

Medtronic PRESTIGE LP User Manual
Instructions for Use
PRESTIGE LP™ CERVICAL DISC
Medtronic Sofamor Danek USA, Inc.
1800 Pyramid Place Memphis, TN 38132
Tel. 800 933 2635 (In U.S.A.)
901 396 3133 (Outside of U.S.A.)
Fax 901 396 0356
CAUTION: Federal (United States) law restricts this device to sale by or on the order of a
Physician.
HOW SUPPLIED
PRESTIGE LP™ Cervical Disc Implants – Sterile
Surgical Instruments – Non-sterile (unless otherwise noted on the package label)
DEVICE DESCRIPTION
The PRESTIGE LP™ Cervical Disc is a two-piece articulating device that is inserted into the
intervertebral disc space as a pre-assembled unit at one or two contiguous cervical levels using an
anterior approach. The device is manufactured from a titanium ceramic composite (Titanium6Aluminum-4Vanadium with 10% Titanium Carbide) and consists of two metal plates which
function via a ball and trough mechanism. The superior component of the implant contains the
ball portion of the mechanism, and the inferior component contains the trough portion. These two
features engage to create an interface designed to allow for motion after implantation. Each
component is affixed to the adjacent vertebral body by two rail geometries incorporating antimigration teeth, which are press fit into two pre-drilled holes in the vertebral bone. The portion
of the flat surface between the rails that contacts the vertebral endplate has a commercially pure
titanium (CP Ti) plasma thermal sprayed coating per ASTM F1580, designed to permit bony ongrowth for additional device incorporation. The remaining portion of the flat surface is titanium
ceramic roughened to enhance fixation. Each component also contains two anterior tab features
designed to aid in device insertion and to minimize the risk of implanting the device too far into
the intervertebral space.
Medtronic Page 1 of 91

Catalog
Number
Size (Height x AP Dimens ion x
ML Dimension)
6972250
5mm x 12mm x 15mm
6972450
5mm x 14mm x 15mm
6972650
5mm x 16 mm x 15mm
6972260
6mm x 12mm x 17.8mm
6972460
6mm x 14mm x 17.8mm
6972660
6mm x 16mm x 17.8mm
6972860
6mm x 18mm x 17.8mm
6972470
7mm x 14mm x 17.8mm
6972670
7mm x 16mm x 17.8mm
6972870
7mm x 18mm x 17.8mm
6972480
8mm x 14mm x 17.8mm
6972680
8mm x 16mm x 17.8mm
6972880
8mm x 18mm x 17.8mm
Figure 1a: PRESTIGE LP™ Cervical Disc Figure 1b: PRESTIGE LP™
Cervical Disc at two contiguous
levels
The PRESTIGE LP™ Cervical Disc is offered in a variety of configurations to accommodate
varied patient anatomy. The available components are shown in Table 1 below.
The PRESTIGE LP™ Cervical Disc is designed to allow a minimum of 10 degrees lateral
bending (from neutral) and a minimum of 10 degrees flexion/extension (from neutral). The
design is also intended to allow unlimited axial rotation (constrained by ligaments and posterior
elements) and translation of ±2mm in the sagittal plane.
Table 1: PRESTIGE LP™ Cervical Disc Device Sizes
The PRESTIGE LP™ Cervical Disc is implanted using instruments specific to the device, as
well as manual surgical instruments. Instruments specifically designed for implanting
Medtronic Page 2 of 91

PRESTIGE LP™ Cervical Disc consist of trials, trial cutter guides, rail punches, and implant
inserters. General purpose instruments include instruments for cervical distraction and
discectomy preparation.
No warranties, express or implied, are made. Implied warranties of merchantability and fitness
for a particular purpose or use are specifically excluded.
INDICATIONS FOR USE
The PRESTIGE LP™ Cervical Disc is indicated in skeletally mature patients for reconstruction
of the disc from C3-C7 following discectomy at one level or two contiguous levels for intractable
radiculopathy (arm pain and/or a neurological deficit) with or without neck pain, or myelopathy
due to abnormality localized to the level of the disc space and at least one of the following
conditions confirmed by imaging (CT, MRI, X-rays): herniated nucleus pulposus, spondylosis
(defined by the presence of osteophytes), and/or visible loss of disc height as compared to
adjacent levels. The PRESTIGE LP™ Cervical Disc is implanted using an anterior approach.
Patients should have failed at least 6 weeks of non-operative treatment or have had the presence
of progressive symptoms or signs of nerve root/spinal cord compression in the face of continued
non-operative management prior to implantation of the PRESTIGE LP™ Cervical Disc.
CONTRAINDICATIONS
The PRESTIGE LP™ Cervical Disc should not be implanted in patients with the following
conditions:
• Active systemic infection or localized infection at the surgical site;
• Osteoporosis or osteopenia defined as a DEXA bone mineral density T-score ≤ -1.0;
• Allergy or sensitivity to titanium, aluminum or vanadium;
• Marked cervical instability on neutral resting lateral or flexion/extension radiographs;
translation >3.5mm and/or >11° rotational difference from that of either level adjacent to
the treated levels;
• Severe spondylosis at the level to be treated, characterized by bridging osteophytes, loss
of disc height >50%, an absence of motion (<2°) as this may lead to a limited range of
motion and may encourage bone formation (e.g. heterotopic ossification, fusion);
• Severe facet joint arthropathy;
• Significant cervical anatomical deformity or clinically compromised vertebral bodies at
the affected level(s) due to current or past trauma (e.g., by radiographic appearance of
fracture callus, malunion or nonunion) or disease (e.g., ankylosing spondylitis,
rheumatoid arthritis); or
• Significant kyphotic deformity or significant reversal of lordosis.
WARNINGS
The PRESTIGE LP™ Cervical Disc should only be used by surgeons who are experienced with
anterior cervical spinal procedures and have undergone adequate hands-on training in the use of
this specific device. Only surgeons who are familiar with the implant components, instruments,
procedure, clinical applications, biomechanics, adverse events, and risks associated with the
PRESTIGE LP™ Cervical Disc should use this device. Medtronic will offer hands-on training to
physicians prior to their first use of the device. A lack of adequate experience and/or training may
lead to a higher incidence of adverse events, including neurological complications.
Medtronic Page 3 of 91

Correct sizing and placement of the device is essential to optimal performance. Information
regarding proper implant size selection, implant site preparation, and the use of instrumentation
before, during and after PRESTIGE LP™ surgery is provided in the PRESTIGE LP™ Cervical
Disc Surgical Technique manual. Users are advised to read and understand the surgical
technique manual and instructions for use prior to surgery.
Due to the proximity of vascular and neurological structures to the implantation site, there are
risks of serious or fatal hemorrhage and risks of neurological damage with the use of this device.
Serious or fatal hemorrhage may occur if the great vessels are eroded or punctured during
implantation or are subsequently damaged due to breakage of implants, migration of implants, or
if pulsatile erosion of the vessels occurs because of close apposition of the implants. Care must
be taken to identify and protect these structures.
Heterotopic Ossification (HO) is a potential complication associated with artificial cervical discs
and could lead to reduced cervical motion.
Devices with metal-on-metal articulating surfaces (such as the Prestige LP Cervical Disc) may
release wear debris, metallic particles or metal ions locally near the device and/or systemically.
The short and long term effects of the wear debris, metallic particles and metal ions on the body
are not known, but certain groups of patients may be at a higher risk including patients who are
pregnant, patients who are planning to get pregnant, and patients who have renal disease.
PRECAUTIONS
The safety and effectiveness of this device has not been established in patients with the following
conditions:
• Axial neck pain as the solitary symptom;
• Skeletally immature patients, pediatric or adolescent children (<21 years old), or those
over the age of 78;
• Prior cervical spine surgery, including prior surgery at the index level or adjacent levels;
• More than two cervical discs or two non-adjacent cervical discs that require surgical
treatment;
• Facet joint pathology of involved vertebral bodies;
• Spinal metastases;
• An endocrine or metabolic disease that affects bones such as Paget’s disease,
osteomalacia, renal osteodystrophy, Ehlers-Danlos Syndrome, or osteogenesis imperfecta;
• Chronic or acute renal failure or history of renal disease;
• Taking medications known to potentially interfere with bone/soft tissue healing (e.g.,
steroids);
• Diabetes mellitus requiring daily insulin management;
• Serious mental illness;
• Being treated for alcohol and/or drug abuse; and
• Pregnant.
Pre-operative
Medtronic Page 4 of 91

Patient selection is extremely important. In selecting patients for a total disc replacement, the
following factors may negatively affect the success of the procedure: the patient’s occupation or
activity level; prior injury or ongoing illness (e.g., Alzheimer’s disease, emphysema); alcoholism
or drug abuse; and certain degenerative diseases (e.g., degenerative scoliosis, ankylosing
spondylitis) that may be so advanced at the time of implantation that the expected useful life of
the device is substantially diminished.
In order to minimize the risk of periprosthetic vertebral fractures, surgeons must consider all comorbidities, past and present medications, previous treatments, etc. Surgeons should screen
patients to determine if a DEXA bone mineral density measurement is necessary. If DEXA is
performed, the patient should not receive the PRESTIGE LP™ Cervical Disc (per the
contraindications listed above) if the DEXA bone mineral density T-score is ≤ -1.0, as the patient
may be osteoporotic or osteopenic.
The patient should be informed of the potential adverse effects (risk/complications) contained in
this insert (see POTENTIAL ADVERSE EFFECTS OF THE DEVICE ON HEALTH).
Preoperative planning may be used to estimate the required implant size and to assure that the
appropriate range of sizes is available for surgery. Specific preoperative planning is necessary
when performing a two-level procedure. The procedure should not take place if the appropriate
range of sizes will not be available.
Inspect all instruments prior to surgery and replace any worn or damaged items. Instruments
which have been used excessively may be more likely to break.
Intra-operative
Correct selection of the appropriate implant size is extremely important to ensure the placement
and function of the disc. When performing a two-level procedure, to ensure sufficient access to
the two affected disc spaces, make the skin incision centered at the middle vertebral body. A
standard incision for the exposure of two levels is required. See the surgical technique manual
for step-by-step instructions on the surgical technique, including determining the correct implant
size.
Use aseptic technique when removing the PRESTIGE LP™ Cervical Disc components from the
innermost packaging. Carefully inspect each component and its packaging for any signs of
damage, including damage to the sterile barrier. Do not use PRESTIGE LP™ Cervical Disc
components if the packaging is damaged or the implant shows signs of damage.
Use care when handling a PRESTIGE LP™ Cervical Disc component to ensure it does not come
in contact with objects that could damage the implant. Exercise care to ensure implantation
instruments do not contact the highly polished articulating surfaces of the endplates. Damaged
implants are no longer functionally reliable. Visual inspection of the PRESTIGE LP™ Cervical
Disc assembly is recommended prior to implantation. If any part of the assembly appears
damaged, do not use the device.
When preparing the disc space, remove anterior or posterior osteophytes as needed, taking care to
Medtronic Page 5 of 91

perform a complete discectomy while minimizing bone removal, as excessive bone removal may
weaken the vertebral endplates or vertebral body.
Correct positioning of the rail punch is critical prior to performing the rail preparation step. Care
should be taken to correctly position the rail punch during this step.
Ensure proper alignment and placement of the PRESTIGE LP™ Cervical Disc as misalignment
may cause excessive wear and/or early failure of the device.
In a two-level procedure, when placing the first implant, pay special attention to implant height
selection. The goal is to balance the discs to achieve normal sagittal balance and disc space
height. Before placing the first implant, it is important to verify normal sagittal balance and disc
space height by using the Implant Trials. Achieve this by preoperative templating, careful trialing
under lateral fluoroscopy, and comparing the facet and intradiscal heights in healthy adjacent
levels. Implant Trials should fit snugly without distracting the disc spaces.
The PRESTIGE LP™ Cervical Disc components should not be used with components or
instruments of spinal systems from other manufacturers. See the surgical technique manual for
step-by-step instructions.
The PRESTIGE LP™ Cervical Disc implants are designed for single patient use only. Do not reuse, re-process, or re-sterilize the implants. Even if the device appears undamaged, re-use, reprocessing, or re-sterilization may compromise the structural integrity of the implant and the
intended function of the device which could result in patient injury.
Post-operative
Patients in the clinical study of the PRESTIGE LP™ Cervical Disc were instructed to use nonsteroidal anti-inflammatory drugs (NSAIDs) for two weeks postoperatively. It has been reported
in the literature that short-term postoperative use of NSAIDs may reduce the instance of
heterotopic ossification (HO). To reduce the instance of HO, it is recommended that the
PRESTIGE LP™ device be implanted in subjects able to tolerate the use of NSAIDs for two
weeks post-operatively.
Patients should be instructed in postoperative care procedures and should be advised of the
importance of adhering to these procedures for successful treatment with the device. Patients
should be advised to avoid any activities that require repeated bending or twisting, heavy lifting,
and challenging activities such as athletic activities. Gradual increase in physical activity will
depend on individual patient progress.
MRI Safety Information
In non-clinical testing the PRESTIGE LP™ Cervical Disc implanted at either a single level or
two contiguous levels was determined to be MR-conditional. A patient with this device can be
scanned safely, immediately after placement, under the following conditions:
• Static magnetic field of 1.5-Tesla and 3.0-Tesla.
• Maximum spatial gradient magnetic field of 3000-Gauss/cm or less.
Medtronic Page 6 of 91

• Maximum whole body average specific absorption rate (SAR) of 4.0 -W/kg or less under
Normal Operating Mode or first level controlled operating mode.
• Body Coil only.
Under the scan conditions defined above, the PRESTIGE LP™ Cervical Disc is expected to
produce a maximum temperature rise of less than 4.0°C after 15 minutes of continuous scanning.
In non-clinical testing, the image artifact caused by the device extends approximately 20 mm
from the PRESTIGE LP™ Cervical Disc when imaged with a gradient echo pulse sequence in a
3.0 Tesla MRI system.
If the PRESTIGE LP™ Cervical Disc is used in connection with any device which is not
MR Conditional, please be advised that this combination has not been tested in the MR
environment and, therefore, higher heating and possible injury to the patient may occur.
The presence of other implants or the health state of the patient may require a modification of the
MR conditions.
POTENTI AL AD VERSE EFFECTS OF THE D EVI CE ON HEALTH
Below is a list of the potential adverse effects (e.g., complications) associated with the use of the
PRESTIGE LP™ Cervical Disc identified from the PRESTIGE LP™ Cervical Disc clinical
study results, approved device labeling for other cervical total disc replacement devices, and
published scientific literature including: (1) those associated with any surgical procedure; (2)
those associated with anterior cervical spine surgery; and (3) those associated with a cervical
artificial disc device, including the PRESTIGE LP™ Cervical Disc. In addition to the risks listed
below, there is also the risk that surgery may not be effective in relieving symptoms, or may
cause worsening of symptoms. Additional surgery may be required to correct some of the adverse
effects.
1. Risks associated with any surgical procedure include:
• Anesthesia complications including an allergic reaction or anaphylaxis;
• Infection (wound, local, and/or systemic) or abscess;
• Wound dehiscence or necrosis;
• Edema;
• Soft tissue damage or fluid collections, including hematoma or seroma;
• Pain/discomfort at the surgical incision and/or skin or muscle sensitivity over the incision
which may result in skin breakdown, pain, and/or irritation;
• Heart or vascular complications including bleeding, hemorrhage or vascular damage
resulting in catastrophic or potentially fatal bleeding, ischemia, myocardial infarction,
abnormal blood pressure, venous thromboembolism including deep vein thrombosis and
pulmonary embolism, thrombophlebitis, or stroke;
• Pulmonary complications including atelectasis or pneumonia;
• Impairment of the gastrointestinal system including ileus or bowel obstruction;
• Impairment of the genitourinary system including incontinence, bladder dysfunction, or
reproductive system complications;
Medtronic Page 7 of 91

• Neurological complications including nerve damage, paralysis, seizures, changes to
mental status, or reflex sympathetic dystrophy;
• Complications of pregnancy including miscarriage or congenital defects;
• Inability to resume activities of daily living; and
• Death.
2. Risks associated with anterior cervical spine surgery include:
• Injury to surrounding organs and structures including the spinal cord, nerve roots, other
neurologic structures adjacent to the spinal column, vocal cords, adjacent vertebrae,
lymphatic vessels, blood vessels, soft tissue, dura, the trachea, the esophagus, the larynx,
or the pharynx;
• Dysphagia, dysphonia, hoarseness, vocal cord paralysis, laryngeal palsy; or sore throat;
• Tracheal, esophageal, or pharyngeal perforation, fistula, recurrent aspiration, or airway
obstruction;
• Neurological complications, including damage to nerve roots, the spinal cord, or other
nerves possibly resulting in muscle weakness or paralysis, changes in sensation (including
numbness, dysesthesias, or paresthesias), bowel/bladder dysfunction, or pain;
• Neck pain, arm pain, or headache;
• Dural tear or leak or cerebrospinal fistula;
• Discitis, arachnoiditis, or other type of inflammation;
• Loss of disc height; loss of anatomic sagittal plane curvature or vertebral listhesis, spinal
stenosis, or spondylolysis; and
• Scarring, herniation or degeneration of adjacent discs.
3. Risks associated with a cervical artificial disc device, including the PRESTIGE LP™
Cervical Disc, include:
• Risks directly related to the device including malposition, migration/displacement,
subsidence/loss of disc height, device breakage, device disassembly, or early or late
loosening of the device. Any of these issues may cause pain or injury to surrounding
organs and structures including the spinal cord, nerve roots, or other neurologic structures
adjacent to the spinal column (which could cause pain, paralysis, or numbness) or blood
vessel damage or erosion (which could cause catastrophic or fatal bleeding);
• Deterioration in neurologic status including muscle weakness or paralysis, changes in
sensation (including numbness, dysesthesias, or paresthesias), decreased reflexes, or loss
of bowel and/or bladder control;
• Development of new radiculopathy, myelopathy, or pain;
• Failure of the device to improve symptoms or function;
• Problems during placement of the device including trouble sizing the device, anatomical
or technical difficulties implanting the device, or issues with the device instruments (e.g.,
bending or breakage) including the possibility that a fragment of a broken instrument may
remain in the patient after implantation;
• Adverse reaction or allergy to the device materials (titanium, aluminum or vanadium),
device wear debris or metal ions which may lead to a systemic reaction or a local adverse
tissue reaction or chronic inflammation which may lead to implant loosening or failure of
Medtronic Page 8 of 91

the device, osteolysis, bone resorption, tumor formation, autoimmune disease, metallosis,
scarring, or other symptoms;
• Change in the alignment of the spine or loss of proper anatomic curvature, correction,
height or reduction of the spine including spondylolisthesis, change in lordosis, or
instability of the spine;
• Degeneration of other parts of the spine including the facet joints or adjacent discs;
• Fracture of the surrounding vertebrae;
• Unintended bone formation (i.e., heterotopic ossification) that may result in bridging
trabecular bone and may reduce spinal motion or result in unintended fusion at either the
treated level or adjacent levels;
• Device failure which may require a subsequent surgical intervention (including removal
of the PRESTIGE LP™ Cervical Disc, revision, re-operation, or supplemental fixation);
and
• Interference with radiographic imaging because of the presence of the implant.
NOTE: Some of the adverse effects listed above were observed in the PRESTIGE LP™
Cervical Disc clinical studies. For detailed information on the specific adverse events that
occurred in the clinical studies of the PRESTIGE LP™ Cervical Disc, please see Tables 8-10b
and 12 for the single-level study and Tables 31-33 and 35 for the two-level study.
PHYSICIAN NOTE: Although the physician is the learned intermediary between the company
and the patient, the important medical information given in this document should be conveyed to
the patient.
CLINICAL STUDIES
Two IDE studies were conducted to support the safety and effectiveness of the PRESTIGE LP™
Cervical Disc for reconstruction of the disc from C3-C7 following discectomy at one level
(G040086) or two contiguous levels (G050202) for the indications outlined above. The IDE
studies are summarized separately below.
Single-Level Clinical Study (G040086)
The clinical investigation of the PRESTIGE LP™ Cervical Disc was conducted under an
approved IDE #G040086. The study was a prospective, multi-center, non-randomized,
unmasked, non-inferiority clinical trial conducted in the United States to compare the safety and
effectiveness of the PRESTIGE LP™ Cervical Disc to the standard of care (a legally marketed
alternative with similar indications for use) anterior cervical discectomy and fusion (ACDF)
using structural allograft and plate stabilization. The control group consisted of a nonrandomized historical control group that received treatment with ACDF for reconstruction of the
disc from C3-C7 following single-level discectomy for intractable radiculopathy and/or
myelopathy in the previous IDE randomized trial of the PRESTIGE® Cervical Disc (#G010188).
The study consisted of 280 patients treated with the investigational device at 20 investigational
centers in the clinical trial, and 265 patients received the control treatment under a previous IDE
study. Fifty-four additional subjects were enrolled at the same investigational sites, including: 30
patients enrolled into a Metal Ion Cohort (MI) for which metal ion analysis was conducted based
on blood draws at each follow-up time point; and, 24 Continued Access (CA) patients.
Medtronic Page 9 of 91

Clinical Inclusion and Exc lusi on C ri te r ia
To qualify for enrollment in the study, subjects met all of the following inclusion criteria and
none of the following exclusion criteria.
Clinical Inclusion Criteria
Enrollment in the PRESTIGE LP™ study was limited to patients who met the following
inclusion criteria:
• Cervical degenerative disc disease defined as: intractable radiculopathy and/or
myelopathy with at least one of the following items producing symptomatic nerve root
and/or spinal cord compression documented by patient history [e.g., pain, func tiona l
deficit, and/ o r n eurologic deficit and radio gra p hic studies (e.g., computed
tomography (CT), magnetic resonance imaging (MRI), x-rays, etc.)]
o Herniated disc;
o Osteophyte formation
• One level requiring surgical treatment;
• C3-C4 disc to C6-C7 disc level of involvement;
• Unresponsive to non-operative treatment for approximately six weeks or has the presence
of progressive symptoms or signs of nerve root/spinal cord compression in the face of
continued non-operative management;
• No previous surgical intervention at involved level or any subsequent, planned/staged
surgical procedure at the involved or adjacent level(s);
• Is at least 18 years of age, inclusive, at the time of the surgery;
• Preoperative Neck Disability Index score of ≥ 30;
• Has a preoperative neck pain score of ≥ 20 on Preoperative Neck and Arm Pain
Questionnaire;
• If a female of child-bearing potential, patient is not pregnant at the time of surgery;
• Is willing to comply with the study plan and sign the Patient Informed Consent Form.
Clinical Exclusion Criteria
Patients were not permitted to enroll in the PRESTIGE LP™ study if any of the following
exclusion criteria were present:
• Has a cervical spinal condition other than symptomatic cervical disc disease requiring
surgical treatment at the involved level;
• Documented or diagnosed cervical instability defined by dynamic (flexion/extension)
radiographs showing sagittal plane translation > 3.5mm or sagittal plane angulation >
20°;
• More than one cervical level requiring surgical treatment;
• Has a fused level adjacent to the level to be treated;
• Has severe pathology of the facet joints of the involved vertebral bodies;
• Previous surgical intervention at the involved level;
• Has previous diagnosis of osteopenia or osteomalacia;
• Has any of the following that may be associated with a diagnosis of osteoporosis (if
“Yes” to any of the below risk factors, a DEXA Scan will be required to determine
Medtronic Page 10 of 91

eligibility):
o Postmenopausal non-Black female over 60 years of age and weighs less than 140
pounds
o Postmenopausal female that has sustained a non-traumatic hip, spine, or wrist
fracture
o Male over the age of 70
o Male over the age of 60 that has sustained a non-traumatic hip or spine fracture
If the level of bone mineral density (BMD) is a T score of -3.5 or a T score of -2.5 with
vertebral crush fracture, the patient is excluded from the study
• Has presence of spinal metastases;
• Has overt or active bacterial infection, either local or systemic;
• Has severe insulin dependent diabetes;
• Has chronic or acute renal failure or prior history of renal disease;
• Has fever (temperature > 101°F oral) at the time of surgery;
• Has a documented allergy or intolerance to stainless steel, titanium, or a titanium alloy;
• Is mentally incompetent (if questionable, obtain psychiatric consult);
• Is a prisoner;
• Is pregnant;
• Is an alcohol and/or drug abuser currently undergoing treatment for alcohol and/or drug
abuse
• Has received drugs which may interfere with bone metabolism within two weeks prior to
the planned date of spinal surgery (e.g., steroids or methotrexate) excluding routing
perioperative anti-inflammatory drugs;
• Has a history of an endocrine or metabolic disorder known to affect osteogenesis (e.g.,
Paget’s Disease, renal osteodystrophy, Ehlers-Danlos Syndrome, or osteogenesis
imperfecta);
• Has a condition that requires postoperative medications that interfere with the stability of
the implant, such as steroids. (This does not include low dose aspirin for prophylactic
anticoagulation), excluding routine perioperative anti-inflammatory drugs;
• Has received treatment with an investigational therapy within 28 days prior to
implantation surgery or such treatment is planned during the 16 weeks following
implantation with the PRESTIGE LP™ device.
Postoperative Care
The recommended post-operative care included avoidance of overhead lifting, heavy lifting,
repetitive bending, and high-impact exercise or athletic activity for 60 days postoperatively.
Avoidance of prolonged (beyond 2 weeks post-op) non-steroidal anti-inflammatory drug
(NSAID) use was specified in the postoperative regimen, although the use of NSAIDs was
recommended for the first two weeks post-operatively. Post-operative bracing requirements were
left to the discretion of the investigators and included the option for use of a soft collar as
needed. The use of electrical bone growth stimulators was not recommended during the 24month follow-up period. However, in a few cases where an electrical bone growth stimulator
was utilized due to specific patient presentation, they were considered a supplemental form of
therapy for spinal fusion surgery, and deemed failures included in the “Supplemental Fixation”
Adverse Event category. Patients who smoked were encouraged to discontinue smoking.
Medtronic Page 11 of 91

Pre-/Peri-Operative
Postoperative
±
24 mo.
±
Beyond
Preoperative Information
Confirm Patient Eligibility
X
Obtain Informed Consent
X
Obtain HIPAA Authorization
X
Case Report Forms
Patient Enrollment
X
Patient Qualification
X
Preoperative Data
X
Prior History Questionnaire
X
Neurological Status
X X X X X X
Preoperative Gait Assessment and
Foraminal Compression Test
X
Preoperative Patient Survey
X
Preoperative Neck Disability Index
X
Preoperative Neck and Arm Pain
Questionnaire
X
Health Status Questionnaire (SF-36)
X X X X
Surgery Data
X
Hospital Discharge
X
Postoperative Data
X X X X X
Postoperative Patient Survey
X X X X X
Neck Disability Index
X X X X X
Postoperative Neck and Arm Pain
Questionnaire
X X X X X
Postoperative Gait Assessment and
Foraminal Compression Test
X X X X X
Adverse Event Form (if any)
X X X X X X
Outstanding (Unresolved) Adverse E v e nt
(if any)
X X X X X X
Patient Disposition
X X X X X
Imaging – Radiographs and Scans*
Anterior/Posterior X-ray
X X X X X X X
Lateral X-ray
X X X X X X X
Right/Left Lateral Bend X-rays
X X X X X X
Flexion/Extension X-rays
X X X X X X
CT and/or MRI
X
DEXA Scan **
X
Follow-Up Schedule
Patients were evaluated preoperatively (within 6 months of surgery), intraoperatively, and
postoperatively at 6 weeks (±2 weeks), 3 months (±2 weeks), 6 months (± one month), 12
months (± two months), 24 months (± two months), and annually thereafter until the last subject
enrolled in the study had been seen for their 24-month evaluation, as shown in Table 2.
Complications and adverse events were evaluated over the course of the clinical trial. At each
evaluation timepoint, the primary and secondary clinical and radiographic outcome parameters
were evaluated. Success was determined from data collected during the initial 24 months of
follow-up.
Table 2: Schedule of Study Assessments
Procedure
Pre-OP
Surgery/
Hospital
Discharge
6 w ks ±2
wks
3 mo. ±2
wks
6 mo.
1 mo.
12 mo. ±
2mo.
2 mo.
&
* Patients w ho sign consent and are screened el igible, but who do not receive the PRESTIGE LP™ device, were not required to have
the preoperative radiographs obtained and forwarded to Medtronic.
Medtronic Page 12 of 91

** A DEXA S can was only required if the patient had a risk factor that may be associated with a diagnosis of osteoporosis.
Clinical Endpoints
The safety of the PRESTIGE LP™ Cervical Disc was assessed by comparison to the historical
control group with respect to the nature and frequency of adverse events, secondary surgical
procedures, as well as maintenance or improvement in neurological status.
The effectiveness of the PRESTIGE LP™ Cervical Disc device was assessed using a composite
definition of study success. The primary endpoint used for assessment of effectiveness was
improvement in Neck Disability Index (NDI) pain/disability scores.
In addition, several radiograph-assisted assessments were considered in evaluating both safety
and effectiveness including device subsidence, functional spinal unit (FSU) height maintenance,
device migration, and device breakage.
According to the final IDE protocol, an individual patient in either treatment group was
considered an overall success if the following criteria were met at 24 months:
1. An improvement (reduction) of at least 15 points from the baseline Neck Disability Index
score;
2. Maintenance or improvement in neurological status;
3. Disc height (Functional Spinal Unit Height) success (FSU success)
4. No severe adverse event classified as implant-associated, surgical procedure-associated, or
implant/surgical procedure-associated; and
5. No additional surgical procedure classified as “Failure”
An alternative analysis of the primary endpoint analysis was also conducted without the addition
of FSU height as a success criterion.
Secondary endpoints, measured in both treatment groups, included Radiographic Success, neck
pain (VAS), arm pain (VAS), quality of life (SF-36 PCS and MCS scores), patient satisfaction,
patient global perceived effort, gait assessment (Nurick’s classification), and foraminal
compression test. Additional measurements recorded were adjacent level stability, return to
work, and doctor’s perception of results. Radiographic Succcess for maintenance of motion is
defined as >4° but <20° of angular motion based on lateral flexion/extension radiographs and no
radiographic evidence of bridging trabecular bone that forms a continuous bony connection with
the vertebral bodies (bridging bone).
Criteria for the success of the control group was defined in a previous IDE study (G010188).
Briefly, the same success criteria for the primary endpoints exist for the control group as the
investigational group, with the exception that the secondary endpoint for radiographic success
was defined by radiographic evidence of bone spanning the two vertebral bodies, existence of
angular motion stability <4°, and no radiolucent lines covering more than 50% of the implant
surface.
Accountability of PMA Cohort
Medtronic Page 13 of 91

Number of Patients:
12 Months
(±2 Months)
24 Months
(±2 Months)
Enrolled
280
265
333
280
265
333
Theoretical FU
280
265
333
280
265
333
Cumulative Deaths 1
0 2 0 0 2
0
Patients Evaluated Early 2
0 0 0 0 0
0
Patients Not Yet Overdue
0 0 0 0 0
0
Expected3
280
263
333
280
263
333
Evaluable for Overall Success
(% of Total Expected)
274
(97.9%)
223
(84.8%)
326
(97.9%)
271
(96.8%)
220
(83.7%)
322
(96.7%)
Evaluable for Overall Success, In
(% of Total Expected)
Percent Follow-up
98.2%
87.1%
98.5%
97.1%
84.0%
97.3%
The subject accountability data are summarized in Table 3. Please note that Continued Access
Cohort (CA) and the Metal Ion Cohort (MI) were enrolled separately from the IDE Cohort at the
same study sites. Safety and effectiveness data were collected for the IDE, Safety
(IDE+CA+MI), and ACDF Control Cohorts while the statistical analyses were performed with
the IDE Cohort in comparison to the control group.
Table 3: Subject Accountability
PRESTIGE LP™
IDE Cohort
ACDF Control
PRESTIGE LP™
Safety Cohort
PRESTIGE LP™
IDE Cohort
ACDF Control
PRESTIGE LP™
Safety Cohort
Window
In addition to the study subjects described above, nineteen (19) subjects were consented but
declined participation in the study prior to receiving the assigned treatment. The demographic
and preoperative characteristics of the subjects who declined to participate in this study were
comparable to the patients included in this study.
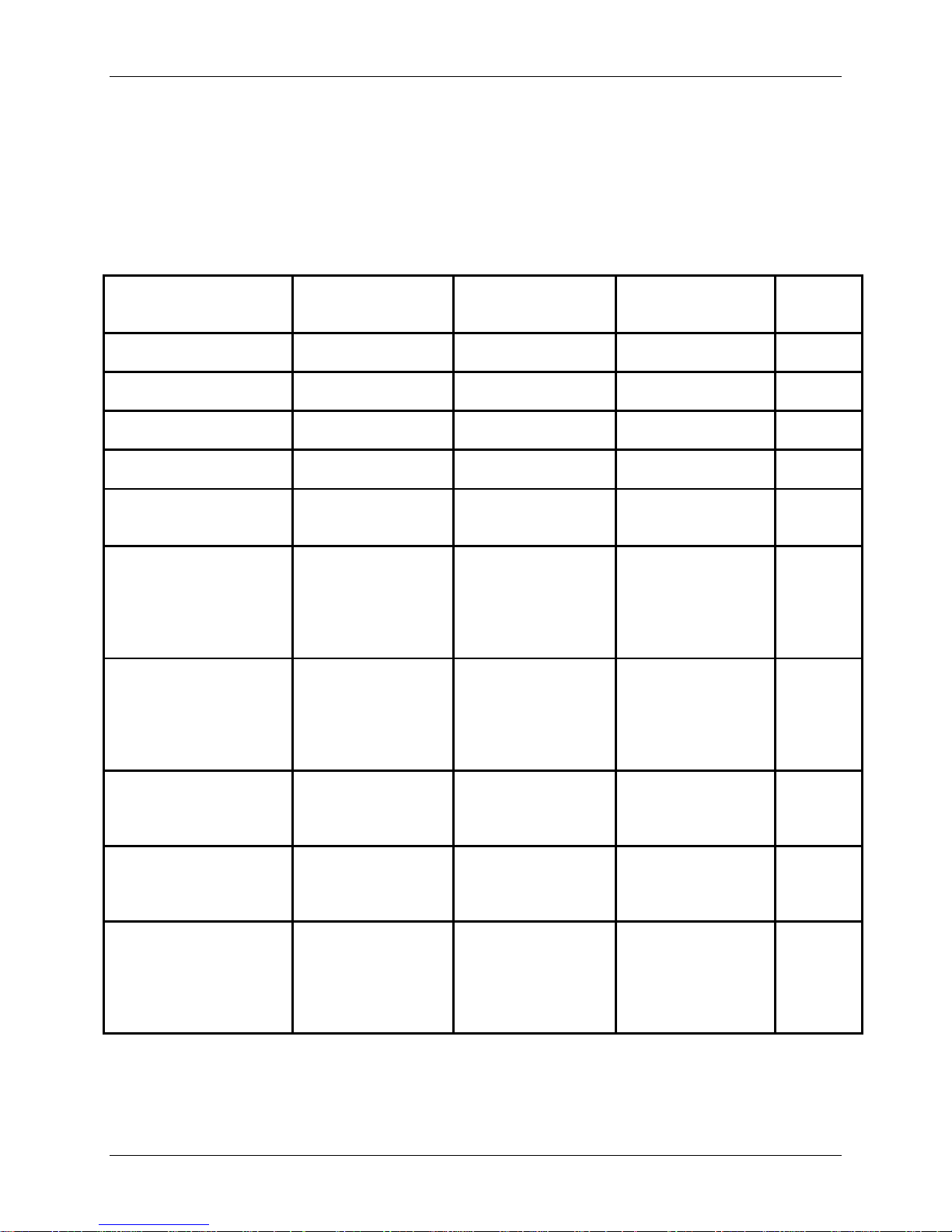
Study Population Demographics and Baseline Parameters
Table 4 presents the summary statistics for demographic and baseline characteristics for the
PRESTIGE LP™ IDE Cohort, the ACDF Control, and PRESTIGE LP™ Safety Cohort. The
demographics of the study population are consistent with the demographics reported for prior
cervical artificial disc studies conducted in the U.S.
The investigational and control treatment groups were very similar demographically, and there
were no statistically significant differences (p<0.05) for any of the variables except for the use of
tobacco and race. Current tobacco use was higher in the control group (34.7% versus 26.4%) as
1
Cumulative deaths are the total number of deaths of study patients at the 12- and 24-month time points. However, none of the
deaths were believed to be in any way related to the study treatment .
2
Patients that completed follow-up visits early before the visit window
3
Expected = Theoretical min us Cumulative Deaths minus Patients Not Yet Overdue plus Patients Evaluated Early for Visit
271
(96.8%)
206
(78.3%)
321
(96.4%)
262
(93.6%)
201
(76.4%)
309
(92.8%)
Medtronic Page 14 of 91
Variables
PRESTIGE LP™ IDE
(N=280)
(N=265)
PRESTIGE LP™
(N=333)
p-value
Control)
Age (years)
44.5 ± 8.8
Range: 23 - 78
43.9 ± 8.8
Range: 22 - 73
43.8 ± 9.0
Range: 23 – 78
Height (inches)
67.7 ± 4.1
Range: 60.0 – 77.0
67.5 ± 4.2
Range: 58.0 – 80.0
67.7 ± 4.0
Range: 60.0 – 77.0
Weight (lbs.)
186.9 ± 45.0
Range: 100.0 – 340.0
184.7 ± 41.5
Range: 98.0 – 328.0
187.3 ± 45.2
Range: 100.0 – 340.0
BMI (kg/m2)
28.5 ± 5.6
Range: 17.2 – 48.2
28.3 ± 5.1
Range: 19.0 – 53.6
28.5 ± 5.6
Range: 17.2 – 48.2
Sex
Female (%)
151 (53.9%)
143 (54.0%)
178 (53.5%)
Race
Other
1 (0.4%)
1 (0.4%)
1 (0.3%)
Marital Status
Widowed
2 (0.7%)
2 (0.8%)
3 (0.9%)
Education Level
> High School
206 (74.1%)
173 (65.5%)
236 (71.3%)
No
277 (98.9%)
263 (99.2%)
330 (99.1%)
Preoperative Medication
Muscle Relaxants
100/279 (35.8%)
114/264 (43.2%)
123/332 (37.0%)
0.095
compared to the IDE Cohort. However, tobacco use was established through use of patient
questionnaires which utilized a binary response (i.e., yes or no), and quantification of the extent
or history of tobacco use was not established. Therefore, it is not possible to definitively ascertain
whether there were any substantial confounding effects from tobacco use on patient outcomes.
Regarding race differences among cohorts, there was a higher percentage of Caucasian subjects
in the IDE Cohort compared to the control group (96.8% versus 91.7%).
Table 4: Study Patient Demographics and Baseline Characteristics
Male (%)
Caucasian
Black
Asian
Hispanic
Single
Married
Divorced
Separated
Cohort
129 (46.1%)
271 (96.8%)
7 (2.5%)
0 (0.0%)
1 (0.4%)
40 (14.3%)
189 (67.5%)
42 (15.0%)
7 (2.5%)
ACDF Control
122 (46.0%)
243 (91.7%)
13 (4.9%)
2 (0.8%)
6 (2.3%)
32 (12.1%)
204 (77.0%)
24 (9.1%)
3 (1.1%)
Safety Cohort
155 (46.5%)
320 (96.1%)
10 (3.0%)
1 (0.3%)
1 (0.3%)
47 (14.1%)
224 (67.3%)
51 (15.3%)
8 (2.4%)
(IDE vs.
0.369
0.622
0.567
0.722
1.000
0.043
0.096
< High School
High School
Previous Ne ck Surgery
Yes
use
Non-Narcotics
Weak Narcotics
Strong Narcotics
Medtronic Page 15 of 91
208/280 (74.3%)
133/279 (47.7%)
15 (5.4%)
57 (20.5%)
3 (1.1%)
62/279 (22.2%)
14 (5.3%)
77 (29.2%)
2 (0.8%)
187/263 (71.1%)
127/263 (48.3%)
58/264 (22.0%)
17 (5.1%)
78 (23.6%)
3 (0.9%)
246/333 (73.9%)
152/332 (45.8%)
68/332 (20.5%)
0.062
1.000
0.441
0.931
1.000

Neck Pain Only
25 (8.9%)
26 (9.8%)
34 (10.2%)
Worker’s Compensation
32/280 (11.4%)
35/365 (13.2%)
54/333 (16.2%)
0.602
Unresolved S pinal
Litigation
Current Tobacco Use
74/280 (26.4%)
92/265 (34.7%)
94/333 (28.2%)
0.041
Current Alcoho l Use
150/280 (53.6%)
141/265 (53.2%)
172/333 (51.7%)
1.000
Preoperative Work Status
188/280 (67.1%)
166/265 (62.6%)
217/333 (65.2%)
0.282
> 6 mos.
173 (61.8%)
161 (60.8%)
212 (63.7%)
Preoperative Pain Status4
Arm and Neck Pain
Arm Pain Only
34/280 (12.1%) 32/265 (12.1%) 61/333 (18.3%) 1.000
Duration of Symptoms
< 6 wks.
6 wks. – 6 mos.
255 (91.1%)
0 (0.0%)
22 (7.9%)
85 (30.4%)
238 (90.2%)
0 (0.0%)
15 (5.7%)
89 (33.6%)
299 (89.8%)
0 (0.0%)
24 (7.2%)
97 (29.1%)
0.769
0.494
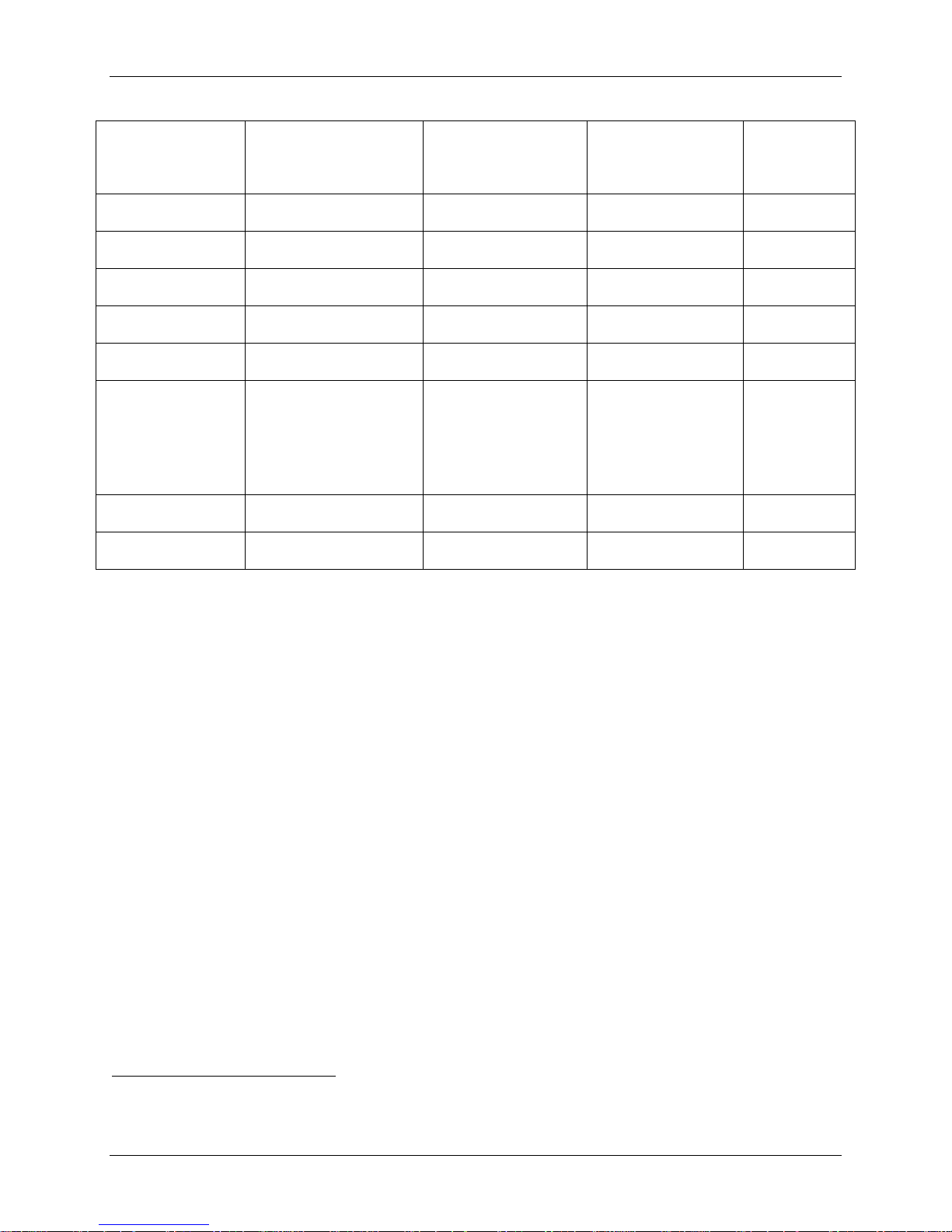
The mean baseline pre-operative assessments for the PRESTIGE LP™ IDE Cohort, Control
Group, and PRESTIGE Safety Cohort are presented in Table 5. There were no statistical
differences between the PRESTIGE LP™ IDE Cohort and Control for NDI, SF-36 PCS, SF-36
MCS, neck pain, and arm pain. There were statistically significant differences in baseline motor
neurologic status (38.2% - PRESTIGE LP™ IDE Cohort; 59.5% - Control) and mean cervical
range of motion (5.67º - PRESTIGE LP™ IDE Cohort; 7.87º - Control). However, after
propensity score adjustments, the variables appeared balanced between groups. Thus, differences
in baseline symptoms were adjusted for in the analysis and are therefore unlikely to have led to
significant bias in the reported results.
4
Arm pain is defined as a subject having an arm pain score ≥20 and neck pain is defined as a subject having a neck pain score
≥20. If a subject has both an arm pain score ≥20 and a neck pain score ≥20, then this subject is considered as having “Arm and
Neck Pain”; if a subject has a neck pain score ≥20 and an arm pain score < 20, then this subject is considered as having “Neck
Pain Only”; if a subject has an arm pain score ≥20 and a neck pain score < 20, then this subject is considered as having “Arm
Pain Only”. Since neck pain score ≥20 is an inclusion criteria, there are no subjects with “Arm Pain Only”.
LP™ Cervical Disc is indicated in skeletally mature patients for reconstruction of the disc at one level from C3-C7 following
single-level discectomy for intractable radiculopathy (arm pain and/or a neurological deficit) with or without neck pain and is
not indicated for treatment of isolated neck pain. No patients were included into the study with neck pain without any other
symptoms.
Medtronic Page 16 of 91
The PRESTIGE
p-value (IDE
Control)
55.5 ± 14.7
Range: 30.0 – 98.0
56.4 ± 15.9
Range: 26.0 – 100.0
56.6 ± 15.0
Range: 30.0 – 98.0
32.2 ± 7.4
Range: 14.3 – 57.9
32.0 ± 7.5
Range: 7.9 – 56.0
32.3 ± 7.1
Range: 14.3 – 57.9
44.5 ± 11.5
Range: 16.5 – 68.3
42.7 ± 12.4
Range: 14.1 – 70.8
43.8 ± 11.9
Range: 16.5 – 68.3
67.0 ± 20.8
Range: 20.0 – 100.0
69.3 ± 21.5
Range: 20.0 – 100.0
68.0 ± 20.8
Range: 20.0 – 100.0
59.6 ± 26.3
Range: 0.0 – 100.0
62.4 ± 28.5
Range: 0.0 – 100.0
59.0 ± 27.1
Range: 0.0 – 100.0
Neurological Status
• Overall5
64/280 (22.9%)
79/264 (29.9%)
73/333 (21.9%)
0.065
Baseline ROM
angulation (º)
5.67 ± 3.69
Range: 0.27 – 18.10
7.87 ± 4.32
Range: 0.74 – 21.34
5.88 ± 3.78
Range: 0.27 – 19.47
Baseline ROM
translation (mm)
0.26 ± 0.25
Range: 0.00 – 1.64
Table 5: Preoperative Evaluation of Endpoints
Variables
NDI
SF-36 PCS
SF-36 MCS
Neck Pain Score
Arm Pain Score
(normal)
• Motor
• Sensory
• Reflexes
PRESTIGE LP™ IDE
Cohort (N=280)
107/280 (38.2%)
117/280 (41.8%)
186/280 (66.4%)
N/A
ACDF Control
(N=265)
157/264 (59.5%)
134/264 (50.8%)
161/264 (61.0%)
PRESTIGE LP™
Safety Cohort
(N=333)
135/333 (40.5%)
147/333 (44.1%)
200/333 (60.1%)
N/A N/A
Cohort vs
ACDF
0.498
0.777
0.079
0.191
0.236
< 0.001
0.039
0.212
< 0.001
Surgery and Hospitalization
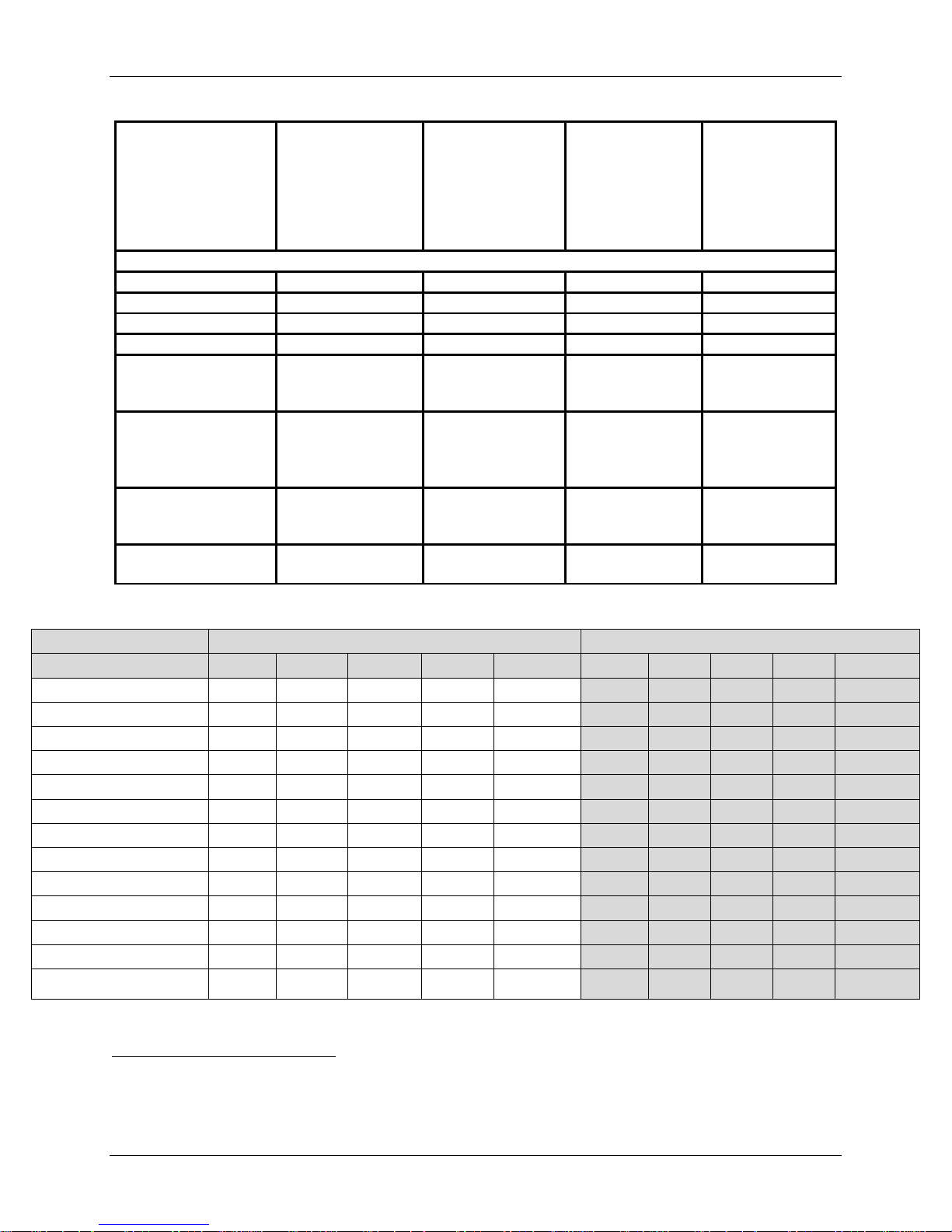
Table 6 summarizes the information related to the surgical procedures and postoperative
hospitalizations of subjects. The most common treated surgical levels were C5-C6 and C6C7.The mean operative times for the IDE and control treatment groups were 1.5 hours and 1.4
hours, respectively, which is a mean difference of 0.1 hours, or 6 minutes and is unlikely to
represent any significant clinical difference. Additionally, investigational subjects were found to
have similar estimated blood loss to the control group subjects (50.5 ml for IDE cohort and 49.4
ml for Safety cohort versus 57.5 ml for control group). The median blood loss was 35 ml for the
IDE cohort versus 50 ml for both the Safety and control groups. The mean hospital stays of
subjects in all treatment groups were similar (1.0 days for all groups). Table 7 summarizes the
PRESTIGE LP™ device implanted by size and level.
5
If at least one of the three components (motor, sensory, reflexes) is not normal, then overall is defined as “not normal”, if all the
components are normal, then overall is defined as “normal”
Medtronic Page 17 of 91
(N=280)
(N=265)
(N=333)
Posterior Mean
(lower, upper)
Spinal Level Treated
C34 (%)
4 (1.4%)
10 (3.8%)
4 (1.2%)
N/A
C45 (%)
21 (7.5%)
15 (5.7%)
28 (8.4%)
N/A
C56 (%)
147 (52.5%)
149 (56.2%)
178 (53.5%)
N/A
C67 (%)
108 (38.6%)
91 (34.3%)
123 (36.9%)
N/A
Operative time (hrs)
1.5 ± 0.6
(n=280)
1.4 ± 0.5
(n=265)
1.4 ± 0.5
(n=333)
Blood Loss (ml)
50.5 ± 73.5
(n=278)
57.5 ± 68.1
(n=263)
49.4 ± 67.9
(n=333)
Hospitalization (days)
1.0 ± 0.5
(n=280)
1.0 ± 0.5
(n=265)
1.0 ± 0.4
(n=333)
Median Return to
Work Time (days)
6mm x 12mm Disc (%)
6mm x 14mm Disc (%)
6mm x 16mm Disc (%)
6mm x 18mm Disc (%)
7mm x 12mm Disc (%)7
7mm x 14mm Disc (%)
7mm x 16mm Disc (%)
7mm x 18mm Disc (%)
8mm x 12mm Disc (%)
8mm x 14mm Disc (%)
8mm x 16mm Disc (%)
8mm x 18mm Disc (%)
4
(1.4%)
21
(7.5%)
147
(52.5%
108
(38.6%)
280
(100.0%)
4
(1.2)
28
(8.4%)
178
(53.5%)
123
(36.9%)
333
(100.0%)
Table 6: Surgical Data
and 95% BCI6 of
the Difference of
Mean between
PRESTIGE LP™
IDE Cohort
ACDF Control
PRESTIGE LP™
Safety Cohort
IDE Cohort and
Control Group
Range: 0.7 – 3.4
Range: 3.0 – 700.0
Median: 35.0
Range: 0.0 – 3.0
Range: 0.6 – 3.4
Range: 0.0 – 700.0
Median: 50.0
Range: 0.0 – 4.0
Range: 0.7 – 3.4
Range: 3.0 – 700.0
Median 50.0
Range: 0.0 – 3.0
0.11 (0.02, 0.22)
-4.7 (-16.8, 7.9)
0.03 (-0.05, 0.11)
40 60 42 N/A
Table 7: All PRESTIGE LP™ Devices Implanted by Size and Level
PRESTIGE LP™ IDE Cohort PRESTIGE LP™ Safety Cohort
C3-C4 C4-C5 C5-C6 C6-C7 Total C3-C4 C4-C5 C5-C6 C6-C7 Total
1 4 15 11
1 10 65 37
2 1 35 23
0 0 0 0
0 0 1 1
0 2 5 8
0 4 16 9
0 0 9 11
0 0 0 0
0 0 0 3
0 0 1 4
0 0 0 1
31 (11.1%)
113 (40.4%)
61 (21.8%)
0 (0.0%)
2 (0.7%)
15 (5.4%)
29 (10.4%)
20 (7.1%)
0 (0.0%)
3 (1.1%)
5 (1.8%)
1 (0.4%)
1 8 28 12
1 13 76 42
2 1 37 23
0 0 0 0
0 0 1 2
0 2 9 11
0 4 17 12
0 0 9 11
0 0 0 0
0 0 0 4
0 0 1 5
0 0 0 1
49(14.7%)
132 (39.6%)
63 (18.9%)
0 (0.0%)
3 (0.9%)
22 (6.6%)
33 (9.9%)
20 (6.0%)
0 (0.0%)
4 (1.2%)
6 (1.8%)
1 (0.3%)
Total (%)
6
BCI = Bayesian HPD Credible Interval
7
Th e 7 mm x 12mm PRESTIGE LP™ Cervical Dis c was a part of the size offerings in the IDE study, but is no t a part o f
the size offerings availabl e for market.
Medtronic Page 18 of 91

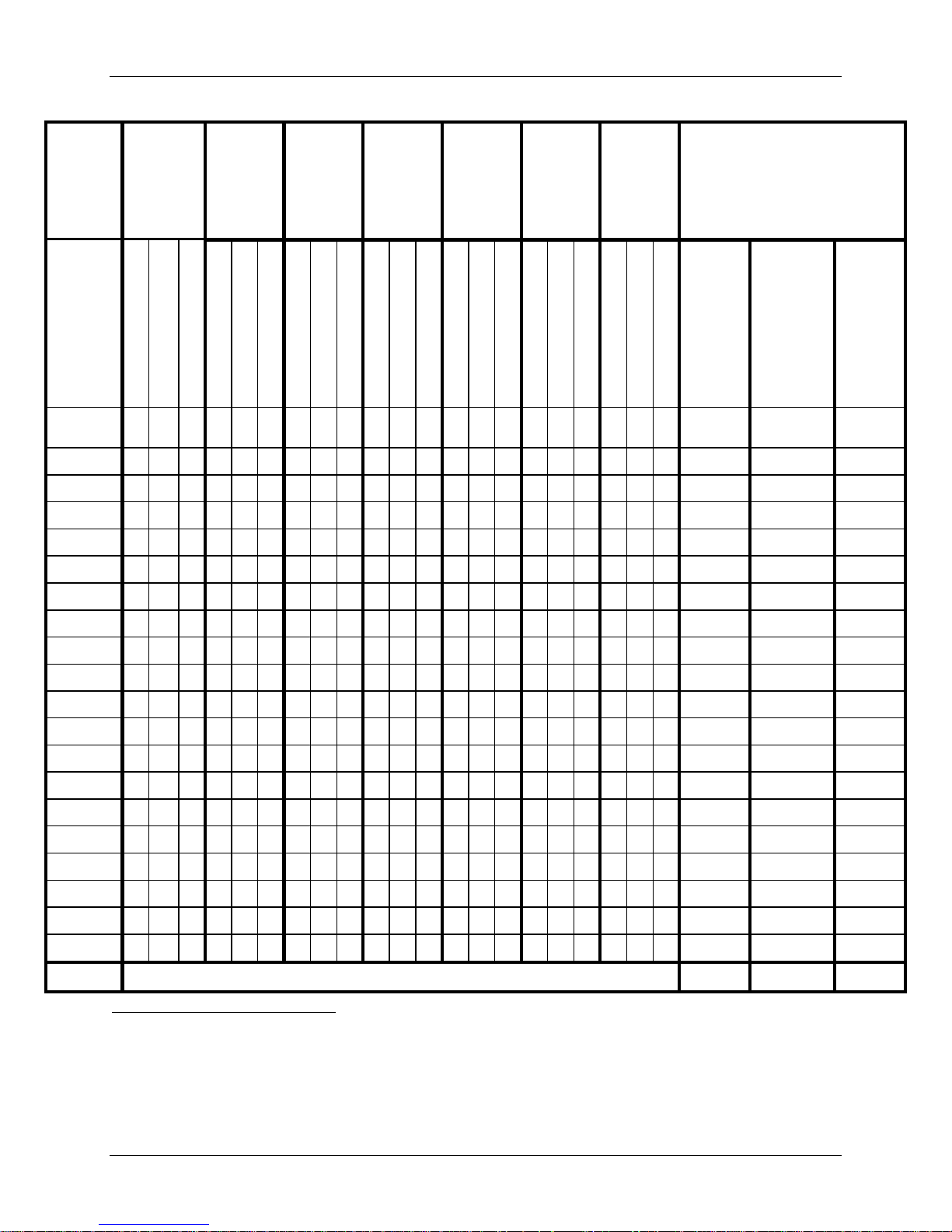
Adverse Event Type
Posterior Mean and
Control10
Patients (%)
257 (91.8%)
219 (82.6%)
306 (91.9%)
Events
(Events/Patient)
Events
(Events/Patient)
Patients (%)
72 (25.7%)
71 (26.8%)
78 (23.4%)
Events
(Events/Patient)
Severe Adverse
(Grade 3 or 4)
Patients (%)
133 (47.5%)
98 (37.0%)
163 (48.9%)
Events
(Events/Patient)
Severe Device or
(Grade 3 or 4)
Events
(Events/Patient)
Safety and Effectiveness Results
Safety Results
The analysis of safety was based on the as-treated cohort of 598 total patients with surgery (333
PRESTIGE LP™ “Safety” subjects consisting of 280 PRESTIGE LP™ IDE Cohort subjects, as
well as 54 subjects from the Continued Access (CA) and Metal Ion (MI) Cohorts8; and 265
ACDF control subjects). This was a non-randomized study and the ACDF group was a historical
control. A summary of the total number of adverse events is shown in Table 8. Adverse events
were classified by the independent Clinical Adjudication Committee (CAC) for severity and
relationship to the device and/or surgical procedure.
Table 8: Summary of Adverse Events Up to the 24-Month Time Interval
All Adverse Events
(AEs)
Device or
Device/Surgical
Procedure Related
AEs
Surgical Procedure
Related AEs Only
Events
Device/ProcedureRelated AEs
PRESTIGE LP™
Measure
Patients (%)
Patients (%) 14 (5.0%) 13 (4.9%) 16 (4.8%)
IDE Cohort
(N=280)
1559 (5.57) 1198 (4.52) 1863 (5.59)
34 (12.1%) 41 (15.5%) 44 (13.2%)
61 (0.22) 60 (0.23) 76 (0.23)
132 (0.47) 121 (0.46) 140 (0.42)
433 (1.55) 267 (1.01) 518 (1.56)
33 (0.12) 22 (0.08) 40 (0.12)
ACDF Control
(N=265)
PRESTIGE LP™
Safety Cohort
(N=333)
95% BCI9 of the
Difference of Event
Rate between IDE
Cohort and ACDF
10.2% (4.1%, 16.2%)
-2.9% (-9.2%, 3.3%)
-0.5% (-8.6%, 7.4%)
13.3% (3.5%, 21.8%)
0.7% (-3.0%, 4.6%)
Table 9 provides summary data on the number of adverse events in each treatment group by
treatment level, including post-hoc statistical anal ysis and comparison between the PRESTIGE
LP™ IDE Cohort and the ACDF control group through the 24-month time point using
Frequentist methods. The percentage of subjects with adverse events was not statistically
different between the two groups for all levels except for C5-C6; however, this difference was
not clinically meaningful.
8
One Metal Ion Cohort subject was also an IDE Cohort subject.
9
BCI = Bayesian HPD Credible Interval
10
95% BCI of the difference of the event rate between the investigational group and control group was only determined for the
“All Adverse Events” category because the analysis was pre-defined. All other analyses were not pre-defined.
Medtronic Page 19 of 91

Level
(N=280)
(N=265)
(N=333)
Point Estimate and 95%
Cohort
C3-C4
4/4 (100%)
9/10 (90.0%)
4/4 (100.0%)
10.0% (-19.9%, 39.9%)
C4-C5
20/21 (95.2%)
12/15 (80.0%)
27/28 (96.4%)
15.2% (-5.6%, 36.1%)
C5-C6
135/147 (91.8%)
124/149 (83.2%)
163/178 (91.6%)
8.6% (1.1%, 16.2%)
C6-C7
98/108 (90.7%)
74/91 (81.3%)
112/123 (91.1%)
9.4% (-0.1%, 19.0%)
Table 9: Summary o f Total Advers e Events by Level Treated thro ug h Month 24- IDE
and Safety Population
Confidence Interval11 of
Difference of Adverse
Treatment
PRESTIGE LP™
IDE Cohort
ACDF Control
PRESTIGE LP™
Safety Cohort
Rate between IDE Cohort
and ACDF Control
Table 10 reports adverse events from all patients to establish the safety profile of the device.
Adverse events are listed in alphabetical order. Adverse event rates are based on the number of
patients having at least one occurrence of an adverse event, divided by the number of patients in
that treatment group. Subjects experiencing adverse events in more than one category are
represented in each category in which they experienced an adverse event.
The overall adverse event rate was higher for subjects treated with the PRESTIGE LP™ device
(IDE Cohort, 91.8%; Safety Cohort, 91.9%) compared to the Control (82.6%) through 24
months. The adverse event rate between the PRESTIGE LP™ IDE Cohort and the Control was
statistically different with the 95% BCI for the difference of adverse events rates between the
PRESTIGE LP™ IDE Cohort and the ACDF Control Cohort being (4.1%, 16.2%), excluding 0.
However, when comparing device-related adverse events, the rates are comparable (see Table
10b below). Although the rate of PRESTIGE LP™ IDE subjects having at least one adverse
event was statistically higher than the control group rate, the difference in adverse event rates
was not considered to be clinically meaningful and this finding may be attributable to the higher
follow-up rates (and potentially, higher reporting of events) for investigational subjects as
compared to the ACDF control subjects. Specifically, note that the 24-month follow-up rates are
97.1% and 84.0% respectively for the PRESTIGE LP™ IDE Cohort and ACDF Control Cohort.
Table 11 lists the brief definitions for all adverse events.
There were a total of three deaths in the investigational group and five deaths in the control
group, of which two deaths occurred in the control group prior to 24 months (at the 12-month
time point) and none in the investigational group prior to 24 months. Deaths were evaluated
based upon available information and none of the deaths were believed to be in any way related
to the study treatment.
11
The 95% CI was provided using Frequentist Farrington and Manning methods
Medtronic Page 20 of 91
<
≥
<
≥
<
≥
<
≥
<
≥
<
PRESTIGE LP™ IDE
Total # Events
Anatomi cal /
Difficulty
3 (1.1) 5 2 (0.8)
2
5 (1.5)
7
Cardiac
Disorders
16 (5.7)
21
18 (6.8)
20
19 (5.7)
24
0 (0.0) 0 0 (0.0)
0
0 (0.0)
0
Dysphagia /
Dysphonia
26 (9.3)
33
22 (8.3)
23
30 (9.0)
38
35 (12.5)
55
38 (14.3)
68
43 (12.9)
68
Heterotopic
Ossification
27 (9.6)
31
15 (5.7)
21
31 (9.3)
35
16 (5.7)
17
5 (1.9)
5
21 (6.3)
22
34 (12.1)
57
27 (10.2)
37
40 (12.0)
63
Neck and / or
Arm Pain
144 (51.4)
275
124 (46.8)
213
184 (55.3)
353
136 (48.6)
242
108 (40.8)
217
162 (48.6)
289
0 (0.0) 0 29 (10.9)
30
0 (0.0)
0
93 (33.2)
177
81 (30.6)
133
110 (33.0)
207
146 (52.1)
278
132 (49.8)
231
175 (52.6)
328
24 (8.6)
34
17 (6.4)
23
27 (8.1)
39
83 (29.6)
172
55 (20.8)
103
106 (31.8)
212
61 (21.8)
71
35 (13.2)
44
69 (20.7)
79
26 (9.3)
42
9 (3.4)
11
31 (9.3)
47
12 (4.3)
13
4 (1.5)
4
13 (3.9)
14
Wound (NonInfectious)
25 (8.9)
34
13 (4.9)
13
27 (8.1)
36
Any Adverse
Event
257 (91.8)
1559
219 (82.6)
1198
306 (91.9)
1863
Table 10: Adverse Events in Pivotal Study Through 24 Months
Surgery
14
l
4 Weeks)
Postoperative
(1 day -
9 Weeks)
6 Weeks
4 Wks (
5 Months)
3 Months
9 Wks (
9 Months)
6 Months
5 Mos(
12 Months
12, 13
19 Months)
9 Mos(
30 Months))
24 Months
19 Mos((
Adverse
Events
ACDF Contro
PRESTIGE LP™ IDE Cohort
Technical
Cancer 0 0 0 1 1 1 0 0 0 0 0 0 1 0 1 3 0 3 0 1 2
Death
2 0 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 2 2 2 2 1 3 0 2 0 3 3 3 4 3 5 10 9 11
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
1 4 1 16 11 17 4 4 6 5 3 5 5 0 5 0 1 1 2 0 3
PRESTIGE LP™ Safety Cohor t
ACDF C ontrol
PRESTIGE LP™ IDE Cohort
PRESTIGE LP™ Safety Cohor t
ACDF C ontrol
PRESTIGE LP™ IDE Cohort
PRESTIGE LP™ Safety Cohor t
ACDF C ontrol
PRESTIGE LP™ IDE Cohort
PRESTIGE LP™ Safety Cohort
ACDF C ontrol
PRESTIGE LP™ IDE Cohort
PRESTIGE LP™ Safety Cohor t
ACDF C ontrol
PRESTIGE LP™ IDE Cohort
PRESTIGE LP™ Safety Cohor t
ACDF C ontrol
PRESTIGE LP™ IDE Cohort
Total adverse events
Total ( Up to 24 Month)
# of Patients Reporting &
Cohort
# Patients
(% of 280)
# Patients
(% of 265)
Total # Events
ACDF Control
PRESTIGE LP™ Safety Cohor t
2 (0.7)
2
0 (0.0)
0
PRESTIGE LP™ Safety Cohort
2 (0.6)
# Patients
(% of 333)
Total # Events
2
Gastrointestinal 2 7 2 8 7 8 1 3 2 4 4 7 3 2 7 20 21 22 17 24 20
0 0 0 2 1 2 6 4 6 1 2 1 4 1 5 7 2 9 11 11 12
Implant Events 6 0 6 1 1 1 2 1 3 3 0 3 1 0 3 0 2 1 4 1 5
Infection 1 2 1 5 3 5 6 5 7 6 2 9 11 1 12 16 10 16 12 14 13
3 1 4 31 16 35 43 19 57 57 49 77 37 40 50 68 50 84 36 38 46
Neurological 2 6 2 18 21 20 34 19 42 47 35 55 31 16 39 73 60 88 37 60 43
Non-Union 0 0 0 0 0 0 0 1 0 0 3 0 0 8 0 0 8 0 0 10 0
Other 5 3 8 19 14 22 12 13 14 24 16 28 29 9 38 55 21 58 33 57 39
Other Pain15 3 6 3 22 19 23 32 11 39 49 45 65 57 30 69 65 57 71 50 63 58
Respiratory 0 0 0 7 4 7 0 4 1 1 1 1 10 5 10 3 1 6 13 8 14
Spinal Event 0 0 0 19 8 22 22 6 34 20 25 25 26 15 31 44 20 53 41 29 47
Trauma 0 0 0 6 2 7 5 4 6 12 12 13 11 5 12 19 9 20 18 12 21
Urogenital 1 0 1 1 0 1 2 0 2 1 1 1 9 3 9 12 5 13 16 2 20
Vascular 3 1 4 1 0 1 1 0 1 0 1 0 2 1 2 2 1 2 4 0 4
0 2 1 14 4 15 3 2 3 4 2 4 4 1 4 5 2 5 4 0 4
12
Based on 24-month cohort.
13
Some adverse events may lead to additional surgeries or interventions. Refer to Table 4 for more information.
14
Control=Single-level anterior interbody fusion procedure with allograft and plate stabilization. Non-randomized control arm
from IDE study of PRESTIGE® Cervical Disc.
15
Back and/or lower extremity (LE) pain adverse events (AEs) and Headache AE’s were classified as “Other Pain ” AEs
for the PRESTIGE LP™ IDE study.
Medtronic Page 21 of 91
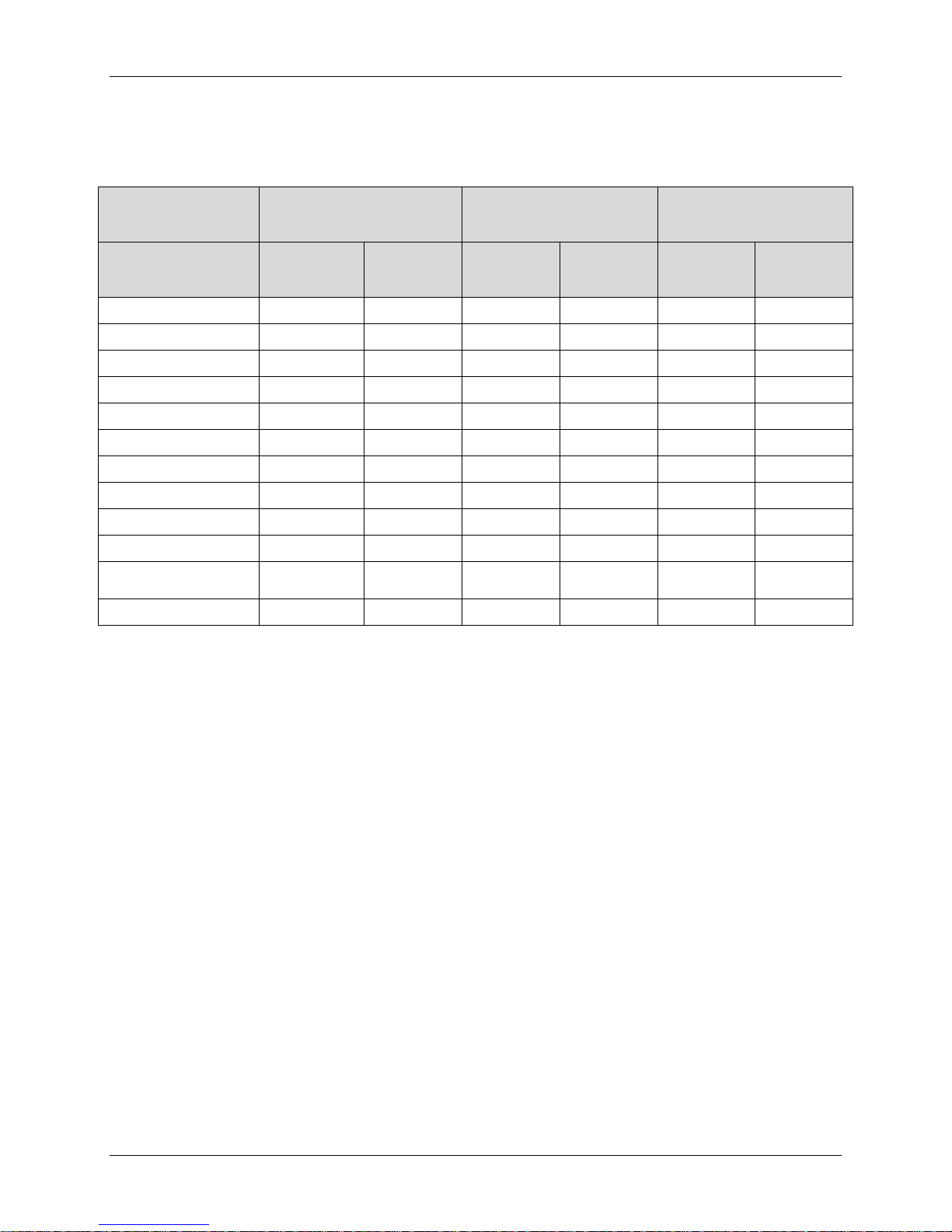
PRESTIGE LP™
(N=280)
PRESTIGE LP™
(N=333)
Device Relationship
Determined by CAC
Wound (NonInfectious)
Table 10b: Adverse Events Classified as Device-Related or Device/Surgical Procedure-
Related According to the Clinical Adjudication Committee through Month 24 – Safety
Population
of Adverse Event
Dysphagia / Dysphonia 0 0 (0. 0) 1 1 (0.4) 0 0 (0.0)
Heterotopic Ossification 4 4 (1.4) 3 2 (0.8) 6 6 (1.8)
Implant Events 16 15 (5.4) 5 5 (1.9) 20 19 (5.7)
Neck and / or Arm Pain 9 7 (2.5) 6 4 (1.5) 13 11 (3.3)
Neurological 11 9 (3.2) 7 7 (2.6) 14 11 (3.3)
Non-Union 0 0 (0.0) 27 27 (10.2) 0 0 (0.0)
Other 2 2 (0.7) 2 2 (0.8) 3 3 (0.9)
Other Pain 5 5 (1.8) 4 3 (1.1) 5 5 (1.5%)
Spinal Event 13 8 (2.9) 4 2 (0.8) 13 8 ( 2.4%)
Trauma 1 1 (0.4) 0 0 (0.0) 2 2 (0.6)
Any Adverse Event 61 34 (12.1) 60 41 (15.5) 76 44 (13.2)
IDE Cohort
Events
N
0 0 (0.0) 1 1 (0.4) 0 0 (0.0)
Patients
N (%)
ACDF Control
(N=265)
Events
N
Patients
N (%)
Safety Cohort
Events
N
Patients
N (%)
Medtronic Page 22 of 91

Adverse Event Category
Definition
Anatomical/Technical Difficulty
– Cervical Study Surgery
Anatomical or technical difficulty encountered during the original implantation of the
PRESTIGE LP™ device or control treatment device
Anatomical or technical difficulty encountered during an additiona l surgery involving
control treatment device
Anatomical/
Cervical
Technical problem encountered during an additional surgery that involved a region
Cancer
A malignancy or malignant tumor/neoplasm
Cardiac Disorders
Any condition of the heart
Death
Termination of life due to any cause
Dysphagia
Difficulty in swallowing
Dysphonia
Difficulty in speaking
Gastrointestinal
Any condition pertaining to the stomach and intestines
Heterotopic Ossification Cervical
Event involving heterotopic ossification at any region of the cervical spine
Heterotopic Ossification - NonCervical
Event involving heterotopic ossification at any region of the spine that is not cervical
or any other region of the body.
Implant Events - Malpositioning
Poor or inappropriate placement of the implant
Implant Events - Displacement
Incomplete or partial dislocation of the implant
Implant Events - Loosening
Wear around the implant and/or loosening o f t he implant surface
Implant Events - Breakage
Breakage of any implant or implant component
Event that is implant-related, but does not meet the definition of malpositioned
implant, implant displacement, implant loosening, or implant breaking
Infection - Superficial
An infection near the surface of the surgical incision
Infection - Deep
An infection below the fascia at the surgical incision
Infection - Other Wound
Infection occurring in other surgical wound not involving the study
Infection - Hematoma
Swelling or mass of blood that has become infected
Infection - CSF Leak
Infection resulting from the leakage of CSF
Infection - Systemic
Infection pertaining to the whole body
Infection - Urinary Tract
Infection of any part of the urinary system
Infection - Other
Any infection not listed above
Pain - Neck
Pain (includ ing stiffness, strain, t ightness) in the neck
Pain - Upper Extremities
Pain (includ ing stiffness, strain, t ightness) in the shoulder, arm, wrist or hand
Pain - Neck and Upper
Extremities
Pain (includ ing stiffness, strain, t ightness) in the neck and shoulder, a rm, wrist, or
hand
Pain (including stiffness, strain, tightness) in an area that is not of cervical spine
etiology (e.g., abdominal pain of unknown etiology, hea dache, flank pai n, bursitis).
Other
Event not associated with any other categories (e.g., weight loss, tinnitus, substance
abuse, insomnia).
Respiratory
Ailments or symptoms associated with respiration or the respirato ry system
Spinal Event – Cervical Study
Surgery
Event involving the treated level of cervical spine
Spinal Event – Cervical NonStudy Surgery
Event involving one or more cervical spine level(s), with the exception of the treated
level
Spinal Event - Non-Cervical
Event involving one or more spine levels other than cervical spine
Trauma
Physical injury caused by a physical force or traumatic event (e.g. motor vehicle
accident, fall, etc.)
Urogenital
Any condition of, relating to, affecting, treating, or being the organs or functions o f
excretion and reproduction
Table 11: Adverse Event Categories
Anatomical/Technical Difficulty
– Cervical Non-Study Surgery
Technical Difficulty Non-
Implant Events - Other
the cervical region, but did not involve the PRESTIGE LP™ device or original
other than the cervical spine
Pain - Other
Medtronic Page 23 of 91

Injury to a vasc ular structure that is sustained during the course of the operative
procedure
Vascular – Vertebral artery
Injury to vertebral artery occurring at any time
Vascular - Other
Disorder or condition in which the vascular system is affected
Wound (Non-Infectious)
Any issue of surgical inci s i on, such as hematoma, excluding infection
Vascular – injury (intraoperative)
Bayesian analyses were conducted on all adverse events using non-informative priors. The
results are presented in Table 12 with 95% Bayesian Credible Intervals (BCI) for the difference
in adverse event rates (PRESTIGE LP™ IDE – ACDF). BCIs that exclude zero indicate
statistical differences in the adverse event rates between the PRESTIGE LP™ IDE cohort and the
ACDF Control group while the BCIs that include zero fail to conclude that this is a statistical
difference in the adverse event rates between the two groups. Based on the BCIs, statistical
differences were noted between groups for the adverse event rates in the following categories:
heterotopic ossification, implant events, neurological, non-union, spinal events, trauma,
urogenital, vascular, and wound (non-infectious). All are statistically higher for the PRESTIGE
LP™ IDE Cohort except for non-union which was statistically higher for the control group.
Medtronic Page 24 of 91
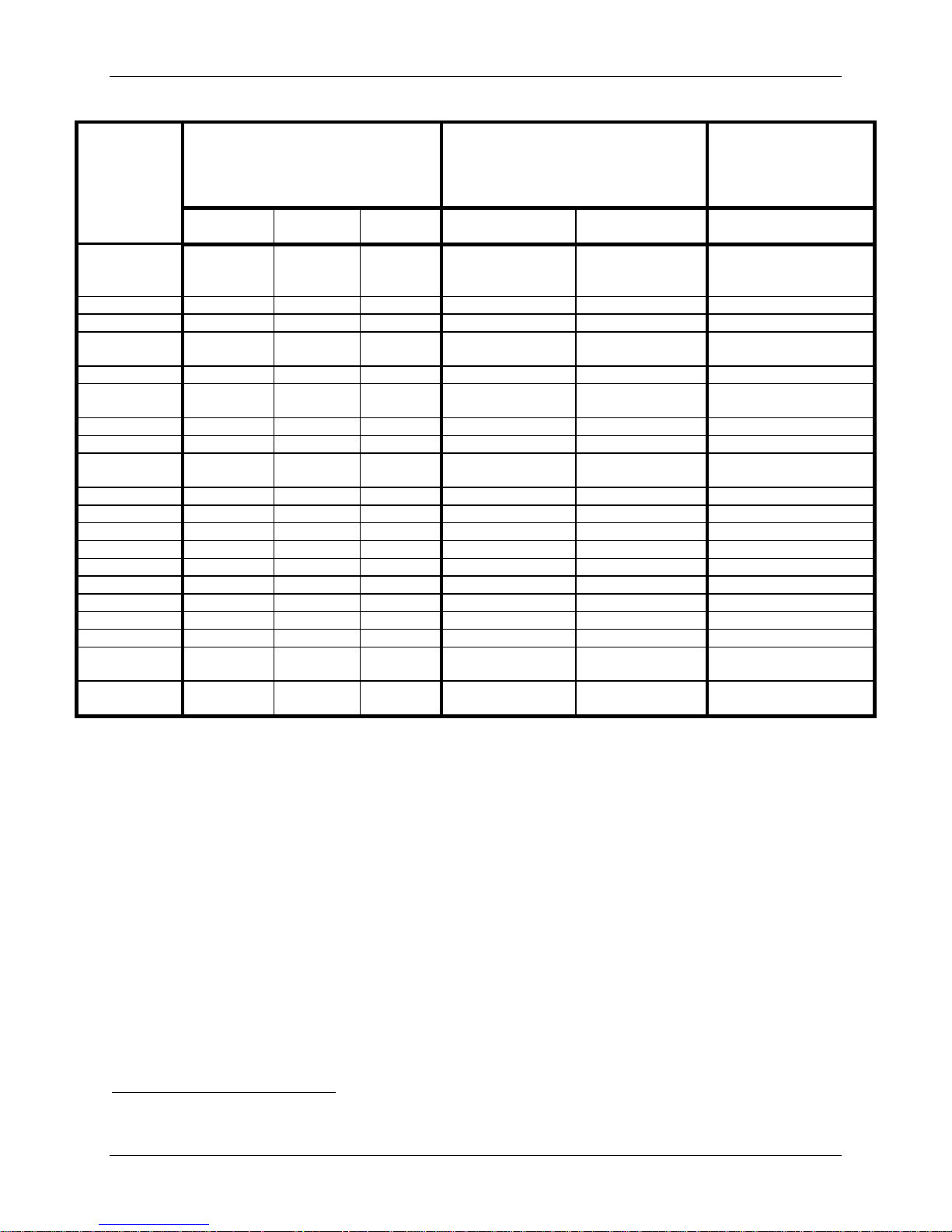
Posterior Mean and 95%
Control
ACDF
Control
Safety
Cohort
Anatomical /
Difficulty
Cancer
3 (1.1%)
2 (0.8%)
5 (1.5%)
1.0 (0.1%, 2.3%)
0.6% (0.0%. 1.6%)
0.4% (-1.2% , 2.0%)
Cardiac Disorders
16 (5.7%)
18 (6.8%)
19 (5.7%)
5.4% (2.8%, 8.2%)
7.0% (4.0%, 10.4%)
-1.6% (-6.0%, 2.9%)
Dysphagia /
Dysphonia
Gastrointestinal
35 (12.5%)
38 (14.3%)
43 (12.9%)
12.9% (9.0% , 17.3%)
13.7% (9.3%, 17.9%)
-0.8% (-7.2%, 5.1%)
Heterotopic
Ossification
Implant Events
16 (5.7%)
5 (1.9%)
21 (6.3%)
5.7% (3.0%, 8.7%)
1.8% (0.4%, 3.4%)
3.9% (0.6%, 7.4%)*
Infection
34 (12.1%)
27 (10.2%)
40 (12.0%)
12.0% (8.2%. 16.1%)
10.3% (6.6%, 14.2%)
1.7% (-3.8%, 7.5%)
Neck and / or
Arm Pain
Neurological
136 (48.6%)
108 (40.8%)
162 (48.6%)
49.4% (43.2%, 55.6%)
39.8% (33.7%, 46.1%)
9.6% (0.6%, 18.9%)*
Non-Union
0 (0.0%)
29 (10.9%)
0 (0.0%)
0.0%
11.4% (7.3%, 15.4%)
-11.3% (-15.4%, -7.3%)*
Other
93 (33.2%)
81 (30.6%)
110 (33.0%)
33.6% (27.9%, 39.5%)
30.1% (24.4%, 36.0%)
3.5% (-4.9%, 12.1%)
Other Pain
146 (52.1%)
132 (49.8%)
175 (52.6%)
51.3% (45.3%, 57.4%)
50.7% (44.2%, 56.8%)
0.6% (-8.5%, 9.7%)
Respiratory
24 (8.6%)
17 (6.4%)
27 (8.1%)
8.3% (4.9%, 11.7%)
6.5% (3.6%, 9.7%)
1.9% (-2.9%, 6.7%)
Spinal Event
83 (29.6%)
55 (20.8%)
106 (31.8%)
31.4% (25.7%, 37.2%)
19.0% (13.8%, 23.8%)
12.4% (4.5%, 20.3%)*
Trauma
61 (21.8%)
35 (13.2%)
69 (20.7%)
21.2% (16.4%, 26.3%)
13.5% (9.4%, 17.9%)
7.6% (0.7%, 14.4%)*
Urogenital
26 (9.3%)
9 (3.4%)
31 (9.3%)
8.7% (5.2%, 12.2%)
3.5% (1.4%, 5.8%)
5.2% (1.1%, 9.9%)*
Vascular
12 (4.3%)
4 (1.5%)
13 (3.9%)
4.6% (2.3%, 7.3%)
1.2% (0.2%, 2.5%)
3.4% (0.6%, 6.5%)*
Wound (NonInfectious)
Any adverse
Event
Table 12: Bayesian Comparison of Posterior Probabilities of Adverse Events
Adverse Event
Technical
Patients Experiencing Adverse Events (%)
IDE Cohort
2 (0.7%) 0 (0.0%) 2 (0.6%) 0.5% (0.0%, 1.4%) 0.0% (0.0%, 0.1%) 0.5% (0.0%, 1.4%)
26 (9.3%) 22 (8.3%) 30 (9.0%) 9.3% (5.9%, 12.9% ) 8.2% (4.8%, 11.6%) 1.0% (-4.2%, 6.1%)
27 (9.6%) 15 (5.7%) 31 (9.3%) 10.2% (6.7%, 14.0%) 5.0% (2.5%, 7.7%) 5.2% (0.5%, 10.1%)*
144 (51.4%) 124 (46.8%) 184 (55.3%) 51.9% (45.9%, 57.9%) 46.2% (39. 7%, 52.3%) 5.7% (-3.3%, 15.0%)
Posterior Mean and 95% HPD of Adverse
Event Rate
IDE Cohort ACDF Control IDE - ACDF
BCI16 of Difference of
Adverse Event Rate between
LP IDE Cohort and ACDF
25 (8.9%) 13 (4.9%) 27 (8.1%) 9.6% (6.0%, 13.1% ) 4.2% (1.9% , 6.6%) 5.3% (0.7%, 9.7%)*
257 (91.8%) 219 (82.6%) 306 (91.9%) 92.3% (89.0%, 95.4%) 82.0% (77. 2%, 86.9%) 10.2% (4.1%, 16.2%)*
*Asterisk denotes statistical difference.
16
BCI = Bayesian HPD Credible Interval
Medtronic Page 25 of 91

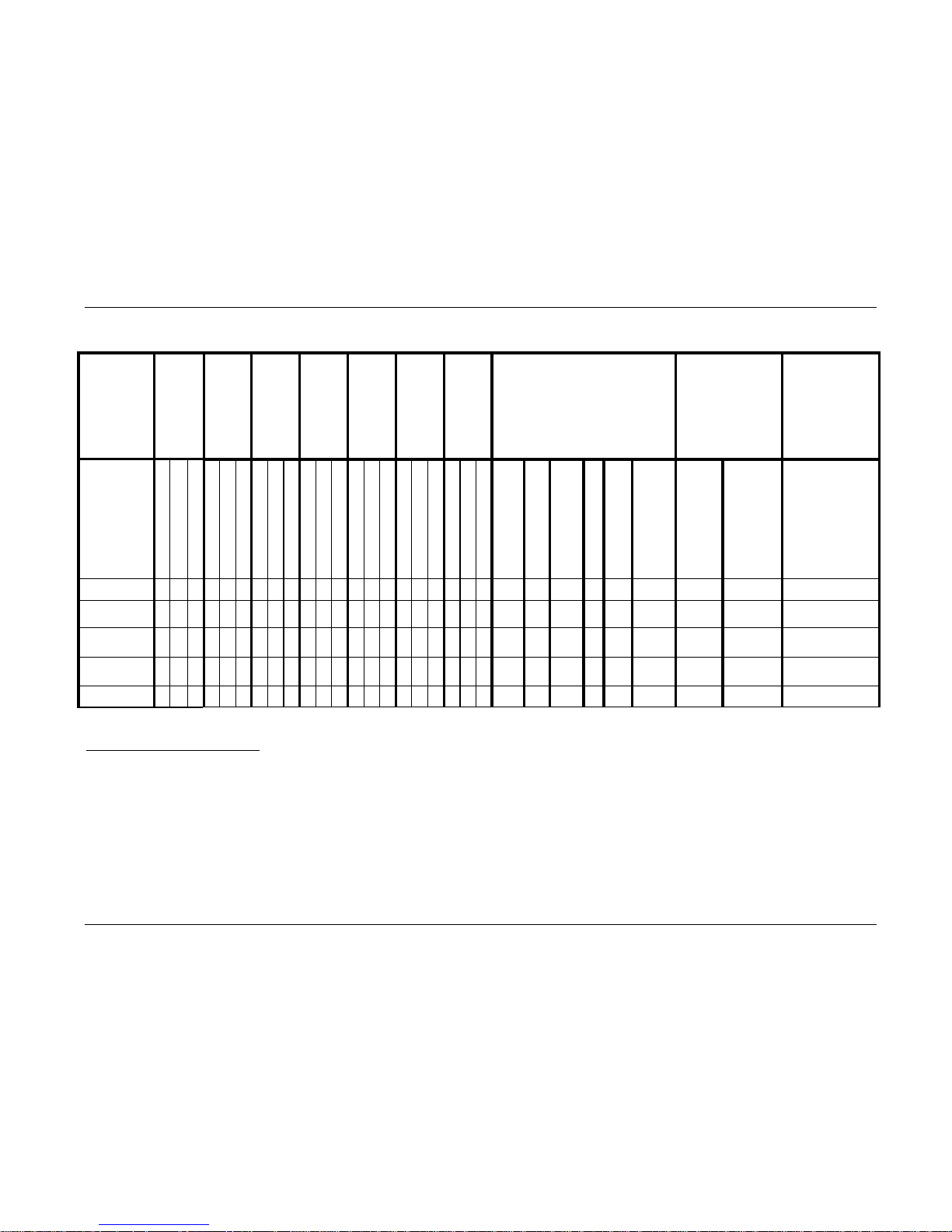
Table 13 summarizes the secondary interventions in the PRESTIGE LP™ device and control
treatment groups that occurred at or before the 24-month post-operative interval. Revisions,
removals, and supplemental fixations were considered second surgery failures in the clinical
study. Reoperations were not considered second surgery failures in the study. Table 13 also
presents the Bayesian statistical comparison of secondary surgeries between the PRESTIGE
LP™ IDE device and control treatment groups.
Overall, there were a greater number of subjects undergoing secondary surgical procedures at the
index level in the ACDF control group [21 (7.9 %)] compared to the PRESTIGE LP™ IDE [14
(5.0%)] and Safety Cohorts [15 (4.5%)]. Bayesian statistical comparison of secondary surgeries
between the PRESTIGE LP™ IDE Cohort and ACDF control treatment groups were performed
(if zero is excluded from the 95% BCI of the difference of the event rates, the event rates are
considered to be statistically different between the two groups). The only statistical difference
between the control and PRESTIGE LP™ Safety Cohort occurred in the Supplemental Fixation
category, with the investigational cohort requiring fewer supplemental fixation procedures than
the control cohort. However, this category also included use of external bone stimulators as
“supplemental fixation,” which may inflate the numbers in the ACDF control group, as all
“supplemental fixation” patients were considered failures due to secondary surgery. Among the
eight ACDF control subjects who had supplemental fixation, two had supplemental fixation
without using any external bone stimulators, one had “supplemental fixation” with and without
using an external bone stimulator and 5 subjects had “supplemental fixation” with external bone
stimulators only. Excluding the 5 subjects only using external bone stimulators, the supplemental
fixation rates are comparable between the two treatment groups.
Medtronic Page 26 of 91
Medtronic Page 27 of 91
Table 13. Secondary Interventions and Surgical Procedures Up to the 24-Month Visit.
Surgery
Postoperative
(1 day -
<
4 Weeks)
6 Weeks
(
≥
4 Wks -
<
9 Weeks)
3 Months
(
≥
9 Wks -
<
5 Months)
6 Months
(
≥
5 Mos-
<
9 Months)
12 Months
(
≥
9 Mos-
<
19 Months)
24 Months
((
≥
19 Mos-
<
30 Months))
Total 24 Month
# of Patients Reporting &
Total adverse events
Posterior Mean and
95% HPD of Secondary
Surgery Rate
Posterior Mean and 95%
BCI
17
of Difference of
Secondary Surgery Rate
between LP IDE Coho rt
and ACDF Control
Complication
IDE
Contro
l
Safety Cohort
IDE
Control
Safety Cohort
IDE
Control
Safety Cohort
IDE
Control
Safety Cohort
IDE
Control
Safety Cohort
IDE
Control
Safety Cohort
IDE
Control
Safety Cohort
IDE # Patients
(% of 280)
IDE Total # Even ts
Control # Patients
(% of 265)
Control Total # Events
Safety Cohort # Patients
(% of 333
Safety Cohort Total #
Events
PRESTIGE
LP™
IDE Cohort
ACDF
Control
PRESTIGE™ LP IDE –
ACDF Control
Revisions18
0 0 0 0 1 0 1 0 1 0 3 0 0 0 0 0 1 0 0 0 0
1
(0.4)
1
5
(1.9)
5
1
(0.3)
1
0.4%
(0.0%, 1.1%)
1.6%
(0.3%, 3.3%)
-1.3%
(-3.2%, 0.4%)
Removals
19
0 0 0 1 0 1 1 0 1 1 1 1 3 2 3 3 6 4 1 2 1
10
(3.6)
10
11
(4.2)
11
11
(3.3)
11
3.7%
(1.6%, 6.0%)
3.8%
(1.6%, 6.2%)
-0.1%
(-3.7%, 3.2%)
Supplemental
Fixations
20
0 0 0 0 0 0 0 0 0 0 0 0 1 3 1 1 5 1 0 1 0
2
(0.7)
2
8
(3.0)
9
2
(0.6)
2
0.5%
(0.0%, 1.3%)
3.2%
(1.3%, 5.5%)
-2.7%
(-5.0%, -0.5%)
Reoperations
21
0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 1 1 1 2 0 2
3
(1.1)
3
2
(0.8)
2
3
(0.9)
3
1.1%
(0.1%, 2.3%)
0.6%
(0.0%, 1.6%)
0.4%
(-1.2%, 2.1%)
Total
0 0 0 1 1 1 2 1 2 1 4 1 4 5 4 5 13 6 3 3 3
14
(5.0)
16
21
(7.9)
27
15
(4.5)
17
5.3%
(2.8%, 8.2%)
7.1%
(4.1%, 10.4%)
-1.9%
(-6.4%, 2.3%)
17
BCI = Bayesian HPD Credible Interval
18
A procedure that adjusts or in any way modifies the original implant configuration (e.g., adjusting position of the original configuration, removal with replacement with the same
type of study implant).
19
A procedure that removes one or more components of the original implant configuration without replacement with the same type of trial implant. Removals include elective
removals.
20
A procedure at the involved level in which additional spinal devices not approved as part of the protocol are placed. This categorization of Supplemental Fixations includes
supplemental therapies (i.e. external bone growth stimulators). There were a total of six (6) external bone growth stimulators used in the ACDF Control group. Three (3) occurred
at six (6) months, and three (3) occurred at 12 months. No external bone growth stimulators were used in the IDE or Safety Cohorts. Please note that since this additional device
was used, and included as a supplemental fixation, these patients were considered failures in the Primary Endpoint.
21
A procedure that involves any surgical procedure at the involved level that does not remove, modify, or add any components and that is not considered a Removal. Revision, or
Supplemental Fixation.
Group
Cause/Adverse Event
Action
Secondary
Category
Surgery
Safety Cohort
C4-C5 displaced device with
scan was positive for osteopenia
C4-C5 explant of PRESTIGE
anterior cervical fusion
Removal
01 Day- <4
(01 day)
Safety Cohort
C4-C5 PRESTIGE LP™ artificial
space secondary to a fall
C4-C5 explant of PRESTIGE
Removal
06 Weeks
Safety Cohort
C5-C6 device extrusion
C5-C6 explant of PRESTIGE
disc; different size
Revision
06 Weeks
Safety Cohort
C6-C7 large recurrent disc herniation
C6-C7 explant of PRESTIGE
and fusion
Removal
03 Months
Safety Cohort
Severe neck pain
C5-C6 explant of PRESTIGE
anterior cervical fusion
Removal
06 Months
Safety Cohort
C6-C7 cervical radiculopathy with
C6-C7 explant PRESTIGE
and fusion
Removal
06 Months
Safety Cohort
C5-C6 artificial disc dislodging
C5-C6 explant of PRESTIGE
anterior cervical fusion
Removal
06 Months
Safety Cohort
C3-C4 foraminal stenosis; possible
disc protrusion
C3-C4 posterior cervical fusion
Supplemental
06 Months
Safety Cohort
C5-C6 herniated disc with right upper
C6-C7 explant PRESTIGE
and fusion
Removal
12 Months
Safety Cohort
Radiating paracervical pain and right
C4-C5 explant of PRESTIGE
by anterior cervical fusion
Removal
12 Months
Safety Cohort
Neck pain radiating to shoulders
C6-C7 explant of PRESTIGE
cervical fusion
Removal
12 Months
Safety Cohort
IDE
C3-C4, C5-C6 foraminal stenosis
C3-C4, C5-C6 left posterior
laminectomy
Reoperation
12 Months
(528 days)
Safety Cohort
Access
C7 subsidence into the vertebral body
event
C6-C7 explant of PRESTIGE
microdiscectomy and fusion
Removal
12 Months
Safety Cohort
C6-C7 cervical radiculopathy with
above.
C5-C7 posterior cervical
Supplemental
12 Months
Table 14: Secondary Surgical Interventions at the Index Level– Procedure Details
IDE
IDE
IDE
IDE
IDE
IDE
fractured vertebrae; subject's bone
disc compressed into the vertebral
body and rotated within the disc
with cord compression and severe
stenosis
cervical stenosis
LP™ artificial disc followed by
LP™ artificial disc followed by
anterior cervical fusion
LP™ artificial disc and
replacement with new
PRESTIGE LP™ artificial
LP™ artificial disc with
anterior cervical discectomy
LP™ artificial disc followed by
LP™ artificial disc,C5-C7
anterior cervical discectomy
Surgical
Intervention
Time to
Index Level
Weeks
(49 days)
(56 days)
(150 days)
(159 days)
(215 days)
IDE
IDE
IDE
IDE
IDE
Continued
IDE
posteriorly
C4 impingement; C4-C5 foraminal
extremity radiculopathy
shoulder pain
and lucency as a result of traumatic
cervical stenosis. Additionally, the
patient had removal due to the same
diagnosis at 215 days as referenced
LP™ artificial disc followed by
LP™ artificial disc, C5-C7
anterior cervical discectomy
LP™ artificial disc, followed
LP™ artificial disc; anterior
LP™ artificial disc, anterio r
fusion; C6-C7 posterior
cervical foraminotomy
Fixation
Fixation
(259 days)
(262 days)
(423 days)
(469 days)
(518 days)
(546 days)
(568 days)
Medtronic Page 28 of 91
 Loading...
Loading...