

Page 1

Endurant™ II/IIs
Stent Graft System
Instructions for Use
Page 2

Medtronic, Medtronic with rising man logo, and Medtronic logo are trademarks of Medtronic. Third-party trademarks (“TM*”) belong to their respective owners. The
following list includes trademarks or registered trademarks of a Medtronic entity in the United States and/or in other countries.
Aptus™, EndoAnchor™, Endurant IIs™, Endurant II™, Endurant™, Heli-FX™, Pacific™, Reliant™, Talent™
Page 3

EXPLANATION OF SYMBOLS ON PRODUCT LABELING
Refer to the device labeling to see which symbols apply to this product.
Contents: One Device
Do not use if package is damaged
Nonpyrogenic
Peel here
MR Conditional
Caution: Federal law (USA) restricts this device to sale by or on the order of a physician.
Sterilized using irradiation
Catalog number
Serial number
Use-by date
Do not reuse
Manufacturer
Manufactured in
Consult instructions for use at this website
3
Page 4

Contents
1 Device description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
1.1 Stent graft . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
1.2 Delivery system . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
1.3 Heli-FX EndoAnchor system . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2 Indications for use . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
3 Contraindications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
4 Warnings and Precautions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
4.1 General . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
4.2 Patient selection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
4.3 Before the implant procedure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
4.4 During the implant procedure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
4.5 Treatment and follow-up . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
4.6 MRI safety information . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
5 Adverse events . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
5.1 Observed adverse events . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
5.2 Potential adverse events . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
5.3 Device-related adverse events reporting . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
6 Summary of clinical studies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
6.1 Endurant stent graft system US clinical study . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
6.2 ENGAGE PAS cohort . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
6.3 ANCHOR Registry short neck cohort . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
7 Patient selection and treatment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
7.1 Individualization of treatment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
8 Patient counseling information . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
9 How supplied . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
9.1 Sterility . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
9.2 Contents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
9.3 Storage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
10 Clinical use information . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
10.1 Physician training requirements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
10.2 Recommended device sizing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
10.3 Device inspection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
10.4 Additional required equipment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
10.5 Additional recommended equipment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
10.6 MRI information . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
11 Implant instructions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
11.1 Vascular access and device preparation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
11.2 Delivery procedure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
12 Bail-Out Techniques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
12.1 Screw gear handle disassembly . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
12.2 Ballooning . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78
12.3 Back-end handle disassembly . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78
12.4 Snare the tapered tip . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78
13 Follow-up imaging recommendations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78
13.1 General . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78
13.2 X-ray . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
13.3 CT with contrast . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
13.4 Noncontrast CT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
4
Page 5

13.5 Duplex ultrasound . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
13.6 MRI or MRA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
13.7 Imaging tests . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
13.8 Supplemental imaging . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
14 Additional Surveillance and Treatment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
15 Device registration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
16 Disclaimer of Warranty . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
5
Page 6

1 Device description
1
1 1
2
1
1 1
2
1
1
1
1
1 1
1
1
5
8
7
4
3
1 1
10
1
1 1
2
9
10
10
1
1 1
2
1
6
1 1
3
The Endurant II/Endurant IIs stent graft system (hereinafter referred to as Endurant II/IIs stent graft system) is intended to treat
infrarenal abdominal aortic or aortoiliac aneurysms using an endovascular approach. When placed within the aneurysm, the stent
graft provides an alternative conduit for blood flow within the patient’s vasculature.
The stent graft system is comprised of 2 main components: the implantable stent graft and the disposable delivery system. The stent
graft is preloaded into the delivery system and advanced to the aneurysm using fluoroscopic guidance. Upon deployment, the stent
graft self-expands to conform to the shape and size of the seal zones above and below the aneurysm.
The Endurant II/IIs stent graft can also be used with the Heli-FX EndoAnchor system (available separately). The Heli-FX EndoAnchor
system is designed to provide fixation and augment sealing between the Endurant II/IIs stent graft and the native artery. The system
consists of an EndoAnchor implant that is delivered using the Heli-FX applier through the steerable Heli-FX guide.
1.1 Stent graft
There are 2 main body stent graft configurations: a bifurcated main body stent graft and an aorto-uni-iliac (AUI) main body stent graft.
First, a bifurcated device is implanted into the patient’s aorta. If a bifurcated device cannot be implanted, an AUI device can be used.
After placement of the bifurcated or AUI device, limb stent graft configurations are introduced separately into the vessel and mated
with the implanted main body stent graft configuration. Depending on the patient’s anatomy, a limb configuration may not be required
in the AUI configuration. If additional distal or proximal coverage is needed, an iliac or aortic extension configuration is introduced
separately into the vessel and mated with the implanted main body stent graft configuration.
All Endurant II/IIs stent graft configurations are composed of nitinol stents sewn to a fabric graft with nonresorbable sutures.
Radiopaque markers are sewn onto the stent graft to aid in visualization and to facilitate accurate placement. The nitinol stents may
also be visible under fluoroscopy.
Stent grafts should be oversized to be larger than the measured vessel inner diameter (aortic components are oversized
approximately 10–20%; limb components are oversized approximately 10–25%). Recommended device sizing (Section 10.2)
contains detailed sizing information for all stent graft components, including available ranges of length and diameter. Table 1 contains
a summary of the stent graft materials.
Figure 1. Stent graft configurations and locations of RO markers
6
Page 7

1 Radiopaque marker
2 ‘e’ marker
3 Radiopaque gate marker
4 Endurant II aortic extension configuration
5 Endurant II bifurcated configuration
Note: This and all other product graphics appearing in this manual are not drawn to scale.
Table 1. Stent graft materials
Component Material
Stents Nickel-titanium (nitinol) alloy
Button radiopaque markers Platinum-iridium alloy
“e” radiopaque marker Platinum
Contralateral gate marker Platinum-iridium alloy
Graft material Polyester
Suture Polyester and polyethylene
The Endurant II/Endurant IIs stent graft system does not contain natural rubber latex. However, during the manufacturing/assembly
process, it may have incidental contact with latex-containing products.
1.1.1 Bifurcated configuration
The bifurcated stent graft is available in 2 configurations: the Endurant II bifurcated configuration and the Endurant IIs bifurcated
configuration. The Endurant II bifurcated configuration is an aortoiliac stent graft that is available in 3 lengths. The Endurant IIs
bifurcated configuration is an aortic configuration available in a single, shorter length (Figure 1). The proximal end of both bifurcated
configurations deploy into the proximal neck and upper section of the aneurysm. The proximal end of the bifurcated configuration is
composed of nitinol stents sewn to a fabric graft. The suprarenal portion of the proximal end is not covered with graft fabric (Figure 1).
The suprarenal stent also contains anchor pins to fix the stent graft in place inside the aorta above the renal arteries without obstructing
them with graft fabric. The diameters of the available proximal aortic section of the bifurcated stent graft configurations range from
23 mm to 36 mm, and the covered length of the bifurcated stent graft configurations range from 124 mm to 166 mm (Endurant II
bifurcated stent graft) or 103 mm (Endurant IIs bifurcated stent graft). The aortic sections of the bifurcated stent graft configurations
should be oversized 10% to 20% in relation to the actual measured innervessel diameter. The available sizes can be used in aortas
with diameters ranging from 19 mm to 32 mm.
The aortic section bifurcates distally into 2 smaller tubes: an ipsilateral leg and a shorter contralateral leg. In the Endurant II bifurcated
configuration, all stents on the ipsilateral leg are sewn to the outside of the fabric creating a smooth inner lumen. In the Endurant IIs
bifurcated configuration, the 4 distal stents are sewn to the inside of the ipsilateral leg graft fabric. For all sizes, the stents on the
contralateral leg are sewn to the inside of the graft fabric (Figure 1). The Endurant II bifurcated stent graft configurations have an
ipsilateral leg that range in diameters from 13 mm to 20 mm and may be used in iliac arteries with diameters ranging from 10 mm to
18 mm. The Endurant II bifurcated stent graft configurations have a contralateral leg that range in diameters from 12 mm to 14 mm.
The Endurant IIs bifurcated stent graft ipsilateral and contralateral legs are available in 14 mm diameter and are not intended to extend
to the iliac arteries.
Note: The bifurcated configurations are always used with the limb configuration (Section 1.1.3).
6 Endurant IIs bifurcated configuration
7 Endurant II iliac extension configuration
8 Endurant II limb configuration
9 Endurant II aorto-uni-iliac configuration
10 Overlap marker
1.1.2 Aorto-Uni-Iliac (AUI) configuration
The AUI device is indicated for the endovascular treatment of infrarenal abdominal aortic or aortoiliac aneurysms only in patients
whose anatomy does not allow the use of a bifurcated device. The proximal end of the aorto-uni-iliac (AUI) configuration is deployed
into the proximal neck and upper section of the aneurysm. The proximal section of the AUI configuration is composed of nitinol stents
sewn to a fabric graft. The suprarenal portion of the proximal end is not covered with fabric (Figure 1). The suprarenal stent includes
anchor pins to fix the stent graft in place inside the aorta above the renal arteries without obstructing them with graft fabric. The AUI
configurations are available in proximal diameters ranging from 23 mm to 36 mm, with a 102 mm covered length. The aortic sections
of the AUI configurations should be oversized 10% to 20% in relation to the actual measured inner vessel diameter. The available sizes
can be used in aortas with diameters ranging from 19 mm to 32 mm. For additional sizing information, see Recommended device
sizing (Section 10.2). Distally, the aortic section tapers down to a smaller diameter tube. In the distal end of the tapered AUI device,
the stents are sewn to the inside of the graft fabric (Figure 1). The distal end has a diameter of 14 mm.
Note: Depending on the patient’s anatomy, a limb configuration may not be required for all AUI device implantations.
Note: A femoral-femoral artery bypass may be performed in conjunction with implantation of the AUI device to maintain perfusion to
the contralateral femoral artery. In order to prevent backflow into the aneurysm sac, most patients will require occlusion of the iliac
artery. This may be accomplished through methods selected by the physician. The Talent occluder system (available separately) is
an optional component that is often used in conjunction with the AUI configuration. It is closed at both ends to stop retrograde blood
7
Page 8

flow into the aneurysm sac. For details on the Talent occluder system, refer to the instructions for use. If complete atherosclerotic
obstruction of the vessel is already present, occluding the iliac artery may not be necessary.
1.1.3 Limb configuration
The proximal end of the limb configuration deploys within the legs of the bifurcated configuration, while the distal end deploys into the
iliac artery. For the AUI device, the proximal end of the limb configuration deploys within the distal end of the AUI configuration while
the distal end deploys into the iliac artery on the ipsilateral side. The proximal end of the limb configuration has an open web
configuration, which contains no graft material in its stent valleys (Figure 1). The distal diameter of limb configurations range from
10 mm to 28 mm with lengths ranging from 82 mm to 199 mm. The proximal diameter is 16 mm for all sizes of limb configuration, so
they can dock with all available bifurcated stent graft configurations. The limb configuration should be oversized 10% to 25% in relation
to the inner vessel diameter and can be used in iliac arteries ranging from 8 mm to 25 mm.
Note: A limb device is implanted on both the ipsilateral and contralateral legs of an Endurant IIs bifurcated configuration. See Deploy
Limb Stent Graft Into Ipsilateral Leg (Endurant IIs bifurcated configuration only) (Section 11.2.14).
1.1.4 Iliac extension configuration
An iliac extension configuration is available if additional distal stent graft length is needed. It has an open web configuration on the
proximal section (Figure 1). The diameters of iliac extension configurations range from 10 mm to 28 mm with a covered length of
82 mm. Similar to the limb configuration, the iliac extension configuration is designed for oversizing 10% to 25% and can be used in
iliac arteries ranging from 8 mm to 25 mm in diameter.
Note: An appropriately sized limb configuration can be used as an iliac extension configuration.
1.1.5 Aortic extension configuration
An aortic extension configuration is available if additional proximal stent graft length is needed. The aortic extension stent graft has
a bare proximal suprarenal stent with anchor pins (Figure 1). The diameters of available aortic extension configurations range from
23 mm to 36 mm with the aortic extension having a covered length from 49 mm to 70 mm. Similar to the bifurcated and AUI
configurations, the aortic extension configuration is designed for oversizing of 10% to 20% in relation to the actual measured inner
vessel diameter. The available sizes can be used in aortas with diameters ranging from 19 mm to 32 mm.
1.2 Delivery system
The Endurant II delivery system, which delivers all stent graft configurations, consists of a single-use, disposable catheter with an
integrated handle to provide controlled deployment. It is available in 14, 16, 18, and 20 Fr graft cover diameters and a working length
of 57 cm ± 2 cm. The catheter assembly is flexible and compatible with a 0.035 in (0.89 mm) guidewire. There are 2 types of delivery
systems: the aortic (Figure 2) and the iliac (Figure 3) delivery systems. The aortic delivery system delivers the bifurcated, aortic
extension, and AUI configurations. The iliac delivery system delivers the limb and iliac extension stent graft configurations. The aortic
delivery system features a tip capture mechanism, which is not present in the iliac delivery system.
8
Page 9

Figure 2. Aortic delivery system
1 Rear handle
2 Back-end wheel
3 Screw gear
4 External slider
5 Trigger
6 Front grip
7 Graft cover
8 Markerband
9 Spindle
10 Sleeve
11 Tapered tip
9
Page 10

Figure 3. Iliac delivery system
1 Rear handle
2 Screw gear
3 External slider
4 Trigger
5 Front grip
6 Graft cover
7 Markerband
8 Tapered tip
1.3 Heli-FX EndoAnchor system
The Endurant II/IIs stent graft can also be used with the Heli-FX EndoAnchor system (available separately). The system consists of
an EndoAnchor implant that is delivered using the Heli-FX applier through the steerable Heli-FX guide. The EndoAnchor implant is
designed to provide fixation and augment sealing between the Endurant II/IIs stent graft and the native artery. The EndoAnchor
implant may be implanted at the time of the initial stent graft implantation or during a secondary (for example, repair) procedure.
Note: For additional information about using this system with the Endurant II/IIs stent graft, refer to Section 11.2.17 . For additional
information about the Heli-FX EndoAnchor system, refer to the instructions for use provided with the system.
2 Indications for use
The Endurant II/IIs bifurcated stent graft is indicated for the endovascular treatment of infrarenal abdominal aortic or aortoiliac
aneurysms. They may be utilized in conjunction with the Heli-FX EndoAnchor system when augmented radial fixation and/or sealing
is required; in particular, in the treatment of abdominal aortic aneurysms with short (≥ 4 mm and < 10 mm) infrarenal necks (see Neck
length definition below). The Endurant II aorto-uni-iliac (AUI) stent graft is indicated for the endovascular treatment of infrarenal
abdominal aortic or aortoiliac aneurysms in patients whose anatomy does not allow the use of a bifurcated stent graft. The Endurant
II/IIs stent graft system is indicated for use in patients with the following characteristics:
• Adequate iliac or femoral access vessel morphology that is compatible with vascular access techniques, devices, or accessories
• Proximal neck length of:
– ≥10 mm; or
– ≥ 4 mm and < 10 mm when used in conjunction with the Heli-FX EndoAnchor system (bifurcated stent graft only)
Note: Neck length is defined as the length over which the aortic diameter remains within 10% of the infrarenal diameter.
• Infrarenal neck angulation of ≤ 60°
• Aortic neck diameters with a range of 19 to 32 mm
• Distal fixation length of ≥15 mm
• Iliac diameters with a range of 8 to 25 mm
• Morphology suitable for aneurysm repair
10
Page 11

3 Contraindications
The Endurant II/IIs stent graft system is contraindicated in:
• Patients who have a condition that threatens to infect the graft
• Patients with known sensitivities or allergies to the device materials listed in Table 1
When used with the Heli-FX EndoAnchor system, the Endurant II/IIs stent graft system is also contraindicated in:
• Patients with known sensitivities to the EndoAnchor implant materials
For contraindications regarding ancillary devices used with the Endurant II/IIs stent graft system, refer to the instructions for use
provided with each device.
4 Warnings and Precautions
Caution: Read all instructions carefully. Failure to properly follow the instructions, warnings, and precautions may lead to serious
consequences or injury to the patient.
4.1 General
• The Endurant II/IIs stent graft system should only be used by physicians and teams trained in vascular interventional techniques,
including training in the use of this device. Specific training expectations are described in Physician Training Requirements
(Section 10.1).
• When the Endurant II/IIs stent graft system is used with the Heli-FX EndoAnchor system, it should only be used by physicians and
teams who are also trained in the use of the EndoAnchor system.
• The Heli-FX EndoAnchor system should be used when augmented radial fixation and/or sealing is required; in particular, in the
treatment of abdominal aortic aneurysms with short (≥ 4 mm and < 10 mm) infrarenal necks.
• Always have a vascular surgery team available during implantation or reintervention procedures in the event that conversion to
open surgical repair is necessary.
4.2 Patient selection
• Inappropriate patient selection may result in poor device performance or device performance not otherwise in accordance with
the specifications.
• Do not use the Endurant II/IIs stent graft system in patients unable to undergo, or who will not be compliant with, the necessary
preoperative and postoperative imaging and implantation procedures (Chapter 10 to Chapter 13).
• The Endurant II/IIs stent graft system is not recommended in patients who cannot tolerate contrast agents necessary for
intraoperative and postoperative follow-up imaging.
• The Endurant II/IIs stent graft system is not recommended in patients exceeding weight or size limits necessary to meet imaging
requirements.
• Key anatomic elements that may affect successful exclusion of the aneurysm include severe proximal neck angulation (>60°);
short proximal aortic neck (<10 mm) without use of the Heli-FX EndoAnchor system; very short proximal aortic neck (<4 mm); and
thrombus and/or calcium formation at the arterial implantation sites, specifically the proximal aortic neck and distal iliac artery
interface. Irregular calcification and/or plaque may compromise the fixation and sealing of the implantation sites. Necks exhibiting
these key anatomic elements may be more conducive to graft migration.
• Methodologies for measurement of neck length vary. Where neck length is defined as the length over which the aortic diameter
remains within 10% of the infrarenal diameter, other definitions may result in an estimation of neck length that is longer than that
obtained using this definition.
• Deploying the stent graft in an area of vessel calcification may lead to abrasion of the stent graft on calcified plaque, potentially
causing development of holes or tears in the graft.
• For infrarenal EVAR procedures using the Heli-FX EndoAnchor system:
– The access vessel diameter and morphology should be compatible for use with the Heli-FX EndoAnchor system.
– The EndoAnchor implant should be implanted only into areas of aortic tissue that are free of or have insignificant calcified
plaque or thrombus, or where such pathology is diffuse and less than 2 mm in thickness.
– Refer to the Heli-FX EndoAnchor system instructions for use for additional information.
• Iliac conduits may be used to ensure the safe insertion of the delivery system if the patient’s access vessels, as determined by
treating physician, preclude safe insertion of the delivery system.
• The long-term safety and effectiveness of the Endurant II/IIs stent graft system has not been established.
• The safety and effectiveness of the Endurant II/IIs Stent Graft System with the Heli-FX EndoAnchor system has not been
evaluated in an appreciable number of patients in ASA Class I-II.
• The safety and effectiveness of the Endurant II AUI device with the Heli-FX EndoAnchor system has not been evaluated.
11
Page 12

• The safety and effectiveness of the Endurant II/IIs stent graft system has not been evaluated in patients who:
– Are less than 18 years old
– Are pregnant or lactating
– Have an aneurysm that is:
Suprarenal
Juxtarenal or pararenal
Isolated iliofemoral
Mycotic
Inflammatory
Pseudoaneurysm
– Have a dominant patent inferior mesenteric artery and an occluded or stenotic celiac or superior mesenteric artery
– Have an untreated thoracic aneurysm >4.5 cm in diameter
– Require emergent aneurysm treatment, eg, trauma or rupture
– Have a history of bleeding diathesis or coagulopathy
– Have had a myocardial infarction (MI) or cerebrovascular accident (CVA) within 3 months prior to implantation
– Have a reversed conical neck, which is defined as a >4 mm distal increase over a 10 mm length
– Have a known hypersensitivity or contraindication to anticoagulants, antiplatelets, or contrast media, which is not amenable to
pre-treatment
– Have significant (typically >25% of vessel circumference of aortic neck and iliac artery, or >50% of the length of the iliac artery)
aortic mural thrombus at either the proximal or distal attachment location that would compromise bilateral fixation and seal of
the device
– Have ectatic iliac arteries requiring bilateral exclusion of hypogastric blood flow
– Have arterial access site that is not expected to accommodate the diameter of the device (14 to 20 Fr) due to size or tortuosity
– Have active infection at the time of the index procedure documented by pain, fever, drainage, positive culture, or leukocytosis
(WBC >11,000/mm3) that is treated with antimicrobial agents (nonprophylactic)
– Have congenital degenerative collagen disease
– Have congenital degenerative collagen disease, eg, Marfan’s Syndrome
– Have a creatinine >2.0 mg/dL
– Are on dialysis
– Have connective tissue disorder
• The safety and effectiveness of the Endurant II/IIs stent graft system with the Heli-FX EndoAnchor system has additionally not
been evaluated in patients who:
– have an infrarenal aortic neck with significant thrombus or calcium that precludes adequate EndoAnchor implant penetration
of the aortic wall
• All patients should be advised that endovascular treatment requires lifelong, regular follow-up to assess their health and the
performance of their endovascular graft. Patients with specific clinical findings (eg, endoleaks, enlarging aneurysms, or changes
in the structure or position of the endovascular graft), or less than the recommended number of EndoAnchor implants when used
in short proximal necks (≥ 4 mm and < 10 mm), should receive enhanced follow-up. Specific follow-up guidelines are described in
Chapter 13.
• Patients experiencing reduced blood flow through the graft limb or leaks may be required to undergo secondary interventions or
surgical procedures.
• Intervention or conversion to standard open surgical repair following initial endovascular repair should be considered for patients
experiencing enlarging aneurysms or endoleak. An increase in aneurysm size or persistent endoleak may lead to aneurysm
rupture.
• The AUI device should be used in patients whose anatomy does not allow the use of a bifurcated device. To maintain perfusion
to the contralateral limb, a femoral-femoral artery bypass may be performed. Timing of the femoral-femoral artery bypass is up to
the discretion of the physician.
• Reduced blood flow through the AUI device may lead to impairment of blood perfusion to the lower half of the body. The physician
should emphasize to the patient that graft occlusion can have life threatening consequences. The patient should be counseled
to seek medical attention immediately if he/she experiences pain in one or both legs, or if one or both legs become pale in color
and/or cool to the touch.
4.3 Before the implant procedure
• Preoperative planning for access and placement should be performed before opening the device packaging.
12
Page 13

• Carefully inspect the Endurant II/IIs stent graft system packaging and device for damage or defects prior to use. Do not use product
if any sign of damage or breach of the sterile barrier is observed. Do not attempt to resterilize the delivery system or stent graft.
• Do not bend, kink, or otherwise alter the delivery system prior to implantation because it may cause deployment difficulties.
• To reduce the risk of thrombotic complications, an additional bolus of IV heparin should be administered before inserting the
device.
4.4 During the implant procedure
• Exercise care in handling and delivery technique to help prevent vessel rupture.
• Studies indicate that the danger of micro-embolization increases with increased procedure duration.
• Renal complications may occur:
– from an excess use of contrast agents
– as a result of embolic or misplaced stent graft
• Exercise care and utilize suitable imaging techniques when deploying the aortic endograft into a short proximal neck, to ensure
accurate placement. Inaccurate placement could result in unsuccessful implantation of EndoAnchor implants, the need to place
a proximal extension, or unintentional artery coverage.
• Do not deploy the stent grafts in a location that will cause an endoleak or occlude arteries necessary to supply blood flow to organs
or extremities. This could necessitate surgical removal of the device.
• Use fluoroscopic guidance to advance the delivery system, detect kinking, or assess alignment problems with the stent graft
devices. Do not use excessive force to advance or withdraw the delivery system when resistance is encountered. If the delivery
system kinks during insertion, do not attempt to deploy the stent graft. Remove the delivery system and insert a new one.
• Do not continue to torque the delivery system if the tip is not rotating with the delivery system.
• Exercise particular care in difficult areas, such as areas of stenosis, intravascular thrombosis, or in calcified or tortuous vessels.
Balloon angioplasty at the site of a narrowed or stenotic vessel may be considered prior to attempting to gently reintroduce the
catheter delivery system.
• An inadequate seal zone may result in increased risk of leakage into the aneurysm or migration of the stent graft.
• Systemic anticoagulation should be used during the implantation procedure based on hospital/physician protocol. If heparin is
contraindicated, an alternative anticoagulant should be considered.
• Stent grafts cannot be replaced or drawn back into the delivery system, even if only partially deployed.
• If the graft cover is accidentally withdrawn, the device will prematurely deploy and may be incorrectly positioned.
• For the limb stent graft overlap criteria with Endurant IIs ipsilateral leg only, please refer to Table 70. As noted in Table 70, for the
limb stent graft configurations that have an overlap criteria of 3 stents only, do not overlap more than 3 stents.
• When deploying the stent graft, be sure to hold the front grip of the delivery system stationary.
• If a balloon catheter is used, do not over-inflate or inflate outside the graft material. Follow all manufacturer instructions regarding
catheter operation.
• Expansion of the balloon outside of the graft material can result in rupture of the aorta, vessel dissection, or graft tears. When
expanding a vascular prosthesis, there is an increased risk of vessel injury or rupture, and possible patient death, if the balloon’s
proximal and distal radiopaque markers are not completely within the covered (graft fabric) portion of the prosthesis.
• High-pressure injections of contrast media made at the edges of the stent graft immediately after implantation can cause
endoleaks.
• For infrarenal EVAR procedures using the Heli-FX EndoAnchor system:
– Always use fluoroscopy for guidance, delivery, and observation of any Heli-FX system components within the vasculature.
– Medtronic recommends that the implantation of EndoAnchor implants be done after the aortic endograft has been placed and
any balloon remodeling of the infrarenal seal zone of the stent graft system has been completed. Exercise care in balloon
remodeling of the stent graft system to avoid moving the main body endograft from its intended implant location.
– EndoAnchor implant locations should be based upon a detailed examination of the preoperative CT imaging in cases involving
irregular or eccentric plaque in the intended sealing zone(s). The EndoAnchor implant should be implanted only into areas of
aortic tissue that are free of or have insignificant calcified plaque or thrombus, or where such pathology is diffuse and less than
2 mm in thickness. Attempting to place EndoAnchor implants into more severe plaque or thrombus may be associated with
implantation difficulty and suboptimal endograft fixation and/or sealing.
– The recommended number of EndoAnchor implants for a bifurcated endograft is based on native vessel diameter and is
independent of the amount of endograft oversizing.
– Stability of the stent graft in short (≥ 4 mm and < 10 mm) infrarenal necks is augmented by the EndoAnchor implants. Ensure
successful deployment of the recommended minimum number of EndoAnchor implants. Where the number of successfully
deployed EndoAnchor implants is below the minimum recommended, there may be greater risk of postoperative Type 1a
endoleak or migration.
13
Page 14

4.5 Treatment and follow-up
• Any endoleak left untreated during the implantation procedure must be carefully monitored after implantation.
• All patients with endovascular aneurysm repair should undergo periodic imaging to evaluate the stent graft, aneurysm size, and
occlusion of vessels in the treatment area. Significant aneurysm enlargement (>5 mm), the appearance of a new endoleak,
evidence of perigraft flow, change in aneurysm pulsatility, or migration resulting in an inadequate seal zone should prompt further
investigation and may indicate the need for additional intervention or surgical conversion.
• Additional treatment including endovascular treatment or surgical conversion should be strongly considered in the following
cases:
– aneurysm growth >5 mm, with or without endoleak, since last follow-up
– change in aneurysm pulsatility, with or without growth or endoleak
– persistent endoleak, with or without aneurysm growth
– stent graft migration resulting in an inadequate seal zone
– decrease in renal function due to renal artery occlusion (migration or poor placement)
• Following endovascular aneurysm repair (EVAR), spinal cord ischemia (SCI) may result in a rare complication of paraplegia or
paraparesis. Cerebrospinal fluid (CSF) drain is advised if spinal cord ischemia is suspected.
4.6 MRI safety information
Nonclinical testing has demonstrated that the Endurant II/IIs stent graft system is MR Conditional. It can be scanned safely in both
1.5 T and 3.0 T MR systems only, with using only the specific testing parameters (Section 10.6). Additional MRI safety information is
found in Section 10.6. For additional MRI safety information about the Heli-FX EndoAnchor system, refer to the instructions for use
provided with the system.
5 Adverse events
5.1 Observed adverse events
Major adverse events observed in the clinical study supporting approval of the bifurcated device are provided in Table 2, Table 3, and
Table 24 (Section 6.1.1 and Section 6.2.1.11). Major adverse events observed in the clinical study supporting approval of the AUI
device are provided in Table 6, Table 7, and Table 42 (Section 6.1.2 and Section 6.2.2.11). Major adverse events observed in the
clinical evidence supporting approval of the short infrarenal neck indication are provided in Table 58 (Section 6.3.6.1).
5.2 Potential adverse events
Adverse events that may occur or require intervention include, but are not limited to:
• Amputation
• Anesthetic complications and subsequent attendant problems (eg, aspiration)
• Aneurysm enlargement
• Aneurysm rupture and death
• Aortic damage, including perforation, dissection, bleeding, rupture, and death
• Arterial or venous thrombosis or pseudoaneurysm
• Arteriovenous fistula
• Bleeding, hematoma, or coagulopathy
• Bowel complications (for example, ileus, transient ischemia, infarction, necrosis)
• Cardiac complications and subsequent attendant problems (for example, arrhythmia, myocardial infarction, congestive heart
failure, hypotension, hypertension)
• Claudication (for example, buttock, lower limb)
• Death
• Edema
• Embolization (micro and macro) with transient or permanent ischemia or infarction
• EndoAnchor (for infrarenal EVAR procedures using the Heli-FX EndoAnchor system): partial implant deployment, inaccurate
implant deployment, implant fracture, implant dislodgement, implant embolization, stent graft damage, modeling balloon damage
• Endoleak
• Femoral-femoral artery bypass thrombosis
• Fever and localized inflammation
• Genitourinary complications and subsequent attendant problems (for example, ischemia, erosion, fistula, incontinence,
hematuria, infection)
• Hepatic failure
14
Page 15

• Impotence
• Infection of the aneurysm, device access site, including abscess formation, transient fever, and pain
• Lymphatic complications and subsequent attendant problems (eg, lymph fistula)
• Neurologic local or systemic complications and subsequent attendant problems (eg, confusion, stroke, transient ischemic attack,
paraplegia, paraparesis, paralysis)
• Occlusion of device or native vessel
• Pulmonary complications and subsequent attendant problems
• Renal complications and subsequent attendant problems (for example, artery occlusion, contrast toxicity, insufficiency, failure)
• Stent graft: improper placement; incomplete deployment; migration; suture break; occlusion; infection; stent fracture; graft
twisting or kinking; insertion and removal difficulties; graft material wear; dilatation; erosion; puncture and perigraft flow
• Surgical conversion to open repair
• Vascular access site complications, including infection, pain, hematoma, pseudoaneurysm, arteriovenous fistula, dissection
• Vascular spasm or vascular trauma (for example, iliofemoral vessel dissection, bleeding, rupture, death)
• Vessel damage
• Wound complications and subsequent attendant problems (eg, dehiscence, infection, hematoma, seroma, cellulitis)
5.3 Device-related adverse events reporting
Any adverse event or clinical incident involving the Endurant II/IIs stent graft system or Heli-FX EndoAnchor system should be
immediately reported to Medtronic. To report an incident in the US, call (800) 465-5533.
6 Summary of clinical studies
The clinical evidence supporting the indications and safety and effectiveness of the Endurant II and Endurant IIs stent graft system is
described below:
Study Objective
Endurant Stent Graft System US Clinical Study Demonstrate that Endurant Bifurcated and Aorto-Uni-Iliac (AUI)
stent graft systems are safe and effective for the endovascular
treatment of infrarenal abdominal aortic or aortoiliac aneurysms.
ENGAGE Post Approval Study (PAS) The objective of the Post Approval Study Evaluating the Long
Term Safety And Effectiveness of the Endurant Stent Graft System (ENGAGE PAS) was to evaluate safety and effectiveness of
Endurant at 5 years through freedom from Aneurysm-Related
Mortality (ARM).
ANCHOR Registry – Short Neck Cohort To evaluate the safety and effectiveness of the Endurant
II/Endurant IIs Stent Graft System used in conjunction with the
Heli-FX EndoAnchor System in the endovascular treatment of
subjects with abdominal aortic aneurysms having an infrarenal
neck length of ≥ 4 mm and < 10 mm.
The premarket Endurant stent graft system US Clinical Study (Endurant US IDE Study; Section 6.1) was conducted on the Endurant
stent graft system, which is the previous version of the currently marketed Endurant II stent graft system and Endurant IIs stent graft
system. The Endurant II stent graft system and Endurant IIs stent graft system were approved without the submission of additional
clinical study data after enrollment for both the Endurant bifurcated and AUI study arms were completed using the original Endurant
stent graft system devices.
As a condition of approval, Medtronic conducted a post-approval study called the ENGAGE Post-Approval Study (PAS) to evaluate
the long-term safety and effectiveness of the Endurant stent graft system (Section 6.2).
The Endurant II stent graft system is based on design modifications to the previously approved Endurant stent graft system.
Specifically, the following changes were made to the Endurant stent graft system as part of the Endurant II stent graft system:
1. Use of the 18 Fr aortic delivery system to deliver the 28 mm aortic stent grafts (previously delivered using a 20 Fr delivery system
for Endurant stent graft system)
2. Addition of longer contralateral limbs (156 mm and 199 mm) to the portfolio and thereby increasing the overall length of the iliac
delivery system
15
Page 16

3. Radiopaque (RO) marker changes:
a. addition of a button marker between the 3rd and 4th stent of the contralateral limb and iliac extension configurations
b. change to the material of the contralateral gate marker (from gold wire to coiled Platinum-Iridium (90%–10%))
c. change to the position of the contralateral stub button marker to the true lateral position
4. Increase in the hydrophilic coating length from 33.0 cm to 50.8 cm for the aortic and iliac delivery systems.
The design modifications for Endurant II were based on feedback from Endurant users. The primary design attributes of the Endurant
stent graft system were retained in the Endurant II stent graft system. The design changes were qualified through in vitro testing. Based
on the similarities of the Endurant II stent graft system to the Endurant stent graft system and the non-clinical testing, the clinical data
obtained on the Endurant stent graft system (Endurant US IDE Study and ENGAGE PAS) is applicable to the Endurant II stent graft
system as well.
The Endurant IIs stent graft system is based on the following design modifications made to the previously approved Endurant II stent
graft system:
1. Shorten the ipsilateral leg for consistent 103 mm of covered length.
2. Have a consistent ipsilateral and contralateral leg diameter of 14 mm.
3. Radiopaque (RO) marker addition on distal end of the ipsilateral leg.
4. Four distal stents are sewn internally in the ipsilateral leg.
5. Additional limb configuration is required to mate with ipsilateral leg.
Additionally, the Endurant II contralateral limb stent graft configuration was renamed the Endurant II limb stent graft configuration.
Medtronic utilized real-world evidence from the ANCHOR registry (Section 6.3) to establish the safety and effectiveness of Endurant
II/IIs stent graft system used in conjunction with Heli-FX in the treatment of abdominal aortic aneurysms (AAAs) with short infrarenal
necks (≥ 4 mm and < 10 mm).
6.1 Endurant stent graft system US clinical study
The Endurant stent graft system US Clinical Study (Endurant US IDE Study) was a controlled, prospective, two-arm (Bifurcated IDE
and AUI), non-randomized, multi-center clinical study to evaluate the safety and effectiveness of the Endurant stent graft system in the
treatment of infrarenal abdominal aortic and aortoiliac aneurysms. The Bifurcated IDE arm (Section 6.1.1) enrolled 150 subjects
across 26 sites in the United States. The AUI arm (Section 6.1.2) enrolled 44 subjects across 15 sites in the United States and Canada.
Subjects enrolled in the Bifurcated IDE and AUI arms were implanted with the original Endurant stent graft system.
The primary safety endpoint for both arms was the proportion of subjects who had no Major Adverse Event (MAE) within 30 days of
the index procedure, where the Bifurcated IDE arm was compared to the dataset of Talent AAA pivotal study (also referred as Talent
control group) and the AUI arm was compared to a performance goal (PG).
The primary effectiveness endpoint analysis for both arms was a composite defined as the proportion of subjects who have a
successful aneurysm treatment as evaluated at the time of the initial index procedure and at 12 months. The successful aneurysm
treatment endpoint included successful delivery and deployment of the stent graft, and freedom from: aneurysm diameter increase,
type I & III endoleaks, aneurysm rupture through 12 months post-implant, stent graft occlusion, conversion to surgery, and migration.
Follow up evaluations were conducted at 1 month, 6 months, 12 months, and annually thereafter for a total of 5 years from the index
procedure. The 1-year clinical data for the Bifurcated IDE arm is provided in Section 6.1.1 and for AUI arm in Section 6.1.2.
Subsequent annual follow-up through 5 years post index procedure for both Bifurcated IDE arm and AUI arm is presented in
Section 6.2 as part of the ENGAGE PAS cohort.
6.1.1 Bifurcated IDE arm
For the Bifurcated IDE arm, the mean subject age was 73.1 years and 40.7% (61/150) of subjects were above age 75. The mean age
and sex/gender distribution were similar between the Bifurcated IDE arm and Talent control group. Subjects were classified based on
the Society for Vascular Surgery (SVS) score. 29.3% of subjects were classified as SVS 3, 54.7% as SVS 2, 16.0% as SVS 1, and 0%
as SVS 0 for the Bifurcated IDE arm. The baseline SVS risk classifications were similar between the Bifurcated IDE arm and Talent
control group, with over 80% of subjects with SVS 2 or above in both study groups. In addition, the baseline medical histories were also
similar with high prevalence of hypertension, chronic obstructive pulmonary disease, and tobacco use in the past 10 years in both
study groups.
Baseline aneurysm and anatomical measurements were similar between the study groups. On average, the maximum aneurysm
diameter was 55.9 mm for Bifurcated IDE arm and 55.0 mm for the Talent control group; mean measurement of proximal neck diameter
was 23.5 mm for Bifurcated IDE arm and 25.3 mm for Talent control group; the mean diameter of right and left iliac neck were 14.2 mm
and 13.9 mm for Bifurcated IDE arm and 14.5 mm and 14.3 mm for Talent control group; proximal neck length was 31.0 mm for
Bifurcated IDE arm and 22.9 mm for Talent control group; infra-renal neck angulation was 35.2° for Bifurcated IDE arm and 30.5° for
Talent control group. The primary safety endpoint is an analysis of the MAEs within 30 days. 96% of subjects in the Bifurcated IDE arm
were MAE-free as compared to 89.2% of subjects in the Talent control group as shown in Table 2 and Table 3.
16
Page 17

Table 2. MAE free-rate within 30 days
MAE free rate within 30 days Bifurcated IDE arm (%m/n) Talent control group (%m/n)
MAEs free-rate within 30 days 96.0% (144/150) 89.2% (148/166)
Table 3. MAEs within 30 days
Major Adverse Event (MAE) within 30
days
a
Bifurcated IDE arm (%m/n) Talent control group (%m/n)
MAE at 30 days 4.0% (6/150) 10.8% (18/166)
All-cause death 0.0% (0/150) 1.8% (3/166)
Myocardial infarction 0.7% (1/150) 1.8% (3/166)
Renal failure 0.7% (1/150) 1.8% (3/166)
Respiratory failure 1.3% (2/150) 3.0% (5/166)
Paraplegia 0.0% (0/150) 0.0% (0/166)
Stroke 0.7% (1/150) 1.2% (2/166)
Bowel ischemia 1.3% (2/150) 0.6% (1/166)
Procedural blood loss ≥1000 cm
a
A subject may report multiple MAEs; hence, number of subjects with any MAE may not be the sum of those in each MAE category.
3
0.7% (1/150) 5.4% (9/166)
The primary effectiveness endpoint demonstrated 97.5% successful aneurysm treatment rate through 12 months for Bifurcated IDE
arm, while the Talent control group demonstrated a rate of 87.1% (Table 4). There were three (3) subjects in the Bifurcated IDE arm
that were considered treatment failures as described: During the index procedure, one subject in the Bifurcated IDE arm had the main
bifurcated body implanted but the physician was not able to cannulate the contralateral gate due to a pre-existing challenging
anatomy. The subject was ultimately converted to aorto-uni-iliac in-situ and a femoral to femoral bypass was performed. The second
subject experienced an aneurysm rupture at the index procedure. The third subject had a stent graft occlusion resulting in a femoral
to femoral bypass.
Table 4. Successful aneurysm treatment
Bifurcated IDE arm % (m/n)
Successful aneurysm treatment
a
Denominator is the number of subjects evaluable for this endpoint
b
Successful aneurysm treatment endpoint includes successful delivery and deployment, and no aneurysm growth, endoleaks, stent graft
occlusion, conversion to surgery, rupture, and migration.
b
97.5% (118/121) 87.1% (108/124)
a
Talent control group % (m/n)
a
During the index procedure, technical success was demonstrated by 99.3% successful delivery and deployment of the Endurant
bifurcated stent graft, compared to 97.6% of the Talent control group (Table 5).
Table 5. Technical success
Bifurcated IDE arm % (m/n) Talent control group % (m/n)
Technical success
a
Defined as the successful delivery and deployment of the stent graft.
a
99.3% (149/150) 97.6% (162/166)
6.1.2 AUI arm
For the AUI arm, the mean subject age was 73.8 years. 34.1% of subjects were classified as SVS 3, 50.0% as SVS 2, 15.9% as SVS
1, and 0% as SVS 0. Baseline medical histories demonstrated high prevalence of hyperlipidemia, hypertension and peripheral
vascular disease.
Baseline aneurysm and anatomical measurements included an average maximum aneurysm diameter was 52.3 mm; mean
measurement of proximal neck diameter was 22.3 mm; the mean diameter of ipsilateral and contralateral iliac diameter were both
11.5 mm; mean ipsilateral and contralateral iliac stenosis were 24.0 mm and 57.0 mm respectively; mean distal diameter of the aorta
or aneurysm above the aortic bifurcation was 22.7 mm; mean proximal neck length was 25.4 mm; mean infra-renal and suprarenal
angulations were 26.3° and 16.1° respectively. Details of the AUI arm demographic data are presented in Section 6.2.2.
The primary safety endpoint analysis demonstrated that 88.6% of subjects in the AUI arm were MAE-free within 30 days of index
procedure. Table 6 and Table 7 provide an analysis of the MAEs within 30 days.
Table 6. Freedom from Major Adverse Events within 30 days
Freedom from Major Adverse Events (MAE) within 30 days Endurant AUI % (m/n)
Freedom from MAEs within 30 days 88.6% (39/44)
17
Page 18

Table 7. Major Adverse Events within 30 days
Major Adverse Events (MAE) within 30 Days
a,b
Endurant AUI (% m/n)
MAEs within 30 days 11.4% (5/44)
All-cause mortality 2.3% (1/44)
Myocardial infarction 6.8% (3/44)
Renal failure 2.3% (1/44)
Respiratory failure 4.5% (2/44)
Paraplegia 0.0% (0/44)
Stroke 2.3% (1/44)
Bowel ischemia 0.0% (0/44)
Procedural blood loss ≥1000 cm
3
4.5% (2/44)
MAE w/o blood loss at 30 Days 11.4% (5/44)
a
A subject may report multiple MAEs; hence, number of subjects with any MAE may not be the sum of those in each MAE category.
b
The % MAE rate at 30 days is 100% - % Freedom from MAEs within 30 days.
The primary effectiveness endpoint was demonstrated by 99.7% successful delivery and deployment of the Endurant AUI stent graft.
Successful aneurysm treatment rate through 12 months in the Endurant AUI arm was 97.2%. One (1) subject in the AUI arm was
considered a treatment failure. This subject experienced a technical failure during the index procedure as the investigator had difficulty
recapturing the tip of the delivery system which engaged the suprarenal stents. While the investigator attempted to disengage the tip
from the suprarenal stent, the stent graft moved proximally, causing the left renal artery to be 50% obstructed by the stent graft fabric.
A left renal artery stent was placed to treat the unintentional coverage.
Table 8. Successful aneurysm treatment
Primary effectiveness endpoint Endurant AUI % (m/n)
Successful aneurysm treatment
a
Denominator is number of subjects evaluable for this endpoint.
b
Two-sided 95% confidence interval was based on a binomial distribution.
c
Successful aneurysm treatment endpoint includes successful delivery and deployment, and no aneurysm growth, endoleaks, stent graft
occlusion, conversion to surgery, rupture and migration.
c
97.2% (35/36) [ 85.5%, 99.9%]
a
95% Confidence interval
b
6.2 ENGAGE PAS cohort
The ENGAGE PAS was a multicenter, post-market, non-controlled, non-randomized, two-arm, prospective post-approval study
evaluating the long-term safety and effectiveness of the Endurant stent graft system. ENGAGE PAS cohort includes All Bifurcated
subjects arm and the AUI arm. The All Bifurcated subjects arm has a total of 328 subjects, of which 150 subjects were previously
enrolled in the Endurant US IDE study (Bifurcated IDE arm; refer Section 6.1.1 above) and 178 subjects were prospectively enrolled
as De Novo subjects in 24 participant sites in the US. The AUI arm consisted of 44 AUI subjects which were previously enrolled as a
separate arm of the Endurant US IDE study (refer Section 6.1.2 above). Clinical data was collected preoperatively to establish
eligibility at baseline, during implantation of the Endurant stent graft system, throughout the hospital stay, and postoperatively at 1
month, 12 months, and annually thereafter for a total of 5 years. In cases where follow-up data was not available for a specific visit,
subsequent follow-up data was used, e.g., when there is evidence of stent graft patency, endoleak, or freedom from migration at
12-months post index procedure. The clinical ethics committee (CEC) reviewed and adjudicated any data point pertaining to stent
graft occlusions, device migrations, type I and III endoleaks, and aneurysm sac enlargement that require further clarification. Events
were reviewed by CEC members and adjudicated as being device related, procedure related, and/or aneurysm related. ENGAGE
PAS primary endpoint was freedom from Aneurysm-Related Mortality Rate (ARM) at 5 years (1826 days), where ARM was defined
as death from rupture of the abdominal aortic aneurysm (AAA) or from any procedure intended to treat the AAA. For All Bifurcated
subjects, this primary endpoint was analyzed by comparing freedom from ARM rate observed in the study to a pre-defined
performance goal (PG) of 92%. For the AUI arm, this primary endpoint was analyzed descriptively by calculating the Kaplan Meir (KM)
estimate of 5-year freedom from ARM along with a 2-sided 95% confidence interval. In addition, technical success, MAE rates, and
other additional measures were collected at specific time intervals for both All Bifurcated subjects and AUI arm.
6.2.1 ENGAGE PAS - All bifurcated subjects
6.2.1.1 Subject accountability and follow-up
For All Bifurcated subjects (Bifurcated IDE arm + De Novo arm), 211 subjects of the total 328 subjects were eligible for 5-year
follow-up, of which 194 (92%) completed clinical followup within the visit window and 174 (82%) completed imaging follow-up.
Detailed subject accountability and follow-up are presented in Table 9.
18
Page 19

Table 9. Subject and imaging accountabilitya – All Bifurcated subjects
Subjects
with imaging
Interval
(analysis
window)
Originally
performed
(site repor-
Subject follow-up
Clini-
Imagcal
fol-
Eligi-
ble
low-
up
ing
fol-
low-
up
b
CT
imag-
ing
ted)
KUB
imag-
ing
328 1
Subjects with adequate
imaging to assess the
parameter (site reported) Subject events occurring before next visit
Aneu
rysm
size
incre
ase
Endo
leak
Migra
tion
enrolled
Events
after
implant
but before
a 1 month
visit
1 month
(day 1-90)
326 323
(99%)
322
(99%)
320
(98%)
276
(85%)
311
(95%)
Events
after 1
month
visit but
before a
12 month
visit
12 month
(day
312 295
(95%)
288
(92%)
285
(91%)
270
(87%)
283
(91%)
278
(89%)
(92%)
305-548)
Events
between
12-month
and
2-year
visit
2 year
(day
293 285
(97%)
263
(90%)
259
(88%)
243
(83%)
265
(90%)
266
(91%)
(88%)
549-913)
Events
between
2-year
and
3-year
visit
3 year
(day
269 236
(88%)
208
(77%)
204
(76%)
187
(70%)
217
(81%)
208
(77%)
(76%)
914-1278
)
286
257
205
c
Tech-
nical
obser
vat-
d
ion
322
(99%)
289
(93%)
271
(92%)
221
(82%)
No
impla
nt
Con-
ver-
sion
to
sur-
gery
Deat
With-
draw
al/ear
ly termina-
h
tion
0 2 0 0
0 12 2 0
0 12 6 1
0 11 13 0
Lost
to fol-
low-
up
Not
due
for
next
visit
0
0
0
0
19
Page 20

Table 9. Subject and imaging accountabilitya – All Bifurcated subjects (continued)
Subjects
with imaging
Interval
(analysis
window)
Events
Subject follow-up
Clini-
Imagcal
fol-
Eligi-
ble
low-
up
ing
fol-
low-
up
performed
(site repor-
CT
imag-
b
ing
ted)
KUB
imag-
ing
Subjects with adequate
imaging to assess the
parameter (site reported) Subject events occurring before next visit
Aneu
rysm
size
incre
ase
Endo
leak
Migra
tion
c
Tech-
nical
obser
vat-
d
ion
No
impla
nt
Con-
ver-
sion
to
sur-
gery
Deat
With-
draw
al/ear
ly termina-
h
tion
Lost
to fol-
low-
up
Not
due
for
next
visit
0 16 13 1 0
between
3-year
and
4-year
visit
4 year
(day
239 215
(90%)
192
(80%)
183
(77%)
173
(72%)
198
(83%)
189
(79%)
191
(80%)
201
(84%)
1279-164
4)
Events
0 18 6 4
0
between
4-year
and
5-year
visit
5 year
(day
211 194
(92%)
174
(82%)
164
(78%)
157
(78%)
173
(82%)
166
(79%)
167
(79%)
179
(85%)
1645-200
9)
a
Data analysis sample size varies for each of the time points above and in the following figures. This variability is due to subject availability for
follow-up, as well as, quantity and quality of images available from specific time points for evaluation. For example, the number and quality of
images available for evaluation of endoleak at 1 month is different than the number and quality of images available at 12 months due to variation
in the number of image exams performed and/or the number of images with acceptable evaluation quality.
b
Imaging Follow-up count includes CT with and without contrast, MRA with and without contrast, ultrasound and x-ray imaging.
c
Category has been expanded to include x-ray as an acceptable imaging method to evaluate migration.
d
Technical observations assessed by imaging include stent graft kinking, stent graft twisting, stent graft wireform fracture, suprarenal bare stent
fracture, anchoring pin fracture, stent graft stenosis, stent graft occlusion and other technical observations.
Note: The deaths of three (3) subjects are not counted above since two (2) occurred after the subjects’ 5-year cutoff dates, and one
(1) subject death is unknown as the site was unable to determine the exact date of death due to insufficient information on the event.
6.2.1.2 Subject demographics and baseline medical history
Baseline medical history for All Bifurcated subjects included high prevalence of hyperlipidemia, hypertension, and coronary artery
disease. Table 10 through Table 14 provide the subject demographics, baseline medical history, baseline aneurysm and anatomical
measurements, and SVS classification for All Bifurcated subjects.
Table 10. Subject demographics – All Bifurcated subjects
Parameter category/statistics All Bifurcated subjects
Age (years)
All
n 328
Mean ± SD 72.3 ± 8.1
Median 72.0
Min, Max 50, 88
Male
20
Page 21

Table 10. Subject demographics – All Bifurcated subjects (continued)
Parameter category/statistics All Bifurcated subjects
n 283
Mean ± SD 72.1 ± 8.1
Median 72.0
Min, Max 50, 87
Female
n 45
Mean ± SD 73.6 ± 8.0
Median 73.0
Min, Max 58, 88
Gender % (m/n)
Male 86.3% (283/328)
Race % (m/n)
White/Caucasian 96.6% (317/328)
Black, non-Hispanic 2.1% (7/328)
Hispanic/Mexican 0.0% (0/328)
Asian/Pacific Islander 0.9% (3/328)
Native American 0.0% (0/328)
Other/Unknown 0.3% (1/328)
Table 11. Baseline medical history - All Bifurcated subjects
All Bifurcated subjects %
Body System / Condition
(m/n)
a
Cardiac
Angina 16.3% (53/326)
Arrhythmia 33.6% (110/327)
Congestive heart failure 12.5% (41/328)
Coronary artery bypass graft 28.4% (93/328)
Coronary artery disease 54.3% (178/328)
Myocardial infarction 30.2% (98/325)
Percutaneous transluminal coronary angioplasty 23.8% (78/328)
Valvular heart disease (VHD) 9.8% (32/328)
Other cardiac 15.2% (50/328)
Pulmonary
Chronic obstructive pulmonary disease 35.2% (115/327)
Renal
Abnormal renal function 19.5% (64/328)
Renal insufficiency 11.6% (38/328)
Cerebrovascular/Neurological
Cerebral vascular accident 8.8% (29/328)
Paraparesis 0.9% (3/328)
Paraplegia 0.0% (0/328)
Transient ischemic attack (TIA) 5.2% (17/327)
Other Cerebrovascular/Neurological 9.8% (32/328)
Vascular
Abdominal aortic aneurysm 3.0% (10/328)
Any thoracic aneurysm 1.5% (5/328)
Peripheral vascular disease 24.7% (81/328)
Thromboembolic event 3.4% (11/328)
Other vascular 11.0% (36/328)
Other conditions
21
Page 22

Table 11. Baseline medical history - All Bifurcated subjects (continued)
All Bifurcated subjects %
Body System / Condition
(m/n)
a
Alcoholism 4.0% (13/328)
Bleeding disorder 0.9% (3/328)
Cancer 26.2% (86/328)
Carotid artery disease 19.6% (64/326)
Diabetes 22.9% (75/327)
Family history of aneurysm 15.9% (52/327)
GI complications 46.0% (151/328)
Hyperlipidemia 85.3% (278/326)
Hypertension 83.8% (275/328)
Liver disease 3.4% (11/328)
Tobacco use in the last 10 years 49.2% (161/327)
b
Other
a
Denominator reflects the number of subjects that selected “yes” or “no” to the corresponding condition. Subjects that selected “unknown” for
certain categories were not counted in the denominator.
b
”Other” was captured only for the Endurant IDE arm. As a result of this, the denominator reflects the total number of subjects enrolled in the
Endurant IDE arm.
87.3% (131/150)
Table 12. Baseline aneurysm and anatomical characteristics (site reported) – All Bifurcated subjects
Dimension All Bifurcated subjects
Maximum aneurysm
diameter (mm)
a
n
328
Mean ± SD 56.6 ± 8.2
Median 54.0
Min, Max 42, 98
Proximal neck diameter
(mm)
a
n
328
Mean ± SD 23.8 ± 3.2
Median 23.0
Min, Max 19, 40
Right iliac diameter
2
(mm)
a
n
327
Mean ± SD 14.1 ± 3.6
Median 13.0
Min, Max 8, 25
Left iliac diameter (mm)
2
a
n
328
Mean ± SD 13.8 ± 3.4
Median 13.0
Min, Max 8, 25
Proximal neck length
(mm)
a
n
328
Mean ± SD 27.0 ± 12.7
Median 24.0
Min, Max 5, 76
Infrarenal neck angle (°)
a
n
328
Mean ± SD 24.6 ± 15.8
Median 25.0
22
Page 23

Table 12. Baseline aneurysm and anatomical characteristics (site reported) – All Bifurcated subjects (continued)
Dimension All Bifurcated subjects
Min, Max 0, 60
Suprarenal neck angle (°)
a
n
327
Mean ± SD 14.1 ± 11.5
Median 10.0
Min, Max 0, 60
a
n = number of subjects with readable scans.
Note: It was noted that four (4) subjects had baseline anatomical characteristics outside the Endurant stent graft system Instructions
for Use. The measurements were entered into the database by investigational sites after the subjects were enrolled into the study.
Medtronic had taken steps to ensure that the investigational sites enrolled and treated subjects following the guidelines of the
Instructions for Use for the Endurant stent graft system. Medtronic conducted retraining to the sites on the Instructions for Use, and
investigational sites were notified of these protocol deviations and they reported them to their IRBs.
Table 13. Baseline maximum aneurysm diameter (site reported) – All Bifurcated subjects
Statistics/category All Bifurcated subjects
Maximum aneurysm
diameter % (m/n)
a
< 40 mm 0.0% (0/328)
40 mm - < 50 mm 5.5% (18/328)
50 mm - < 60 mm 72.9% (239/328)
60 mm - < 70 mm 13.4% (44/328)
70 mm - < 80 mm 5.5% (18/328)
80 mm - < 90 mm 1.8% (6/328)
90 mm - < 100 mm 0.9% (3/328)
≥ 100 mm 0.0% (0/328)
a
n = number of subjects with readable scans.
Table 14. SVS classification – All Bifurcated subjects
SVS classification All Bifurcated subjects % (m/n)
SVS 0 0.3% (1/328)
SVS 1 18.3% (60/328)
SVS 2 51.8% (170/328)
SVS 3 29.6% (97/328)
6.2.1.3 Devices implanted
Table 15 provides the number of Endurant stent graft devices implanted (which includes the bifurcated stent graft as well as aortic and
iliac extensions) at the index procedure per subject. Table 16 shows the distribution of sizes of the bifurcated stent graft used and
Table 17 provide the devices implanted by type.
Table 15. Total number of devices implanted at index procedure – All Bifurcated subjects
Number of devices implanted All Bifurcated subjects % (m/n)
a
1 0.3% (1/327)
2 38.8% (127/327)
3 32.7% (107/327)
4 25.7% (84/327)
5 2.1% (7/327)
6 0.3% (1/327)
≥ 7 0.0% (0/327)
a
Denominator includes all subjects who were implanted with at least one test device.
23
Page 24

Table 16. Devices implanted by size at index procedure – All Bifurcated subjects
Stent graft proximal diameter (main bifurcated, mm) All Bifurcated subjects % (m/n)
a
23 8.87% (29/327)
25 24.46% (80/327)
28 35.17% (115/327)
32 21.10% (69/327)
36 10.40% (34/327)
a
Denominator includes all subjects who received the main bifurcated test device.
Table 17. Devices implanted by type - All Bifurcated subjects
Device type All Bifurcated subjects % (m/n)
a
Main bifurcated 100.0% (327/327)
Contralateral limb 99.7% (326/327)
Extension - any type 31.2% (102/327)
Extension - iliac 28.4% (93/327)
Extension - aorta 3.1% (10/327)
a
Denominator includes all subjects who received the test device. A subject may receive multiple device types
6.2.1.4 Acute procedural data
Acute procedural data for All Bifurcated subjects is summarized in Table 18. The median procedure duration was 83 minutes and
84.8% (278/328) of the subjects received general anesthesia. The median blood loss reported for all subjects was 150 cm3.
Table 18. Acute procedural data – All Bifurcated subjects
Acute procedural data All Bifurcated subjects
Duration of procedure (min)
n 328
Mean ± SD 93.5 ± 45.2
Median 83.0
Min, Max 26, 318
Subjects receiving general anesthesia 84.8% (278/328)
Estimated blood loss (cm3)
n 327
Mean ± SD 170.4 ± 146.6
Median 150.0
Min, Max 0, 1450
Subjects requiring blood transfusion 1.2% (4/328)
Time in ICU (hour)
n 328
Mean ± SD 8.9 ± 24.0
Median 0.0
Min, Max 0, 216
Overall hospital stay (day)
n 328
Mean ± SD 2.2 ± 2.4
Median 1.0
Min, Max 1, 22
Note: Only available data were included in the calculation.
6.2.1.5 Technical success
During the index procedure, 99.1% of All Bifurcated subjects were recorded as having successful delivery and deployment of the
Endurant bifurcated stent graft as shown in Table 19. One (1) subject in the Bifurcated IDE arm and two (2) subjects in the De Novo
arm experienced a technical failure at the time of implant. The IDE subject experienced a deployment failure as there was difficulty
cannulating the gate due to challenging anatomy. There were no device malfunctions associated with the technical failure. This
subject was followed until expiration on day 721 post implant; the death was adjudicated as not related to the aneurysm, device, or
procedure. One (1) De Novo subject experienced unintentional coverage of both renal arteries that resulted in a right renal artery
24
Page 25

occlusion. This was treated with renal stenting and blood flow was restored to the right renal artery. Blood flow was restored to the left
renal artery using a pull-down technique to the stent graft. This subject has reported no additional clinical sequelae. The other De Novo
subject did not receive an Endurant device due to unsuitable anatomy at the time of the index procedure and was enrolled as intent
to treat (ITT) subject based on the protocol definition of enrollment.
Table 19. Technical success – All Bifurcated Subjects
Outcome All Bifurcated subjects % (m/n)
Technical success
a
Technical success of the Endurant stent graft system (assessed intra operatively) is successful delivery and deployment of the Endurant stent graft
system in the planned location and with no unintentional coverage of both internal iliac arteries or any visceral aortic branches and with the removal
of the delivery system.
a
99.1% (325/328)
6.2.1.6 Aneurysm-Related Mortality (ARM)
ARM is defined as a death of any cause within 30 days of the initial procedure or a death adjudicated as caused by an aneurysm
rupture. Additionally, if a death occurred within 30 days of any procedure intended to treat the AAA, then it is presumed to be aneurysm
related unless there is evidence to the contrary. For All Bifurcated subjects, the overall freedom from ARM at 5 years was 99.0% and
is presented in the Kaplan-Meier estimates in Table 20 and Figure 4.
There have been three (3) reports of ARM. One (1) De Novo subject died 4 days post-procedure due to a GI bleed. The other De Novo
subject died 6 days post-procedure due to cardiopulmonary failure. One (1) IDE subject died between 1097-1461 days
post-procedure due to aneurysm expansion due to a Type I endoleak. All three (3) deaths were adjudicated as aneurysm-related by
the CEC. The lower bound of 1-sided 95% confidence limit (98%) is higher than the Performance Goal of 92%. As a result, at a
one-sided significance level of 0.05, the pre-specified Performance Goal was met.
Table 20. Kaplan-Meier estimates for Freedom from Aneurysm-Related Mortality – All Bifurcated subjects
No. at risk
732-1096
0-30 days 31-365 days 366-731 days
a
328 326 310 292 267 234
days
1097-1461
days
1462-1826
days
No. of events 2 0 0 0 1 0
No. censored
Kaplan-Meier estimate
Standard error
b
c
c
0 16 18 25 32 73
0.994 0.994 0.994 0.994 0.990 0.990
0.004 0.004 0.004 0.004 0.006 0.006
95% CI lower limit 0.985 0.985 0.985 0.985 0.979 0.979
95% CI upper limit 1.000 1.000 1.000 1.000 1.000 1.000
a
Number of subjects at risk at the beginning of the interval.
b
Subjects are censored because their last follow-up has not reached the end of the time interval or because they withdraw, are lost to follow-up or
die from non aneurysm-related death.
c
Estimate along with its standard error using Greenwood method were made at end of time interval.
25
Page 26

Figure 4. Kaplan-Meier curve for freedom from Aneurysm-Related Mortality – All Bifurcated subjects
6.2.1.7 All-Cause mortality
There has been a total of 74 deaths reported for All Bifurcated subjects. Freedom from all-cause mortality through 5 years is 76.8%,
as presented in the Kaplan-Meier estimates below in Table 21 and Figure 5.
Table 21. Kaplan-Meier estimates for freedom from all-cause mortality – All Bifurcated subjects
No. at risk
732-1096
0-30 days 31-365 days 366-731 days
a
328 326 310 292 267 234
days
1097-1461
days
1462-1826
days
No. of events 2 12 12 12 19 13
No. censored
Kaplan-Meier estimate
Standard error
b
c
c
0 4 6 13 14 60
0.994 0.957 0.920 0.881 0.816 0.768
0.004 0.011 0.015 0.018 0.022 0.024
95% CI lower limit 0.985 0.935 0.890 0.845 0.773 0.720
95% CI upper limit 1.000 0.979 0.949 0.917 0.859 0.816
a
Number of subjects at risk at the beginning of the interval.
b
Subjects are censored because their last follow-up has not reached the end of the time interval or because they withdraw or are lost to follow-up.
c
Estimate along with its standard error using Greenwood method were made at end of time interval.
Note: Two (2) De Novo subjects are not included above as the date of death is unknown.
Note: One (1) De Novo subject and one (1) Bifurcated IDE subject are not included above as the date of death was after 5-year cutoff.
26
Page 27

Figure 5. Kaplan-Meier curve for freedom from all-cause mortality – All Bifurcated subjects
6.2.1.8 Aneurysm rupture
Table 22 presents rate of aneurysm rupture reported for All Bifurcated subjects. No aneurysm rupture was reported for De Novo
subjects while two (2) were reported for IDE subjects. One (1) subject experienced an intra-operative rupture, which was successfully
treated. The other subject experienced an aneurysm rupture on day 1212 post-index procedure, due to a Type I endoleak causing
aneurysm expansion. The subject refused intervention and subsequently died on day 1212 post-implant.
Table 22. Aneurysm ruptures – All Bifurcated subjects
Category
0-30 days %
(m/n)
a
31-365 days
% (m/n)
a
0-365 days
% (m/n)
a
366-731
days %
(m/n)
a
732-1096
days %
a
(m/n)
1097-1461 days
% (m/n)
a
1462-1826
days %
a
(m/n)
Aneurysm rupture 0.3% (1/328) 0.0% (0/314) 0.3% (1/314) 0.0% (0/301) 0.0% (0/279) 0.4% (1/251) 0.0% (0/220)
a
m = number of subjects in category, n = number of subjects who experienced the corresponding event during that interval or who were followed
at least until the lower endpoint of the analysis window without experiencing the corresponding event.
6.2.1.9 Conversion to open repair
There have been no conversions to open repair reported for All Bifurcated subjects.
6.2.1.10 Secondary endovascular procedures
Table 23 presents the rate of secondary endovascular procedures for All Bifurcated subjects. A total of 39 secondary endovascular
procedures have been performed on 31 subjects. Of the 39 reported secondary endovascular procedures, 21 were performed on De
Novo subjects and 18 were performed on IDE subjects. Reasons for secondary endovascular procedure included endoleaks (18),
migrations (2), occlusions or thrombosis (15), stent graft kinking (1), stenosis (1), ischemia (1), and aneurysm (1).
27
Page 28

Table 23. Secondary endovascular procedures – All Bifurcated subjects
366-731
0-30 days %
Category
Secondary endovas-
cular procedure
a
m = number of subjects in category, n = number of subjects who experienced the corresponding event during that interval or who were followed
at least until the lower endpoint of the analysis window without experiencing the corresponding event.
b
Subjects having multiple secondary procedures in the same timeframe will be counted only once therefore the number of secondary procedures
performed are higher than the number of subjects undergoing secondary procedures.
b
(m/n)
1.8% (6/328) 3.2%
31-365 days
a
% (m/n)
(10/314)
0-365 days
a
% (m/n)
5.1%
(16/314)
days %
a
(m/n)
1.0% (3/301) 1.8% (5/279) 1.2% (3/250) 3.6% (8/220)
a
732-1096
days %
a
(m/n)
1097-1461 days
% (m/n)
a
1462-1826
days %
a
(m/n)
6.2.1.11 Major adverse events
Table 24 summarizes major adverse events (MAEs) for All bifurcated subjects within 30 days post-procedure and annually thereafter.
Table 24. Major adverse events at time points – All Bifurcated subjects
Category
One or more major
adverse events
b
(MAE)
All-cause mortality
0-30 days %
(m/n)
6.1%
(20/328)
31-365 days
a
% (m/n)
6.8%
(22/325)
0.6% (2/328) 3.7%
(12/325)
0-365 days
a
% (m/n)
11.6%
(38/327)
4.3%
(14/327)
a
366-731
days %
(m/n)
4.2%
(13/308)
3.9%
(12/308)
a
732-1096
days %
a
(m/n)
5.6%
(16/284)
4.2%
(12/284)
1462-1826
1097-1461 days
% (m/n)
a
days %
(m/n)
8.9% (23/258) 8.8%
(20/226)
7.4% (19/258) 5.8%
(13/226)
a
Bowel ischemia 0.9% (3/328) 0.0% (0/325) 0.9% (3/327) 0.0% (0/308) 0.0% (0/284) 0.0% (0/258) 0.0% (0/226)
Myocardial infarc-
tion
2.1% (7/328) 1.2% (4/325) 3.4%
(11/327)
1.0% (3/308) 1.1% (3/284) 0.8% (2/258) 1.8% (4/226)
Paraplegia 0.0% (0/328) 0.0% (0/325) 0.0% (0/327) 0.0% (0/308) 0.0% (0/284) 0.0% (0/258) 0.0% (0/226)
Procedural blood
loss ≥ 1000 cm
0.6% (2/328) NA 0.6% (2/327) NA NA NA NA
3
Renal failure 1.2% (4/328) 1.5% (5/325) 2.4% (8/327) 0.6% (2/308) 0.7% (2/284) 0.8% (2/258) 1.3% (3/226)
Respiratory failure 2.4% (8/328) 0.9% (3/325) 3.1%
1.0% (3/308) 0.4% (1/284) 1.6% (4/258) 1.3% (3/226)
(10/327)
Stroke 0.6% (2/328) 1.5% (5/325) 1.8% (6/327) 0.3% (1/308) 0.7% (2/284) 1.2% (3/258) 1.3% (3/226)
a
m = number of subjects in category, n = number of subjects who experienced the corresponding event during that interval or who were followed
at least until the lower endpoint of the analysis window without experiencing the corresponding event. For example, for column “0-30 days”,
“31-365”, “0-365”, “366-731”, “732-1096”, “1097-1461”, “1462-1826”, a subject has to be followed respectively for at least 0 day, 305 days, 305
days, 548 days, 914 days, 1279 days and 1645 days in order to be included in the denominator if he/she does not experience the event in the
corresponding time interval. For MAE subcategories, the same denominator would be used as that for “One or more major adverse events (MAE)”
to allow comparison between subcategories within the MAE.
b
The CEC adjudicated MAEs through 30 days for the Engage De Novo arm and through 1 year for the Endurant IDE arm. Once the CEC no longer
adjudicated MAEs, then the MAEs are site reported.
Note: A subject having an event at different time points is counted more than once.
6.2.1.12 Endoleaks
Details regarding the site reported endoleaks for All Bifurcated subjects are presented in Table 25. Type I endoleaks were observed
on 8 subjects. Eight (8) subjects underwent a secondary endovascular procedure to treat a Type I endoleak with the intervention
resolving the Type I endoleak in seven (7) of the subjects. One (1) subject died following the secondary endovascular procedure due
to aneurysm enlargement and refused another secondary procedure. There were no Type III, IV, or V endoleaks. Type II endoleaks
were seen in 62 subjects and accounted for a majority of endoleaks reported. Type II endoleaks are dependent on subject anatomy
and are not indicative of device failure.
28
Page 29

Table 25. All Endoleaks by visit – All Bifurcated subjects (site reported)
Endoleaks
1 month (1-90
days) %
a
(m/n)
12 month
(305-548
days) %
(m/n)a,
b
2 year
(549-913
days) %
a
(m/n)
3 year
(914-1278
days) %
a
(m/n)
4 year
(1279-1644
days) %
a
(m/n)
5 year (1645-2009
days) % (m/n)
a
Type I 0.3% (1/311) 0.7% (2/279) 0.8% (2/266) 0.5% (1/208) 1.6% (3/189) 0.0% (0/166)
Type II 11.3%
(35/311)
9.3% (26/279) 9.4% (25/266) 10.1%
(21/208)
11.6%
(22/189)
9.6% (16/166)
Type III 0.0% (0/311) 0.0% (0/279) 0.0% (0/266) 0.0% (0/208) 0.0% (0/189) 0.0% (0/166)
Type IV 0.0% (0/311) 0.0% (0/279) 0.0% (0/266) 0.0% (0/208) 0.0% (0/189) 0.0% (0/166)
Type V 0.0% (0/311) 0.0% (0/279) 0.0% (0/266) 0.0% (0/208) 0.0% (0/189) 0.0% (0/166)
Indeterminate 0.3% (1/311) 0.0% (0/279) 0.8% (2/266) 0.0% (0/208) 0.0% (0/189) 0.6% (1/166)
Subjects had endoleaks
of any type
a
Denominator is the number of subjects who had readable images at the time of assessment. If there are two or more assessments in the same time
window, then the assessment closest to the target day will be used in the analysis.
b
Events that happened between 91-304 days will be counted in the column of 1 year.
c
A subject may have more than one type of endoleak; hence, number of subjects who had endoleaks of any type may not be the sum of those in
each type.
c
11.6%
(36/311)
9.3% (26/279) 10.2%
(27/266)
10.6%
(22/208)
12.7%
(24/189)
10.2% (17/166)
6.2.1.13 Aneurysm expansion
Aneurysm expansion is defined as a greater than 5 mm increase in maximum aneurysm diameter as measured on CT/MRI as
compared to the 1-month follow-up scan. A subject experiencing growth in any time point will continue to be counted in subsequent
time points if aneurysm expansion of more than 5 mm is reported at that time point. Table 26 shows aneurysm expansion as reported
by the sites. A total of 14 subjects have had aneurysm expansion of > 5 mm compared to the 1-month follow-up imaging. The majority
of subjects reported stable or decreased aneurysm diameter through 5 years.
Table 26. Aneurysm expansion – change in aneurysm diameter during follow-up - All Bifurcated subjects (site reported)
Changea in aneurysm diam-
eter from baseline (mm)
1 year (305-548
days) % (m/n)
2 year (549-913
b
days) % (m/n)
3 year (914-1278
b
days) % (m/n)
4 year
(1279-1644
b
days) % (m/n)
b
5 year
(1645-2009
days) % (m/n)
b
Increase > 5 mm 0.4% (1/283) 1.9% (5/265) 2.8% (6/217) 2.5% (5/198) 5.8% (10/173)
c
Stable
50.2% (142/283) 35.8% (95/265) 31.8% (69/217) 34.8% (69/198) 28.3% (49/173)
Decrease greater than 5 mm 49.5% (140/283) 62.3% (165/265) 65.4% (142/217) 62.6% (124/198) 65.9% (114/173)
a
Change in aneurysm diameter is based on 1-month imaging. When 1-month imaging was not available, the pre-discharge imaging was used as
the baseline.
b
m = number of subjects in category, n = number of subjects with known value of AAA diameter at both 1-month visit (or pre-discharge) and the
corresponding follow-up visit.
c
"Stable" refers to no change (increase or decrease) of more than 5 mm.
6.2.1.14 Stent graft migration
Stent graft migration is defined as evidence of movement of the stent graft relative to fixed anatomic landmarks, which is not due to
remodeling of the subject’s vasculature. Proximal migration is observed when the stent graft covers a renal artery or movement is >
10 mm. Distal migration is observed when the stent graft moves > 10 mm relative to fixed anatomic landmarks. Two (2) De Novo
subjects have been reported with proximal stent graft limb migration of > 10 mm. One (1) subject experienced proximal left iliac limb
migration 1499 days post-procedure and one (1) subject experienced proximal right iliac limb migration 1479 days post-procedure.
Both subjects underwent a secondary endovascular procedure to perform placement of an extension; both events were resolved at
the conclusion of the procedure. There were no reports of stent graft migration > 10 mm for any IDE subjects.
6.2.1.15 Technical observations
Table 27 summarizes the site reported technical observations for All Bifurcated subjects, with 67 events observed in 53 subjects. A
total of 20 secondary endovascular procedures were performed on 16 subjects to treat technical observations. Of those secondary
procedures, eleven (11) were performed to treat stent graft occlusion, three (3) were performed to treat thrombosis, two (2) were
performed to treat migration, one (1) was performed to treat aneurysm expansion, one (1) was performed to treat ischemia and
embolus, one (1) was performed to treat stenosis, and one (1) was performed to treat kinking. Nineteen (19) of the secondary
endovascular procedures performed resulted in resolution of the technical observation while one (1) resulted in an additional
secondary intervention to resolve the event.
29
Page 30

Table 27. Technical observations at follow-up – All Bifurcated subjects (site reported)
Category
1 month (1-90
days) %
a
(m/n)
1 year
(305-548
days) %
(m/n) a,
b
2 Year
(549-913
days) %
a
(m/n)
3 year
(914-1278
days) %
a
(m/n)
4 year
(1279-1644
days) %
a
(m/n)
5 year (1645-2009
days) % (m/n)
a
Anchoring pin fracture 0.0% (0/314) 0.0% (0/275) 0.0% (0/252) 0.0% (0/201) 0.0% (0/184) 0.0% (0/163)
Stent graft wireform frac-
0.0% (0/314) 0.4% (1/279) 0.0% (0/250) 0.0% (0/200) 0.0% (0/184) 0.0% (0/164)
ture
Suprarenal bare stent
0.0% (0/314) 0.0% (0/279) 0.0% (0/252) 0.0% (0/201) 0.0% (0/185) 0.0% (0/164)
fracture
c
Other
3.1% (10/319) 1.4% (4/278) 0.4% (1/258) 0.5% (1/210) 1.0% (2/191) 0.6% (1/169)
Stent graft kinking 1.6% (5/319) 1.4% (4/276) 0.8% (2/249) 0.5% (1/195) 0.6% (1/179) 0.0% (0/160)
Stent graft twisting 0.0% (0/320) 0.0% (0/279) 0.0% (0/255) 0.0% (0/205) 0.0% (0/189) 0.0% (0/169)
Stent graft stenosis 1.3% (4/315) 1.1% (3/280) 0.8% (2/255) 0.0% (0/198) 0.0% (0/179) 0.0% (0/164)
Stent graft occlusion
a
Denominator is the number of subjects evaluable for the corresponding events at each time interval, excluding subjects with events in any of the
previous time periods. For each corresponding event, each subject was counted only once. However, a subject who experienced one event can
still be counted for another different event as well as under any Technical Observations in a different time period.
b
Events that happened between 91-304 days will be counted in the column of 1 year.
c
The “other” events category includes, but is not limited to air in sac, thrombus in left iliac artery, and air noted within the stent.
d
Stent graft occlusion refers to loss of patency of the stent graft.
d
2.5% (8/316) 0.0% (0/275) 0.4% (1/254) 0.0% (0/201) 0.5% (1/183) 0.0% (0/164)
6.2.2 ENGAGE PAS - AUI Arm
6.2.2.1 Subject accountability and follow-up
For the AUI arm, 29 subjects out of the total 44 subjects were eligible for 5-year follow-up. Of the eligible subjects, 27 (93%) completed
clinical follow-up within the visit window and 20 (69%) completed imaging follow-up. Detailed subject accountability and follow-up are
presented in Table 28.
Table 28. Subject and imaging accountabilitya –AUI Arm
Subjects
with imaging
Interval
(analysis
window)
Originally
performed
(site repor-
Subject follow-up
Clini-
Imagcal
fol-
Eligi-
ble
low-
up
ing
fol-
low-
up
b
CT
imag-
ing
ted)
KUB
imag-
ing
44 0
Subjects with adequate
imaging to assess the
parameter (site reported) Subject events occurring before next visit
Aneu
rysm
size
incre
ase
enrolled
Events
after
implant
but before
a 1 month
visit
1 month
(day 1-90)
43 43
(100
%)
43
(100
%)
43
(100
%)
40
(93%)
Events
after 1
month
visit but
before a
12 month
visit
Endo
leak
43
(100
%)
Migra
tion
c
Tech-
nical
obser
vat-
d
ion
43
(100
%)
No
impla
nt
Con-
ver-
sion
to
sur-
gery
Deat
With-
draw
al/ear
ly termina-
h
tion
0 1 0 0
0 5 0 0
Lost
to fol-
low-
up
Not
Due
for
Next
Visit
0
0
30
Page 31

Table 28. Subject and imaging accountabilitya –AUI Arm (continued)
Subjects
with imaging
Interval
(analysis
window)
12 month
(day
305-548)
Events
Subject follow-up
Clini-
Imagcal
fol-
Eligi-
ble
low-
up
38 38
(100
(89%)34(89%)32(84%)36(95%)36(95%)35(92%)36(95%)
%)
ing
fol-
low-
up
34
performed
(site repor-
CT
imag-
b
ing
ted)
KUB
imag-
ing
Subjects with adequate
imaging to assess the
parameter (site reported) Subject events occurring before next visit
Aneu
rysm
size
incre
ase
Endo
leak
Migra
tion
c
Tech-
nical
obser
vat-
d
ion
No
impla
nt
Con-
ver-
sion
to
sur-
Deat
gery
0 1 0 0
With-
draw
al/ear
ly termina-
h
tion
Lost
to fol-
low-
up
Not
Due
for
Next
Visit
0
between
12-month
and
2-year
visit
2 year
(day
37 36
(97%)34(92%)34(92%)32(86%)33(89%)33(89%)33(89%)35(95%)
549-913)
Events
0 2 1 0
0
between
2-year
and
3-year
visit
3 year
(day
34 33
(97%)29(85%)29(85%)28(82%)29(85%)28(82%)28(82%)30(88%)
914-1278
)
Events
0 2 0 0
0
between
3-year
and
4-year
visit
4 year
(day
32 30
(94%)27(84%)27(84%)22(69%)27(84%)26(81%)25(78%)28(88%)
1279-164
4)
Events
0 3 0 0
0
between
4-year
and
5-year
visit
5 year
(day
29 27
(93%)20(69%)20(69%)13(45%)24(83%)20(69%)16(55%)24(83%)
1645-200
9)
a
Data analysis sample size varies for each of the time points above and in the following figures. This variability is due to subject availability for
follow-up, as well as, quantity and quality of images available from specific time points for evaluation. For example, the number and quality of
images available for evaluation of endoleak at 1 month is different than the number and quality of images available at 12 months due to variation
in the number of image exams performed and/or the number of images with acceptable evaluation quality.
b
Imaging Follow-up count includes CT with and without contrast, MRA with and without contrast, ultrasound and x-ray imaging.
31
Page 32

c
Category has been expanded to include x-ray as an acceptable imaging method to evaluate migration.
d
Technical observations assessed by imaging include stent graft kinking, stent graft twisting, stent graft wireform fracture, suprarenal bare stent
fracture, anchoring pin fracture, stent graft stenosis, stent graft occlusion, and other technical observations.
6.2.2.2 Subject demographics and baseline medical history
Baseline medical history included high prevalence of hyperlipidemia, hypertension, and peripheral vascular disease. Table 29
through Table 33 provide the subject demographics, baseline medical history, baseline aneurysm and anatomical measurements,
and SVS classification for the AUI arm.
Table 29. Subject demographics – AUI Arm
Parameter category/statistics AUI Arm
Age (years)
All
n 44
Mean ± SD 73.8 ± 7.6
Median 73.5
Min, Max 59, 89
Male
n 30
Mean ± SD 73.5 ± 7.5
Median 74.0
Min, Max 59, 89
Female
n 14
Mean ± SD 74.5 ± 8.0
Median 72.0
Min, Max 65, 87
Gender % (m/n)
Male 68.2% (30/44)
Race % (m/n)
White/Caucasian 95.5% (42/44)
Black, non-Hispanic 2.3% (1/44)
Hispanic/Mexican 0.0% (0/44)
Asian/Pacific Islander 0.0% (0/44)
Native American 0.0% (0/44)
Other/Unknown 2.3% (1/44)
Table 30. Baseline medical history -- AUI Arm
Body system / condition AUI Arm
Cardiac
Angina 25.0% (11/44)
Arrhythmia 34.1% (15/44)
Congestive heart failure 15.9% (7/44)
Coronary artery bypass graft 27.3% (12/44)
Coronary artery disease 59.1% (26/44)
Myocardial infarction 31.8% (14/44)
Percutaneous transluminal coronary angioplasty 29.5% (13/44)
Valvular heart disease (VHD) 31.8% (14/44)
Other cardiac 29.5% (13/44)
Pulmonary
Chronic obstructive pulmonary disease 59.1% (26/44)
Renal
Abnormal renal function 31.8% (14/44)
Renal insufficiency 15.9% (7/44)
32
Page 33

Table 30. Baseline medical history -- AUI Arm (continued)
Body system / condition AUI Arm
Cerebrovascular/Neurological
Cerebral vascular accident 9.1% (4/44)
Paraparesis 0.0% (0/44)
Paraplegia 0.0% (0/44)
Transient ischemic attack (TIA) 6.8% (3/44)
Other cerebrovascular/neurological 25.0% (11/44)
Vascular
Abdominal aortic aneurysm 0.0% (0/44)
Any thoracic aneurysm 0.0% (0/44)
Peripheral vascular disease 75.0% (33/44)
Thromboembolic event 4.5% (2/44)
Other vascular 50.0% (22/44)
Other conditions
Alcoholism 15.9% (7/44)
Bleeding disorder 0.0% (0/44)
Cancer 18.2% (8/44)
Carotid artery disease 40.9% (18/44)
Diabetes 25.0% (11/44)
Family history of aneurysm 11.4% (5/44)
GI complications 72.7% (32/44)
Hyperlipidemia 88.6% (39/44)
Hypertension 86.4% (38/44)
Liver disease 6.8% (3/44)
Tobacco use in the last 10 years 65.9% (29/44)
Other 90.9% (40/44)
Table 31. Baseline aneurysm and anatomical characteristics (site reported) – AUI Arm
Dimension AUI Arm
Maximum aneurysm
diameter (mm)
a
n
44
Mean ± SD 52.3 ± 9.0
Median 52.5
Min, Max 37, 78
Proximal neck diameter
(mm)
a
n
44
Mean ± SD 22.3 ± 2.7
Median 23.0
Min, Max 19, 30
Ipsilateral iliac diameter
(mm)
a
n
44
Mean ± SD 11.5 ± 3.2
Median 10.0
Min, Max 8, 23
Contralateral iliac diameter (mm)
a
n
26
Mean ± SD 11.5 ± 2.6
Median 11.0
33
Page 34

Table 31. Baseline aneurysm and anatomical characteristics (site reported) – AUI Arm (continued)
Dimension AUI Arm
Min, Max 8, 21
Ipsilateral iliac stenosis
(%)
a
n
44
Mean ± SD 24.0 ± 26.0
Median 17.5
Min, Max 0, 100
Contralateral iliac stenosis (%)
a
n
44
Mean ± SD 57.0 ± 40.1
Median 60.0
Min, Max 0, 100
Distal diameter of the
aorta or aneurysm above
the aortic bifurcation
(mm)
a
n
44
Mean ± SD 22.7 ± 9.1
Median 20.5
Min, Max 10, 48
Proximal neck length
(mm)
a
n
44
Mean ± SD 25.4 ± 11.9
Median 24.0
Min, Max 10, 58
Infrarenal angle (°)
a
n
44
Mean ± SD 26.3 ± 16.2
Median 24.5
Min, Max 0, 60
Suprarenal angle (°)
a
n
44
Mean ± SD 16.1 ± 12.7
Median 12.5
Min, Max 0, 42
a
n = number of subjects with readable scans.
Table 32. Distribution of maximum aneurysm diameters (site reported) – AUI Arm
Statistics/category AUI Arm
Maximum aneurysm diameter % (m/n)
< 30 mm 0.0% (0/44)
30 mm - < 40 mm 9.1% (4/44)
40 mm - < 50 mm 25.0% (11/44)
50 mm - < 60 mm 47.7% (21/44)
60 mm - < 70 mm 15.9% (7/44)
70 mm - < 80 mm 2.3% (1/44)
≥ 80 mm 0.0% (0/44)
34
Page 35

Table 33. Baseline SVS classification - AUI Arm
SVS classification AUI Arm % (m/n)
SVS 0 0.0% (0/44)
SVS 1 15.9% (7/44)
SVS 2 50.0% (22/44)
SVS 3 34.1% (15/44)
6.2.2.3 Devices implanted
Table 34 provides the number of Endurant stent graft devices implanted (which includes the AUI stent graft as well as aortic and iliac
extensions) at the index procedure per subject and Table 35 provides the number of devices implanted by type. Table 36 provides the
number of devices implanted by size.
Table 34. Number of devices implanted at Index procedure - AUI Arm
Number of devices implanted AUI Arm (% m/n)
a
1 2.3% (1/44)
2 56.8% (25/44)
3 36.4% (16/44)
4 4.5% (2/44)
≥ 5 0.0% (0/44)
a
Denominator includes all subjects who were implanted with at least one test device.
Table 35. Devices implanted by type at Index procedure – AUI Arm
Device type AUI Arm % (m/n)
a
Main AUI 100.0% (44/44)
Iliac extension
b
97.7% (43/44)
Extension - any type 6.8% (3/44)
Extension - iliac
b
2.3% (1/44)
Extension - aorta 4.5% (2/44)
Talent Occluder 22.7% (10/44)
a
Denominator includes all subjects who received the test device. A subject may receive multiple device types
b
’Iliac Extension’ refers to the Endurant Contralateral Iliac Limb component as the Endurant AUI stent graft device does not have a contralateral iliac
limb. ’Extension - iliac’ refers to the Endurant Iliac Extension component.
Table 36. Devices implanted by size – AUI Arm
Stent graft proximal diameter (mm) AUI Arm (% m/n)
a
23 18.2% (8/44)
25 31.8% (14/44)
28 29.5% (13/44)
32 15.9% (7/44)
36 4.5% (2/44)
a
Denominator includes all subjects who were implanted with at least one (1) test device.
6.2.2.4 Acute procedural data
Acute procedural data for the AUI arm is summarized in Table 37. The median procedure duration was 91.5 minutes and 79.5% (35/44)
of the subjects received general anesthesia. The median blood loss reported for all subjects was 200 cm3.
Table 37. Acute procedural data – AUI Arm
Acute procedural data AUI Arm
Duration of procedure (min)
n 44
Mean ± SD 117.7 ± 66.6
Median 91.5
Min, Max 33, 295
Subjects receiving general anesthesia 79.5% (35/44)
35
Page 36

Table 37. Acute procedural data – AUI Arm (continued)
Acute procedural data AUI Arm
Estimated blood loss (cm3)
n 44
Mean ± SD 264.2 ± 221.2
Median 200.0
Min, Max 5, 1200
Subjects requiring blood transfusion 6.8% (3/44)
Time in ICU (hour)
n 44
Mean ± SD 16.6 ± 39.3
Median 0.0
Min, Max 0, 186
Overall hospital stay (day)
n 44
Mean ± SD 3.2 ± 2.7
Median 2.5
Min, Max 1, 13
6.2.2.5 Technical success
For the AUI arm, technical success was achieved in 97.7% (43/44) subjects, as shown in Table 38. One (1) technical failure was
reported. This subject experienced a technical failure during the index procedure as the investigator had difficulty recapturing the tip
of the delivery system which engaged the suprarenal stents. While the investigator attempted to disengage the tip from the suprarenal
stent, the stent graft moved proximally, causing the left renal artery to be 50% obstructed by the stent graft fabric. A stent was placed
in the left renal artery to treat the unintentional coverage.
Table 38. Technical success – AUI Arm
Outcome AUI Arm % (m/n)
Technical success
a
Technical success of the Endurant stent graft system (assessed intra-operatively) is successful delivery and deployment of the Endurant stent graft
system in the planned location and with no unintentional coverage of both internal iliac arteries or any visceral aortic branches and with the removal
of the delivery system.
a
97.7% (43/44)
6.2.2.6 Aneurysm-Related Mortality (ARM)
The overall freedom from ARM at 5 years for the AUI arm is 97.7% as presented in the Kaplan-Meier estimates in Table 39 and
Figure 6. There has been one (1) report of ARM. The subject died on Day 1 post procedure. The autopsy report noted “severe
atherosclerosis with unremarkable aneurysm repair and fem-fem bypass” and “no signs of thrombus or external hemorrhage in the
endovascular stent graft or bypass.” The CEC adjudicated as not related to the aneurysm or the device but related to the procedure.
However, the death is counted as aneurysm-related in this table per definition of ARM, which is defined as a death of any cause within
30 days of the initial procedure.
Table 39. Kaplan-Meier estimates for Freedom from Aneurysm-Related Mortality – AUI Arm
No. at risk
732-1096
0-30 days 31-365 days 366-731 days
a
44 43 38 37 34 32
days
1097-1461
days
1462-1826
days
No. of events 1 0 0 0 0 0
No. censored
Kaplan-Meier estimate
Standard error
b
c
c
0 5 1 3 2 13
0.977 0.977 0.977 0.977 0.977 0.977
0.022 0.022 0.022 0.022 0.022 0.022
95% CI lower limit 0.933 0.933 0.933 0.933 0.933 0.933
95% CI upper limit 1.000 1.000 1.000 1.000 1.000 1.000
a
Number of subjects at risk at the beginning of the interval.
b
Subjects are censored because their last follow-up has not reached the end of the time interval or because they withdraw, are lost to follow-up or
die.
c
Estimate along with its standard error using Greenwood method were made at end of time interval.
36
Page 37

Note: CEC adjudication result on the one Endurant AUI Subject death is not device-related, not aneurysm-related, but
procedure-related based on the autopsy results available to the CEC. However, the death is counted as aneurysm related above per
definition of ARM.
Figure 6. Kaplan-Meier curve for Freedom from Aneurysm-Related Mortality – AUI Arm
6.2.2.7 All-cause mortality
For the AUI arm, there have been a total of 14 deaths reported, ranging from 1 day to 1791 days post-index procedure. Freedom from
all-cause mortality through 5 years is 67.6%, as presented in the Kaplan-Meier estimates in Table 40 and Figure 7.
Table 40. Kaplan – Meier Estimates for Freedom from All-Cause Mortality – AUI Arm
No. at risk
732-1096
0-30 days 31-365 days 366-731 days
a
44 43 38 37 34 32
days
1097-1461
days
1462-1826
days
No. of events 1 5 1 2 2 3
No. censored
Kaplan-Meier estimate
Standard error
b
c
c
0 0 0 1 0 10
0.977 0.864 0.841 0.795 0.748 0.676
0.022 0.052 0.055 0.061 0.066 0.072
95% CI lower limit 0.933 0.762 0.733 0.675 0.619 0.535
95% CI upper limit 1.000 0.965 0.949 0.914 0.877 0.816
a
Number of subjects at risk at the beginning of the interval.
b
Subjects are censored because their last follow-up has not reached the end of the time interval or because they withdraw or are lost to follow-up.
c
Estimate along with its standard error using Greenwood method were made at end of time interval.
37
Page 38

Figure 7. Kaplan-Meier curve for Freedom from All-Cause Mortality – AUI Arm
6.2.2.8 Aneurysm rupture
No aneurysm ruptures have been reported for subjects enrolled in the AUI arm.
6.2.2.9 Conversion to open surgery
No conversion to open surgery has been reported for subjects enrolled in the AUI arm.
6.2.2.10 Secondary endovascular procedures
A total of two (2) secondary endovascular procedures were reported for the AUI arm as shown in Table 41. On day 182 post-procedure,
one (1) subject experienced stenosis of the stent graft and was taken to the operating room for placement of a covered stent graft to
treat the stent graft stenosis. Patency was restored to the left lower extremity and into the left to right femoral-femoral bypass graft as
a result. On day 1672 post-procedure, one (1) subject experienced an arterial occlusion and emergency thrombectomy of the aorta,
right iliac, and AUI device with a right common iliac stent graft placement. The subject recovered from the procedure but died four (4)
months later of lung cancer.
Table 41. Secondary endovascular procedures – AUI Arm
Category
Secondary endovas-
366-731
0-30 days %
(m/n)
31-365 days
a
% (m/n)
0-365 days
a
% (m/n)
days %
a
(m/n)
0.0% (0/44) 2.6% (1/39) 2.6% (1/39) 0.0% (0/37) 0.0% (0/35) 0.0% (0/33) 3.2% (1/31)
a
732-1096
days %
a
(m/n)
1097-1461 days
% (m/n)
a
1462-1826
days %
a
(m/n)
cular procedure
a
m = number of subjects in category, n = number of subjects who experienced the corresponding event during that interval or who were followed
at least until the lower endpoint of the analysis window without experiencing the corresponding event.
6.2.2.11 Major adverse events
Table 42 summarizes major adverse events (MAEs) for all subjects in the AUI arm within 30 days post-procedure and annually
thereafter.
38
Page 39

Table 42. Major adverse events at time points – AUI Arm
Category
One or more major
adverse events
b
(MAE)
All-cause mortal-
366-731
0-30 days %
(m/n)
11.4% (5/44) 18.6% (8/43) 27.3%
31-365 days
a
% (m/n)
0-365 days
a
% (m/n)
days %
a
(m/n)
18.4% (7/38) 11.1% (4/36) 8.8% (3/34) 12.5% (4/32)
(12/44)
2.3% (1/44) 11.6% (5/43) 13.6% (6/44) 2.6% (1/38) 5.6% (2/36) 5.9% (2/34) 9.4% (3/32)
a
732-1096
days %
a
(m/n)
1097-1461 days
% (m/n)
a
1462-1826
days %
a
(m/n)
ity
Bowel ischemia 0.0% (0/44) 2.3% (1/43) 2.3% (1/44) 0.0% (0/38) 0.0% (0/36) 0.0% (0/34) 0.0% (0/32)
Myocardial infarc-
6.8% (3/44) 0.0% (0/43) 6.8% (3/44) 10.5% (4/38) 0.0% (0/36) 2.9% (1/34) 3.1% (1/32)
tion
Paraplegia 0.0% (0/44) 0.0% (0/43) 0.0% (0/44) 0.0% (0/38) 0.0% (0/36) 0.0% (0/34) 0.0% (0/32)
Procedural blood
loss ≥ 1000 cm
4.5% (2/44) NA 4.5% (2/44) NA NA NA NA
3
Renal failure 2.3% (1/44) 4.7% (2/43) 6.8% (3/44) 2.6% (1/38) 2.8% (1/36) 0.0% (0/34) 0.0% (0/32)
Respiratory failure 4.5% (2/44) 7.0% (3/43) 11.4% (5/44) 10.5% (4/38) 2.8% (1/36) 0.0% (0/34) 0.0% (0/32)
Stroke 2.3% (1/44) 2.3% (1/43) 4.5% (2/44) 2.6% (1/38) 2.8% (1/36) 0.0% (0/34) 0.0% (0/32)
a
m = number of subjects in category, n = number of subjects who experienced the corresponding event during that interval or who were followed
at least until the lower endpoint of the analysis window without experiencing the corresponding event. For example, for column “0-30 days”,
“31-365”, “0-365”, “366-731”, “732-1096”, “1097-1461”, and “1462-1826”, a subject has to be followed respectively for at least 0 day, 305 days, 305
days, 548 days, 914 days, 1279 days and 1645 days in order to be included in the denominator if he/she does not experience the event in the
corresponding time interval. For MAE subcategories, the same denominator would be used as that in the row of “One or more major adverse events
(MAE)” to allow comparison between subcategories within the MAE.
b
The CEC adjudicated MAEs through 1 year for the Endurant IDE arm. Once the CEC no longer adjudicates MAEs, then the MAEs above are as
site reported.
Note: A subject having an event at different time points is counted more than once.
6.2.2.12 Endoleaks
Details regarding the site reported endoleaks for the AUI subjects are presented in Table 43. No Type I, Type III, Type IV, or Type V
endoleaks have been reported by the investigational sites at any time point. A total of 16 Type II endoleaks were reported. Type II
endoleaks, which are dependent on subject anatomy and are not indicative of device failure were the only type of endoleaks reported.
Table 43. All Endoleaks by visit – AUI Arm
Endoleaks
1 month (1-90
days) %
a
(m/n)
12 month
(305-548
days) %
(m/n)a,
b
2 year
(549-913
days) %
a
(m/n)
3 year
(914-1278
days) %
a
(m/n)
4 year
(1279-1644
days) %
a
(m/n)
5 year (1645-2009
days) % (m/n)
a
Type I 0.0% (0/43) 0.0% (0/36) 0.0% (0/33) 0.0% (0/28) 0.0% (0/26) 0.0% (0/20)
Type II 7.0% (3/43) 13.9% (5/36) 6.1% (2/33) 10.7% (3/28) 7.7% (2/26) 5.0% (1/20)
Type III 0.0% (0/43) 0.0% (0/36) 0.0% (0/33) 0.0% (0/28) 0.0% (0/26) 0.0% (0/20)
Type IV 0.0% (0/43) 0.0% (0/36) 0.0% (0/33) 0.0% (0/28) 0.0% (0/26) 0.0% (0/20)
Type V 0.0% (0/43) 0.0% (0/36) 0.0% (0/33) 0.0% (0/28) 0.0% (0/26) 0.0% (0/20)
Indeterminate 2.3% (1/43) 0.0% (0/36) 0.0% (0/33) 0.0% (0/28) 0.0% (0/26) 0.0% (0/20)
Subjects had endoleaks
of any type
a
Denominator is the number of subjects who had readable images at the time of assessment. If there are two or more assessments in the same time
window, then the assessment closest to the target day will be used in the analysis.
b
Events that happened between 91-304 days will be counted in the column of 1 year.
c
A subject may have more than one type of endoleak; hence, number of subjects who had an endoleak of any type may not be the sum of those
in each type.
c
9.3% (4/43) 13.9% (5/36) 6.1% (2/33) 10.7% (3/28) 7.7% (2/26) 5.0% (1/20)
6.2.2.13 Aneurysm expansion
Aneurysm expansion is defined as a greater than 5 mm increase in maximum aneurysm diameter as measured on CT/MRI as
compared to the 1-month follow-up scan. A subject experiencing growth in any time point will continue to be counted in subsequent
time points if aneurysm expansion of more than 5 mm is reported at that time point. Table 44 shows the aneurysm expansion for the
AUI arm as reported by the sites. At 1 year, for all patients, the aneurysm expansion was either stable or decreased greater than 5 mm.
39
Page 40

At the subsequent annual follow-up visits, at least 95.8% of the Endurant AUI subjects had aneurysm decrease greater than 5 mm or
were stable.
Table 44. Aneurysm expansion - changes in aneurysm diameter during follow-up – AUI Arm
Changea in aneurysm diam-
eter from baseline (mm)
1 year (305-548
days) % (m/n)
2 year (549-913
b
days) % (m/n)
3 year (914-1278
b
days) % (m/n)
4 year
(1279-1644
b
days) % (m/n)
b
5 year
(1645-2009
days) % (m/n)
b
Increase > 5 mm 0.0% (0/36) 0.0% (0/33) 0.0% (0/29) 3.7% (1/27) 4.2% (1/24)
c
Stable
52.8% (19/36) 30.3% (10/33) 24.1% (7/29) 22.2% (6/27) 25.0% (6/24)
Decrease greater than 5 mm 47.2% (17/36) 69.7% (23/33) 75.9% (22/29) 74.1% (20/27) 70.8% (17/24)
a
Change in aneurysm diameter is based on 1-month imaging. When 1-month imaging was not available, the pre-discharge imaging was used as
the baseline.
b
m = number of subjects in category, n = number of subjects with known value of AAA diameter at both 1-month visit (or pre-discharge) and the
corresponding follow-up visit.
c
"Stable" refers to no change (increase or decrease) of more than 5 mm.
6.2.2.14 Stent graft migration
Stent graft migration is defined as evidence of movement of the stent graft relative to fixed anatomic landmarks, which is not due to
remodeling of the subject’s vasculature. Proximal migration is observed when the stent graft covers a renal artery or movement is >
10 mm. Distal migration is observed when the stent graft moves > 10 mm relative to fixed anatomic landmarks. There have been no
reports of stent graft migration > 10 mm for any subject in the AUI arm.
6.2.2.15 Technical observations
Table 45 summarizes the site reported technical observations for the AUI arm. Two (2) subjects reported stent graft kinking at the
1-year visit. One (1) secondary endovascular procedure was performed to treat stent graft kinking. The kinking was successfully
resolved at the conclusion of the procedure.
Table 45. Technical observations (site reported) – AUI Arm
Category
1 month (1-90
days) %
a
(m/n)
1 year
(305-548
days) %
(m/n)a,
b
2 Year
(549-913
days) %
a
(m/n)
3 year
(914-1278
days) %
a
(m/n)
4 year
(1279-1644
days) %
a
(m/n)
5 year (1645-2009
days) % (m/n)
a
Anchoring pin fracture 0.0% (0/42) 0.0% (0/34) 0.0% (0/33) 0.0% (0/28) 0.0% (0/23) 0.0% (0/14)
Stent graft wireform frac-
0.0% (0/42) 0.0% (0/34) 0.0% (0/33) 0.0% (0/29) 0.0% (0/23) 0.0% (0/14)
ture
Suprarenal bare stent
0.0% (0/42) 0.0% (0/34) 0.0% (0/33) 0.0% (0/29) 0.0% (0/23) 0.0% (0/14)
fracture
c
Other
0.0% (0/41) 5.9% (2/34) 0.0% (0/32) 0.0% (0/27) 0.0% (0/27) 0.0% (0/24)
Stent graft kinking 0.0% (0/42) 5.9% (2/34) 0.0% (0/32) 0.0% (0/27) 0.0% (0/21) 0.0% (0/12)
Stent graft twisting 0.0% (0/42) 0.0% (0/34) 0.0% (0/33) 0.0% (0/29) 0.0% (0/22) 0.0% (0/13)
Stent graft stenosis 0.0% (0/43) 0.0% (0/35) 0.0% (0/30) 0.0% (0/29) 0.0% (0/24) 5.0% (1/20)
Stent graft occlusion
a
Denominator is the number of subjects evaluable for the corresponding events at each time interval, excluding subjects with events in any of the
previous time periods. For each corresponding event, each subject was counted only once. However, a subject who experienced one event can
still be counted for another different event as well as under any Technical Observations in a different time period.
b
Events that happened between 91-304 days will be counted in the column of 1 year.
c
The “other” events category includes but is not limited to increased velocities in iliac artery and increased velocities in limb of graft.
d
Stent graft occlusion refers to loss of patency of the stent graft.
d
0.0% (0/43) 0.0% (0/36) 0.0% (0/32) 0.0% (0/29) 0.0% (0/24) 0.0% (0/20)
6.2.3 The ENGAGE PAS strengths and weakness
The main strengths of the ENGAGE PAS study are the commitment by the study site teams, efficient Medtronic study management
processes, and patients willing to comply with the visit schedule, which was key in making it a high follow-up compliance rate study
of 92%. This led to optimized data collection, robustness of data that resulted in 100% monitoring of clinical data, and independent
CEC adjudication of all deaths. This provided data on long term safety and efficacy of the Endurant stent graft system and provided
an understanding of where the device could be improved in the future. The limitation of this study was the image compliance rate of
approximately 82%. The compliance rate increased from the 3-year time point through the final 5-year time point due to the efforts of
Medtronic to maximize image compliance throughout the study by working closely with sites to collect this data. However, attempts
40
Page 41

by the sites to bring subjects back for long term follow-up visits were complicated by patient relocation, health status, and patient
willingness to return.
6.2.4 Conclusion
Based on the current ENGAGE PAS cohort data, no new concerns regarding safety or effectiveness have been observed in the
Endurant Bifurcated IDE subjects, De Novo subjects, and the Endurant AUI subjects through 5 years. The results of the ENGAGE PAS
cohort demonstrate the long-term safety and effectiveness of the Endurant stent graft system in the treatment of infrarenal AAA and
aortoiliac aneurysms.
6.3 ANCHOR Registry short neck cohort
The ANCHOR Registry is a prospective, observational, international, multi-center, post-market registry.
Subjects enrolled in the Primary Group (utilization of Heli-FX EndoAnchor implants during initial endovascular treatment) of the
registry with neck lengths of ≥4 mm and <10 mm and treated with Endurant or Endurant II/IIs stent graft system comprise the study
cohort, referred to as the “Short Neck Cohort”. The Short Neck Cohort included 70 subjects from 22 centers in the US (19) and the
EU (3). A prospectively defined, retrospective analysis of clinical outcomes was performed. Information on subjects with neck lengths
<4 mm and treated with any endovascular graft (the “Very Short Neck Cohort”) and those with neck lengths of >10 mm and treated
with Endurant or Endurant II/Endurant IIs stent graft system (the “On-Label Neck Cohort”) is also presented to provide context for the
Short Neck Cohort outcomes.
Clinical outcomes utilized to evaluate the safety and effectiveness of the “short neck” treatment included morbidity and mortality
measures for safety and technical success and rates of reintervention and Type 1a endoleak for effectiveness, among others. All
outcomes were analyzed descriptively and no formal hypothesis test was planned, therefore success/failure criteria were not applied.
Results through 1 year are presented. Subjects in the Short Neck Cohort will be followed for a total of 5 years from the index procedure.
6.3.1 Subject accountability and follow-up
The ANCHOR Registry protocol collected data from standard of care follow-up visits and imaging. Visit windows were not
prospectively defined. For the purposes of the Short Neck Cohort evaluation, statistical analysis windows were defined broadly in
order to include as many subjects as possible and present more complete follow-up information.
Clinical follow up of the Short Neck cohort was greater than 90% at both the 1-month and 12- month time points. 88% (58/66) of
subjects had 12-month imaging follow-up, while 54/66 (82%), 53/66 (80%), and 41/66 (62%) subjects had adequate imaging to
assess aneurysm size increase, Type Ia endoleaks, and migration, respectively.
Table 46. Subject and imaging accountability
Subjects with ade-
quate imaging to
Interval
(Analy-
sis Win-
dow)
Origi-
Subjects with imaging
Subject follow-up
performed
Sub-
jects-
with
Eligi-
ble
Clinical
Follow-
b
up
Imaging
Followup
fol-
low-u
p
pend-
ing
Duple
CT
Imag-
c
ing
KUB
Imag-
d
ing
Ultra-
sound
70 0 0 0
assess the parame-
Aneur
ysm
x
size
increaseEndo-
ter
leak
a
Migra-
tion
Subject events occurring
before next visit
With-
drawa
No
Impla
nt Death
l/Early
Termination
Lost
to Fol-
low-
up
nally
Enrolled
1 Month
(Day 1-1
70 64
(91%)62(89%)
0 (0%) 60
(86%)12(17%)19(27%)
59
(84%)
4 0 0
83)
12 Mont
hs
66 61
(92%)58(88%)
0 (0%) 43
(65%)13(20%)32(48%)54(82%)53(80%)41(62%)
8 3 1
(Day 184
-913)
Based on number of all enrolled subjects within the Short Neck Cohort with available data.
a
Not the number of subjects with these reported events, but rather, the number with adequate imaging, such as a paired size data to evaluate
aneurysm growth
b
Eligible for follow-up = eligible for follow-up from the previous interval − (death + withdrawn + lost to follow-up) from the previous interval. All subjects
that had an endovascular graft implanted are eligible for follow-up for the operative row.
41
Page 42

c
Subjects still within the follow-up window, but data not yet available
d
CT count includes CT and MRA
6.3.2 Study demographics and baseline medical history
The medical history and the risk factors were noted to be similar across the Short Neck Cohort, the Very Short Neck Cohort and the
On-Label Neck Cohort. Cardiovascular risk factors, including hypertension, past or current tobacco use and hyperlipidemia, occurred
frequently in the populations. A large percentage of subjects were diagnosed with cardiac disease. The baseline ASA risk
classifications were also similar, with well over 80% of subjects in ASA Class III or IV across all three study groups.
Table 47 and Table 48 provide the demographics, baseline medical history and ASA risk classification of the Short Neck, Very Short
Neck and On-Label Neck Cohorts.
Table 47. Baseline characteristics and risk factors
ANCHOR supportive
ANCHOR Short Neck
(N = 70) % (m/n)
a
cohort: Very Short
Neck < 4 mm (all stent
grafts) (N = 32) %
(m/n)
a
ANCHOR supportive
cohort: On-Label Neck
≥ 10 mm (Endurant)
(N = 100) % (m/n)
a
Age (year)
n 70 32 100
Mean ± SD 71.31 ± 8.13 75.66 ± 8.37 71.95 ± 8.41
Gender %(m/n)
Male 72.9% (51/70) 53.1% (17/32) 79.0% (79/100)
Female 27.1% (19/70) 46.9% (15/32) 21.0% (21/100)
Tobacco Use (past or current) 80.0% (56/70) 87.5% (28/32) 78.0% (78/100)
Hypertension 84.3% (59/70) 87.5% (28/32) 78.0% (78/100)
Hyperlipidemia 74.3% (52/70) 68.8% (22/32) 71.0% (71/100)
Diabetes 22.9% (16/70) 12.5% (4/32) 19.0% (19/100)
Cardiac disease
Coronary artery disease 8.6% (6/70) 12.5% (4/32) 8.0% (8/100)
CHF 7.1% (5/70) 15.6% (5/32) 4.0% (4/100)
Prior MI 34.3% (24/70) 28.1% (9/32) 14.0% (14/100)
Chronic Obstructive Pulmonary Disease (COPD) 44.3% (31/70) 43.8% (14/32) 27.0% (27/100)
Renal disease
Renal insufficiency 11.4% (8/70) 15.6% (5/32) 14.0% (14/100)
Dialysis-dependent renal 0.0% (0/70) 0.0% (0/32) 0.0% (0/100)
Failure (ESRD)
Stroke/Cerebral vascular accident 10.0% (7/70) 6.3% (2/32) 9.0% (9/100)
Bleeding disorder 2.9% (2/70) 6.3% (2/32) 1.0% (1/100)
Gastrointestinal disease 34.3% (24/70) 37.5% (12/32) 39.0% (39/100)
PAD 18.6% (13/70) 12.5% (4/32) 11.0% (11/100)
Thoracic aneurysm 2.9% (2/70) 0.0% (0/32) 5.0% (5/100)
a
m = number of subjects in category, n = number of all enrolled subjects with non-missing values.
Table 48. Baseline ASA classification
ANCHOR supportive
cohort: On-Label Neck
ANCHOR Short Neck
(Core laboratory
reported) (N=70) %
(m/n)
a
ANCHOR Short Neck
(Core laboratory
reported) (N=32) %
(m/n)
a
≥ 10 mm (Endurant)
(Core laboratory
reported) (N=100) %
ASA Classification
Class I 2.9% (2/70) 0.0% (0/32) 1.0% (1/100)
Class II 4.3% (3/70) 0.0% (0/32) 13.0% (13/100)
Class III 67.1% (47/70) 65.6% (21/32) 67.0% (67/100)
Class IV 25.7% (18/70) 34.4% (11/32) 19.0% (19/100)
a
m = number of subjects in category, n = number of all enrolled subjects with non-missing values.
42
(m/n)
a
Page 43

6.3.3 Baseline aneurysm characteristics
Table 49 provides the baseline aneurysm and anatomical measurements of the three cohorts. The mean maximum aneurysm
diameter reported as per core lab was 57.70 ± 12.74 mm (range: 34.1 mm to 112.0 mm). The mean core lab-reported neck length
(defined as that length over which the aortic diameter remains within 10% of the infrarenal diameter) was 6.86 ± 1.59 mm (range:
4.1 mm to 10.0 mm) for the Short Neck Cohort. Mean site-reported neck length (typically measured from the lowest main renal artery
to where the neck visually dilates) was 12.07 ± 5.58 mm (range: 4.0 mm to 33.0 mm). The core lab methodology should be applied
when determining whether EndoAnchor implants should be used with the endograft.
Table 49. Anatomical and other measurements (core laboratory-reported)
ANCHOR supportive
cohort: Very Short
ANCHOR Short Neck
(core laboratory
Measurement
Proximal neck diameter at renals
n 70 32 100
Mean ± SD 25.74 ± 4.04 25.55 ± 5.20 25.69 ± 4.13
Median 25.90 24.85 24.75
Min, Max 19.0, 36.5 16.9, 35.7 17.4, 39.8
Proximal neck length
n 70 32 100
Mean ± SD 6.86 ± 1.59 2.95 ± 0.82 22.37 ± 11.82
Median 6.69 3.03 18.20
Min, Max 4.1, 10.0 1.1, 4.0 10.0, 62.0
Distal aortic diameter
n 70 32 100
Mean ± SD 28.80 ± 4.79 28.34 ± 5.82 27.84 ± 4.53
Median 28.40 26.35 27.50
Min, Max 19.3, 40.5 19.1, 49.3 18.4, 43.2
Maximum aortic diameter
n 69 32 100
Mean ± SD 57.70 ± 12.74 56.46 ± 10.33 55.91 ± 11.06
Median 55.00 53.70 53.05
Min, Max 34.1, 112.0 41.7, 92.5 28.5, 101.0
Suprarenal angulation
n 70 32 100
Mean ± SD 14.07 ± 8.28 13.31 ± 8.71 15.70 ± 10.34
Median 12.00 13.50 14.00
Min, Max 1.0, 38.0 2.0, 39.0 2.0, 49.0
Infrarenal angulation
n 69 32 100
Mean ± SD 20.59 ± 14.44 23.16 ± 15.98 26.14 ± 18.55
Median 18.00 18.50 22.00
Min, Max 2.0, 69.0 1.0, 68.0 1.0, 80.9
Neck thrombus average thickness
n 64 29 94
Mean ± SD 0.85 ± 1.06 1.23 ± 1.42 0.89 ± 1.48
Median 0.00 1.30 0.00
Min, Max 0.0, 3.5 0.0, 5.3 0.0, 8.9
Neck thrombus circumference > 1 mm
n 64 29 94
Mean ± SD 61.41 ± 86.33 62.94 ± 80.45 46.63 ± 72.63
Median 0.00 19.60 0.00
Min, Max 0.0, 320.0 0.0, 298.0 0.0, 320.0
reported) (N = 70)
Neck < 4 mm (all stent
grafts) (core labora-
tory reported) (N = 32)
ANCHOR supportive
cohort: On-Label Neck
≥ 10 mm (Endurant)
(core laboratory
reported) (N = 100)
43
Page 44

Table 49. Anatomical and other measurements (core laboratory-reported) (continued)
Measurement
ANCHOR Short Neck
(core laboratory
reported) (N = 70)
ANCHOR supportive
cohort: Very Short
Neck < 4 mm (all stent
grafts) (core labora-
tory reported) (N = 32)
ANCHOR supportive
cohort: On-Label Neck
≥ 10 mm (Endurant)
(core laboratory
reported) (N = 100)
Neck calcium average thickness
n 70 32 100
Mean ± SD 1.31 ± 1.23 1.77 ± 1.28 1.03 ± 1.11
Median 1.30 1.94 1.00
Min, Max 0.0, 4.0 0.0, 4.3 0.0, 4.6
Neck calcium circumference > 1 mm
n 70 32 100
Mean ± SD 23.39 ± 29.83 38.50 ± 43.62 18.26 ± 24.91
Median 16.25 20.40 10.00
Min, Max 0.0, 155.0 0.0, 180.0 0.0, 114.0
6.3.4 Devices implanted
Endurant II/IIs stent graft system: Endurant stent grafts were utilized in 47.1% (33/70) of subjects in the Short Neck Cohort and
Endurant II/Endurant IIs stent grafts were utilized in 52.9% (37/70) of the subjects. The most frequently used proximal stent graft size
in the Short Neck Cohort was 36 mm, which was used in 34.3% (24/70) of subjects. Most subjects were implanted with an Endurant
or Endurant II/Endurant IIs stent graft with a proximal diameter of 28 mm or larger.
Heli-FX EndoAnchor implants: The average number of EndoAnchor implants per subject was 5.49 ± 2.08, with a median of 5.00,
minimum of 2.0 and maximum of 12.0 (n=70). Endurant and Endurant II/IIs stent graft system and Heli-FX EndoAnchor system:
Table 50 shows the proximal diameter of the endografts used in the Short Neck Cohort, the recommended number of EndoAnchor
implants (as per the Heli-FX EndoAnchor instructions for use) and the actual number of EndoAnchor implants used, by percentage
of subjects.
Table 50. Endurant stent graft sizing (proximal diameter) and Heli-FX EndoAnchor system
Proximal size of the Endurant
stent graft 23 mm (m/n)% 25 mm (m/n)% 28 mm (m/n)% 32 mm (m/n)% 36 mm (m/n)%
7.1% (5/70) 8.6% (6/70) 25.7% (18/70) 24.3% (17/70) 34.3% (24/70)
Recommended number of
EndoAnchor implants
a
4 4 4 4 6
Number of EndoAnchor implants
used
2 0.0% (0/5) 0.0% (0/6) 5.6% (1/18) 5.9% (1/17) 0.0% (0/24)
3 20.0% (1/5) 16.7% (1/6) 0.0% (0/18) 0.0% (0/17) 8.3% (2/24)
4 40.0% (2/5) 16.7% (1/6) 44.4% (8/18) 41.2% (7/17) 33.3% (8/24)
5 20.0% (1/5) 16.7% (1/6) 11.1% (2/18) 5.9% (1/17) 0.0% (0/24)
6 20.0% (1/5) 33.3% (2/6) 27.8% (5/18) 17.6% (3/17) 29.2% (7/24)
> 6 0.0% (0/5) 16.7% (1/6) 11.1% (2/18) 29.4% (5/17) 29.2% (7/24)
Based on number of all enrolled subjects within the Short Neck Cohort with available data.
m = number of subjects in category, n = number of all enrolled subjects within the Short Neck Cohort with non-missing values.
a
The Heli-FX EndoAnchor system instructions for use provides a minimum recommended number of EndoAnchor implants based on aortic neck
diameter, not device size. The recommended number of EndoAnchor implants used in an aortic neck of ≤ 29 mm is 4, and a minimum of
6 EndoAnchor implants are recommended in an aortic neck >29. The numbers provided in this table are based on the most likely aortic neck
diameter for a given device size, based on the Endurant II/IIs stent graft system instructions for use recommended device sizing relative to the aortic
vessel inner diameter.
Heli-FX EndoAnchor system: Table 51 provides information regarding the appliers and guides utilized for the Short Neck Cohort.
Table 51. Heli-FX EndoAnchor system usage
Short Neck Cohort (N = 70)
Number of Heli-FX appliers used
n 49
Mean ± SD 1.02 ± 0.14
44
Page 45

Table 51. Heli-FX EndoAnchor system usage (continued)
Short Neck Cohort (N = 70)
Median 1.00
Min, Max 1.0, 2.0
Size of Heli-FX applier(s) used (m/n)%
SA-85 Heli-FX Applier 98.0% (48/49)
HA-18-114 Heli-FX Applier 2.0% (1/49)
Number of Heli-FX guides used
n 49
Mean ± SD 1.04 ± 0.20
Median 1.00
Min, Max 1.0, 2.0
Size of Heli-FX guide(s) used (m/n)%
Heli-FX Guide 22 mm − SG-64 73.5% (36/49)
Heli-FX Guide 22 mm − HG-18-90-22 0.0% (0/49)
Heli-FX Guide 42 mm − HG-18-90-42 0.0% (0/49)
Heli-FX Guide 28 mm − HG-16-62-28 26.5% (13/49)
Heli-FX Guide 32 mm − HG-18-90-32 2.0% (1/49)
Based on number of all enrolled subjects within the Short Neck Cohort with available data.
m = number of subjects in category, n = number of all enrolled subjects within the Short Neck Cohort with non-missing values.
Accessory Device Usage: Twenty-five of 70 subjects in the Short Neck Cohort received at least one accessory device during the index
procedure. The type of accessory device used and number of subjects is as follows: Stent (11 subjects), balloon (10 subjects), coil
(4 subjects), cuff (3 subjects), vascular plug (2 subjects), and vascular graft (1 subject). Note that subjects may have received more
than one accessory device.
6.3.5 Acute procedural data
Acute procedural data for the ANCHOR Short Neck Cohort are shown in Table 52. The mean duration of the procedure was
148.00 ± 80.03 minutes, with average time of EndoAnchor implant being 17.12 ± 11.54 minutes. The mean overall intensive care unit
(ICU) stay was 0.84 ± 1.63 days, and the mean overall hospital stay was 3.73 ± 4.31 days.
Table 52. Initial procedural data − Short Neck cohort
Measurement Short Neck cohort (N = 70)
Type of procedure (m/n)%
Elective 69.4% (34/49)
Urgent (investigator assessed) 22.4% (11/49)
Emergent (investigator assessed) 8.2% (4/49)
Procedure entry sitea (m/n)%
Left femoral artery 82.9% (58/70)
Right femoral artery 91.4% (64/70)
Left iliac artery 0.0% (0/70)
Right iliac artery 0.0% (0/70)
Left: Other - Brachial percutaneous 1.4% (1/70)
Right: Other - Brachial 1.4% (1/70)
Type of access (m/n)%
Open 62.9% (44/70)
Percutaneous 37.1% (26/70)
Duration of implant procedure (min)
n 70
Mean ± SD 148.00 ± 80.03
Median 127.50
Min, Max 38.0, 423.0
Type of anesthesia used (m/n)%
General 84.3% (59/70)
Spinal 2.9% (2/70)
45
Page 46

Table 52. Initial procedural data − Short Neck cohort (continued)
Measurement Short Neck cohort (N = 70)
Epidural 1.4% (1/70)
Local 11.4% (8/70)
Volume of contrast (cc)
n 26
Mean ± SD 126.12 ± 80.76
Median 102.50
Min, Max 30.0, 399.0
Total fluoroscopic time (mins)
n 44
Mean ± SD 35.34 ± 21.98
Median 30.00
Min, Max 7.0, 123.0
Subjects Receiving EndoAnchor implants (m/n)% 100.0% (70/70)
Time to implant EndoAnchor implants (mins)
b
n 68
Mean ± SD 17.12 ± 11.54
Median 13.50
Min, Max 4.0, 60.0
Hospital stay (days)
c
n 70
Mean ± SD 3.73 ± 4.31
Median 2.00
Min, Max 1.0, 24.0
Procedure stay (days)
d
n 70
Mean ± SD 3.03 ± 3.17
Median 2.00
Min, Max 1.0, 23.0
Duration of ICU stay (days)
e
n 69
Mean ± SD 0.84 ± 1.63
Median 0.00
Min, Max 0.0, 11.0
Based on number of all enrolled subjects within the Short Neck Cohort with available data.
m = number of subjects in category, n = number of all enrolled subjects within the Short Neck Cohort with non-missing values.
a
Note that more than one procedure entry site per subject may be reported if multiple entry sites were used.
b
EndoAnchor implant total implant time includes all EndoAnchor implants for one subject
c
Overall hospital stay (days) = Date of Hospital Discharge − Date of Hospital Admission. In the case where Date of Hospital Discharge = Date of
Hospital Admission, Overall hospital stay will be considered to be 0.5 day
d
Procedural hospital stay (days) = Date of Hospital Discharge − Date of Initial Procedure. In the case where Date of Hospital Discharge = Date of
Initial Procedure, procedural hospital stay will be considered to be 0.5 day
e
ICU stay: days collected as 0, 1, 2…10 days or 10+days. 10+days set to 11 days for summary statistics
6.3.6 Study results: Safety outcomes
Safety outcomes are reported in terms of the prospectively identified safety-related measures shown in Table 53, with further detail
on ARM in Table 54 and other safety outcomes included in Table 55 to Table 59.
Aneurysm-related mortality is defined as any death within 30 days of the index procedure or secondary procedure to address
aneurysm, or death from any rupture. Four of 70 subjects (5.7%) died within 30 days of the index procedure. It is important to note that
the adverse events leading to death were assessed by the investigator, and no events were noted as related to the aneurysm.
No subjects experienced aneurysm rupture through one year. 15.7% (11/70) of Short Neck Cohort subjects experienced at least one
major adverse event through one month. One subject experienced renal insufficiency within 30 days of the index procedure. No renal
46
Page 47

failure was reported by sites through one month; however, one subject with a history of renal insufficiency died on Day 13, and the
investigator-determined cause of death included renal failure.
Table 53. Summary of safety-related measures
Safety-related measures
ANCHOR Short Neck
(N = 70)
ANCHOR supportive
cohort: Very Short
Neck < 4 mm (all stent
grafts) (N = 32)
ANCHOR supportive
cohort: On-Label Neck
≥ 10 mm (Endurant)
(N = 100)
Aneurysm-related mortality through 30 days 5.7% (4/70) 6.3% (2/32) 0.0% (0/100)
Aneurysm-related mortality through 12 months 5.9% (4/68) 7.1% (2/28) 0.0% (0/91)
Aneurysm rupture through 30 days 0.0% (0/70) 0.0% (0/32) 0.0% (0/100)
Aneurysm rupture through 12 months 0.0% (0/64) 0.0% (0/26) 0.0% (0/91)
Major adverse events through 30 days 15.7% (11/70) 9.4% (3/32) 4.0% (4/100)
Renal insufficiency through 30 days
Renal failure through 30 days
c
a
0.0% (0/70)
0.0% (0/70)
b
d
6.3% (2/32) 0.0% (0/100)
0.0% (0/32) 0.0% (0/100)
m = number of subjects in category, n = number of subjects with available imaging assessments for aneurysm expansion and
migration and endoleak at 12 months, and number of subjects who had an event in question or reached the lower time window of the
time period for site-reported events.
a
A rise in creatinine greater than 50% above the pre-procedure level resulting in a creatinine level above the upper limit of normal (site reported)
b
Subject 138-012 experienced renal insufficiency through 30 days from the index procedure. The site entered the start date of this event as a partial
date of March 2014 in the database. Due to the partial date entry, the days from index for this event was imputed per the statistical analysis plan.
The imputation of this event resulted in the event populating at -2 days from index procedure. Although the exact start date is unknown, it is known
that the event occurred within the same month of the index procedure. The renal insufficiency category in this table captures events between
Day 0-30. Due to this, the renal insufficiency event for this subject at -2 days is not populating in this table.
c
Defined as when the need for dialysis is required, an increase in serum creatinine of 2x the baseline value or new need for hemodialysis (site
reported)
d
Subject 109-031 died on Day 13 post-index procedure and the investigator-determined cause of death included acute renal failure. Acute renal
failure was captured under multi-system organ failure in Table 58.
Table 54. Summary of aneurysm-related deaths through 12 months
Subject number Days to death
Death within
30 days of the ini-
tial procedure
Death within
30 days of a
re-intervention
Cause of death (investigator deter-
mined)
172-014 6 Yes No Cardiac arrest, congestive heart failure
109-009 9 Yes No Cardiac and respiratory arrest
172-026 5 Yes No Cardiac arrest
109-031 13 Yes No Acute alcoholic hepatitis, acute renal fail-
ure, acute pancreatitis
Based on number of all enrolled subjects within the Short Neck Cohort with available data.
6.3.6.1 Additional safety outcomes
Key additional safety outcomes of all-cause mortality, major adverse events through 12 months and serious adverse events through
12 months are shown in Table 55 to Table 59. Of the 70 subjects in the Short Neck Cohort, five died within the first 12 months of the
index procedure. No subjects died on the day of the index procedure. Four of 70 subjects (5.7%) died within the 30 days of the index
procedure. One additional subject died on Day 353 post-index procedure due to septic shock secondary to pneumonia/ septicemia.
This event was not determined to be related to the aneurysm, device, or index procedure.
Kaplan-Meier survival estimates for all-cause mortality (ACM) were made through 365 days post-implant as shown in Table 55 and
Figure 8. The Kaplan-Meier one-year survival estimate for ACM was 92.6%.
Table 55. Kaplan-Meier estimates for All-cause mortality − Short Neck cohort subjects
Day 0 Day 30 Day 182 Day 365
No. at risk
a
70 66 61 53
No. of events 0 4 4 5
No. censored
b
0 0 5 12
47
Page 48

Table 55. Kaplan-Meier estimates for All-cause mortality − Short Neck cohort subjects (continued)
Day 0 Day 30 Day 182 Day 365
Kaplan-Meier estimate
c
1.000 0.943 0.943 0.926
Peto standard error 0.000 0.028 0.028 0.034
a
Number of subjects at risk at each timepoint. Based on number of all enrolled subjects within the Short Neck Cohort with available data.
b
Subjects are censored because no event was observed by the time point, including those not yet reached the correspondent time point or lost to
follow-up.
c
Estimate made at each timepoint.
Note: All numbers except standard errors are cumulative.
Figure 8. Freedom from All-Cause Mortality
A total of 18 MAEs were reported among 15 subjects between 0 and 365 days. Among those, 13 MAEs were reported in 11 subjects
between 0 and 30 days, and five MAEs were reported in five subjects between 31 and 365 days. Three subjects had more than one
MAE within the first year.
There were no reports of paraplegia, or renal failure through 12 months. However, as mentioned previously, one subject with a history
of renal insufficiency died on Day 13 post-index procedure and the investigator-determined cause of death included acute renal
failure.
Table 56. Major Adverse Events (MAEs) through 12 months
Event 0-30 days % (m/n) 31-365 days % (m/n) 0-365 days % (m/n)
One or more major adverse events (MAE)
15.7% (11/70) 7.6% (5/66) 21.4% (15/70)
Total number of MAEs 13 5 18
All-Cause mortality 5.7% (4/70) 1.5% (1/66) 7.1% (5/70)
Bowel ischemia 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Myocardial infarction 2.9% (2/70) 1.5% (1/66) 2.9% (2/70)
Paraplegia 0.0% (0/70) 0.0% (0/66) 0.0% (0/70)
Procedural blood loss
Renal failure 0.0% (0/70)
a
7.1% (5/70) 0.0% (0/66) 7.1% (5/70)
b
0.0% (0/66) 0.0% (0/70)
b
Respiratory failure 1.4% (1/70) 3.0% (2/66) 4.3% (3/70)
Stroke 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
48
Page 49

Table 56. Major Adverse Events (MAEs) through 12 months (continued)
Event 0-30 days % (m/n) 31-365 days % (m/n) 0-365 days % (m/n)
Based on number of all enrolled subjects within the Short Neck Cohort with available data.
m = number of subjects in category, n = number of subjects are at risk at the beginning of the time period.
a
Volume of blood loss was not collected, therefore, procedural blood loss/hemorrhage reported as an SAE at the time of the index procedure or
secondary procedure has been reported as an MAE
b
Subject 109-031 died on Day 13 post-index procedure and the investigator-determined cause of death included acute renal failure. Acute renal
failure was captured under multi-system organ failure in Table 57.
Thirty of 70 subjects (42.9%) experienced one or more serious adverse event between 0 and 365 days. Of those, 16 of 70 subjects
(22.9%) experienced one or more serious adverse event between 0 and 30 days, and 19 out of 66 subjects (28.8%) experienced one
or more serious adverse event between 31 and 365 days.
The most common type of SAEs were cardiac disorders, reported in 9 out of 70 subjects (12.9%). The second most common type of
SAEs were vascular disorders and gastrointestinal disorders, reported in 7 out of 70 subjects (10.0%).
Table 57. Serious Adverse Events (SAEs) through 12 months
Category 0 to 30 Days % (m/n) 31 to 365 Days % (m/n) 0 to 365 Days % (m/n)
Number of subjects eligible for each follow-up
70 66 70
window
Subjects experiencing one or more SAEs
a
22.9% (16/70) 28.8% (19/66) 42.9% (30/70)
Blood and lymphatic system disorders 2.9% (2/70) 1.5% (1/66) 4.3% (3/70)
Anemia 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Anemia postoperative 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Neutropenic fever 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Cardiac disorders 7.1% (5/70) 7.6% (5/66) 12.9% (9/70)
Arrhythmia 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Cardiac arrest 2.9% (2/70) 0.0% (0/66) 2.9% (2/70)
Congestive cardiac failure aggravated 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Congestive heart failure 0.0% (0/70) 4.5% (3/66) 4.3% (3/70)
Coronary artery disease aggravated 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Myocardial infarction 2.9% (2/70) 1.5% (1/66) 2.9% (2/70)
Ventricular tachycardia 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Gastrointestinal disorders 2.9% (2/70) 7.6% (5/66) 10.0% (7/70)
Diverticulitis 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
GI bleed 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Hernia inguinal 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Ischemic colitis 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Pancreatic cancer 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Retroperitoneal hematoma 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Retroperitoneal mass 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
General disorders and administration site con-
4.3% (3/70) 0.0% (0/66) 4.3% (3/70)
ditions
Device occlusion 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Multi organ failure 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Stent-graft endoleak 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Infections and infestations 1.4% (1/70) 6.1% (4/66) 7.1% (5/70)
Bronchitis 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Pneumonia 0.0% (0/70) 3.0% (2/66) 2.9% (2/70)
Sepsis 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Septicaemia 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Urinary tract infection 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Device occlusion 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Multi organ failure 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Stent-graft endoleak 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
49
Page 50

Table 57. Serious Adverse Events (SAEs) through 12 months (continued)
Category 0 to 30 Days % (m/n) 31 to 365 Days % (m/n) 0 to 365 Days % (m/n)
Injury, poisoning and procedural complica-
4.3% (3/70) 1.5% (1/66) 5.7% (4/70)
tions
Femoral artery injury 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Post procedural bleeding 2.9% (2/70) 0.0% (0/66) 2.9% (2/70)
Procedural bleeding 2.9% (2/70) 0.0% (0/66) 2.9% (2/70)
Scalp laceration 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Vascular access site bleeding 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Metabolism and nutrition disorders 1.4% (1/70) 3.0% (2/66) 4.3% (3/70)
Respiratory failure 1.4% (1/70) 3.0% (2/66) 4.3% (3/70)
Neoplasms benign, malignant and unspecified
0.0% (0/70) 3.0% (2/66) 2.9% (2/70)
(including cysts and polyps)
Bladder cancer recurrent 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Hepatic cancer 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Subarachnoid hemorrhage 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Psychiatric disorders 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Mental status changes 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Renal and urinary disorders 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Renal insufficiency 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Respiratory, thoracic and mediastinal disor-
0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
ders
COPD exacerbation 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Skin and subcutaneous tissue disorders 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Lower extremities ulcers of 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Vascular disorders 5.7% (4/70) 6.1% (4/66) 10.0% (7/70)
Aneurysm 1.4% (1/70) 1.5% (1/66) 2.9% (2/70)
Deep vein thrombosis 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Hypertension 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Peripheral artery dissection 0.0% (0/70) 1.5% (1/66) 1.4% (1/70)
Peripheral ischemia 1.4% (1/70) 1.5% (1/66) 2.9% (2/70)
Thrombosis 1.4% (1/70) 1.5% (1/66) 2.9% (2/70)
Based on number of all enrolled subjects within the Short Neck Cohort with available data.
m = number of subjects experiencing one or more serious adverse events in a category, n = number of subjects at risk at the
beginning of the time period.
a
A subject may report multiple adverse events and in different subcategories; hence, number of subjects in each category may not be the sum of
those in each subcategory. Each subject was only counted once in each subcategory and category.
Investigators were asked to assess relatedness of reported SAEs to device, index or re- intervention procedure, or AAA disease,
results of which are shown in Table 58. All events determined to be related to device, procedure, or AAA occurred within 30 days of
the index procedure, many of which occurred on the day of the index procedure. There were no reintervention-related SAEs or
unanticipated adverse device effects (UADEs) reported through 365 days. There were no device-related SAEs related to EndoAnchor
implants.
Two out of 70 subjects (2.9%) were reported to have an AAA-related SAE between 0 and 30 days, both occurred on Day 0. One subject
experienced a femoral artery injury and bleeding at the vascular access site. The other subject experienced post-procedural bleeding.
One out of 70 subjects was reported to have a device-related SAE between 0 and 30 days, which was an endograft limb occlusion.
An additional subject had a device-related event at 59 days, which was peripheral ischemia. There were no device-related SAEs
related to EndoAnchor implants.
Table 58. Relatedness of Serious Adverse Events through 12 months
0-30 Days % (m/n) 31-365 Days % (m/n) 0-365 Days % (m/n)
Index procedure related SAEs 12.9% (9/70) 0.0% (0/66) 12.9% (9/70)
Re-intervention related SAEs 0.0% (0/70) 0.0% (0/66) 0.0% (0/70)
AAA-related SAEs 2.9% (2/70) 0.0% (0/66) 2.9% (2/70)
50
Page 51

Table 58. Relatedness of Serious Adverse Events through 12 months (continued)
0-30 Days % (m/n) 31-365 Days % (m/n) 0-365 Days % (m/n)
Device related SAE 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Related to Endograft 1.4% (1/70) 0.0% (0/66) 1.4% (1/70)
Related to Heli-FX 0.0% (0/70) 0.0% (0/66) 0.0% (0/70)
Related to both 0.0% (0/70) 0.0% (0/66) 0.0% (0/70)
UADEs 0.0% (0/70) 0.0% (0/66) 0.0% (0/70)
Based on number of all enrolled subjects within the Short Neck Cohort with available data.
m = number of subjects in category, n = number of subjects who are at risk at beginning of the time period.
6.3.7 Study results: Effectiveness outcomes
The analysis of effectiveness was based on the Short Neck Cohort of 70 patients available for the 1-month evaluation and 66 patients
available for the 12-month evaluation.
The primary effectiveness outcomes were technical success rate, Type Ia endoleak rate at 1 month and 12 months and reintervention
rate through 12 months.
Technical success was defined as:
• Successful delivery, defined as:
– Access to the targeted aortic site was achieved
– Successful delivery of the main body to the intended landing zone was achieved
• Successful and accurate deployment of the stent graft, defined as:
– successful deployment of the endovascular stent-graft at the intended implantation site was achieved
– successful and accurate deployment of EndoAnchor implants was achieved
– absence of unintentional covering of the renal arteries was achieved
The overall technical success rate for both endograft and EndoAnchor implants was 88.6% (62/70). Delivery and deployment of the
main body stent graft at the intended landing zone was achieved in 94.3% (66/70) and 100% (70/70), respectively, with absence of
unintentional coverage of the renal arteries achieved in 97.1% (68/70) of subjects. Delivery and deployment of the EndoAnchor
implants at the target aortic site was achieved in 100% (70/70) and 92.9% (65/70), respectively. Overall procedural success, as noted
by investigators, was determined to be 97.1% (68/70).
Reasons for not achieving technical success:
• Inadequate penetration of at least one EndoAnchor implant into the aortic wall: 4 subjects
• Unsuccessful delivery of the endograft main body into the intended landing zone: 3 subjects Endografts were delivered slightly
distal to the intended landing zone. One subject had unintentional coverage of the renal artery due to cuff placement. Bilateral
renal stents were placed to maintain renal perfusion.
• Factors related to both the Endograft and EndoAnchor implants: 1 subject Endograft main body required additional proximal stent
graft extension, resulting in unintentional coverage of the left accessory renal artery. 7 EndoAnchor implants were implanted
successfully but Type II endoleak was observed at the proximal neck.
Type Ia endoleak was reported by the core lab in 6.8% (4/59) of subjects at the 1-month follow- up visit and 1.9% (1/53) of subjects
at the 12-month follow-up visit. Only one subject required re-intervention to treat a Type Ia endoleak.
Three of 64 subjects (4.7%) had one or more re-interventions through 12 months. Thrombosis, occlusion, and Type Ia endoleak were
the reasons for reintervention. There were no conversions to open surgical repair through 12 months.
A summary of primary outcomes for the Short Neck Cohort, Very Short Neck Cohort and On- Label Neck Cohort is provided in
Table 59, below. This is followed by a discussion of the impact of the number of EndoAnchor implants used on primary outcomes.
Table 59. Summary of Primary Effectiveness Outcomes
ANCHOR Short
Event
Type Ia Endoleak at 1-month
Type Ia Endoleak at 1-year
a
a
Neck (N = 70)
6.8% (4/59) [1.9%, 16.5%] 16.0% (4/25) 0.0% (0/91)
1.9% (1/53) [0.0%, 10.1%] 0.0% (0/20) 0.0% (0/73)
95% Confidence
Interval
51
ANCHOR suppor-
tive cohort: Very
Short Neck < 4 mm
(all stent grafts)
(N = 32)
ANCHOR suppor-
tive cohort:
On-Label Neck
≥ 10 mm
(Endurant)
(N = 100)
Page 52

Table 59. Summary of Primary Effectiveness Outcomes (continued)
ANCHOR suppor-
Event
ANCHOR Short
Neck (N = 70)
95% Confidence
Interval
ANCHOR suppor-
tive cohort: Very
Short Neck < 4 mm
(all stent grafts)
(N = 32)
tive cohort:
On-Label Neck
≥ 10 mm
(Endurant)
(N = 100)
Secondary procedures through 1 year 4.7% (3/64) [1.0%, 13.1%] 7.7% (2/26) 2.2% (2/91)
Secondary endovascular procedures
4.7% (3/64) [1.0%, 13.1%] 7.7% (2/26) 2.2% (2/91)
through 1 year
Conversion to open surgical repair
0.0% (0/64) [0.0%, 5.6%] 0.0% (0/26) 0.0% (0/91)
through 1 year
Other secondary open surgical proce-
1.6% (1/64) [0.0%, 8.4%] 0.0% (0/26) 0.0% (0/91)
dures through 1 year
Technical success rate at index proce-
88.6% (62/70) [78.7%, 94.9%] 84.4% (27/32) 94.9% (94/99)
dureb:
Successful delivery:
Access to the targeted aortic site was
100.0% (70/70) [94.9%, 100.0%] 100.0% (32/32) 100.0% (100/100)
achieved by the EndoAnchor system
Successful delivery of the main body
94.3% (66/70) [86.0%, 98.4%] 96.9% (31/32) 99.0% (99/100)
to the intended landing zone
Successful and accurate deployment of
the Endurant II/Endurant IIs stent graft
and the Aptus Heli-FX EndoAnchor system
Successful deployment of the endovascular stent graft at the intended
implantation site
c
Successful and accurate deployment
of EndoAnchor implants was
acheived
d
Absence of unintentional coverage of
100.0% (70/70) [94.9%, 100.0%] 100.0% (32/32) 98.0% (97/99)
92.9% (65/70) [84.1%, 97.6%] 87.5% (28/32) 96.0% (96/100)
97.1% (68/70) [90.1%, 99.7%] 96.9% (31/32) 100.0% (100/100)
the renal arteries
Based on number of all enrolled subjects within the Short Neck Cohort with available data.
m = number of subjects in category, n = number of subjects with available values. For secondary procedures through 1 year, the
denominator includes subjects who had an event or had been followed for at least 184 days.
a
Core laboratory reported
b
Technical success was defined as: successful delivery and deployment of the stent graft, without unintentional coverage of the renal arteries, and
successful implantation of the EndoAnchor implants at the target aortic site.
c
Absence of misdeployment of the main body proximal to the intended landing zone
d
Each EndoAnchor implant adequately penetrated the aortic wall (investigator’s assessment) and, in the investigator’s opinion, the implantation of
the EndoAnchor implants was successful.
18 subjects did not have the recommended number of EndoAnchor devices implanted per the Heli-FX EndoAnchor system
instructions for use. 15 of these achieved technical success, that is, successful delivery and deployment of the endograft and
EndoAnchor implants without unintentional coverage of the renal arteries. Three subjects did not achieve technical success due to
inadequate penetration of at least 1 EndoAnchor implant into the aortic wall. Three of the four subjects who reported Type Ia endoleak
at 1-month follow-up had less than the recommended number of implanted EndoAnchor implants. Two of the three Short Neck Cohort
subjects who underwent secondary procedures through 12 months were implanted with less than the recommended number of
EndoAnchor implants, however, one of the two subjects had an intervention that was entirely unrelated to the quantity of EndoAnchor
implants.
Table 60. Outcomes in subjects where the recommended number of EndoAnchor implants were not used
Subject num-
ber
Recommended
number of
EndoAnchor
implants
a
Number of
EndoAnchor
implants used
Technical suc-
cess
(Yes/No/UNK)
Type Ia Endo-
leak at 30 days
(Yes/No/UNK)
Type Ia Endo-
b
leak at
12 months
(Yes/No/UNK)
Re-intervention
b
through
365 days
(Yes/No/UNK)
100-052 4 3 No No UNK UNK
109-041 6 2 Yes No No No
52
Page 53

Table 60. Outcomes in subjects where the recommended number of EndoAnchor implants were not used (continued)
Subject num-
ber
Recommended
number of
EndoAnchor
implants
a
Number of
EndoAnchor
implants used
Technical suc-
cess
(Yes/No/UNK)
Type Ia Endo-
leak at 30 days
(Yes/No/UNK)
Type Ia Endo-
b
leak at
12 months
(Yes/No/UNK)
Re-intervention
b
through
365 days
(Yes/No/UNK)
109-002 6 4 Yes Yes Yes Yes
109-048 6 3 Yes Yes No Yes
109-057 6 4 Yes No No No
109-016 6 4 Yes No No No
109-042 4 2 Yes No No No
109-039 6 4 Yes No UNK No
135-004 6 4 Yes No No No
135-006 6 4 No No No No
155-002 6 4 Yes UNK UNK No
168-010 6 4 Yes No No No
172-005 6 4 Yes No No No
172-024 6 4 Yes No No No
178-009 6 4 Yes No No No
178-011 6 4 No Yes UNK No
178-005 6 4 Yes No No No
189-003 6 3 Yes No No No
Based on number of all enrolled subjects within the Short Neck Cohort with available data.
a
Based on the IFU, the minimum recommended number of EndoAnchor implants used in an aortic neck of ≤ 29 mm is 4, and a minimum of
6 EndoAnchor implants are recommended in an aortic neck > 29 mm
b
Core laboratory reported
6.3.7.1 Supportive effectiveness outcomes
Supportive effectiveness outcomes included initial implantation outcomes, device-related measures (loss of endograft or
EndoAnchor device integrity) and maintenance of EndoAnchor implants in the aortic wall.
Investigators reported an overall procedure success of 97.1% (68/70); unsuccessful procedures were reported in one subject due to
a Type II endoleak observed at the proximal neck and in a second subject due to a persistent Type Ia endoleak.
Table 61. Initial implantation outcomes
Event Short Neck Cohort (N = 70) (m/n)%
Overall procedure was successful (investigator assessed) 97.1% (68/70)
Endograft components misdeployed proximally to intended landing zone
Stent graft main body
a
0.0% (0/70)
Left iliac extension 0.0% (0/70)
Right iliac extension 0.0% (0/70)
Aortic cuff 1.4% (1/70)
Endoleak present at end of procedure
Type Ia 12.9% (9/70)
Type Ib 1.4% (1/70)
Type II 20.0% (14/70)
Type III 0.0% (0/70)
Type IV 7.1% (5/70)
Based on number of all enrolled subjects within the Short Neck Cohort with available data.
m = number of subjects in category, n = number of subjects with available values
a
Misdeployment of the main body component proximally to the intended landing zone is a component of technical success
There was no loss of endograft or EndoAnchor device integrity reported within the 1-month or 12-month follow-up window for the Short
Neck Cohort.
EndoAnchor implants maintained adequate penetration through the aortic wall in 98% (50/51) of the subjects at 30 days and in 94.9%
(37/39) of subjects at 12 months. See Table 62.
53
Page 54
Table 62. Maintenance of adequate penetration by EndoAnchor implants
% (m/n)
Did EndoAnchor implants maintain adequate penetration into aortic wall
30 days 98.0% (50/51)
12 months 94.9% (37/39)
Based on number of all enrolled subjects within the Short Neck Cohort with available data.
m = number of subjects in category, n = number of subjects with available values
6.3.7.2 Other effectiveness-related measures
Other effectiveness-related measures of interest are summarized in Table 63. Three of 70 Short Neck Cohort subjects underwent
secondary procedures through 1 month. No subjects underwent conversion to open surgical repair through 12 months. Type III
endoleaks were reported by core lab in 1.7% (1/59) of subjects at 1-month follow-up and 1.9% (1/53) of subjects at 12-month
follow-up. No subjects experienced endograft migration at one year.
Table 63. Summary of effectiveness-related measures
Effectiveness-related measures
Aneurysm expansion at 12 months
Migration at 12 months
a,c
a,b
ANCHOR supportive
cohort: Very Short
ANCHOR Short Neck
(N = 70)
Neck < 4 mm (all stent
grafts) (N = 32)
0.0% (0/54) 5.3% (1/19) 0.0% (0/73)
0.0% (0/41) 0.0% (0/16) 0.0% (0/58)
ANCHOR supportive
cohort: On-Label Neck
≥ 10 mm (Endurant)
(N = 100)
Secondary procedures through 30 days 4.3% (3/70) 0.0% (0/32) 1.0% (1/100)
Conversion to open surgical repair through 12
0.0% (0/64) — —
months
Type III endoleaks at 30 days
Type III endoleaks at 12 months
a
a
1.7% (1/59) — —
1.9% (1/53) — —
m = number of subjects in category, n = number of subjects with available imaging assessments for aneurysm expansion and
migration and endoleak at 12 months, and number of subjects who had an event in question or reached the lower time window of the
time period for site-reported events.
a
Based on core laboratory related data.
b
Aneurysm expansion (alternatively noted as AAA diameter increase) is defined as a > 5 mm increase in maximum diameter as compared to 1
month post-implantation measurement.
c
Migration is defined as a > 10 mm movement of the aortic endograft from its position at the 1 month post-implantation measurement.
6.3.8 Data post 12 months
As of the data cut-off for this summary, one core lab-reported Type Ia endoleak was identified in one subject on Day 1271. The subject
did not undergo re-intervention to treat it. One subject underwent a secondary endovascular procedure and conversion to open
surgical repair on Day 900 to treat a Type II endoleak and aneurysm enlargement. One subject underwent a secondary endovascular
procedure on Day 758 to treat a Type Ib endoleak and aneurysm enlargement. No aneurysm ruptures had occurred beyond the
12-month time point as of the data cut-off. One subject experienced renal failure on Day 888, which was assessed by the investigator
as not related to the aneurysm, procedure or device and the event has been reported as resolved. Seven subjects experienced MAEs
beyond the 12-month time point. A total of nine deaths occurred after Day 365, none of these deaths were determined to be
aneurysm-related
7 Patient selection and treatment
7.1 Individualization of treatment
Each Endurant II/IIs stent graft system must be ordered in the appropriate size to fit the patient’s anatomy. Proper sizing of the device
is the responsibility of the physician. The stent graft should be oversized to be larger than the vessel inner diameter (aortic
configurations are recommended to be oversized 10 to 20%; iliac configurations are recommended to be oversized 10 to 25%). Refer
to Section 10.2 for further details. The stent graft configurations cover aortic diameters ranging from 19 to 32 mm and iliac diameters
from 8 to 25 mm. The recommended overall length of the stent graft including multiple deployed devices should extend from the lowest
renal artery to just above the internal iliac or hypogastric artery. All stent graft lengths and diameters necessary to complete the
procedure should be available to the physician, especially when pre-operative case planning measurements (treatment
diameters/lengths) are not certain. Use of this approach allows for greater intraoperative flexibility to achieve optimal procedural
outcomes.
54
Page 55

Medtronic may consult with physicians to determine proper stent graft dimensions based on the physician’s assessment of the
patient’s anatomical measurements. The benefits and risks previously described should be carefully considered for each patient
before use of the stent graft system.
Use of the AUI device may require occlusion of the contralateral iliac artery in conjunction with a standard femoral-femoral bypass
procedure. Occlusion of the iliac artery can be performed using a Talent occluder system. For details on use and implantation refer
to the Talent occluder system instructions for use. Patients treated with an AUI device must be carefully evaluated prior to the
procedure to determine the need and timing for closure of an iliac artery to prevent backflow into the aneurysm sac.
Caution: Vessel over-distension and damage, or partial stent graft infolding, may be caused by excessive oversizing of the stent graft
in relation to the diameter of the blood vessel. Also, due to the nature of the design and the flexibility of the Endurant II/IIs stent graft
system, the overall length of each stent graft component may be shorter when deployed.
The Endurant II/IIs stent graft system may be utilized in conjunction with the Heli-FX EndoAnchor system when augmented radial
fixation and/or sealing is required; in particular, in the treatment of abdominal aortic aneurysms with short (≥ 4 mm and < 10 mm)
infrarenal necks
For infrarenal EVAR procedures using the Heli-FX EndoAnchor system:
• EndoAnchor implant locations should be based upon a detailed examination of the preoperative CT imaging in cases involving
irregular or eccentric plaque in the intended sealing zone(s). The EndoAnchor implant should be implanted only into areas of
aortic tissue that are free of or have insignificant calcified plaque or thrombus, or where such pathology is diffuse and less than
2 mm in thickness. Attempting to place EndoAnchor implants into more severe plaque or thrombus may be associated with
implantation difficulty and suboptimal endograft fixation and/or sealing.
• The EndoAnchor implant should not be used in patients with bleeding diathesis or with a known hypersensitivity or allergy to the
device materials.
• See Section 11.2.17 for information regarding the recommended number of EndoAnchor implants.
• Refer to the Heli-FX EndoAnchor system instructions for use for additional information.
8 Patient counseling information
The physician should review the following risks and benefits when counseling the patient about this endovascular device and
procedure:
• patient age and life expectancy
• risks and benefits related to open surgical repair
• risks and benefits related to endovascular repair
• risks related to noninterventional treatment or medical management
• risks of aneurysm rupture compared to endovascular repair
• possibility that subsequent endovascular or open surgical repair of the aneurysm may be required
• the long-term safety and effectiveness of the Endurant II/IIs stent graft system or the Endurant II/IIs stent graft system used in
conjunction with the Heli-FX EndoAnchor system has not been established
• long-term, regular follow-up care is needed to assess patient health status and stent graft performance
• patients with specific clinical findings (eg, endoleaks, enlarging aneurysms) should be monitored closely
• symptoms of aneurysm rupture
• risks and benefits related to femoral-femoral artery bypass, if applicable
Medtronic recommends that the physician disclose to the patient, in written form, all risks associated with treatment using the
Endurant II/IIs stent graft system. Details regarding risks occurring during and after implantation of the device are provided in Adverse
Events (Chapter 5). Additional counseling information can be found in the Patient Information Booklet.
9 How supplied
For information on the supply of the Heli-FX EndoAnchor system, please see the instructions for use supplied with the device.
9.1 Sterility
Each stent graft configuration (bifurcated, AUI, limb, aortic extension, and iliac extension) is individually contained within a delivery
system. It is sterilized using electron beam and is supplied sterile for single use only.
• This device was designed for single use only. Do not reuse, reprocess, or resterilize this device. Reuse, reprocessing, or
resterilization may compromise the structural integrity of the device or create a risk of contamination, which could result in patient
injury, illness, or death.
• If the device is damaged or the integrity of the sterile barrier has been compromised, do not use the product and contact your
Medtronic representative for return information.
55
Page 56

9.2 Contents
• One Endurant II/IIs Stent Graft System
• One set of patient tracking materials
9.3 Storage
Store the system at room temperature in a dark, dry place.
10 Clinical use information
10.1 Physician training requirements
All physicians should complete in-service training prior to using the Endurant II/IIs stent graft system.
Note: Endurant and Endurant II/IIs stent graft systems are considered technically and clinically equivalent. Physicians who have
completed in-service training for Endurant do not need additional training for Endurant II/IIs.
Caution: The Endurant II/IIs stent graft system should only be used by physicians and teams trained in vascular interventional
techniques, and in the use of this device. The following are the knowledge and skill requirements for physicians using the
Endurant II/IIs stent graft system:
• Natural history of abdominal aortic aneurysms (AAA), aortoiliac aneurysms, and comorbidities associated with AAA repair
• Radiographic, fluoroscopic, and angiographic image interpretation
• Appropriate use of radiographic contrast material
• Arterial cutdown, arteriotomy, and repair or percutaneous access and closure techniques
• Nonselective and selective guidewire and catheter techniques
• Embolization
• Angioplasty
• Endovascular stent placement
• Snare techniques
• Techniques to minimize radiation exposure
• Device selection and sizing
Caution: The Heli-FX EndoAnchor system should only be used by physicians and teams trained in the use of this device. For
physician training recommendations for the Heli-FX EndoAnchor system, please see the instructions for use provided with the device.
10.2 Recommended device sizing
10.2.1 Endurant II/IIs stent graft system
The Endurant II/IIs stent graft systems are available in the sizes described in Table 64 through Table 69. For questions about device
sizing, refer to contact information in the back of this Instructions for Use.
Table 64. Sizing chart — Endurant II bifurcated configuration
OD (Fr)
20
18
Proximal x distal diameter
(mm x mm)
36x20
36x16
32x20
32x16
28x20
28x13
25x16
25x13
23x16
23x13
Covered length
(mm)
145, 166 29-32
124, 145, 166
Vessel inner diameter (mm)
26-28
23-2528x16
21-22
19-20
56
Page 57

Table 65. Sizing chart — Endurant IIs bifurcated configuration
OD (Fr)
20
Proximal x distal diameter
(mm x mm)
36x14
32x14 26-28
28x14 23-25
18
25x14 21-22
23x14 19-20
Table 66. Sizing chart — Aorto-Uni-Iliac (AUI) configuration
OD (Fr)
20
Proximal x distal diameter
(mm x mm)
36x14
32x14 26-28
28x14 23-25
18
25x14 21-22
23x14 19-20
Table 67. Sizing chart — limb configuration
OD (Fr)
Proximal x distal diameter
(mm x mm)
16x28
82, 93, 124, 146, 156, 199
16
16x24 19-22
16x20 15-18
16x16
16x13 10-11
16x10 8-9
16x16
14
16x13 10-11
16x10 8-9
Covered length
(mm)
103
Covered length
Covered length
(mm)
146, 156, 199
82, 93, 124
(mm)
102
Vessel inner diameter (mm)
29-32
Vessel inner diameter (mm)
29-32
Vessel inner diameter (mm)
23-25
12-14
12-14
Table 68. Sizing chart — iliac extension configuration
OD (Fr)
Proximal x distal diameter
(mm x mm)
18 28x28
16
14
24x24 19-22
20x20 15-18
13x13 10-11
10x10 8-9
Covered length
(mm)
82
Vessel inner diameter (mm)
23-25
Table 69. Sizing chart — Aortic extension configuration
OD (Fr)
20
18
Proximal x distal diameter
(mm x mm)
36x36
32x32 26-28
28x28 23-25
25x25 21-22
Covered length
(mm)
49, 70
Vessel inner diameter (mm)
29-32
23x23 19-20
Caution: Proper sizing of the Endurant II/IIs stent graft is the responsibility of the physician. This stent graft sizing incorporates the
recommended device oversizing for anatomical dimensions and was based on in-vitro test data.
10.2.2 Heli-FX EndoAnchor system
For information on pre-implant planning for the Heli-FX EndoAnchor system, including the recommended number of EndoAnchor
implants to be used, refer to the Heli-FX EndoAnchor system instructions for use. See also Section 11.2.17, below.
57
Page 58

10.3 Device inspection
Inspect the Endurant II/IIs device and packaging to verify that no damage or defects exist. If the “Use by” date has elapsed, the device
is damaged, or the sterile barrier has been compromised, do not use the device and contact a Medtronic representative for return or
replacement information.
10.4 Additional required equipment
• Additional Endurant II/IIs stent graft systems of various lengths and diameters
• For repair of short proximal neck aneurysms, additional Heli-FX EndoAnchor systems and compatible introducer sheaths.
• Fluoroscope with digital angiographic capabilities (C-arm or fixed unit). Fluoroscopic imaging and the ability to record and recall
all imaging.
• Assorted guidewires of adequate length
• Heparinized saline solution
10.5 Additional recommended equipment
• Introducer sheaths
• Power injector
• Radiopaque ruler with centimeter increments
• Assorted balloon catheters
• Compliant balloon catheters
• Radiopaque contrast media
• Sterile silicone lubricant or sterile mineral oil
• Interventional snare devices
• Endovascular coils and vascular plugs
10.6 MRI information
Nonclinical testing has demonstrated that the Endurant II/IIs stent graft is MR Conditional. A patient with this device can be safely
scanned in an MR system meeting the following conditions:
• Static magnetic field of 1.5 or 3.0 T only
• Maximum spatial gradient magnetic field of 2500 gauss/cm or less
• Maximum MR system reported, whole body averaged specific absorption rate (SAR) of 4 W/kg (First Level Controlled Mode)
Under the scan conditions defined above, the Endurant II/IIs stent graft is expected to produce:
• A maximum temperature rise of 1.00°C after 15 minutes of continuous scanning in a 1.5 T scanner
• and a maximum temperature rise of 3.27°C after 15 minutes of a continuous scanning in a 3.0 T scanner
The image artifact extends approximately 5 mm and 8 mm from the device, both inside and outside the device lumen when scanned
in nonclinical testing using the sequence: spin echo and gradient echo, respectively, in a 3.0 T Siemens TrioTim (VB 13 Software)
MR system with a whole-body coil.
MRI safety and compatibility information for the Heli-FX EndoAnchor system can be found in the instructions for use provided with the
device.
11 Implant instructions
11.1 Vascular access and device preparation
Correct sizing of the aorta and iliac vessels must be determined before implantation of the aortic and iliac stent graft configurations
using contrast-enhanced computer-aided tomography (CT), as well as angiograms of both the iliac arteries and aorta. 3D imaging
may also be beneficial. Refer to Recommended Device Sizing (Section 10.2). These images should be available for review during the
procedure. Vascular instruments and other surgical supplies necessary to gain access to the artery should also be available.
To reduce the risk of thromboembolism, it is recommended that the patient be heparinized for the duration of the procedure.
Caution: Do not retract the graft cover of the delivery system until it is accurately placed within the vasculature and ready for
deployment.
Caution: Never advance or retract equipment from the vasculature without the use of fluoroscopy.
58
Page 59

11.1.1 Vascular access
1. Following aseptic procedure, perform a vascular access at the femoral arteries.
2. Place a guidewire in the ipsilateral femoral artery and advance it above the renal arteries.
3. When implanting a bifurcated stent graft, place a second guidewire via the contralateral side femoral artery and direct to the
abdominal aorta.
4. Over this guidewire, place an angiography catheter above the renal arteries.
5. Take an angiogram.
Note: An additional incision might be necessary to access the common iliac artery.
11.1.2 Device preparation
1. Prior to insertion, view the delivery system under fluoroscopy to visualize the radiopaque markers on the stent graft. The
radiopaque markers indicate the position of the proximal and distal edges of the graft material.
2. Turn the graft cover to align the radiopaque gate marker on the short leg of the bifurcated configuration with the patient’s
contralateral iliac artery.
3. Flush the guidewire lumen with heparinized saline solution.
4. Prior to insertion into the vessel, activate the hydrophilic coating by wiping the outer surface of the graft cover with a sterile gauze,
saturated in saline, until the graft cover is slippery to touch.
11.2 Delivery procedure
Medtronic recommends using an appropriate caliber introducer sheath to perform diagnostic tests. No sheath is necessary for the
introduction of the delivery system or deployment of the stent graft. For infrarenal EVAR procedures using the Heli-FX EndoAnchor
system, the access vessel diameter and morphology should be compatible for use with the device and should accommodate a 16 Fr
introducer sheath.
Caution: Do not remove the guidewire while the delivery system is in the patient.
Warning: To prevent thrombotic problems, a second bolus of IV heparin is recommended before inserting the device.
Note: Section 11.2.1 is applicable to the Endurant II bifurcated, Endurant IIs bifurcated, and Endurant II AUI configurations. Sections
pertinent to only individual main body stent graft configurations will be noted in parentheses.
11.2.1 Introduction of main body configuration
Warning: Do not advance the delivery system without placing a guidewire.
1. Slowly insert the delivery system.
2. Advance over the guidewire so that the proximal most stents and the radiopaque markers are visualized in the target proximal
aortic neck (Figure 9).
3. Inject contrast media through an angiographic (pigtail) catheter into the abdominal aorta and mark the position of the target
location, either on the imaging screen or on the patient’s body.
4. Adjust the position of the stent graft so that the top edge of the graft fabric is just below the lowest renal artery.
Note: The edge of the graft fabric is 0.5 to 1.0 mm above the top edge proximal radiopaque markers.
Note: If the top edge of the graft fabric is to be placed very close to the renal arteries, contrast media may be injected to identify the
location of the lower renal artery and verify the position before full deployment.
Caution: Once proximal position has been identified, do not move the patient or imaging equipment, as it may compromise accuracy
of stent graft placement.
Caution: The angiographic catheter can be removed prior to deployment. However, if the angiographic catheter is not removed until
after deployment, ensure that the tip is straightened (such as with a pigtail catheter) with a guidewire before removal so that the stent
graft is not pulled down.
Caution: When aligning the position of stent graft, be sure the fluoroscope is angled perpendicular to the center line of the infrarenal
aorta to avoid parallax or other sources of visualization error. Some cranial caudal angulation of the image intensifier (I-I) tube may be
necessary, especially if there is anterior angulation of the aneurysm neck.
59
Page 60

Figure 9. Introduction of aortic delivery system
11.2.2 Confirm position (Endurant II and Endurant IIs bifurcated configurations only)
1. Ensure that the distal portion of the contralateral stub leg is above the aortic bifurcation and within the aneurysmal sac, and not
within the iliac vessel.
2. Rotate the handle until the radiopaque marker on the distal stent of the contralateral stub leg is aligned with the contralateral iliac
artery.
Note: When attempting to rotate the system, if the tip does not rotate with the handle, pull back the system and reposition until the
intended position is achieved.
11.2.3 Deploy proximal end of the stent graft configuration
1. With 1 hand on the front grip, hold the delivery system stationary.
2. With the other hand, slowly withdraw the graft cover by rotating the external slider counterclockwise (in the direction of the slider
arrow), until the constrained suprarenal stent is exposed and 2 to 3 of the covered stents have been fully deployed (Figure 10).
3. Use angiography to verify position of the stent graft in relation to the renal arteries.
4. If needed, gently push the entire delivery system proximally or pull distally until the proximal end of the graft material is even with
the distal edge of the lowest renal artery.
Note: In the unlikely event of delivery system failure that results in partial stent graft deployment due to graft cover severance, the
“handle disassembly” technique may permit successful deployment of the stent graft. Refer to Bail-Out Techniques (Chapter 12).
Caution: Do not rotate the graft cover during deployment as this may torque the device and cause it to rotate during deployment.
Caution: If the graft cover is accidentally withdrawn, the stent graft will prematurely deploy and may be incorrectly positioned.
Warning: Failure to properly align the radiopaque markers may result in improper deployment of the stent graft.
60
Page 61

Figure 10. Deploying proximal end of the stent graft configuration
11.2.4 Deploy contralateral leg (Endurant II and Endurant IIs bifurcated configurations only)
Continue holding the front grip of the delivery system stationary and then rotate the slider handle counterclockwise, stopping
immediately after the contralateral leg is released from the graft cover or delivery sheath (Figure 11).
61
Page 62

Figure 11. Deploy the contralateral leg (Endurant II and Endurant IIs bifurcated configurations only)
11.2.5 Deploy covered portion (AUI configuration only)
Continue holding the front grip of the delivery system stationary. Either continue to rotate the slider handle counterclockwise or use
thumb to pull the trigger on the external slider and pull it back until the covered portion of the AUI configuration is completely released
from the graft cover or delivery sheath.
Note: Retract the graft cover past the flexible stent stop tip (approximately 10 mm) to ensure that the graft cover edge does not disturb
the graft position during forward advancement of the catheter for tip recapture.
Caution: When using the trigger to rapidly deploy the stent graft, the delivery system must remain stationary. Do not rotate the delivery
system during stent graft deployment.
62
Page 63

Figure 12. Deploy covered portion (AUI configuration only)
11.2.6 Release proximal end of suprarenal stent
1. Use angiography to verify the position of the stent graft in relation to the renal arteries.
2. Continue to hold the delivery system stationary with 1 hand on the front grip.
3. With the other hand, rotate the back-end wheel clockwise (in the direction of the arrow), moving the tapered tip forward to release
the proximal end of the suprarenal stent (Figure 13).
4. Observe the release of the suprarenal stent under fluoroscopy and continue turning the back-end wheel until it is completely clear
of the delivery system spindle.
Note: In the unlikely event that the proximal end of the suprarenal stent cannot be released, refer to Bail-Out Techniques
(Chapter 12).
Note: As observed in the Endurant IIs bench testing, tip advancement responsiveness may decrease due to excessive bending of the
delivery system in highly tortuous anatomy.
Caution: In the unlikely event that the back-end wheel separates during wheel rotation, remove the wheel. Manually advance the
exposed tabs forward on the screw gear until all of the suprarenal stents release from the spindle. Refer to Bail-Out Techniques
(Chapter 12).
63
Page 64

Figure 13. Release proximal end of suprarenal stent
1 Endurant II bifurcated configuration or Endurant IIs bifurcated configuration
2 Endurant II AUI configuration
11.2.7 Deploy distal end of the stent graft configuration (Endurant II and Endurant IIs bifurcated configurations only)
Either continue to rotate the external slider counterclockwise or while holding the front grip of the delivery system stationary, use thumb
to pull the trigger on the external slider and pull it back until the bifurcated stent graft is completely deployed.
Note: Retract the graft cover past the flexible stent stop tip (approximately 10 mm) to ensure that the graft cover edge does not disturb
the graft position during forward advancement of the catheter for tip recapture.
Caution: When using the trigger to rapidly deploy the stent graft, the delivery system must remain stationary. Do not rotate the delivery
system during stent graft deployment.
64
Page 65

Figure 14. Deploy the distal end of the bifurcated configuration
1 Endurant II bifurcated configuration
2 Endurant IIs bifurcated configuration
11.2.8 Recapture spindle in tapered tip (Endurant II bifurcated and AUI configurations only)
Note: For the Endurant IIs bifurcated configuration, leave the delivery system in situ while deploying the limb stent graft into the
contralateral leg. Skip to Deploy Limb Stent Graft into Contralateral Leg (Section 11.2.10).
1. Continue to hold the delivery system stationary with 1 hand on the front grip.
2. Confirm the spindle has fully separated from the suprarenal stent; gently torque the delivery system if it has not fully separated.
3. Gently rotate the delivery system while pushing the entire delivery system approximately 3 cm proximally so that the tapered tip
and spindle are completely clear of the suprarenal stent.
4. With the other hand, rotate the back-end wheel counterclockwise to recapture the spindle in the tapered tip (Figure 15).
5. Observe the recapture of the spindle within the sleeve of the tapered tip under fluoroscopy.
6. Continue turning the back-end wheel counterclockwise until the spindle has been completely recaptured and the back-end
wheel is at the bottom (Figure 15).
Note: When pushing the delivery system forward, be careful not to displace the distal end of the ipsilateral limb.
Note: Ensure that the suprarenal stent is fully disengaged from the spindle before pushing the delivery system forward.
Note: If the spindle catches on the suprarenal stent during advancement, completely advance the back-end wheel clockwise. Using
a gentle in-and-out motion with the delivery system, rotate the delivery system until the spindle slips past the suprarenal stent. Then
continue with the withdrawal process.
Caution: Stop rotating the back-end wheel when the bottom of the back-end screw gear is reached.
Warning: Failure to adequately advance the delivery system to recapture the spindle can result in the trapping of a suprarenal apex
within the tapered tip sleeve. This will alter the proximal landing zone during delivery system withdrawal.
65
Page 66

Figure 15. Recapture the spindle in the tapered tip
11.2.9 Remove delivery system (Endurant II bifurcated and AUI configurations only)
1. Continue to hold the delivery system stationary with 1 hand on the front grip and the other hand on the external slider.
2. Gently torque and withdraw the delivery system until the spindle is retracted into the fabric portion of the stent graft.
3. Pull back the external slider trigger and hold it stationary while bringing the front grip to the slider (Figure 16).
4. Use continual fluoroscopy and watch the top of the stent graft while slowly pulling back the tapered tip into the graft cover of the
delivery system.
5. Gently remove the delivery system. Use fluoroscopy to ensure that the stent graft does not move during withdrawal.
Note: Maintain vessel access and wire placement until all stent graft configurations are in place.
66
Page 67

Figure 16. Remove delivery system
11.2.10 Deploy limb stent graft configuration Into contralateral leg (Endurant II and Endurant IIs bifurcated configurations only)
1. Prepare the iliac stent graft system as described in Device Preparation (Section 11.1.2).
2. On the patient’s contralateral side, insert a guidewire through the contralateral leg and aortic neck of the previously placed
bifurcated configuration.
3. Place the delivery system over the guidewire and into the contralateral leg of the bifurcated stent graft.
4. Insert the limb stent graft into the contralateral leg of the bifurcated stent graft. The proximal radiopaque marker of the limb should
be aligned to the radiopaque marker at the bifurcation of the bifurcated stent graft configuration (Figure 17). The bifurcated
overlap is shown below for reference purposes only.
5. Ensure there is a 3-stent overlap (Figure 18).
6. With 1 hand on the front grip, hold the delivery system stationary.
7. With the other hand, slowly withdraw the graft cover by rotating the external slider counterclockwise.
8. At any point, pull the slider trigger and pull the external slider all the way back to finish deploying the limb configuration.
9. Remove the delivery system.
67
Page 68

Figure 17. Introduction of the iliac delivery system
1 Endurant II bifurcated configuration
2 Endurant IIs bifurcated configuration
Note: In the unlikely event of delivery system failure that results in partial stent graft deployment, the “handle disassembly” technique
may permit the successful deployment of the stent graft configuration. Refer to Bail-Out Techniques (Chapter 12).
Caution: Do not torque the iliac delivery system while in the patient.
68
Page 69
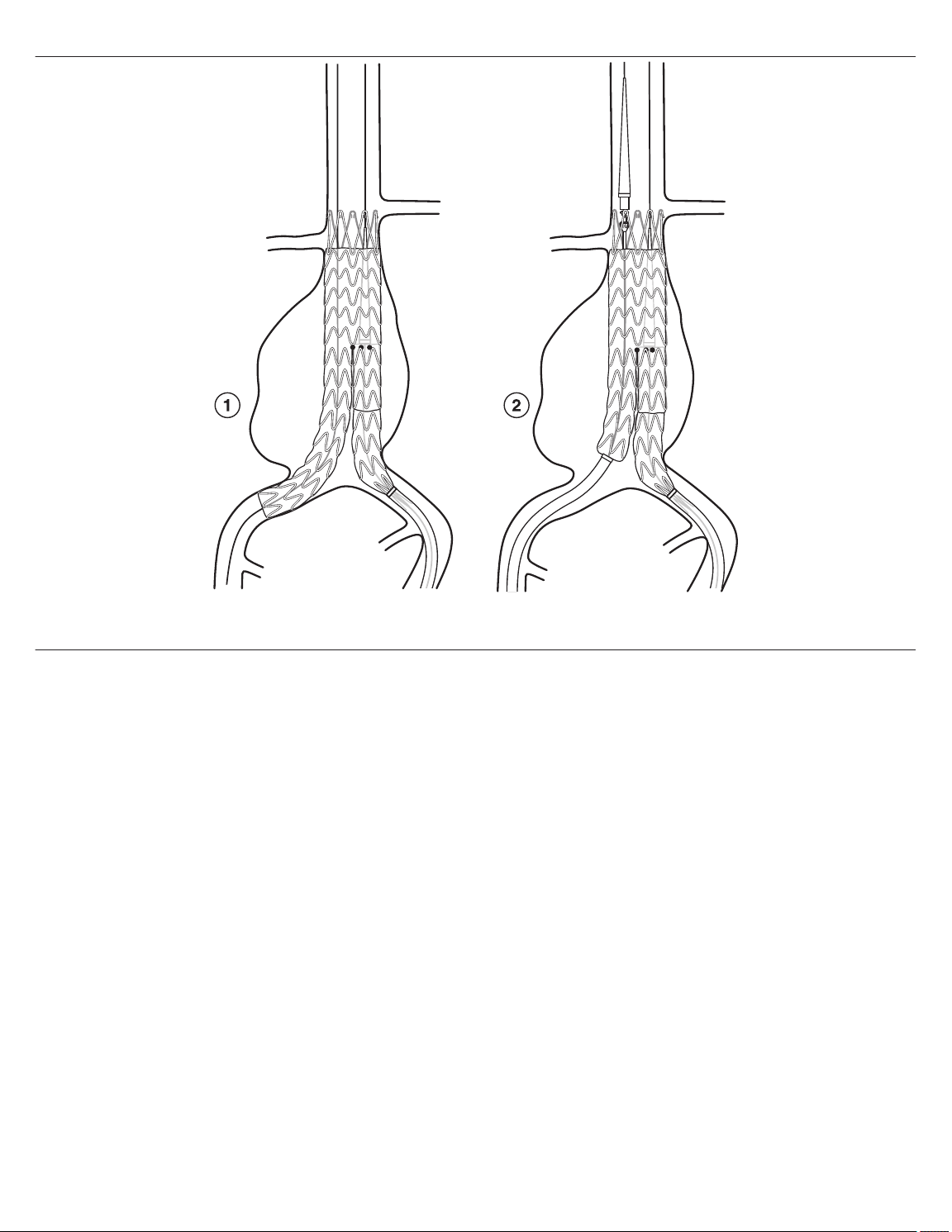
Figure 18. Deploy limb configuration
1 Endurant II bifurcated configuration
2 Endurant IIs bifurcated configuration
11.2.11 Deploy limb stent graft configuration (AUI configuration only)
1. Prepare the iliac stent graft system as described in Device Preparation (Section 11.1.2).
2. Place the delivery system over the guidewire and into the AUI stent graft. The proximal radiopaque marker of the limb stent graft
should be aligned to the radiopaque marker on the AUI stent graft configuration (Figure 19). The AUI and limb stent graft overlap
is shown below for reference purposes only.
69
Page 70

Figure 19. Introduction of iliac delivery system
3. Ensure there is a 3-stent overlap (Figure 20).
4. With 1 hand on the front grip, hold the delivery system stationary.
5. With the other hand, slowly withdraw the graft cover by rotating the external slider counterclockwise.
6. At any point, pull the slider trigger and pull the external slider all the way back to finish deploying the limb configuration.
7. Remove the delivery system.
Note: In the unlikely event of delivery system failure that results in partial stent graft deployment, the “handle disassembly” technique
may permit the successful deployment of the stent graft configuration. Refer to Bail-Out Techniques (Chapter 12).
Caution: Do not torque the iliac delivery system while in the patient.
70
Page 71

Figure 20. Deploy limb configuration
11.2.12 Recapture spindle in tapered tip for delivery system in the ipsilateral leg (Endurant IIs bifurcated configuration only)
1. Hold the delivery system stationary with 1 hand on the front grip.
2. Confirm the spindle has fully separated from the suprarenal stent; gently torque the delivery system if it has not fully separated.
3. Gently rotate the delivery system while pushing the entire delivery system approximately 3 cm proximally so that the tapered tip
and spindle are completely clear of the suprarenal stent.
4. With the other hand, rotate the back-end wheel counterclockwise to recapture the spindle in the tapered tip (Figure 21).
5. Observe the recapture of the spindle within the sleeve of the tapered tip under fluoroscopy.
6. Continue turning the back-end wheel counterclockwise until the spindle has been completely recaptured and the back-end
wheel is at the bottom (Figure 21).
Note: When pushing the delivery system forward, be careful not to displace the distal end of the ipsilateral limb.
Note: Ensure that the suprarenal stent is fully disengaged from the spindle before pushing the delivery system forward.
Note: If the spindle catches on the suprarenal stent during advancement, completely advance the backend wheel clockwise. Using
a gentle in-and-out motion with the delivery system, rotate the delivery system until the spindle slips past the suprarenal stent. Then
continue with the withdrawal process.
Caution: Stop rotating the back-end wheel when the bottom of the back-end screw gear is reached.
Warning: Failure to adequately advance the delivery system to recapture the spindle can result in the trapping of a suprarenal apex
within the tapered tip sleeve. This will alter the proximal landing zone during delivery system withdrawal.
71
Page 72

Figure 21. Recapture spindle in tapered tip
11.2.13 Remove delivery system (Endurant IIs bifurcated configuration only)
1. Continue to hold the delivery system stationary with 1 hand on the front grip and the other hand on the external slider.
2. Gently torque and withdraw the delivery system until the spindle is retracted into the fabric portion of the stent graft.
3. Pull back the external slider trigger and hold it stationary while bringing the front grip to the slider (Figure 22).
4. Use continual fluoroscopy and watch the top of the bifurcated stent graft while slowly pulling back the tapered tip into the graft
cover of the delivery system.
5. Gently remove the delivery system. Use fluoroscopy to ensure that the bifurcated configuration does not move during withdrawal.
Note: Maintain vessel access and wire placement until all stent graft configurations are in place.
72
Page 73

Figure 22. Remove the delivery system
11.2.14 Deploy limb stent graft into ipsilateral leg (Endurant IIs bifurcated configuration only)
1. Prepare the iliac stent graft system as described in Device Preparation (Section 11.1.2).
2. On the patient’s ipsilateral side, track the delivery system over the guidewire and into the ipsilateral leg of the previously placed
bifurcated configuration. The limb stent graft has 2 markers on its proximal edge, 2 markers on its distal edge, and 1 overlap
marker approximately 25 mm distal of the proximal markers.
3. Position the device. Overlap criteria between the limb stent graft and the ipsilateral leg of the bifurcated stent graft is dependent
on limb selection. See Table 70 for recommended device overlap.
Table 70. Recommended device overlap — limb stent graft and ipsilateral leg of Endurant IIs bifurcated stent graft
Proximal diameter Distal diameter Length Overlap Deployment reference
82
93
82
93
82
93
82
93
82
93
3 stents only Refer to step a
16
10
13
20
24
28
73
Page 74

Table 70. Recommended device overlap — limb stent graft and ipsilateral leg of Endurant IIs bifurcated stent graft (continued)
Proximal diameter Distal diameter Length Overlap Deployment reference
124
10
146
156
199
124
13
146
156
199
82
93
16
16
124
146
156
199
3 to 5 stents Refer to step b
124
20
146
156
199
124
24
146
156
199
124
28
146
156
199
a. 3 stent overlap: Align the overlap marker on the limb stent graft with the 2 markers on the distal end of the ipsilateral leg of
the Endurant IIs bifurcated stent graft (Figure 23).
Warning: For the limb stent graft overlap criteria with Endurant IIs ipsilateral leg only, please refer to Table 70. As noted in
Table 70, for the limb stent graft configurations that have an overlap criteria of 3 stents only, do not overlap more than 3 stents.
74
Page 75

Figure 23. Introduction of the iliac delivery system - 3 stent overlap
b. 3 to 5 stent overlap: For the limb stent grafts that can have an overlap between 3 to 5 stents, minimum 3 stent overlap can
be achieved by following the instruction in step a (Figure 23) or the maximum 5 stent overlap can be achieved by aligning the
proximal edge markers on the limb stent graft to the bifurcation marker on the Endurant IIs bifurcated stent graft (Figure 24).
75
Page 76

Figure 24. Introduction of the iliac delivery system - 5 stent overlap
4. With 1 hand on the front grip, hold the delivery system stationary.
5. With the other hand, slowly withdraw the graft cover by rotating the external slide counterclockwise.
6. At any point, pull the slider trigger and pull the external slider all the way back to finish deploying the limb configuration.
7. Remove the delivery system (Section 11.2.13).
Note: In the unlikely event of delivery system failure that results in partial stent graft deployment, the “handle disassembly” technique
may permit the successful deployment of the stent graft configuration. Refer to Chapter 12, Bail-Out Techniques.
Caution: Do not torque the iliac delivery system while in the patient.
11.2.15 Iliac or aortic extension stent graft configurations
1. If an aortic extension stent graft configuration is needed, ensure that there is a minimum 3-stent overlap between the aortic
extension stent graft and the main body stent graft configuration.
2. Follow the main body stent graft deployment process, except rotate the handle to open the extension component entirely before
releasing the proximal end of the suprarenal stent of the aortic configuration.
3. If an iliac extension stent graft configuration is needed, ensure that there is a minimum 3-stent overlap between the iliac extension
and the configuration it is being mated with. This is achieved by aligning the limb overlap marker with the most distal marker on
the configuration with which the extension is being mated.
4. Follow the limb stent graft configuration deployment process (Section 11.2.10).
11.2.16 Smoothing stent graft fabric and modeling stent graft
The Reliant stent graft balloon catheter (packaged separately) can be used to assist in stent graft implantation by modeling the
covered portion of the stent graft and removing wrinkles and folds from the graft material, as needed. Use the balloon catheter to model
the proximal and distal seal zones as well as any overlapping connection (or junction) areas between the stent graft components.
Sub-optimal expansion of the self-expanding stent graft components may also be improved by use of the balloon catheter. Refer to
the Reliant stent graft balloon catheter IFU for specific instructions.
Note: The Reliant stent graft balloon catheter is recommended for use with the Endurant II/IIs stent graft system. Data is not available
for the use of other balloon catheters in remodeling stent grafts.
76
Page 77

Note: When using the Heli-FX EndoAnchor system, Medtronic recommends that the implantation of EndoAnchor implants be done
after the aortic endograft has been placed and any balloon remodeling of the infrarenal seal zone of the stent graft system has been
completed. Exercise care in balloon remodeling of the stent graft system to avoid moving the main body endograft from its intended
implant location.
Note: Care should be taken when inflating the balloon, especially with calcified, tortuous, stenotic, or otherwise diseased vessels.
Inflate slowly. It is recommended that a backup balloon be available.
Caution: Expansion of the balloon outside of the graft material can result in rupture of the aorta, vessel dissection, or graft tears.
Warning: When expanding a vascular prosthesis, there is an increased risk of vessel injury or rupture, and possible patient death, if
the balloon’s proximal and distal radiopaque markers are not completely within the covered (graft fabric) portion of the prosthesis.
Warning: Do not use the Reliant stent graft balloon catheter in the treatment of dissections.
11.2.17 Additional fixation and sealing with the Heli-FX EndoAnchor system (infrarenal neck ≥ 4 mm and < 10 mm in length)
The Heli-FX EndoAnchor system is required when treating short infrarenal necks (≥ 4 mm and < 10 mm in length). For bifurcated
endografts, the recommended minimum number of EndoAnchor implants is based on native vessel diameter and is independent of
the amount of endograft oversizing. For aortic neck diameters ≤29 mm, the recommended minimum number of EndoAnchor implants
is 4. For aortic neck diameters 30 − 32 mm, the recommended minimum number of EndoAnchor implants is 6.
Medtronic recommends that the implantation of EndoAnchor implants be done after the aortic endograft has been placed and any
balloon remodeling of the infrarenal seal zone of the stent graft system has been completed.
1. Prepare the system, per the Heli-FX EndoAnchor system instructions for use.
Note: The access vessel diameter and morphology should be compatible for use with the Heli-FX EndoAnchor system.
2. Implant the EndoAnchor implants per the Heli-FX EndoAnchor system instructions for use.
Note: Stability of the stent graft in short (≥ 4 mm and < 10 mm) infrarenal necks is augmented by the EndoAnchor implants.
Ensure successful deployment of the recommended minimum number of EndoAnchor implants. Where the number of
successfully deployed EndoAnchor implants is below the minimum recommended, there may be greater risk of postoperative
Type 1a endoleak or migration.
Note: Always use fluoroscopy for guidance, delivery, and observation of any Heli-FX system components within the vasculature.
Note: The EndoAnchor implant should be implanted only into areas of aortic tissue that are free of or have insignificant calcified
plaque or thrombus, or where such pathology is diffuse and less than 2 mm in thickness. It should not be used in patients with
bleeding diathesis or with a known hypersensitivity or allergy to the device materials.
3. Remove the system, per the Heli-FX EndoAnchor system instructions for use.
11.2.18 Verify placement and seal
1. At the completion of the procedure, perform angiography to assess the stent graft for proximal and distal endoleaks, to verify
position of the implanted stent graft in relation to the aneurysm and renal arteries, to evaluate implantation of EndoAnchor
implants (if EndoAnchor implants are used) and, if applicable, to assess patency of femoral-femoral bypass graft.
2. Leaks at the attachment or connection sites should be treated using the balloon to remodel the stent graft against the vessel wall.
Note: For an infrarenal EVAR procedure using the Heli-FX EndoAnchor system, additional EndoAnchor implants may be used
to augment the seal.
3. Major leaks that cannot be corrected by re-ballooning may be treated by adding aortic or iliac extension components to the
previously placed stent graft components.
Caution: Any leak left untreated during the implantation procedure must be carefully monitored after implantation.
11.2.19 Seal entry sites
1. Remove ancillary devices prior to repairing the entry site.
2. Repair the entry site with standard closure technique.
12 Bail-Out Techniques
In the unlikely event of Endurant II/IIs delivery system failure, the following bail-out techniques may be used. For information on Heli-FX
EndoAnchor system failure, see the instructions for use provided with the device.
12.1 Screw gear handle disassembly
If partial stent graft deployment due to graft cover severance occurs, the screw gear handle disassembly technique may permit
successful deployment of the stent graft.
77
Page 78

1. Pull back the trigger and fully retract the slider.
2. Stabilize the delivery system.
3. Insert the tips of a pair of hemostats into each of the screw gear handle disassembly ports on the front grip.
4. Disengage the front grip from the screw gear by pressing the tips of the hemostats into the handle disassembly ports
simultaneously advancing the front grip away from the screw gear.
5. Advance the front grip until it fully clears the screw gear.
6. Separate the screw gear halves in order to identify the location of graft cover severance.
7. Manually retract the graft cover with your fingers or with hemostats until the stent graft is fully deployed.
8. Follow the instructions for tip capture deployment and delivery system removal.
12.2 Ballooning
If the captured proximal tip of the suprarenal stent cannot be deployed and the back-end wheel section still works, the ballooning
technique may permit successful deployment of the suprarenal stent.
1. Use a compliant or semi-compliant balloon (Reliant balloon recommended).
2. Insert the balloon and move it to the bifurcated configuration’s aortic section.
3. Inflate the balloon inside the stent graft to vessel size to stabilize stent graft.
4. Follow the instructions for tip capture deployment and delivery system removal.
12.3 Back-end handle disassembly
If no or partial deployment of the proximal end of the suprarenal stent occurs due to back-end wheel failure, the back-end handle
disassembly technique may permit the successful deployment of the suprarenal stent.
1. Use hemostats to depress the exposed tabs to disassemble the back-end wheel.
2. Insert the tips of hemostats into each of the rear handle disassembly ports.
3. Disengage the rear handle by pressing the tips of the hemostats into the handle disassembly ports simultaneously retracting the
rear handle from the delivery system.
4. Stabilize the delivery system.
5. Manually push the exposed tabs of the back-end T-tube to release the suprarenal stent from the spindle.
6. Manually pull back the exposed tabs of the back-end T-tube to recapture the tapered tip after deployment.
7. Follow the instructions for delivery system removal.
8. Hold the exposed tabs of the back-end T-tube so that it remains retracted and the tapered tip recaptured during delivery system
removal.
12.4 Snare the tapered tip
If the back-end handle disassembly technique is unsuccessful due to an excessively high deployment force, the snare the tapered tip
technique may permit successful deployment of the suprarenal stent.
1. Use a snare device.
2. Advance the snare device to the delivery system tapered tip section through upper torso access (for example, brachial).
3. Utilize fluoroscopy to snare the edge of the delivery system tapered tip.
4. Stabilize the delivery system, especially the back-end section.
5. Pull the snare device to separate the suprarenal stent from the tip capture.
6. Manually pull back the back-end T-tube to recapture the tapered tip after deployment.
7. Follow the instructions for delivery system removal.
8. Ensure that the back-end T-tube remains retracted and the tapered tip recaptured during delivery system removal.
13 Follow-up imaging recommendations
13.1 General
Current imaging of stent graft patients includes abdominal X-ray and CT, with and without contrast medium. Alternative imaging
modalities such as magnetic resonance imaging should be used in patients with impaired renal function or intolerance to contrast
media. Determination of imaging techniques should be based on the physician’s clinical assessment of the patient and stent graft
implant, including any adjunct procedures that may have been performed in conjunction with the endovascular stent graft procedure.
After endovascular graft placement, patients should be regularly monitored for perigraft flow, aneurysm growth or changes in the
structure or position of the endovascular graft. Annual imaging is recommended, including 1) abdominal radiographs to examine
device integrity (stent fracture, separation between bifurcated device and proximal cuffs or limb extensions, if applicable), and 2)
78
Page 79

contrast and non-contrast CT to examine aneurysm changes, perigraft flow, patency, tortuosity and progressive disease. If renal
complications or other factors preclude the use of image contrast media, abdominal radiographs and duplex ultrasound may provide
similar information.
Medtronic recommends consideration of an enhanced follow-up schedule for patients with short infrarenal necks (≥ 4 mm and
< 10 mm in length), where the minimum recommended number of EndoAnchor implants were not used.
Additional imaging guidelines for Heli-FX EndoAnchor implants can be found in the instructions for use provided with the device.
13.2 X-ray
Abdominal X-rays should be used to assess the presence of stent graft fracture. Four-view kidney, ureter, bladder (KUB) X-rays should
be taken. Posterior/anterior (PA) and lateral images are recommended for visualization of the stent graft. Ensure the entire device is
captured on images for device assessment.
13.3 CT with contrast
Contrast-enhanced CT should be used to assess stent graft fixation, deformation, apposition to the vessel wall at proximal and distal
fixation sites, stent graft migration, stent graft patency, AAA size, occlusion of branch vessels, and endoleak (including source and
type if present).
A pre-contrast scan of 5 mm thick slices is suggested to determine if there are calcifications or areas where metal artifacts may be
misinterpreted as endoleak. An arterial phase with <3 mm slice thickness and overlapping images with coverage from the celiac artery
to the external iliac is recommended. In aneurysms that are not shrinking and have no apparent endoleak or fixation problems, a late
venous phase scan may be performed. The venous phase scan may also be performed with thicker collimation (5 mm). It is
recommended that the source data set be archived in case specialized evaluation is needed later (volume measurements,
3-dimensional reconstruction, or computer-aided measurement software). If the aneurysm is not shrinking by more than 5 mm within
the first year, volume measurements may be obtained as a more sensitive indicator of AAA size using 3-dimensional software. The
physician will determine the requirement pre-operative care for patients with allergies to contrast.
13.4 Noncontrast CT
For patients with impaired renal function or those who are allergic to contrast medium, a spiral CT without contrast may be considered
to assess stent graft fixation, deformation, apposition to the vessel wall at proximal and distal fixation sites, stent graft migration,
occlusion of vessels, and size of the AAA diameter and volume measurements.
13.5 Duplex ultrasound
For patients with impaired renal function or those who are allergic to contrast medium, a color-duplex ultrasound may be considered
to assess size of AAA diameter, endoleaks, and stent graft occlusion and stenosis. Use duplex ultrasound to assess the patency of
any femoral-femoral artery bypass graft used in conjunction with the AUI device.
13.6 MRI or MRA
Patients with impaired renal function, ie, renal insufficiency, may also be considered for magnetic resonance imaging or angiography
(MRI, MRA) in facilities that have expertise in this area. Artifact may occur related to the stent, and care should be used to insure
adequate imaging of the outer aneurysm wall to assess AAA size. Volume measurement may be helpful if the aneurysm is not clearly
shrinking. If there are concerns regarding imaging of calcified areas, fixation sites, or the outer wall of the aneurysm sac, adjunctive
CT without contrast may be needed.
13.7 Imaging tests
Refer to Table 71 for the recommended follow-up imaging schedule after stent graft implant.
Table 71. Imaging recommendations
Interval Angiogram CTa, b[Contrast & noncon-
trast] Abdominal radiographs
Preprocedure X
Procedural X
1 month X
12 months (annually thereafter) X X
a
CT evaluation may include “3-phase technique,” volume studies, 3-D reconstruction, or computer-aided measurements.
b
If the patient is unable to tolerate a CT with contrast, then a CT without contrast and/or a duplex ultrasound may be considered to assess the
aneurysm and stent graft integrity.
c
Based on the quality of the imaging obtained, an abdominal radiograph may be performed in addition if the imaging was unable to assess stent
graft integrity.
c
79
Page 80

13.8 Supplemental imaging
Note: Additional radiological imaging may be necessary to further evaluate the stent graft in situ based on findings revealed by one
of the surveillance programs. The following recommendations may be considered.
• If there is evidence of poor or irregular position of the stent graft, severe angulation, kinking or migration of the stent graft on
abdominal X-rays, a spiral CT should be performed to assess aneurysm size and the presence or absence of an endoleak.
• If a new endoleak or increase in AAA size is observed by spiral CT, adjunctive studies such as 3-D reconstruction or angiographic
assessment of the stent graft and native vasculature may be helpful in further evaluating any changes of the stent graft or
aneurysm.
• Spiral CT without contrast, MRI or MRA may be considered in select patients who cannot tolerate contrast media or who have
renal function impairment. For centers with appropriate expertise, gadolinium or CO2 angiography may be considered in patients
with renal function impairment requiring angiographic assessment.
14 Additional Surveillance and Treatment
Additional endovascular repair or open surgical aneurysm repair should be considered for patients with evidence of suboptimal stent
graft fixation, proximal endoleak, distal endoleak, junction endoleak, unknown origin of persistent perigraft flow, or increase in AAA
size > 5 mm.
15 Device registration
The Endurant II/IIs stent graft system is packaged with additional specific information which includes:
• Temporary Device Identification Card that includes both patient and stent graft information. Physicians should complete this
card and instruct the patient to keep it in their possession at all times. The patients should refer to this card anytime they visit
additional health practitioners, particularly for additional diagnostic procedures (eg, MRI). This temporary identification card
should only be discarded when the permanent identification card is received.
• Device Tracking Form to be completed by the hospital staff and forwarded to Medtronic for the purposes of tracking all patients
who received an Endurant II/IIs stent graft (as required by Federal Regulation). The hospital’s submission of the device tracking
form to Medtronic is also required for a patient to receive the permanent identification card. Upon receipt of the device tracking
form, Medtronic will mail the patient a permanent device identification card. This card includes important information regarding the
implanted stent graft. Patients should refer to this card anytime they visit health practitioners, particularly for any diagnostic
procedures (eg, MRI). Patients should carry this card with them at all times. If a patient does not receive their permanent device
identification card, or requires changes to the card, call 1-800-551-5544. In addition a patient information booklet (PIB) will be
provided to the physicians during training and additional copies will be available upon request. The PIB will also be available online
on the Medtronic website (www.medtronic.com). This booklet provides patients with basic information on abdominal aortic
aneurysms and endovascular repair therapy.
16 Disclaimer of Warranty
ALTHOUGH THE MEDTRONIC VASCULAR ENDURANT II/ENDURANT IIs STENT GRAFT AND DELIVERY SYSTEM,
HEREAFTER REFERRED TO AS THE ’PRODUCT’, HAS BEEN MANUFACTURED UNDER CAREFULLY CONTROLLED
CONDITIONS, MEDTRONIC, INC., MEDTRONIC VASCULAR, INC. AND THEIR RESPECTIVE AFFILIATES, (COLLECTIVELY
“MEDTRONIC”) HAVE NO CONTROL OVER THE CONDITIONS UNDER WHICH THIS PRODUCT IS USED. THE WARNINGS
CONTAINED IN THE PRODUCT LABELING PROVIDE MORE DETAILED INFORMATION AND ARE CONSIDERED AN INTEGRAL
PART OF THIS DISCLAIMER OF WARRANTY. MEDTRONIC, THEREFORE, DISCLAIMS ALL WARRANTIES, BOTH EXPRESSED
AND IMPLIED, WITH RESPECT TO THE PRODUCT, INCLUDING, BUT NOT LIMITED TO, ANY IMPLIED WARRANTY OF
MERCHANTABILITY OR FITNESS FOR A PARTICULAR PURPOSE. MEDTRONIC SHALL NOT BE LIABLE TO ANY PERSON OR
ENTITY FOR ANY MEDICAL EXPENSES OR ANY DIRECT, INCIDENTAL OR CONSEQUENTIAL DAMAGES CAUSED BY ANY
USE, DEFECT, FAILURE OR MALFUNCTION OF THE PRODUCT, WHETHER A CLAIM FOR SUCH DAMAGES IS BASED UPON
WARRANTY, CONTRACT, TORT OR OTHERWISE. NO PERSON HAS ANY AUTHORITY TO BIND MEDTRONIC TO ANY
REPRESENTATION OR WARRANTY WITH RESPECT TO THE PRODUCT.
The exclusions and limitations set out above are not intended to, and should not be construed so as to contravene mandatory
provisions of applicable law. If any part or term of this Disclaimer of Warranty is held to be illegal, unenforceable or in conflict with
applicable law by a court of competent jurisdiction, the validity of the remaining portions of this Disclaimer of Warranty shall not be
affected, and all rights and obligations shall be construed and enforced as if this Disclaimer of Warranty did not contain the particular
part or term held to be invalid.
80
Page 81

Page 82

Medtronic, Inc.
*M052195T001*
710 Medtronic Parkway
Minneapolis, MN 55432
USA
www.medtronic.com
+1 763 514 4000
LifeLine Technical Support, 24-hour consultation service:
1 877 526 7890
© 2020 Medtronic
M052195T001 AA
2020-10-21
 Loading...
Loading...