

Medtronic SYNERGY 7427, SYNERGY VERSITREL 7427V Technical Manual

SYNERGY™
SYNERGY VERSITREL
Dual-Program Neurostimulators for Spinal Cord
Stimulation (SCS)
Technical Manual
™
7427
7427V
Rx Only


Synergy™ Model 7427
Synergy Versitrel™ Model
7427V
Technical Manual

The following are trademarks of Medtronic®:
DualScreen
Octad
Z Quad Compact
Specify
, Itrel, Mattrix, MemoryMod, N’Vision™, Pisces-
, Pisces-Quad, Pisces-Quad Plus, Pisces Z Quad™, Pisces
™
, SoftStart™, Synergy™, Synergy Versitrel™, and X-trel.
™
, Pisces Z Quad Plus™, Resume TL, Resume II,

Table of Contents
Introduction 5
Overview of Manual 5
Device Description 5
Package Contents 6
Patient Selection 7
Indications 7
Contraindications 7
Warnings 8
Precautions 11
Synergy and Synergy Versitrel Clinical Summary 16
Adverse Events 16
Neurostimulator Hardware Description 27
Identification 28
Power ON Reset 29
Battery Depletion 29
Neurostimulator Software Description 30
Programs 30
Programmable Functions 32
Lead-Extension Options 34
Screening Procedure 34
Synergy and Synergy Versitrel Systems Eligibility 35
Neurostimulator Implantation or Replacement 38
Neurostimulator Implantation 39
Neurostimulator Replacement 45
Explanted Component Disposal 51
Patient Counseling Information 51
Theft Detectors and Security Screening Devices 52
Patient Registration 53
3

Resterilization 54
Specifications 56
Conformance to Standards 61
Appendix A: Battery Longevity Reference 62
Appendix B: Synergy EZ Patient Programmer 76
Keypad 77
Control Switches 78
Programmer Battery 79
Symbols and Indicator Lights 80
Patient Programmer Specifications 83
FCC Information 83
Special Notice 84
Warranty 85
Glossary 88
4

Introduction
Introduction
Overview of Manual
This manual describes specifications and operation of the Synergy
Model 7427 and Synergy Versitrel Model 7427V Neurostimulators. It
includes information about the control equipment used with each
neurostimulator. You will find instructions for handling, storing,
implanting, replacing, and explanting the neurostimulator. General
resterilization guidelines are also provided for the neurostimulator. In
addition, this manual describes some items to discuss with your
patient.
Device Description
The Medtronic Synergy Model 7427 and Synergy Versitrel Model
7427V Neurostimulators are multiprogrammable devices designed
for use in spinal cord stimulation. They accommodate one or two
extensions and can function in either dual-program mode or singleprogram mode. In dual-program mode, the amplitude, pulse width
and up to 8 electrodes are set independently for each program. In
single-program mode, the amplitude, pulse width, and rate are
delivered to the selected electrodes; up to 8 electrodes can be
selected.
The operation of the neurostimulator is supported by a clinician
programmer and patient programmer.
The neurostimulator is powered by a sealed battery and directed by
electronic circuitry to send pulses of controlled electrical stimulation
through the implanted lead-extensions to target sites.
For a complete list of model numbers and components compatible
with the neurostimulator, see the system components sheet
packaged with this manual in the neurostimulator shelf box.
5
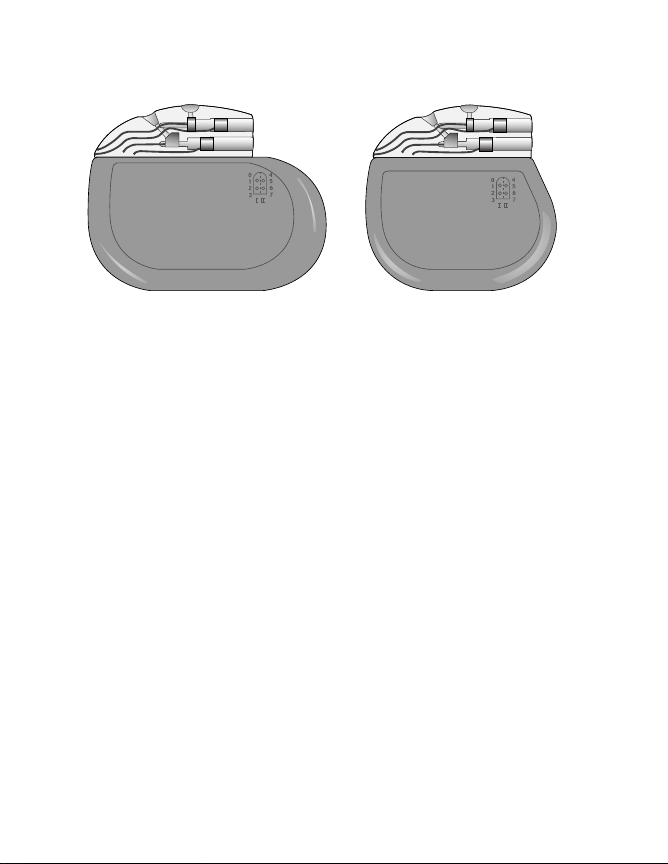
Introduction
Synergy Model 7427 Synergy Versitrel Model 7427V
Figure 1. Synergy Model 7427 and Synergy Versitrel Model 7427V
Neurostimulators.
Notes:
■
Throughout this manual, the Model 7427 Neurostimulator is
typically depicted within figure art.
■
The functionality of the Synergy Versitrel and Synergy
Neurostimulators is identical; however, the longevity of the
Synergy Neurostimulator is longer than that of the Synergy
Versitrel Neurostimulator. Refer to Table 10 on page 60 in
“Specifications” for an overview of physical differences
between the two models. Figure 1 is a comparison of the size
and shape of the Synergy and Synergy Versitrel
Neurostimulators.
Package Contents
■
One Synergy Model 7427 or one Synergy Versitrel
Model 7427V Neurostimulator
■
One hex wrench
■
Product literature
6

Patient Selection
Patient Selection
The Synergy and Synergy Versitrel Neurostimulation Systems are
designed to aid in the management of pain via pulsed electrical
stimulation through nerve structures in the dorsal aspect of the spinal
cord. Activation of these structures produces nerve impulses that can
inhibit the transmission of pain.
Indications
The Synergy Model 7427 and Synergy Versitrel Model 7427V
Neurostimulators are part of dual-program systems for spinal cord
stimulation. The systems are indicated as an aid in the management
of chronic, intractable pain of the trunk or limbs.
Patients should be carefully selected to assure that their pain is of
physiological origin. Also, patients must be appropriate candidates
for surgery.
Contraindications
Patients are contraindicated for internalization if they are clearly
unsuccessful in receiving pain relief during trial stimulation, or if they
are unable to properly operate the system.
After implantation of any system component, the following
contraindication applies:
Diathermy – Do not use shortwave diathermy, microwave diathermy
or therapeutic ultrasound diathermy (all now referred to as
diathermy) on patients implanted with a neurostimulation system.
Energy from diathermy can be transferred through the implanted
system and can cause tissue damage at the location of the implanted
electrodes, resulting in severe injury or death.
Diathermy is further prohibited because it can also damage the
neurostimulation system components resulting in loss of therapy,
requiring additional surgery for system explantation and
replacement. Injury or damage can occur during diathermy treatment
7

Warnings
whether the neurostimulation system is turned “on” or “off.” Advise
your patients to inform all their health care professionals that they
should not be exposed to diathermy treatment.
Warnings
Case Damage – If the neurostimulator case is ruptured or pierced
due to outside forces, severe burns could result from exposure to the
battery chemicals.
Electrocautery – In certain circumstances, electrocautery can
cause the neurostimulator to stop functioning which will require the
neurostimulator to be replaced. It could also change the programmed
parameters to “Power ON Reset” settings which include changing the
output to “off” and the amplitude to zero which would require the
neurostimulator to be reprogrammed with the Medtronic clinician
programmer.
Based on tests to date, if use of electrocautery is necessary, follow
these precautions:
■
Turn off the neurostimulator before performing electrocautery.
■
Only bipolar cautery is recommended.
■
If unipolar cautery is necessary: Do not use high voltage
modes; keep the power setting as low as possible, and keep
the current path (ground plate) as far away from the
neurostimulator, extension, and lead as possible.
■
Confirm the neurostimulator function after
electrocauterization.
The effect of electrocautery on patients with implanted
neurostimulators is unknown.
Equipment Operation – Patients should not operate potentially
dangerous equipment such as power tools or automobiles during
stimulation.
8

Warnings
Magnetic Resonance Imaging (MRI) – Medtronic recommends
physicians not prescribe an MRI for a patient who has any
component of an implanted neurostimulation system for Spinal Cord
Stimulation (SCS). Exposing a patient with an implanted SCS
neurostimulation system to an MRI may potentially injure the patient
and/or damage the neurostimulator. The known potential risks are as
follows:
■
Induced electrical currents from the MRI to the
neurostimulation system may cause heating, especially at the
lead electrode site, resulting in tissue damage. Induced
electrical currents may also stimulate or shock the patient.
Note: This warning applies even if only a lead and/or extension
is implanted in the body.
Heating risks are affected by a number of factors involving the
MRI equipment and the implanted neurostimulation system.
Factors that increase the risks of heating and patient injury
include but are not limited to the following:
– High MRI Specific Absorption Rate (SAR) Radio
Frequency (RF) power levels
– Lower impedance leads and/or extensions (Medtronic
product names or model numbers designated with a “Z”, an
“LZ”, or “Low Impedance”)
– MRI RF transmit coil that is near or extends over the
implanted lead system
– Implanted lead systems with small surface area electrodes
– Short separation distances between lead electrodes and
thermally sensitive tissue
■
An MRI may permanently damage the neurostimulator, which
may require explant or possible replacement.
■
An MRI may affect the functional operation of the
neurostimulator. The MRI may also reset the neurostimulator
parameters to its default settings, which requires
reprogramming with the clinician programmer.
9

Warnings
■
The neurostimulator may move within the implant pocket and
align itself with the MRI field, which may cause patient
discomfort or open a recent neurostimulator implant incision.
In addition, the MRI image details may be degraded, distorted, or
blocked from view by the implanted neurostimulation system.
Pediatric Use – Safety and effectiveness of this system has not
been established for pediatric use.
Postural Changes – Postural changes or abrupt movements may
cause an increase or decrease in the perceived level of stimulation.
Higher levels of stimulation have been described as uncomfortable
(“jolting” or “shocking”) by some patients.
Pregnancy – Safety for use during pregnancy or delivery have not
been established.
Tel emetr y – Do not send a patient home with “????” displayed on
the clinician programmer screen for any programmable value. This
indicates that the parameter or mode is invalid and must be
reprogrammed.
Theft Detectors and Screening Devices – Theft detectors found in
retail stores, public libraries, etc., and security screening devices
found in airports, government buildings, etc., occasionally may cause
intermittent stimulation or a momentary increase in stimulation
intensity. When they pass through these devices, some patients may
perceive intermittent stimulation as switching their neurostimulation
system on and off. It is also possible that patients, especially those
with low stimulation thresholds, may experience a momentary
increase in their perceived stimulation when they pass through these
devices. Higher levels of stimulation have been described as
uncomfortable (“jolting” or “shocking”) by some patients. For
information on how to minimize these interactions when passing
through theft detectors and security screening devices, see “Patient
Counseling Information” on page 51.
10

Precautions
Precautions
Physician Training
Implanting Physicians – Implanting physicians should be
experienced in spinal procedures and review the procedures
described in this technical manual prior to implant.
Prescribing Physicians – Prescribing physicians should be
experienced in the diagnosis and treatment of chronic intractable
pain of the trunk or limbs and should be familiar with the use of the
Synergy and Synergy Versitrel Neurostimulation Systems.
Storage and Sterilization
Resterilization Considerations – All implantable components
are supplied sterile. If resterilization is necessary, refer to
“Resterilization” on page 54 for further information.
Sterilization Method – The neurostimulator was sterilized with
ethylene oxide before shipment.
Storage Temperature – Store the neurostimulator between 0° F
(-18° C) and 125° F (52° C). Temperatures outside this range can
damage components.
System and Therapy
Component Failures – The physician should be aware that all
neurostimulation systems may unexpectedly cease to function. A
system may fail at any time due to random failures of the system
components or the battery (prior to depletion). These events, which
can include electrical short or open circuits and insulation breaches,
cannot be predicted.
Components – The use of non-Medtronic components with this
system may result in damage to Medtronic components, less than
adequate stimulation, or increased risks to the patient.
11

Precautions
Patient Detoxification – It is recommended that patients undergo
detoxification from narcotics prior to implant.
Patient Management – To help ensure maximum benefits from the
neurostimulation system, long-term postsurgical management of
patients is recommended.
Implantation / Explantation
Component Disposal – If explanting a Synergy or Synergy Versitrel
Neurostimulation System component, please remember the
following guidelines:
■
Do not incinerate the neurostimulator; explosion can result if a
neurostimulator is subjected to incineration or cremation
temperatures.
■
Return all explanted components to Medtronic for analysis and
safe disposal.
Component Handling – Handle the implanted components of this
system with extreme care. These components may be damaged by
excessive traction or sharp instruments.
Etched Identification – Place the neurostimulator with the etched
identification side facing outward, away from the muscle layer of the
body.
Extension-Neurostimulator Connection – Wipe off any body
fluids from the extension connector pins or connector block before
connecting them. Contamination of connections can affect
neurostimulation. Do not tighten setscrews without the extension
inserted. This can damage the connector block. Do not insert an
extension into the neurostimulator connector block without visual
verification that the setscrews are sufficiently retracted to allow
insertion.
Implant Considerations – Do not implant a device when the
storage package has been pierced or altered, potentially rendering it
non-sterile; the component shows signs of damage; or the Use By
12
Precautions
date has expired, because this can adversely affect storage package
sterility and battery longevity.
Neurostimulator Handling – Be extremely careful when using
sharp instruments around the neurostimulator to avoid nicking or
damaging the neurostimulator case or the connector block. The
neurostimulator can be damaged if dropped from a height of 12
inches (30 cm) or more onto a hard surface (i.e., a concrete floor). If
this happens, do not implant the neurostimulator.
Lead-Extension Connection – Wipe off any body fluids from the
lead or extension contacts before connecting. Contamination of
connections can affect neurostimulation.
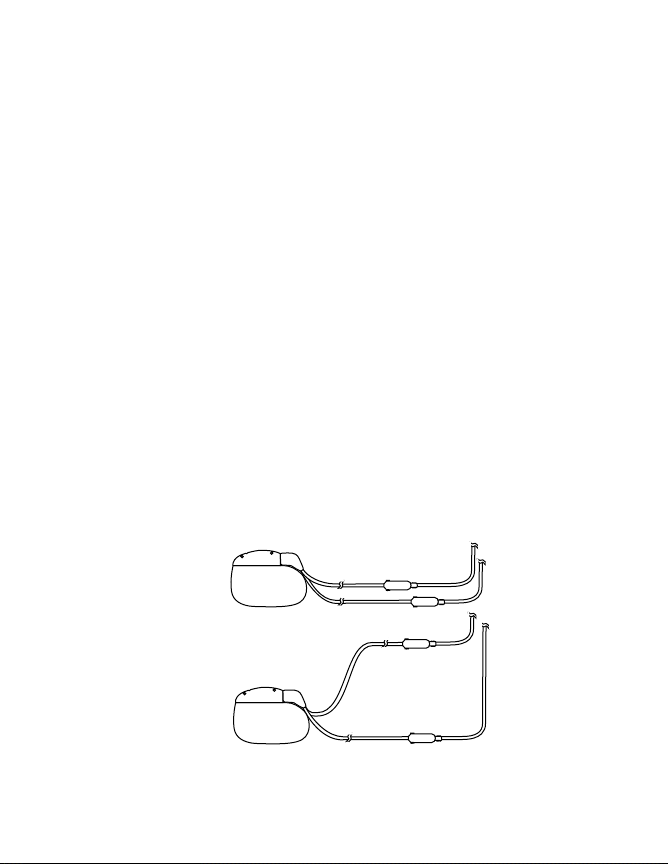
Lead-Extension Routing – It is recommended that the implanted
lead-extensions in dual lead-extension systems be routed so they do
not form a “loop.” When exposed to some theft detectors, looped
lead-extensions increase the potential for patients to experience a
momentary increase in their perceived level of stimulation. Higher
levels of stimulation have been described as uncomfortable (“jolting”
or “shocking”) by some patients as they pass through these devices.
Figure 2 illustrates proper and improper routing methods for dual
lead-extensions.
Proper
Improper
Figure 2. Proper and improper dual lead-extension routing
(use fluoroscopy to verify).
13

Precautions
Single Use – The neurostimulator is intended for Single Use Only.
DO NOT REUSE.
Medical Environment
Most routine diagnostic procedures, such as fluoroscopy and x-rays,
are not expected to affect system operation. However, the following
precautions should be noted.
Effects on Other Medical Devices – The neurostimulation system
may affect the operation of other implanted devices, such as cardiac
pacemakers and implantable defibrillators. Possible effects include
sensing problems and inappropriate device responses. If the patient
requires concurrent implantable pacemaker and/or defibrillator
therapy, careful programming of each system may be necessary to
optimize the patient’s benefit from each device.
External Defibrillators – Safety for use of external defibrillatory
discharges on patients with neurostimulation systems has not been
established. External defibrillation may damage a neurostimulator.
If external defibrillation is necessary, follow these precautions to
minimize current flowing through the neurostimulator and leadextension system:
■
Position defibrillation paddles as far from the neurostimulator
as possible.
■
Position defibrillation paddles perpendicular to the
neurostimulation system.
■
Use the lowest clinically appropriate energy output (watt
seconds).
■
Confirm neurostimulation system function following any
external defibrillation.
Lithotripsy – Use of high output ultrasonic devices, such as an
electrohydraulic lithotriptor, is not recommended for patients with an
implanted neurostimulation system. While there is no danger to the
patient, exposure to high output ultrasonic frequencies may result in
14

Precautions
damage to the neurostimulator circuitry. If lithotripsy must be used,
do not focus the beam near the neurostimulator.
Radiation Therapy – Radiation therapy can cause damage to the
electronic components of a neurostimulator. It is not recommended
to use radiation therapy directly over a neurostimulation device.
Home or Occupational Environment
Cellular Phones – Based on tests to date, cellular phones appear to
have no effect on the Synergy and Synergy Versitrel
Neurostimulation Systems. However, the effect of all cellular phones
on neurostimulation systems is unknown and patients should avoid
placing cellular phones directly over the device.
Electromagnetic Interference – Patients should exercise
reasonable caution in avoidance of devices which generate a strong
electric or magnetic field. Close proximity to high levels of
electromagnetic interference (EMI) may cause a neurostimulator to
unexpectedly cease to function or cause sensitive patients to
experience a momentary increase in their perceived level of
stimulation. Also, severe EMI can permanently erase the
neurostimulator serial number, causing “????” to be displayed in
place of the serial number.
High/Low Pressure Effects – The effects of high/low pressure on
patients with an implanted neurostimulation system are unknown.
Home Appliances – Home appliances that are in good working
order and properly grounded do not usually produce enough
electromagnetic interference (EMI) to interfere with neurostimulator
operation.
Occupational Environments – Commercial electrical equipment
(arc welders, induction furnaces, resistance welders),
communication equipment (microwave transmitters, linear power
amplifiers, high-power amateur transmitters), and high voltage power
lines may generate enough EMI to interfere with neurostimulator
operation if approached too closely.
15

Synergy and Synergy Versitrel Clinical Summary
Synergy and Synergy Versitrel Clinical
Summary
The clinical use of Synergy and Synergy Versitrel Neurostimulation
Systems is supported by Medtronic clinical studies of the Mattrix and
Itrel 3 Spinal Cord Stimulation Systems. All patients were implanted
to aid in the management of chronic, intractable pain of the trunk or
limbs. Following is a summary of the data from those studies that
supports the clinical use of Synergy and Synergy Versitrel Systems.
The Medtronic-sponsored Mattrix study was a retrospective
evaluation of 89 patients. All patients implanted with dual-program
radio frequency Mattrix Systems before April 1996 at the four
participating centers were included in the study, which allowed each
patient to be followed for at least 12 months. A total of 1,261 device
months of experience were acquired for this study.
The Medtronic-sponsored Itrel 3 study was a prospective, open label
evaluation of 84 patients implanted at 11 centers. A total of 823
device months of experience were acquired for this study.
Combined adverse event data for these two studies represents a total
of 2,084 device months of experience collected from 173 patients.
The adverse event experience reported here represents this
combined experience.
Adverse Events
Eighty-three of the 173 patients experienced 116 device-related and
nondevice-related adverse events. Eighty-seven of the adverse
events in 65 patients were considered to be device related. Devicerelated adverse events reported in at least 2 patients each are
provided in Table 1.
16

Synergy and Synergy Versitrel Clinical Summary
Table 1. Mattrix and Itrel 3 Study Combined Device-Related Adverse
Events Experienced By At Least Two Patients Each
a
.
Adverse Event Number of Patients
Reporting
Loss of Pain Relief 22
Lead Migration 16
Infection 8
Pain at Pocket Site 5
Receiver Migration
Antenna Placement Problem
b
b
4
4
Programmer/Telemetry Problem 3
Transmitter Malfunction
b
2
Threshold Rise 2
a
Patients may have experienced more than one event.
b
These device-related adverse events are applicable only to the radio frequency Mattrix
System and are not applicable to a fully implantable system.
Other device-related adverse events experienced in 1 patient each
included receiver malfunction, seroma at receiver site, allergic/
immune response, undesirable change in stimulation, “pain relief no
better than single lead stimulation/stimulation drives patients pain,”
CSF leak, pocket hypersensitivity, loss of electronic serial number
and Power ON Reset condition after MRI, electrode failure, lead
erosion, radicular chest wall stimulation, high electrode impedance
due to lead fracture, suspected lead breakage, and lead/extension
infection.
17

Synergy and Synergy Versitrel Clinical Summary
Potential Adverse Events
Anticipated adverse events which may potentially occur, but were not
reported in the referenced clinical trials, include:
■
Neurostimulator erosion
■
Extension erosion/migration
■
Patients on anticoagulant therapies may be at greater risk for
postoperative complications such as hematomas that can
result in paralysis
■
Placement of the epidural lead-extension is a surgical
procedure that may expose patient to risks of epidural
hemorrhage, hematoma, and/or paralysis
■
Hematoma at the neurostimulator site
■
Undesirable change in stimulation, possibly related to cellular
changes around the electrode(s), shifts in electrode position,
loose electrical connections, or lead-extension fractures, which
has been described as uncomfortable (“jolting” or “shocking”)
by some patients
Stimulation Parameter Use
Stimulation parameters used during the Itrel 3 study are provided in
Ta bl e 2 .
18

Synergy and Synergy Versitrel Clinical Summary
Table 2. Summary of Itrel 3 Study Stimulation Parameters.
Stimulation
Parameter
Amplitude (V)
Average
Standard Deviation
n
Pulse Width (µsec)
Average
Standard Deviation
n
Pulse Rate (Hz)
Average
Standard Deviation
n
1 Month
(82 systems)
3.2
1.6
79
275
76
81
79
25
81
3 Months
(78 systems)
3.2
1.8
73
283
82
78
79
24
78
6 Months
(69 systems)
3.1
1.7
61
280
88
66
81
25
67
12 Months
(63 systems)
3.0
1.8
58
278
81
64
85
32
64
Mattrix Study Design
The Mattrix 12-Month Study was a retrospective review of 12 months
of implant and follow-up experience from a consecutive series of U.S.
patients with Mattrix systems. All patients were implanted to aid in the
management of chronic, intractable pain of the trunk or limbs.
Mattrix Patients Studied
Four investigational sites in the United States participated in this
study. A total of 89 patients were enrolled in the study at these four
sites. Fifty-two percent (46/89, 52%) of study patients were male. The
mean age of study patients was 51 years (range: 29 – 84 years).
Mattrix Study Methods
This study was a retrospective review of at least 12 months of implant
and follow-up experience from a consecutive series of U.S. patients
with Mattrix systems.
Most data was collected retrospectively from clinic records of the last
follow-up for patients with at least 12 months of Mattrix follow-up
experience. Data was gathered prospectively for patients who had
less than 12 months of Mattrix follow-up data in clinic records.
19

Synergy and Synergy Versitrel Clinical Summary
Mattrix Study Results
Average follow-up per patient was 14.2 ± 4.5 months, with a range of
2.3 – 22.9 months. At least one year of follow-up was obtained for
82% (73/89) of the study patients. Proper device operation was
reported by physicians for 93% of patients (83/89).
Sixty-nine patients (69/89, 78%) were using their Mattrix devices at
their last known follow-up. Of the 73 patients who had at least one
year follow-up completed, 64 patients (64/73, 88%) were using their
Mattrix devices at their last known follow-up.
There were 14 system explants (10 device related), 4 patients for
whom therapy was discontinued but no explant occurred (3 device
related), and one patient death (not related to the device). Use
information was not available for one patient. A system survival curve
is presented in Figure 3 that examines the impact of these 13 devicerelated events on device use throughout the clinical study follow-up
period. This standard Kaplan Meier survival curve with a 95%
confidence limit presents the time to system not in use for devicerelated adverse events only. This graph indicates 88.3% (plus or
minus 3.5%) of patients had devices in use at approximately 1 year
of follow-up, where 3.5% represents the 95% confidence limit. At 1.5
years, this number drops to about 83.9%.
The system survival curve presented in Figure 4 examines the
impact of all adverse events on device use throughout the clinical
study follow-up period. This standard Kaplan Meier survival curve
with a 95% confidence limit presents the time to system not in use for
all adverse events. This graph indicates 82.6% (plus or minus 4.1%)
of patients had devices in use at approximately 1 year of follow-up. At
1.5 years, this number drops to about 78.4%.
20
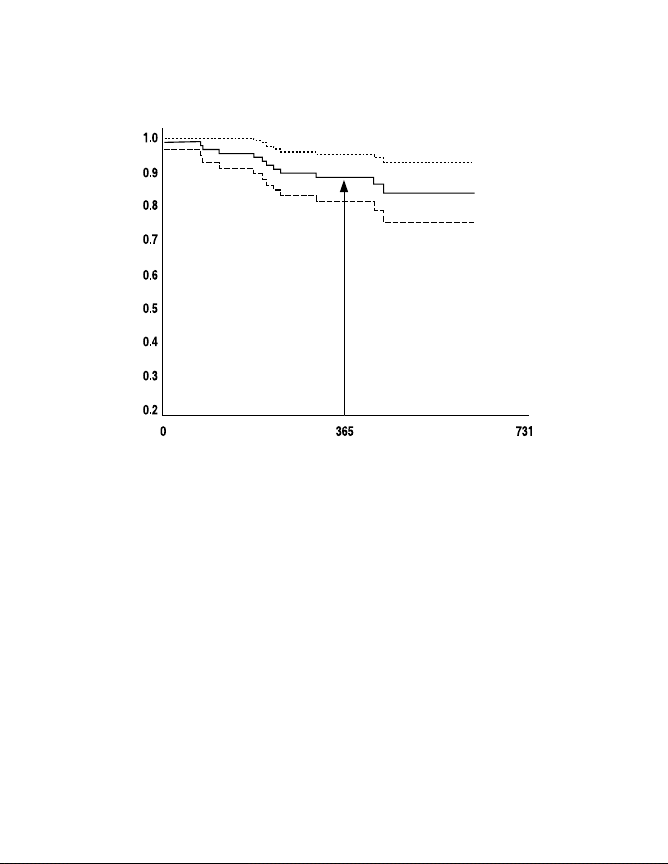
Synergy and Synergy Versitrel Clinical Summary
Device-in-Use Kaplan Meier Survival Curve with 95% Confidence Limits
Survival Probability
Mattrix Retrospective Study—Device-Related Events Only
One-year survival
estimate = 88.3 ± 3.5%
Time Until Not in Use (Days)
Figure 3. Device-in-use survival curve for device-related events.
21
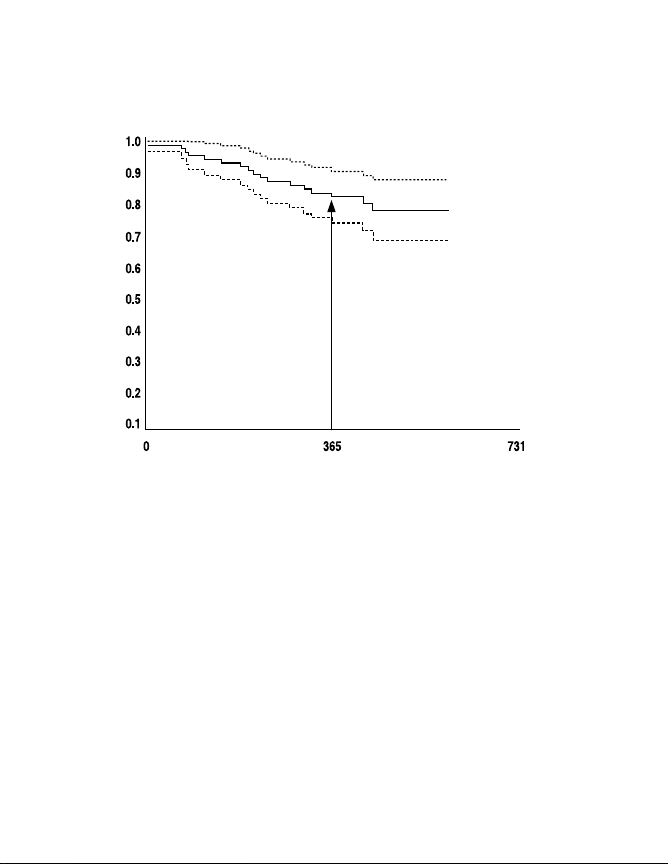
Synergy and Synergy Versitrel Clinical Summary
Device-in-Use Kaplan Meier Survival Curve with 95% Confidence Limits
Mattrix Retrospective Study—All Reported Adverse Events
One-year survival
estimate = 82.6 ± 4.1%
Survival Probability
Time Until Not in Use (Days)
Figure 4. Device-in-use survival curve for all adverse events.
Patient satisfaction with the pain relief provided by the stimulation
system after one year of use was determined by asking the following
question: “Is the patient satisfied with the pain relief provided by the
Mattrix System?” Responses to patient satisfaction are summarized
in Table 3. Investigators indicated that 62% of all study patients
(55/89) were satisfied with pain relief. Of the 73 patients completing
one year of follow-up, this satisfaction with pain relief rose to
70% (51/73 patients). “Other” responses to this satisfaction question
included: “patient would not commit” (3), partial pain relief (2), pain
controlled until patient fell (1), inadequate relief (1), skin irritation (1),
complication before fully optimized (1), and system explanted (1).
22

Synergy and Synergy Versitrel Clinical Summary
Table 3. Investigator Evaluation of Patient Satisfaction.
Patient
Group
Patient with
≥ 12 month
follow-up
(n=73) 51 (70%) 14 (19%) 0 (0%) 8 (11%)
All patients
(n=89) 55 (62%) 22 (25%) 2 (2%) 10 (11%)
Patient
Satisfied
n(%)
Patient
Not Satisfied
n(%)
Missing
Responses
n(%)
Other
Responses
n(%)
Forty-nine of the 89 patients experienced a total of 75 device-related
and nondevice-related adverse events. A total of 65 of these adverse
events were considered to be device related. There were no
unanticipated adverse device events reported. Device-related
adverse events reported by at least 2 patients each in this study are
presented in Table 4.
23

Synergy and Synergy Versitrel Clinical Summary
Table 4. Mattrix Study Device-Related Adverse Events Experienced By
Adverse Event Number of Patients Reporting
Loss of Pain Relief 21
Lead Migration 12
Infection (Receiver Site) 5
Receiver Migration
Antenna Placement Problem
Pain at Receiver Site 3
Transmitter Malfunction
Pain at Pocket Site 2
a
These device-related adverse events are applicable only to the radio frequency Mattrix
System and are not applicable to a fully implantable system.
At Least Two Patients Each.
a
a
a
4
4
2
Other device-related adverse events experienced in 1 patient each
(1%) included receiver malfunction, seroma at receiver site, allergic/
immune response, undesirable change in stimulation, pain relief no
better than single lead stimulation/stimulation drives patients pain,
and CSF leak.
Itrel 3 Study Design
The Itrel 3 study was a prospective, open-label study of 84 spinal
cord stimulation patients followed for 1, 3, 6, and 12 months. All
patients were implanted to aid in the management of chronic,
intractable pain of the trunk or limbs.
Itrel 3 Patients Studied
A total of 11 centers throughout Europe participated in this study,
enrolling 84 patients. One patient received two Itrel 3 systems;
24

Synergy and Synergy Versitrel Clinical Summary
therefore, 85 systems were evaluated. Fifty-four percent (45/84,
54%) of the study patients were male. Average age at study
enrollment was 52.6 years (range: 22 – 86 years).
Itrel 3 Study Methods
Data was prospectively collected in this study for a set of 84
European patients. Follow-up visits were scheduled at 1, 3, 6, and 12
months. Safety data collected in the study included adverse event
experiences and reasons for study discontinuation.
Itrel 3 Study Results
Average daily duration of pain was significantly reduced (p<0.0001)
at each follow-up interval when compared to the duration at baseline.
Patients reported an average of at least 63% pain relief throughout
the follow-up evaluation.
Of the 62 patients completing 12-month follow-up, 97% (60/62, 97%)
indicated stimulation was beneficial and 95% (59/62, 95%) indicated
they would agree to stimulation again for the same benefits.
A total of 34 patients experienced 41 device-related and nondevicerelated adverse events. Twenty-two of the adverse events in 21
patients were considered to be device related. There were no
unanticipated adverse device events reported. Device-related
adverse events reported in at least 2 patients each are provided in
Ta bl e 5 .
25

Synergy and Synergy Versitrel Clinical Summary
Table 5. Itrel 3 Study Device-Related Adverse Events Experienced By
Adverse Event Number of Patients Reporting
Lead Migration
Infection 3
Programmer/Telemetry Problem 3
Threshold Rise 2
a
One patient reported two lead migrations.
At Least Two Patients Each.
a
4
Other device-related adverse events experienced in one patient each
included pocket hypersensitivity, loss of electronic serial number and
Power ON Reset condition after MRI, electrode failure, loss of pain
relief, lead erosion, radicular chest wall stimulation, high electrode
impedance due to lead fracture, suspected lead breakage, and lead/
extension infection.
Individualization of Treatment
Best results are achieved when the patient is fully informed about the
therapy risks and benefits, surgical procedure, follow-up
requirements, and self-care responsibilities. Implantation of the
Synergy or Synergy Versitrel Neurostimulation System may be
appropriate for patients who meet the following criteria:
■
Patients should have chronic, intractable pain of physiological
origin.
■
Patients should be appropriate candidates for surgery.
Before the Synergy or Synergy Versitrel Neurostimulation System is
implanted, the following conditions should be met:
■
Satisfactory paresthesia coverage should be demonstrated by:
– intraoperative screening, or
– pre-existing spinal cord stimulation system.
26

Neurostimulator Hardware Description
■
Ensure that satisfactory paresthesia coverage can be obtained
for patients within the parameter limits of the Synergy or
Synergy Versitrel device. Refer to Table 7 on page 38 for a
summary of maximum stimulation voltage available from the
Synergy or Synergy Versitrel device given set values for pulse
width and rate.
– If the pulse rate is greater than 130 Hz, trial stimulation
should be repeated using a pulse rate of 130 Hz or lower.
– If the pulse width is greater than 450 µsec, trial stimulation
should be repeated using a pulse width of 450 µsec and
increasing the pulse amplitude by 5-10%.
– If both pulse rate and pulse width are within the parameter
capabilities of Synergy or Synergy Versitrel, but the trial
stimulation amplitude is greater than the corresponding
amplitude shown in Table 7, trial stimulation should be
repeated using a lower pulse rate to achieve the necessary
amplitude. If this cannot be accomplished, the high-energy
use patient should be evaluated for treatment with a Mattrix
System.
■
Ensure that unipolar mode stimulation is not required to obtain
satisfactory paresthesia coverage (unipolar mode stimulation
is not available with the Synergy or Synergy Versitrel
Neurostimulation Systems).
Neurostimulator Hardware Description
The neurostimulator is powered by a hermetically sealed silver
vanadium oxide cell and uses an integrated circuit to generate
electrical stimulation pulses. To protect the neurostimulator
components from body fluids, the electronics and power source are
hermetically sealed within an oval-shaped titanium shield.
27
 Loading...
Loading...