CAFR was applied (superiority test).
The increase of 7.0% ± 8.7% in effective CRT pacing during AF from CAFR to CAFRPlus was
further tested for superiority using the paired t-test (Table 6). The p-value from this test was
<0.0001.
The secondary objective of superiority was met.
Table 6: Secondary Objective Result - Superiority Test of CAFRPlus vs. CAFR
Measurement Subjects1 Mean
Difference
95.86% Confidence
Interval2
Nominal
One-sided
P-value3
Nominal
One-sided
P-value3
Difference in % Effective CRT Pacing
during AF (CAFRPlus - CAFR)
1
Subjects with a complete dataset of paired CAFR and CAFRPlus measurements (AF >= 2 hours in each arm).
2
Confidence interval coverage probability dictated by the Hwang-Shih-DeCani alpha spending function.
3
P-value from the paired t-test.
SAS Program: V:\CRTee\Reports\Final_Report\Programs\SecondaryObjective.sas
54 7.0 (4.5, 9.5) <0.0001
Ancillary Objectives:
There were 5 ancillary objectives pre-specified by the study protocol.
AncillaryObjective #1
Objective:
To confirm the safety and proper function of CAFRPlus via the following two metrics:
• Number of times that Power On Reset (POR) occurred
• Number of times that the CAFRPlus algorithm reached the CAFR programmable
maximum heart rate and suspended
This objective was descriptive in nature and there was no pre-specified hypothesis to be tested.
Results:
Number of PORs
9

IDE Number: G140092
There were no occurrences of PORs throughout the course of the study, thus confirming the
overall safety of the CAFRPlus algorithm operation in this aspect.
Number of CAFRPlus suspensions
CIP recommended that the same maximum heart rate was programmed for CAFR and
CAFRPlus. In addition to the maximum heart rate CAFRPlus has a safety feature that will
suspend the algorithm from operating if the programmable max heart rate is reached without
significant amounts of effective CRT (4 of 5 beats ineffective).
Of the 63 subjects randomized, 13 had one or more instances where the CAFRPlus algorithm
reached maximum heart rate and suspended while in the CAFRPlus arm. There were a total of
386 suspensions observed in these subjects. The overall incidence rate was 2.92 suspensions per
subject-month of follow-up with CAFRPlus turned ON.
Ancillary Objective #2
Objective:
To characterize any differences in mean heart rate during AF between when CAFRPlus was
applied and when CAFR was applied. This ancillary objective was descriptive in nature and
there was no pre-specified hypothesis to be tested.
Results:
The average heart rate during AF was 77.0 ± 9.9 when CAFR was ON and 79.5 ± 9.7 when
CAFRPlus was ON. The mean difference (CAFRPlus - CAFR) was 2.5, with a 95% confidence
interval of (1.4, 3.5).
Ancillary Objective #3
Objective:
To characterize the value of the different diagnostic pieces of information available in CRTee.
This ancillary objective was descriptive in nature and there was no pre-specified hypothesis to be
tested.
Results
Clinician’s feedback was collected on how they conducted trouble shooting of ineffective CRT
and what diagnostic tools were helpful to them. A total of 18 cases of ineffective CRT were
identified, mostly via the in-office LV capture efficacy test (67%) and/or real time wave form
markers (56%). These same tools were also indicated as most helpful to understand the problem
(78% and 61%).
Ancillary Objective #4
Objective:
To track the incidence of cardiovascular-related adverse events. This ancillary objective was
descriptive in nature and there was no pre-specified hypothesis to be tested.
Results
The most frequently reported type of cardiovascular event was cardiac failure, which occurred 8
times in 7 subjects (9.9%).
10
IDE Number: G140092
# Non CV Related
Of these 16 CV-related events, 10 occurred during the randomized follow-up periods. The
incidence rate appeared to be slightly higher when CAFRPlus was ON; however the rates were
not statistically different (p=0.37, Exact Poisson Rate Ratio Test). Refer to Table 7 for a
summary of the incidence rates.
The incidence rates of non-cardiovascular related AEs between the two arms during the crossover follow-up periods were also compared. In this case, a slightly higher rate of non CV-related
events occurred during CAFR (p=0.32). Refer to Table 8 for the summary of the incidence rates.
Table 7: Cardiovascular Related Adverse Events Incidence Rate by Algorithm
CRTee
Algorithm
CAFR 3 3.76 0.80
CAFRPlus 7 3.88 1.80
Overall 10 7.64 1.31
SAS Program: V:\CRTee\Reports\Final_Report\Programs\AncillaryObjective4_CVevents.sas
# CV Related
Adverse Events
Person-Years of
Follow-up
Event Rate (per
Person-Year)
Table 8: Non Cardiovascular Related Adverse Events Incidence Rate by
Algorithm
CRTee
Algorithm
CAFR 7 3.76 1.86
CAFRPlus 3 3.88 0.77
Overall 10 7.64 1.31
SAS Program: V:\CRTee\Reports\Final_Report\Programs\AncillaryObjective4_NonCVevents.sas
Adverse Events
Person-Years of
Follow-up
Event Rate (per
Person-Year)
Ancillary Objective #5
Objective:
To characterize the occurrence of spontaneous VT/VF episodes. This ancillary objective was
descriptive in nature and there was no pre-specified hypothesis to be tested.
Results
A total of 30 spontaneous VT/VF episodes plus one VT/VF storm were detected during the
study. Eleven of the episodes were classified as monomorphic ventricular tachycardia, 13
episodes were classified as dual tachycardia, and 6 episodes were classified as atrial
fibrillation/atrial flutter. There was no significant difference in VT/VF event rates between
CAFR and CAFRPlus arms.
11
IDE Number: G140092
5 ADVERSE EVENTS SUMMARY
All adverse events were collected throughout the study duration, starting at the time of signing
the consent form. All reported adverse events were adjudicated by the CEC to determine
relatedness of the event to the procedure, system, or CRTee software, as well as cardiovascular
(CV) relatedness. At the time of the final database lock, 100% of the reported AEs had been
adjudicated by the CEC.
Of the 71 enrolled subjects, there were 36 adverse events reported for 21 subjects. Table 9
provides a summary of all 36 adverse events with regard to seriousness and relatedness to the
CRTee system or CV relatedness. Of these 36 adverse events, none were deemed to be related to
the procedure or CRTee software, and 16 were deemed to be CV related. A total of 11 events
met the criteria for seriousness, primarily (82%) due to in-patient or prolonged hospitalization
(Table 10).
Table 9: Adverse Events Summary
Enrolled
Number of Events (Number, % Subjects)
Total Adverse Events 36 (21, 29.6%) 36 (21, 31.8%)
Serious Adverse Event
Yes 11 (7, 9.9%) 11 (7, 10.6%)
No 25 (16, 22.5%) 25 (16, 24.2%)
Unanticipated Adverse Device Effect
Yes 0 (0, 0.0%) 0 (0, 0.0%)
No 36 (21, 29.6%) 36 (21, 31.8%)
Unanticipated Serious Adverse Device Effect
Yes 0 (0, 0.0%) 0 (0, 0.0%)
No 36 (21, 29.6%) 36 (21, 31.8%)
Procedure Relatedness
Related 0 (0, 0.0%) 0 (0, 0.0%)
Not Related 36 (21, 29.6%) 36 (21, 31.8%)
Unknown 0 (0, 0.0%) 0 (0, 0.0%)
System Relatedness
(N = 71)
Randomized
(N = 66)
Related 0 (0, 0.0%) 0 (0, 0.0%)
Not Related 35 (20, 28.2%) 35 (20, 30.3%)
Unknown 1 (1, 1.4%) 1 (1, 1.5%)
CRTee Algorithm Relatedness
Related 0 (0, 0.0%) 0 (0, 0.0%)
Not Related 36 (21, 29.6%) 36 (21, 31.8%)
12

IDE Number: G140092
SAS Program: V:\CRTee\Reports\Final_Report\Programs\AESummary.sas
Enrolled
Number of Events (Number, % Subjects)
Unknown 0 (0, 0.0%) 0 (0, 0.0%)
Cardiovascular Relatedness
Related 16 (14, 19.7%) 16 (14, 21.2%)
Not Related 19 (12, 16.9%) 19 (12, 18.2%)
Unknown 1 (1, 1.4%) 1 (1, 1.5%)
SAS Program: V:\CRTee\Reports\Final_Report\Programs\AESummary.sas
(N = 71)
Table 10: Seriousness Criteria Met
Serious Adverse Events
Seriousness Criterion (not Mutually Exclusive)
Led to death 1 (9.1%)
Led to life threatening illness or injury 2 (18.2%)
Led to permanent impairment of a body structure or a body function 0 (0.0%)
Led to in-patient or prolonged hospitalization 9 (81.8%)
Led to medical or surgical intervention to prevent life-threatening
illness or injury or permanent impairment to body structure or body
function
4 (36.4%)
Randomized
(N = 66)
(N = 11)
Led to foetal distress, foetal death, or a congenital abnormality or
birth defect
0 (0.0%)
6 DEATH SUMMARY
There was one subject death during the study.
The subject was a 64-year old male in Saudi Arabia. Reported subject’s medical history
included: ischemic cardiomyopathy, hypertension, left ventricular hypertrophy, valve
dysfunction (mitral and tricuspid), prior balloon angioplasty, AF (paroxysmal), type 2 diabetes,
dyslipidemia, renal dysfunction, and hepatitis. Reported subject’s baseline medications included:
cardiac glycosides, aldosterone antagonists, beta-blocker, and other cardiovascular meds
(ivabradine). Reported subjects baseline symptoms included edema and fatigue/weakness.
The subject was consented into the study on 02 Aug 2015, and completed phase 1, phase 2 and
phase 3 visits within 3 weeks of enrollment. The subject expired at home on 29 Oct 2015 (site
awareness 10 Nov 2015). Per site, family indicated the subject died naturally; however, despite
multiple attempts via queries and onsite monitors, no documentation could be obtained from the
site confirming any medical issues during the time just prior to death. Due to minimal
information available, the CEC adjudicated the primary cause of death, death classification,
relatedness to the CRT-D system, and cardiovascular relatedness as unknown.
13

IDE Number: G140092
7 CLINICAL STUDY CONCLUSION
The CRTee feature set was designed to evaluate whether or not CRT paces are effective, and
contains an algorithm that, depending on the pattern of ineffective CRT paces, effective CRT
paces, and senses occurring during AF, increases, decreases or maintains the ventricular pacing
rate in order to increase the amount of effective CRT delivered with the minimum increase in
heart rate. This algorithm is referred to as CAFRPlus and only runs during AF.
The CRTee Study evaluated the CRTee feature set and passed its primary and secondary
objectives, demonstrating safety and efficacy of the CRTee feature set. The study results
demonstrated the following:
• The CAFRPlus algorithm is not inferior to the CAFR algorithm currently included in
Medtronic devices as demonstrated by the percent effective CRT pacing during AF while
CAFRPlus was programmed on not being less than the percent effective pacing during
AF while CAFR was programmed on.
• The CAFRPlus algorithm is superior to the CAFR algorithm currently included in
Medtronic devices as demonstrated by the magnitude of improvement in percent effective
CRT pacing during AF:
An increase of 7.0% ± 8.7% in effective CRT pacing during AF was
observed with the 54 complete paired datasets (p-value of <0.0001)
95.86% Confidence interval: (4.5%, 9.5%)
• The study results were robust to missing data.
• The study results were poolable from different geographical regions.
• There were no unanticipated adverse device effects.
• There were no adverse events related to the CRTee feature set.
The ancillary objectives focused on other clinically relevant outcomes for heart failure patients
and were intended to support the results of the primary and secondary objectives. Furthermore,
adverse events had similar rates between the two study groups. Additionally, the trial
demonstrated that VT/VF detection in this study was not affected by the CAFRPlus algorithm.
There was no significant difference in VT/VF event rates between CAFR and CAFRPlus arms.
The study has demonstrated the safety and effectiveness of the CRTee feature set in the clinical
environment.
14


Medtronic, Inc.
710 Medtronic Parkway
Minneapolis, MN 55432
USA
www.medtronic.com
+1 763 514 4000
Medtronic USA, Inc.
Toll-free in the USA (24-hour technical consultation
for physicians and medical professionals)
Bradycardia: 1-800-505-4636
Tachycardia: 1-800-723-4636
Europe/Middle East/Africa
Medtronic International Trading Sàrl
Route du Molliau 31
Case Postale 84
CH-1131 Tolochenaz
Switzerland
+41 21 802 7000
Authorized Representative in the European Community
Medtronic B.V.
Earl Bakkenstraat 10
6422 PJ Heerlen
The Netherlands
+31 45 566 8000
Technical manuals
www.medtronic.com/manuals
© 2016 Medtronic
M971035A001 A
2016-12-08
*M971035A001*
 Loading...
Loading...