

Page 1

CoreValve™ System
Transcatheter Aortic Valve
Delivery Catheter System
Compression Loading System
Caution: Implantation of the Medtronic CoreValve™ system should be performed only by
physicians who have received Medtronic CoreValve™ training.
These devices are supplied sterile for single use only. After use, dispose of the delivery
catheter system and the compression loading system in accordance with local regulations and
hospital procedures. Do not resterilize.
Instructions for Use
Caution: Federal (USA) law restricts this device to sale by or on the order of a physician.
Page 2

Trademarks may be registered and are the property of their respective owners.
Page 3

Sterile LC: Device has been sterilized using Liquid Chemical
Sterilants according to EN/ISO 14160.
Explanation of symbols on package labeling
Use By
Consult Instructions for Use at this Website
Do Not Reuse
Do Not Resterilize
Size
Serial Number
Reorder Number
Lower Limit of Temperature
Quantity
Lot Number
Sterilized Using Ethylene Oxide
Manufactured In
Nonpyrogenic
MR Conditional
Do Not Use if Package is Damaged
Manufacturer
Date of Manufacture
For US Audiences Only
Model
1
Page 4

CoreValve™ Evolut™ bioprosthesis
1.0 Device description
The Medtronic CoreValve™ system consists of 3 components: the transcatheter aortic valve
(bioprosthesis)a, the delivery catheter system (catheter), and the compression loading system
(CLS).
1.1 Transcatheter aortic valve (bioprosthesis)
Figure 1
The bioprosthesis is manufactured by suturing 3 valve leaflets and a skirt, made from a single
layer of porcine pericardium, onto a self-expanding, multi-level, radiopaque frame made of
Nitinol. It is designed to replace the native or surgical bioprosthetic aortic heart valve without
open heart surgery and without concomitant surgical removal of the failed valve. The
bioprosthesis is processed with alpha-amino oleic acid (AOA™), which is an
antimineralization treatment derived from oleic acid, a naturally occurring long-chain fatty
acid.
The bioprosthesis is available for a range of aortic annulus and ascending aorta diameters as
shown in Table 1.
Table 1: Patient anatomical diameters
Bioprosthesis model Size Aortic annulus
diameter
MCS-P4-23-AOA-US 23 mm 17b/18 mm–20 mm ≤34 mm
CoreValve™ bioprosthesis
MCS-P3-26-AOA-US 26 mm 20 mm–23 mm ≤40 mm
MCS-P3-29-AOA-US 29 mm 23 mm–26 mm ≤43 mm
MCS-P3-31-AOA-US 31 mm 26 mm–29 mm ≤43 mm
Ascending aorta
diameter
1.2 Delivery catheter system (catheter)
The catheter with AccuTrak™ stability layer is compatible with a 0.035 in (0.889 mm)
guidewire. The distal (deployment) end of the system features an atraumatic, radiopaque tip
and a capsule that covers and maintains the bioprosthesis in a crimped position. The handle is
a
The terms “bioprosthesis” and “transcatheter aortic valve” are synonymous terms and are used interchangeably
throughout the document to refer to the CoreValve™ device.
b
17 mm for surgical bioprosthetic aortic annulus
2
Page 5

MCS-P3-26-AOA-US,
MCS-P3-31-AOA-US
located on the proximal end of the catheter and is used to load and deploy the bioprosthesis.
The handle includes a macro slider to open and close the capsule and micro knob to facilitate
precise bioprosthesis placement. The micro knob is turned counterclockwise to load the
bioprosthesis and clockwise to deploy the bioprosthesis.
The AccuTrak™ stability layer is fixed at the handle and extends down the outside of the
catheter shaft approximately 91 cm. It provides a barrier between the retractable delivery
catheter system, introducer sheath, and vessel walls, thus enabling the catheter to retract
freely and providing a more stable platform for deployment. The outer diameter of the
catheter is 15 Fr (AccuTrak™ stability layer) and 12 Fr, and the outer diameter of the valve
capsule is 18 Fr (Figure 2). The catheter can be used for femoral, subclavian/axillary, or
ascending aortic (direct aortic) access sites. The catheter is available in 2 different models
(Table 2).
Table 2: Catheter models and system compatibility
Catheter model Corresponding CLS
DCS-C4-18F-23US CLS-3000-18F-US MCS-P4-23-AOA-US
DCS-C4-18F-US CLS-3000-18F-US
1. 112.5 cm
2. 90.9 cm
3. 15 Fr
4. 12 Fr
model
Figure 2
Corresponding
bioprosthesis model(s)
MCS-P3-29-AOA-US,
5. 18 Fr
6. 7.3 cm (Model DCS-C4-18F-US); 6.9 cm (Model DCS-C4-18F-23US)
3
Page 6

1.3 Compression loading system (CLS)
The CLS compresses the bioprosthesis into the catheter. The CLS comprises the following:
Figure 3
1. Inflow tube (straight tube)
2. Outflow cone
3. Outflow cap
4. Outflow tube (tube with flared ends)
5. Inflow cone
4
Page 7

2.0 Indications
The Medtronic CoreValve™ system is indicated for relief of aortic stenosis in patients with
symptomatic heart disease due to severe native calcific aortic stenosis who are judged by a
heart team, including a cardiac surgeon, to be at intermediate or greater risk for open surgical
therapy (i.e., predicted risk of surgical mortality ≥ 3% at 30 days, based on the Society of
Thoracic Surgeons (STS) risk score and other clinical comorbidities unmeasured by the STS
risk calculator).
The Medtronic CoreValve™ system is indicated for use in patients with symptomatic heart
disease due to failure (stenosed, insufficient, or combined) of a surgical bioprosthetic aortic
valve who are judged by a heart team, including a cardiac surgeon, to be at high or greater
risk for open surgical therapy (i.e., STS predicted risk of operative mortality score ≥8% or at
a ≥15% risk of mortality at 30 days).
5
Page 8

3.0 Contraindications
The CoreValve™ system is contraindicated for patients presenting with any of the following
conditions:
• Known hypersensitivity or contraindication to aspirin, heparin (HIT/HITTS) and
bivalirudin, ticlopidine, clopidogrel, Nitinol (Titanium or Nickel), or sensitivity to
contrast media, which cannot be adequately premedicated
• Ongoing sepsis, including active endocarditis
• Preexisting mechanical heart valve in aortic position
6
Page 9

4.0 Warnings and precautions
4.1 Warnings
4.1.1 General
• Implantation of the Medtronic CoreValve™ system should be performed only by
physicians who have received Medtronic CoreValve™ training.
• The transcatheter aortic valve is to be used only in conjunction with the delivery catheter
system and the compression loading system.
• This procedure should only be performed where emergency aortic valve surgery can be
performed promptly.
• Do not use any of the Medtronic CoreValve™ system components if any of the
following has occurred:
• It has been dropped, damaged, or mishandled in any way
• The Use By date has elapsed
• Mechanical failure of the delivery catheter system and/or accessories may result in
patient complications.
4.1.2 Transcatheter aortic valve (bioprosthesis)
• Do not use the bioprosthesis if any of the following conditions is observed:
• There is any damage to the container (e.g., cracked jar or lid, leakage, broken or
missing seals)
• The serial number tag does not match the container label
• The freeze indicator in the secondary package has activated
• The storage solution does not completely cover the bioprosthesis
• Accelerated deterioration of the bioprosthesis may occur in patients presenting with an
altered calcium metabolism.
4.2 Precautions
4.2.1 General
• Do not contact any of the Medtronic CoreValve™ system components with cotton or
cotton swabs.
• Do not expose any of the Medtronic CoreValve™ system components to organic
solvents, such as alcohol.
• Do not introduce air into the catheter.
7
Page 10

• The safety and effectiveness of the Medtronic CoreValve™ system have not been
evaluated in the pediatric population.
• The safety and effectiveness of the bioprosthesis for aortic valve replacement have not
been evaluated in the following patient populations:
• Patients who do not meet the criteria for symptomatic severe native aortic stenosis
as defined below:
• Symptomatic severe high-gradient aortic stenosis: aortic valve area
≤1.0 cm2 or aortic valve area index ≤0.6 cm2/m2, a mean aortic valve
gradient ≥40 mmHg, or a peak aortic-jet velocity ≥4.0 m/s
• Symptomatic severe low-flow/low-gradient aortic stenosis: aortic valve
area ≤1.0 cm2 or aortic valve area index ≤0.6 cm2/m2; a mean aortic valve
gradient <40 mmHg; and a peak aortic-jet velocity <4.0 m/s
• Who are at low surgical risk (predicted perioperative mortality risk of <3%)
• With untreated, clinically significant coronary artery disease requiring
revascularization
• With a preexisting prosthetic heart valve with a rigid support structure in either
the mitral or pulmonic position if either the preexisting prosthetic heart valve
could affect the implantation or function of the bioprosthesis or the implantation
of the bioprosthesis could affect the function of the preexisting prosthetic heart
valve
• With cardiogenic shock manifested by low cardiac output, vasopressor
dependence, or mechanical hemodynamic support
• The safety and effectiveness of a CoreValve™ bioprosthesis implanted within a failed
preexisting transcatheter bioprosthesis have not been demonstrated.
• Implanting a CoreValve™ bioprosthesis in a degenerated surgical bioprosthesis
(transcatheter aortic valve in surgical aortic valve [TAV in SAV]) should be avoided in
the following conditions. The degenerated surgical bioprosthesis presents with a:
• Significant concomitant perivalvular leak (between the prosthesis and the native
annulus), is not securely fixed in the native annulus, or is not structurally intact
(e.g., wireform frame fracture)
• Partially detached leaflet that in the aortic position may obstruct a coronary
ostium
• Stent frame with a manufacturer’s labeled inner diameter <17 mm
• The safety and effectiveness of the bioprosthesis for aortic valve replacement have not
been evaluated in patient populations presenting with the following:
• Blood dyscrasias as defined: leukopenia (WBC <1000 cells/mm3),
thrombocytopenia (platelet count <50,000 cells/mm3), history of bleeding
diathesis or coagulopathy, or hypercoagulable states
8
Page 11

• Congenital bicuspid or unicuspid valve verified by echocardiography
• Mixed native aortic valve disease (aortic stenosis and aortic regurgitation with
predominant aortic regurgitation [3-4+])
• Moderate to severe (3-4+) or severe (4+) mitral or severe (4+) tricuspid
regurgitation
• Hypertrophic obstructive cardiomyopathy
• New or untreated echocardiographic evidence of intracardiac mass, thrombus, or
vegetation
• Native aortic annulus size <18 mm or >29 mm per the baseline diagnostic
imaging or surgical bioprosthetic aortic annulus size <17 mm or >29 mm
• Transarterial access not able to accommodate an 18 Fr sheath
• Sinus of valsalva anatomy that would prevent adequate coronary perfusion
• Moderate to severe mitral stenosis
• Severe ventricular dysfunction with left ventricular ejection fraction (LVEF)
<20% as measured by resting echocardiogram
• Symptomatic carotid or vertebral artery disease
• Severe basal septal hypertrophy with an outflow gradient
• Do not expose the bioprosthesis to solutions other than the storage and rinse solutions.
• Do not add antibiotics or any other substance to either the storage or rinse solutions. Do
not apply antibiotics or any other substance to the bioprosthesis.
• Do not allow the bioprosthesis to dry. Maintain tissue moisture with irrigation or
immersion.
• Do not attempt to repair a damaged bioprosthesis.
• Do not handle or use forceps to manipulate the bioprosthesis leaflet tissue.
• Do not deform the bioprosthesis in excess of what is experienced during crimping,
loading, and implantation.
4.2.2 Prior to use
• Exposure to glutaraldehyde may cause irritation of the skin, eyes, nose, and throat. Avoid
prolonged or repeated exposure to the vapors. Use only with adequate ventilation. If skin
contact occurs, immediately flush the affected area with water (minimum of 15 minutes).
In the event of eye contact, flush with water for a minimum of 15 minutes and seek
medical attention immediately.
• The bioprosthesis and the glutaraldehyde storage solution are sterile. The outside of the
bioprosthesis container is nonsterile and must not be placed in the sterile field.
• Damage may result from forceful handling of the catheter. Prevent kinking of the catheter
when removing it from the packaging.
9
Page 12

• This device was designed for single patient use only. Do not reuse, reprocess, or
resterilize this product. Reuse, reprocessing, or resterilization may compromise the
structural integrity of the device and/or create a risk of contamination of the device,
which could result in patient injury, illness, or death.
• The bioprosthesis size must be appropriate to fit the patient’s anatomy. Proper sizing of
the device is the responsibility of the physician. Refer to Table 1 for available sizes.
Failure to implant a device within the sizing matrix could lead to adverse effects such as
those listed in Section 5.0.
• Patients must present with access vessel diameters of ≥6 mm or an ascending aortic
(direct aortic) access site ≥60 mm from the basal plane.
• Implantation of the bioprosthesis should be avoided in patients with aortic root angulation
(angle between plane of aortic valve annulus and horizontal plane/vertebrae) of >30° for
right subclavian/axillary access or >70° for femoral and left subclavian/axillary access.
• Use caution when using the subclavian/axillary approach in patients with a patent Left
Internal Mammary Artery (LIMA) graft (for left subclavian/axillary approach only) or
patent Right Internal Mammary Artery (RIMA) graft (for right subclavian/axillary
approach only).
• For direct aortic access, ensure the access site and trajectory are free of patent RIMA or a
preexisting patent RIMA graft.
4.2.3 During use
• Adequate rinsing of the bioprosthesis with sterile saline, as described in the Instructions
for Use, is mandatory before implantation. No other solutions, drugs, chemicals, or
antibiotics should ever be added to the glutaraldehyde or rinse solutions as irreparable
damage to the leaflet tissue, which may not be apparent under visual inspection, may
result.
• During rinsing, do not touch the leaflets or squeeze the bioprosthesis.
• With the exception of attaching the bioprosthesis frame loops to the catheter tabs, do not
touch the capsule or the transition between the capsule and the catheter shaft. To protect
the capsule, handle the catheter using the catheter shaft or, during loading, the loading
tools.
• If a capsule becomes damaged during loading or the capsule fails to close, replace the
entire system (bioprosthesis, catheter, and CLS). Do not use a catheter with a damaged
capsule.
• Prevent contamination of the bioprosthesis, its storage solution, the catheter, and the CLS
with glove powder.
• After a bioprosthesis has been inserted into a patient, do not attempt to reload that
bioprosthesis on the same or any other catheter.
• During implantation, if resistance to deployment is encountered (e.g., the micro knob
starts clicking or is tight or stuck), apply upward pressure to the macro slider while
10
Page 13

turning the micro knob. If the bioprosthesis still does not deploy, remove it from the
patient and use another system.
• While the catheter is in the patient, ensure the guidewire is extending from the tip. Do not
remove the guidewire from the catheter while the catheter is inserted in the patient.
• Once deployment is initiated, retrieval of the bioprosthesis from the patient (e.g., use of
the catheter) is not recommended. Retrieval of a partially deployed valve using the
catheter may cause mechanical failure of the delivery catheter system, aortic root
damage, coronary artery damage, myocardial damage, vascular complications, prosthetic
valve dysfunction (including device malposition), embolization, stroke, and/or emergent
surgery.
• During deployment, the bioprosthesis can be advanced or withdrawn as long as annular
contact has not been made. Once annular contact is made, the bioprosthesis cannot be
advanced in the retrograde direction; if necessary, and the frame has only been deployed
≤2/3 of its length, the bioprosthesis can be withdrawn (repositioned) in the antegrade
direction. However, use caution when moving the bioprosthesis in the antegrade
direction.
• Use the handle of the delivery system to reposition the bioprosthesis. Do not use the outer
catheter sheath.
• Once deployment is complete, repositioning of the bioprosthesis (e.g., use of a snare
and/or forceps) is not recommended. Repositioning of a deployed valve may cause aortic
root damage, coronary artery damage, myocardial damage, vascular complications,
prosthetic valve dysfunction (including device malposition), embolization, stroke, and/or
emergent surgery.
• Do not attempt to retrieve a bioprosthesis if any one of the outflow struts is protruding
from the capsule. If any one of the outflow struts has deployed from the capsule, the
bioprosthesis must be released from the catheter before the catheter can be withdrawn.
• Ensure the capsule is closed before catheter removal. If increased resistance is
encountered when removing the catheter through the introducer sheath, do not force
passage. Increased resistance may indicate a problem and forced passage may result in
damage to the device and/or harm to the patient. If the cause of resistance cannot be
determined or corrected, remove the catheter and introducer sheath as a single unit over
the guidewire, and inspect the catheter and confirm that it is complete.
• Clinical long-term durability has not been established for the bioprosthesis. Evaluate
bioprosthesis performance as needed during patient follow-up.
• Postprocedure, administer appropriate antibiotic prophylaxis as needed for patients at risk
for prosthetic valve infection and endocarditis.
• Postprocedure, administer anticoagulation and/or antiplatelet therapy per hospital
protocol.
• Excessive contrast media may cause renal failure. Preprocedure, measure the patient’s
creatinine level. During the procedure, monitor contrast media usage.
11
Page 14

• Conduct the procedure under fluoroscopy. Fluoroscopic procedures are associated with
the risk of radiation damage to the skin, which may be painful, disfiguring, and longterm.
• The safety and efficacy of a CoreValve™ bioprosthesis implanted within a transcatheter
bioprosthesis have not been demonstrated. However, in the event that a CoreValve™
bioprosthesis must be implanted within a transcatheter bioprosthesis to improve valve
function, valve size and patient anatomy must be considered before implantation of the
CoreValve™ bioprosthesis to ensure patient safety (e.g., to avoid coronary obstruction).
• In the event that valve function or sealing is impaired due to excessive calcification or
incomplete expansion, a postimplant balloon dilatation of the bioprosthesis may improve
valve function and sealing. To ensure patient safety, valve size and patient anatomy must
be considered when selecting the size of the balloon used for dilatation. The balloon size
chosen for dilatation should not exceed the diameter of the native aortic annulus or, for
surgical bioprosthetic valves, the manufacturer’s labeled inner diameter. Refer to the
specific balloon catheter manufacturer’s compliance chart to ensure that the applied
inflation pressure does not result in a balloon diameter that exceeds the indicated annulus
range for the bioprosthesis. Refer to the specific balloon catheter manufacturer’s labeling
for proper instruction on the use of balloon catheter devices. Note: Bench testing has only
been conducted to confirm compatibility with NuMED Z-MED II™ Balloon Aortic
Valvuloplasty catheters where CoreValve™ bioprosthesis device performance was
maintained after dilatation. Data on file.
4.3 Magnetic resonance imaging (MRI)
MRI may be used on the bioprosthesis only under specific conditions. See Section 6.2 MRI
safety information for more information.
12
Page 15

5.0 Potential adverse events
Potential risks associated with the implantation of the Medtronic CoreValve™ transcatheter
aortic valve may include, but are not limited to, the following:
• Death
• Cardiac arrest
• Coronary occlusion, obstruction, or vessel spasm (including acute coronary closure)
• Emergent surgery (e.g., coronary artery bypass, heart valve replacement, valve explant)
• Multi-organ failure
• Heart failure
• Myocardial infarction
• Cardiogenic shock
• Respiratory insufficiency or respiratory failure
• Cardiovascular injury (including rupture, perforation, or dissection of vessels, ventricle,
myocardium, or valvular structures that may require intervention)
• Ascending aorta trauma
• Cardiac tamponade
• Cardiac failure or low cardiac output
• Prosthetic valve dysfunction including, but not limited to, fracture; bending (out-of-round
configuration) of the valve frame; under-expansion of the valve frame; calcification;
pannus; leaflet wear, tear, prolapse, or retraction; poor valve coaptation; suture breaks or
disruption; leaks; mal-sizing (prosthesis-patient mismatch); malposition (either too high
or too low)/malplacement; regurgitation; stenosis
• Thrombosis/embolus (including valve thrombosis)
• Valve migration/valve embolization
• Ancillary device embolization
• Emergent percutaneous coronary intervention (PCI)
• Emergent balloon valvuloplasty
• Major or minor bleeding that may or may not require transfusion or intervention
(including life-threatening or disabling bleeding)
• Allergic reaction to antiplatelet agents, contrast medium, or anesthesia
• Infection (including septicemia and endocarditis)
• Stroke, transient ischemic attack (TIA), or other neurological deficits
• Permanent disability
13
Page 16

• Renal insufficiency or renal failure (including acute kidney injury)
• Mitral valve regurgitation or injury
• Tissue erosion
• Vascular access related complications (e.g., dissection, perforation, pain, bleeding,
hematoma, pseudoaneurysm, irreversible nerve injury, compartment syndrome,
arteriovenous fistula, stenosis)
• Conduction system disturbances (e.g., atrioventricular node block, left-bundle branch
block, asystole), which may require a permanent pacemaker
• Cardiac arrhythmias
• Encephalopathy
• Pulmonary edema
• Pericardial effusion
• Pleural effusion
• Myocardial ischemia
• Peripheral ischemia
• Bowel ischemia
• Heart murmur
• Hemolysis
• Cerebral infarction-asymptomatic
• Non-emergent reoperation
• Inflammation
• Fever
• Hypotension or hypertension
• Syncope
• Dyspnea
• Anemia
• Angina
• Abnormal lab values (including electrolyte imbalance)
14
Page 17

6.0 Patient information
6.1 Registration information
A patient registration form is included in each bioprosthesis package. After implantation,
please complete all requested information. The serial number is located on both the package
and the identification tag attached to the bioprosthesis. Return the original form to the
Medtronic address indicated on the form and provide the temporary identification card to the
patient prior to discharge.
Medtronic will provide an Implanted Device Identification Card to the patient. The card
contains the name and telephone number of the patient’s physician as well as information
that medical personnel would require in the event of an emergency. Patients should be
encouraged to carry this card with them at all times.
6.2 MRI safety information
Nonclinical testing and modeling have demonstrated that the Medtronic CoreValve™
bioprosthesis is MR Conditional. A patient with this device can be safely scanned in an MR
system meeting the following conditions:
• Static magnetic field of 1.5 T and 3.0 T
• Maximum spatial gradient magnetic field of 2500 gauss/cm (25 T/m)
• Maximum MR system reported, whole body averaged specific absorption rate (SAR) of
≤2.0 W/kg (Normal Operating Mode)
Based on nonclinical testing and modeling, under the scan conditions defined above, the
Medtronic CoreValve™ bioprosthesis is expected to produce a maximum in vivo
temperature rise of less than 3.6˚C after 15 minutes of continuous scanning. Based on
nonclinical data, the image artifact caused by the device will extend no greater than 7 mm
from the Medtronic CoreValve™ bioprosthesis when imaged with a gradient echo pulse
sequence and a 3.0 T MRI system.
Scanning under the conditions defined above may be performed immediately after
implantation.
The presence of other implants or medical circumstances of the patient may require lower
limits on some or all of the above parameters. For deployment of a Medtronic CoreValve™
bioprosthesis inside of a failed surgical bioprosthetic valve, consult the MRI labeling
pertaining to the failed valve for additional artifact information.
15
Page 18

7.0 How supplied
7.1 Packaging
The bioprosthesis is supplied sterile and nonpyrogenic in a sealed container made of glass
and a screw cap with a liner. The outside of the container is nonsterile and must not be
placed in the sterile field. A freeze indicator is placed inside the labeled carton. If the freeze
indicator has been activated, do not use the bioprosthesis.
The catheter is packaged in a double-pouch configuration and sterilized with ethylene oxide
gas. The catheter is sterile if the pouches are undamaged and unopened. The outer surfaces of
the outer pouch are nonsterile and must not be placed in the sterile field.
The CLS is packaged in a double-pouch configuration. The CLS is sterile if the pouches are
undamaged and unopened. The outer surfaces of the outer pouch are nonsterile and must not
be placed in the sterile field. The CLS is sterilized with ethylene oxide gas.
7.2 Storage
Store the bioprosthesis at room temperature. Avoid exposing to extreme fluctuations of
temperature. Avoid freezing. Appropriate inventory control should be maintained so that
bioprostheses with earlier Use By dates are implanted preferentially. Store the catheter and
CLS in a cool, dry environment.
16
Page 19

8.0 Additional equipment
Note: While extensive, this equipment list is not meant to cover all possible scenarios.
Transesophogeal echocardiogram (TEE) or transthoracic echocardiography (TTE) on
standby
Temporary pacer insertion
• Temporary pacemaker catheter (4 Fr or 5 Fr), per hospital protocol
• Sterile sleeve for pacemaker catheter
• Hemostatic vessel introducer sheath
• Temporary pacemaker generator
• Sterile temporary pacemaker-to-generator cable
If indicated, pulmonary artery catheter insertion
• Standard pulmonary artery catheter
• Hemostatic vessel introducer sheath
• Saline flush line connected to pressure transducer
Baseline aortography via radial, brachial, or femoral approach
• 5 Fr or 6 Fr pigtail angiographic catheter
• 6 Fr hemostatic vessel introducer sheath
• 2-port manifold with saline flush line and pressure tubing or transducer
• Power injector syringe
• Contrast media
• High-pressure power injector tubing
Predilatation of implant site
• 2-port manifold with saline flush and transducer
• 9 Fr and 18 Fr hemostatic vessel introducer sheaths
• Standard length 0.035 in (0.889 mm) straight guidewire
• Appropriate suture-mediated closure system, if applicable
• Angiographic catheter
• 0.035 in (0.889 mm) × 260 cm standard high-support guidewire to be shaped with a
pigtail loop
• Balloon valvuloplasty catheters, ≤4 cm length × 18 mm, 20 mm, 22 mm or 23 mm, and
25 mm diameters
• Inflation device or syringe and diluted 1:5 contrast media
17
Page 20

Bioprosthesis implantation
• 18 Fr hemostatic vessel introducer sheath
Standby supplies (must be available in the room)
• Pericardiocentesis tray
• 35 mm × 120 cm single loop snare
• Standard percutaneous coronary intervention (PCI) equipment
• 14 Fr and 16 Fr hemostatic vessel introducer sheaths
• Standard cardiac catheterization lab equipment
• Intra-aortic balloon pump (IABP)
18
Page 21

9.0 Instructions for use
Figure 4
1. Catheter tip
2. Capsule
3. Catheter shaft
4. Tube flush port
5. AccuTrak™ stability layer
6. Macro slider
7. Micro knob
8. Luer-lock connection flush port
9.1 Inspection and bioprosthesis loading procedure
Caution: Once the bioprosthesis is removed from its container and the catheter and CLS are
removed from their packaging, ensure all subsequent procedures are performed in a sterile
field.
9.1.1 Inspection prior to use
1. Before removing the bioprosthesis, catheter, or CLS from its primary packaging,
carefully inspect the packaging for any evidence of damage that could compromise
the sterility or integrity of the device (e.g., cracked jar or lid, leakage, broken or
missing seals, torn or punctured pouch).
Caution: Do not use the product if there is evidence of damage.
2. Inspect the temperature indicator located within the packaging for the bioprosthesis to
ensure it has not been activated.
Caution: Do not use the bioprosthesis if the temperature indicator has been activated.
9.1.2 Preparation of the catheter
3. Wipe the length of the catheter with a moist (saline) gauze.
4. Use the micro knob and macro slider on the handle to open and close the catheter
(Figure 4).
19
Page 22

5. Attach a stopcock to the first flush port. Attach a 10 mL syringe filled with saline to
the stopcock on the first flush port and flush. Repeat step for the second flush port on
the catheter (Figure 5).
Figure 5
6. Verify no catheter leakage is observed during any of the flushing steps. If leakage is
observed, use a new system.
7. Attach a 10 mL syringe filled with saline to the third flush port on the handle on the
catheter (Figure 6) and flush.
Figure 6
8. Fill a loading bath with cold, sterile saline (0°C to 8°C [32°F to 46°F]), and place the
CLS components in the bath.
9.1.3 Bioprosthesis rinsing procedure
9. Fill each of 3 rinsing bowls with approximately 500 mL of fresh, sterile saline at
ambient temperature (15°C to 25°C [59°F to 77°F]).
10. Confirm the integrity of the primary bioprosthesis container. Open the container and
remove the bioprosthesis by carefully grasping one of the frame loops. Let any
remaining solution drain from the bioprosthesis completely.
Caution: The bioprosthesis should not be handled or manipulated with sharp or
pointed objects. Use atraumatic blunt-tipped forceps only. Do not use the forceps to
grasp the tissue portion of the bioprosthesis.
Note: Retain the container with the original solution. It may be needed to store and
return a rejected bioprosthesis.
11. Compare the serial number on the container with the serial number on the tag
attached to the bioprosthesis.
Caution: If the serial numbers do not match, Do not use the bioprosthesis.
20
Page 23

12. Carefully remove the serial number tag from the bioprosthesis and retain the tag.
Ensure that the suture that was used to secure the serial number tag to the
bioprosthesis is completely removed from the bioprosthesis.
13. Immerse the entire bioprosthesis in a sterile rinsing bowl.
14. Gently agitate the bioprosthesis by hand for 2 minutes to remove the glutaraldehyde
from the bioprosthesis.
15. Repeat steps 13 and 14 in each of the 2 remaining rinsing bowls to ensure complete
removal of glutaraldehyde from the bioprosthesis.
16. Leave the bioprosthesis submerged in sterile saline until it is ready to be loaded.
9.1.4 Bioprosthesis loading procedure
Caution: Rapid capsule advancement can contribute to difficulties with loading the valve.
Slowly advancing the capsule helps facilitate successful loading.
Caution: With the exception of attaching the bioprosthesis frame loops to the catheter tabs,
do not touch the capsule or the transition between the capsule and the catheter shaft. To
protect the capsule, handle the catheter using the catheter shaft or, during loading, the loading
tools.
Note: If a capsule becomes damaged during loading or the capsule fails to close, replace the
entire system (bioprosthesis, catheter, and CLS). Do not use a catheter with a damaged
capsule.
Perform the bioprosthesis loading procedure while the bioprosthesis, CLS, capsule, and
catheter tip are immersed in cold, sterile saline (0°C to 8°C [32°F to 46°F]).
17. To open the capsule, activate the macro slider and slide back.
18. Submerge and cool the bioprosthesis in a bath filled with cold, sterile saline.
19. Advance the outflow tube (tube with flared ends) over the catheter shaft toward the
handle (Figure 7).
Figure 7
20. Gently squeeze the outflow part of the cold bioprosthesis frame and insert it into the
outflow cone (Figure 8).
Note: As applicable, all subsequent bioprosthesis loading steps should be performed
under chilled (0°C to 8°C [32°F to 46°F]) saline.
21
Page 24

Figure 8
21. Slowly continue to insert the frame into the outflow cone.
22. Once the bioprosthesis is fully inserted, secure the outflow cap onto the outflow cone
(Figure 9).
Figure 9
23. Carefully insert the inflow tube (straight tube) into the outflow cap (Figure 10).
Figure 10
24. Gently continue to advance the inflow tube until the bioprosthesis frame loops begin
to separate.
25. Insert the distal catheter tip into the inflow tube (Figure 11).
Figure 11
Note: The distal end of the catheter (Figure 11) may look slightly different from the
figures in Section 9.0. The functionality of the catheter is the same.
26. Carefully withdraw the inflow tube and attach the exposed frame loops to the catheter
tabs (Figure 12).
22
Page 25

Figure 12
27. Rotate the micro knob to advance the capsule to cover the bioprosthesis frame loops
and the top of the outflow struts (Figure 13).
Note: Ensure that the capsule has covered all of the outflow struts and the
bioprosthesis frame loops are securely attached to the catheter tabs.
Figure 13
28. Advance the outflow tube over the radiopaque marker band of the capsule prior to
advancing the capsule further (Figure 14).
Figure 14
29. Remove the outflow cap and inflow tube from the outflow cone (Figure 15).
Figure 15
30. Move the outflow cone away from the bioprosthesis over the catheter toward the
handle.
31. Advance the inflow cone over the bioprosthesis using the outflow tube (Figure 16).
23
Page 26

Figure 16
Note: Ensure the bioprosthesis frame axis is visually aligned (coaxial) with the inflow
cone axis during the insertion of the bioprosthesis into the inflow cone (Figure 17).
Complete the insertion of the bioprosthesis into the inflow cone in one uninterrupted
movement.
Figure 17
1. Inflow cone axis
2. Bioprosthesis frame axis
32. Continue to advance the bioprosthesis into the inflow cone until the outflow tube
contacts the inside of the inflow cone (Figure 18).
Figure 18
33. Visually inspect the bioprosthesis within the inflow cone to verify there is no crease
or infold in the frame beyond the second node from the inflow end. Ensure inspection
is performed circumferentially around the entire bioprosthesis.
Caution: If a crease or infold greater than 2 nodes long is noticed, do not use the
bioprosthesis or catheter. Prepare a new bioprosthesis to load into a new catheter.
24
Page 27

Figure 19
Figure 20
34. Hold the inflow cone against the outflow tube while slowly advancing the capsule
over the bioprosthesis until the capsule comes within approximately 5 mm of the
catheter tip (Figure 21). If the micro knob clicks, apply upward pressure to the macro
slider and continue turning the micro knob (Figure 22).
Figure 21 Figure 22
35. With the catheter tip submerged in cold saline, flush both tube flush ports with saline.
36. Slowly advance the capsule over the bioprosthesis until the capsule contacts the
catheter tip.
37. If the micro knob has fully advanced the capsule and a small gap remains between the
end of the capsule and the catheter tip, stabilize the handle with one hand; position the
other hand on the blue catheter shaft and gently advance the capsule manually to
close the gap between the capsule and the catheter tip (Figure 23).
Figure 23
38. Remove the outflow cone and outflow tube from the catheter (Figure 24).
Figure 24
25
Page 28

39. Conduct a final visual inspection of the loaded bioprosthesis to make sure the frame is
free of creases or infolds beyond the second node from the inflow end. Ensure
inspection is performed circumferentially around the entire bioprosthesis.
Caution: If a crease or infold greater than 2 nodes long is noticed, do not use the
bioprosthesis or catheter. Prepare a new bioprosthesis to load into a new catheter.
Figure 25
40. Leave the loaded bioprosthesis submerged in cold saline until implantation.
Figure 26
9.2 Bioprosthesis implantation
9.2.1 Vascular access
Note: Vascular access should be achieved per hospital protocol (either percutaneously or via
surgical cutdown).
Note: The primary access artery will be used to introduce the CoreValve™ system and, if
predilatation is performed, the balloon catheter; the secondary access artery will be used to
introduce the reference pigtail.
1. Establish a central venous line. Insert a 4 Fr or 5 Fr temporary pacemaker catheter via
the right internal jugular vein (or other appropriate access vessel) per hospital
protocol.
2. Insert a 6 Fr introducer sheath into the secondary access artery.
3. Insert an 18 Fr introducer sheath into the primary access artery.
4. Administer anticoagulant according to hospital protocol. If heparin is administered as
an anticoagulant, check the activated clotting time (ACT) after initial bolus of heparin
and recheck every 30 minutes thereafter. Maintain ACT ≥250 seconds.
Note: Anticoagulant may be administered at any time prior to this point, but avoid
delaying beyond this point.
9.2.2 Crossing the valve
5. Advance the graduated pigtail catheter to the ascending aorta and position the distal
tip in the noncoronary cusp of the aortic valve.
6. Identify the ideal annular viewing plane using contrast injections at various
angiographic angles.
Note: It is recommended that a dedicated individual prepare and operate the contrast
injector.
26
Page 29

7. Insert an angiographic catheter over a standard J-tip guidewire into the primary access
sheath and advance to the ascending aorta.
8. Exchange the J-tip guidewire for a 0.035 in (0.889 mm) straight-tip guidewire.
Advance the straight-tip guidewire across the aortic valve into the left ventricle (LV).
9. After crossing the aortic valve with the guidewire, advance the angiographic catheter
into the LV.
10. Exchange the straight-tip guidewire for an exchange-length J-tip guidewire.
11. Exchange the angiographic catheter for a 6 Fr pigtail catheter.
12. Remove the guidewire and connect the catheter to the transducer. Using both
catheters, record the aortic pressure gradient.
13. Using a right anterior oblique (RAO) projection, advance the previously pigtail-
shaped, 0.035 in (0.889 mm) high-support guidewire through the pigtail catheter and
position in the apex of the LV.
14. Remove the pigtail catheter while maintaining guidewire position in the LV.
9.2.3 Predilatation of the implant site
Note: The need for predilatation of the native valve is determined by the heart team.
Information for failed surgical bioprostheses: Balloon predilatation of a stenotic surgical
aortic bioprosthesis has not been evaluated. In cases where there is severe stenosis,
predilatation of the surgical aortic bioprosthesis may be done at the discretion of the heart
team and the steps used are identical to native valve predilatation.
15. Insert the valvuloplasty balloon through the 18 Fr introducer sheath and advance it to
the ascending aorta.
16. Reposition the angiographic equipment to the ideal viewing plane. Position the
valvuloplasty balloon across the valve, while maintaining strict fluoroscopic
surveillance of the distal tip of the guidewire in the LV.
17. Perform balloon valvuloplasty per hospital protocol and remove the valvuloplasty
balloon while maintaining guidewire position across the aortic valve.
9.2.4 Deployment
18. Insert the device over the 0.035 in (0.889 mm) guidewire and into the introducer
sheath with the macro slider facing upward. Advance the device while maintaining
strict fluoroscopic surveillance of the guidewire in the LV.
19. When crossing the aortic arch, it is critical that the guidewire is controlled to prevent
it from moving forward. Without proper management of the distal tip of the
guidewire, the guidewire could move forward and cause trauma to the LV.
20. Advance the device through the valve. Perform an angiogram to confirm that the
pigtail catheter is in position within the noncoronary cusp of the aortic root.
Fluoroscopically identify the appropriate landmarks.
27
Page 30

Figure 27
21. Note the radiopaque bands (Figure 28). Follow the diagrams in Figure 29 and
Figure 32 for the optimal placement of the bioprosthesis. The bioprosthesis should be
placed so that the skirt is within the aortic annulus (approximately 4 mm to 6 mm
below the annulus). In native anatomy, the annulus is defined as the angiographic
floor of the aortic root. For surgical bioprostheses, consider the features of the valve
when determining the optimal placement.
Figure 28 Figure 29
22. After attaining optimal catheter position, slowly turn the micro knob and begin to
deploy the bioprosthesis. As the inflow aspect of the bioprosthesis starts to flare
outward, monitor bioprosthesis position under fluoroscopy.
Caution: During implantation, if resistance to deployment is encountered (e.g., the
micro knob starts clicking or is tight or stuck), apply mild upward pressure to the
macro slider while turning the micro knob (Figure 22). If the bioprosthesis still does
not deploy, remove it from the patient and use another system.
Figure 30
23. Perform an angiogram. Once annular contact is made, the bioprosthesis should not be
advanced into a lower position.
28
Page 31

Note: The force required to move the bioprosthesis into a higher position becomes
noticeably greater once annular contact is made.
24. Continue deploying rapidly to the 2/3 deployment point; stop turning the micro knob.
Note: Shortly after annular contact, the blood pressure will be reduced until the
2/3 deployment point, when the bioprosthesis leaflets are exposed and are
functioning.
Figure 31
25. Perform an angiogram to assess the location of the bioprosthesis. Refer to Figure 29
and Figure 32 for the optimal placement of the bioprosthesis skirt within the aortic
annulus (approximately 4 mm to 6 mm below the annulus).
Figure 32
26. If the bioprosthesis is positioned low, slight repositioning of a partially deployed
bioprosthesis (≤2/3 of the bioprosthesis length) can be achieved by carefully
withdrawing the catheter.
27. When satisfactory position is achieved, withdraw the reference pigtail catheter to the
ascending aorta. Continue to turn the micro knob until both frame loops disengage.
Use orthogonal views under fluoroscopy to confirm that the frame loops have
detached from the catheter tabs. If a frame loop is still attached to a catheter tab, do
not pull on the catheter. Under fluoroscopy, advance the catheter slightly and, if
necessary, gently rotate the handle clockwise (<180°) and then counterclockwise
(<180°) to disengage the loop from the catheter tab.
29
Page 32

9.2.5 Postdeployment
28. Under fluoroscopic guidance, confirm that the catheter tip is coaxial with the inflow
portion of the bioprosthesis.
29. Withdraw the catheter to the aorta, while maintaining guidewire position.
Note: For transfemoral access, withdraw the catheter until the catheter tip is
positioned in the descending aorta. For direct aortic access and subclavian access,
withdraw the catheter until the catheter tip is close to the distal tip of the introducer
sheath.
30. Close the capsule and remove the catheter through the 18 Fr introducer sheath.
Note: If the capsule does not close properly, gently rotate the catheter clockwise
(<180°) and then counterclockwise (<180°) until the capsule closes.
Caution: Ensure the capsule is closed before catheter removal. If increased resistance
is encountered when removing the catheter through the introducer sheath, do not
force passage. Increased resistance may indicate a problem and forced passage may
result in damage to the device and/or harm to the patient. If the cause of resistance
cannot be determined or corrected, remove the catheter and introducer sheath as a
single unit over the guidewire, and inspect the catheter and confirm that it is
complete.
31. Dispose of the device in accordance with local regulations and hospital procedures.
32. Advance a 6 Fr pigtail catheter over the guidewire into the LV.
33. Remove the guidewire and connect the pigtail catheter to the transducer.
34. Using both pigtail catheters, record aortic pressure gradient.
35. Remove the 6 Fr pigtail over a standard, J-tip guidewire.
36. Perform a postimplant aortogram with the reference pigtail to ensure coronary
patency and assess aortic regurgitations.
Note: In the event that valve function or sealing is impaired due to excessive
calcification or incomplete expansion, a postimplant balloon dilatation of the
bioprosthesis may improve valve function and sealing. To ensure patient safety, valve
size and patient anatomy must be considered when selecting the size of the balloon
used for dilatation. The balloon size chosen for dilatation should not exceed the
diameter of the native aortic annulus or, for surgical bioprosthetic valves, the
manufacturer’s labeled inner diameter. Refer to the specific balloon catheter
manufacturer’s compliance chart to ensure that the applied inflation pressure does not
result in a balloon diameter that exceeds the indicated annulus range for the
bioprosthesis. Refer to the specific balloon catheter manufacturer’s labeling for
proper instruction on the use of balloon catheter devices. Note: Bench testing has only
been conducted to confirm compatibility with NuMED Z-MED II™ Balloon Aortic
Valvuloplasty catheters where CoreValve™ bioprosthesis device performance was
maintained after dilatation. Data on file.
30
Page 33

37. Remove the 18 Fr introducer sheath and complete the puncture site closure per
hospital protocol.
38. Perform contrast angiography to verify the absence of any vascular complications.
39. Remove the reference pigtail catheter over a standard guidewire. Remove the 6 Fr
introducer and close the access site per hospital protocol.
40. Administer anticoagulation and/or antiplatelet therapy as required according to
hospital protocol.
31
Page 34

10.0 Return of explanted bioprostheses
Medtronic is interested in obtaining recovered bioprostheses. Specific pathological studies of
the explanted bioprosthesis will be conducted under the direction of a consulting pathologist.
A written summary of the findings will be returned to the physician. To obtain a product
return kit, contact a Medtronic distribution center or a Medtronic Representative. If a kit is
not available, place the explanted bioprosthesis in a container of glutaraldehyde or 10%
buffered formalin immediately after excision. For further instructions on the return of an
explanted device, contact a Medtronic Representative.
32
Page 35

11.0 Summary of clinical study
The Medtronic CoreValve™ U.S. Pivotal Trial was designed and executed to evaluate the
safety and effectiveness of the CoreValve™ system to treat symptomatic severe aortic
stenosis in subjects necessitating aortic valve replacement. The trial was divided into
2 cohorts—patients who were determined by a heart team to be at high risk for surgery
(predicted operative mortality of ≥15% [and predicted operative mortality or serious,
irreversible morbidity risk of <50%]) or those who were determined to be at extreme risk for
surgery (irreversible morbidity risk of ≥50% at 30 days). Section 11.2 presents the results of
the High Risk cohort, and Section 11.3 presents the results of the Extreme Risk cohort.
The Medtronic CoreValve™ SURTAVI Trial was designed and executed to evaluate the
safety and efficacy of transcatheter aortic valve implantation (TAVR) in subjects with severe,
symptomatic aortic stenosis (AS) at intermediate surgical risk (heart team agreement of
predicted risk of operative mortality is ≥3% and <15% at 30 days) by randomizing subjects
to either surgical aortic valve replacement (SAVR) or TAVR. Section 11.1 presents the
results of the SURTAVI Trial.
11.1 Intermediate Risk trial (SURTAVI)
The Surgical Replacement and Transcatheter Aortic Valve Implantation (SURTAVI) trial is
a prospective, randomized, unblinded, multi-center investigational study. The purpose of this
trial is to investigate the safety and efficacy of transcatheter aortic valve implantation
(TAVR) in subjects with severe, symptomatic aortic stenosis (AS) at intermediate surgical
risk by randomizing subjects to either surgical aortic valve replacement (SAVR) or TAVR.
A total of 1746 subjects were randomized in this study (879 subjects were randomized to
TAVR and 867 subjects were randomized to surgical aortic valve replacement [SAVR]) at
87 activated centers. Severe aortic stenosis was defined as an aortic valve area of ≤0.8 cm2 or
aortic valve area index ≤0.5 cm2, a mean aortic valve gradient of >40 mmHg or jet velocity
>4 m/sec. The primary objective of the study was to demonstrate that the safety and
effectiveness of the Medtronic CoreValve™ system (TAVR), as measured by all-cause
mortality or disabling stroke at 24 months, is non-inferior to surgical aortic valve
replacement (SAVR) in the treatment of symptomatic severe aortic stenosis in subjects who
have a predicted intermediate risk for aortic valve surgery.
Of the 879 subjects randomized to TAVR, 864 received an attempted implant and comprise
the primary analysis cohort (the modified intention-to-treat [mITT] cohort) TAVR set, while
796 of the 867 randomized to SAVR received an attempted implant and comprise the mITT
SAVR cohort. The implanted population (863 TAVR and 794 SAVR) consists of all subjects
who were implanted with a valve. Of the 863 subjects in the Implanted TAVR group, 724
were attempted with the CoreValve™ system, 139 with the CoreValve™ Evolut™ R system.
The following data summarize the results from the SURTAVI trial.
11.1.1 Patient population
The demographics of the study population are shown in Table 3. The treatment arms were
generally well balanced (i.e., no statistically significant differences were identified between
the treatment arms) with respect to age, gender, baseline NYHA classification, and aggregate
33
Page 36

Age (years)
79.9 ± 6.2 (864)
79.7 ± 6.1 (796)
(-0.37, 0.81)
Male
57.6% (498/864)
55.0% (438/796)
(-2.15%, 7.37%)
NYHA Class
II
39.8% (344/864)
41.8% (333/796)
(-6.71%, 2.72%)
III
54.6% (472/864)
51.6% (411/796)
(-1.80%, 7.78%)
IV
5.6% (48/864)
6.5% (52/796)
(-3.30%, 1.31%)
STS Score (risk of
mortality, %)
Logistic EuroScore (%)
11.9 ± 7.6 (864)
11.6 ± 8.0 (795)
(-0.44, 1.06)
Coronary artery disease
62.6% (541/864)
64.2% (511/796)
(-6.20%, 3.05%)
Previous MI
14.5% (125/864)
13.9% (111/796)
(-2.84%, 3.88%)
Previous reintervention
Coronary artery bypass
surgery
Percutaneous coronary
intervention
Cerebrovascular disease
17.5% (151/864)
16.3% (130/796)
(-2.47%, 4.73%)
Peripheral vascular
disease
Prior stroke
6.6% (57/864)
7.2% (57/796)
(-3.04%, 1.87%)
Chronic lung
disease/COPD
Home oxygen
2.1% (18/864)
2.6% (21/795)
(-2.09%, 0.92%)
Creatinine level >2 mg/dl
1.6% (14/864)
2.1% (17/796)
(-1.90%, 0.81%)
Atrial fibrillation/atrial
flutter
Permanent pacemaker
implantation
History of hypertension
92.7% (801/864)
90.3% (719/796)
(-0.30%, 5.10%)
Cirrhosis of the liver
0.5% (4/863)
0.6% (5/795)
(-0.99%, 0.60%)
Echocardiographic findings—Implanted Population
Effective orifice area
(cm2)
Mean gradient (mmHg)
47.2 ± 14.3 (856)
47.8 ± 13.8 (786)
(-2.03, 0.70)
1
Continuous measures - Mean ± SD (Total no.); categorical measures - % (no./Total no.)
BCI: Bayesian credible interval
indicators of surgical risk (STS score and EuroSCORE). Most the subjects were in NYHA
class II and III.
Table 3: Subject Demographics and Clinical Characteristics – mITT Set
Demographics and
Baseline
Characteristics
Summary Statistics1
Difference
TAVR SAVR
(TAVR – SAVR)
(95% BCI)2
4.4 ± 1.5 (864) 4.5 ± 1.6 (796) (-0.28, 0.03)
16.0% (138/864) 17.2% (137/796) (-4.83%, 2.34%)
21.3% (184/864) 21.2% (169/796) (-3.88%, 3.99%)
30.8% (266/864) 29.9% (238/796) (-3.54%, 5.29%)
35.4% (305/862) 33.5% (267/796) (-2.74%, 6.39%)
28.1% (243/864) 26.5% (211/796) (-2.68%, 5.89%)
9.7% (84/864) 9.0% (72/796) (-2.14%, 3.47%)
0.8 ± 0.2 (790) 0.8 ± 0.2 (727) (-0.01, 0.03)
2
34
Page 37

Number of Index Procedures
863
Total delivery catheter in the body time (min)
15.0 ± 15.9
Type of Anesthesia
General
75.7% (653/863)
Conscious Sedation
24.3% (210/863)
Respiratory Support Required
69.8% (602/863)
Access Site
Femoral
93.2% (804/863)
Percutaneous
81.3% (654/804)
Surgical cut-down
18.7% (150/804)
Iliac
0.5% (4/863)
Percutaneous
75.0% (3/4)
Surgical cut-down
25.0% (1/4)
Subclavian axillary
2.3% (20/863)
Direct Aortic
4.1% (35/863)
Other
0.0% (0/863)
Total Time in Cath Lab or OR (min)
190.8 ± 61.3
Total Procedure Time (min)
52.3 ± 32.7
Pre-TAVR balloon valvuloplasty performed
47.2% (407/863)
Post-TAVR balloon valvuloplasty performed
29.0% (250/863)
1
Continuous measures - Mean ± SD; categorical measures - % (no./Total no.). Data include subjects with the
index procedure defined as the first procedure that the delivery catheter is introduced.
11.1.2 Procedure data
As shown in Table 4, total time the delivery catheter was in the body was approximately
15 minutes. A majority of TAVR subjects were administered general anesthesia while the
remaining subjects underwent the procedure with conscious sedation. A substantial majority
of the subjects (greater than 90%) has the valve delivered via iliofemoral access and
percutaneous access was more common than surgical cut-down. Balloon predilatation was
performed in approximately half of the subjects and postdilatation was performed in
approximately 30%.
Table 4: Procedural Data Summary for TAVR Subjects – mITT Set
Summary Statistics1
Assessment
N=864
11.1.3 Safety and effectiveness results
11.1.3.1 Primary safety and effectiveness endpoint
The primary objective was to demonstrate that the safety and effectiveness of TAVR using
the Medtronic CoreValve™ and CoreValve™ Evolut™ R systems, as measured by the allcause mortality or disabling stroke rate during a fixed follow-up of 24 months, is non-inferior
to SAVR in the treatment of symptomatic severe aortic stenosis in subjects who were
determined by the heart team to be at intermediate surgical risk.
The “early win” assessment of the primary endpoint included all subjects in the mITT
population (N = 1660). The median of the posterior distribution for the primary endpoint
35
Page 38

Posterior Median (95% BCI)
12.6% (10.2%, 15.3%)
14.0% (11.4%, 17.0%)
Difference (TAVR-SAVR) Posterior
Median (95% BCI)
Primary Objective – Non-Inferiority
Posterior Probability
,.
data
> 0.9999
Posterior Threshold for Non-Inferiority
0.971
Non-inferiority test
Passed
event rate was 12.6% for the TAVR arm and 14.0% for the SAVR arm, with a median of the
posterior distribution of the difference in the primary endpoint event rate (TAVR – SAVR) of
-1.4% and a 95% Bayesian credible interval (BCI) of (-5.2%, 2.3%), as summarized in
Table 5. The posterior probability of non-inferiority with a margin of 7% was > 0.9999,
which is greater than the pre-specified threshold of 0.971, thus the primary endpoint noninferiority could be concluded.
Table 5: Primary Endpoint: All-Cause Mortality or Disabling Stroke at 24 Months -
mITT Set
TAVR
N=864
-1.4% (-5.2%, 2.3%)
SAVR
N=796
Figure 33 shows K-M rates of all-cause mortality or disabling stroke in the mITT set for both
treatment arms up to 24 months follow-up.
36
Page 39

Figure 33: Primary Endpoint: All-Cause Mortality or Disabling Stroke Kaplan-Meier
Event Rate – mITT Set
Note: The confidence intervals were calculated without multiplicity adjustment. The adjusted
confidence intervals could be wider than presented here. As such, confidence intervals are
provided to illustrate the variability only and should not be used to draw any statistical
conclusion.
11.1.3.2 Key secondary safety and effectiveness endpoints
Hierarchical testing of secondary endpoints
Hypothesis testing was performed on pre-specified secondary endpoints using a hierarchical
test procedure, as shown in Table 6. TAVR was found to be non-inferior to SAVR within the
pre-specified non-inferiority margins in terms of mean gradient and EOA at 12 months, the
NYHA functional classification change from baseline to 12 months, and the KCCQ score
change from baseline to 30 days. TAVR was determined to be superior to SAVR with respect
to length of index procedure hospital stay, the mean pressure gradient at 12 months, EOA at
12 months, and the KCCQ score change from baseline to 30-days.
TAVR was not found to be superior to SAVR with respect to days alive and out of hospital at
12 months. The remaining secondary endpoints were not tested.
37
Page 40

#1 Mean
months
#2 EOA at 12
months
2.2 ± 0.6
(545)
1.8 ± 0.6
(455)
#3 NYHA
months)
#4 KCCQ
baseline)
#5 Length of
hospital stay
#6 Mean
months
#7 EOA at 12
months
2.2 ± 0.6
(545)
1.8 ± 0.6
(455)
#8 KCCQ
baseline)
Note: The Implanted population was used for the mean gradient and EOA, and the mITT population for the rest.
Table 6: Secondary Endpoints: Hierarchical Testing
Secondary
Endpoint
Non-inferiority testing
gradient at 12
change
(baseline – 12
summary
score change
(30 day –
Superiority testing
index
procedure
TAVR
Mean ±
SD (N)
8.3 ± 4.0
(590)
1.3 ± 0.8
(604)
18.4 ±
22.8
(819)
5.8 ± 4.9
(863)
SAVR
Mean ± SD
(N)
11.7 ± 5.6
(500)
1.3 ± 0.8
(508)
5.9 ± 27.0
(700)
9.8 ± 8.0
(795)
Difference
(TAVR-SAVR)
(95% BCI)
(-4.0, -2.8) 1.00 0.95 Passed
(0.3, 0.5) 1.00 0.95 Passed
(-0.1, 0.1) 1.00 0.95 Passed
(10.0, 15.1) 1.00 0.95 Passed
(-4.7, -3.4) 1.00 0.975 Passed
Posterior
Probability
Pr(HA | data)
Threshold
Test
Result
gradient at 12
summary
score change
(30 day –
8.3 ± 4.0
(590)
18.4 ±
22.8
(819)
11.7 ± 5.6
(500)
5.9 ± 27.0
(700)
(-4.0, -2.8) 1.00 0.975 Passed
(0.3, 0.5) 1.00 0.975 Passed
(10.0, 15.1) 1.00 0.975 Passed
11.1.3.3 Additional effectiveness data
Valve performance
Effective orifice area (EOA) and mean gradient for TAVR and SAVR subjects are shown in
Figure 34 and Figure 35.
38
Page 41

Figure 34: TAVR and SAVR EOA by Visit (Implanted Population)
Note: Line plot with mean and standard deviation.
Figure 35: TAVR and SAVR Mean Gradient by Visit (Implanted Population)
Note: Line plot with mean and standard deviation.
Figure 36 shows total aortic regurgitation (AR) severity over time for both treatment arms.
Figure 37 shows paravalvular aortic regurgitation.
39
Page 42

Figure 36: TAVR and SAVR Total Aortic Regurgitation by Visit (Implanted Population)
Note: Values < 1.0% are not labeled.
Figure 37: Paravalvular Aortic Regurgitation by Visit (Implanted Population)
Note: Values < 1.0% are not labeled.
40
Page 43

NYHA functional class
NYHA functional classification was evaluated for subjects at each interval for the TAVR and
SAVR treatment arms. NYHA classification data for subjects at each interval are shown in
Figure 38.
Figure 38: TAVR and SAVR NYHA Classification by Visit (mITT Population)
Note: Values < 1.0% are not labeled.
Health status/QoL change
QoL was measured using the Kansas City Cardiomyopathy Questionnaire (KCCQ), the SF36 Health Status Questionnaire, and the EuroQoL (EQ-5D) measure.
The KCCQ overall and clinical summary scores for the two treatment arms are shown in
Figure 39 and Figure 40, respectively.
41
Page 44
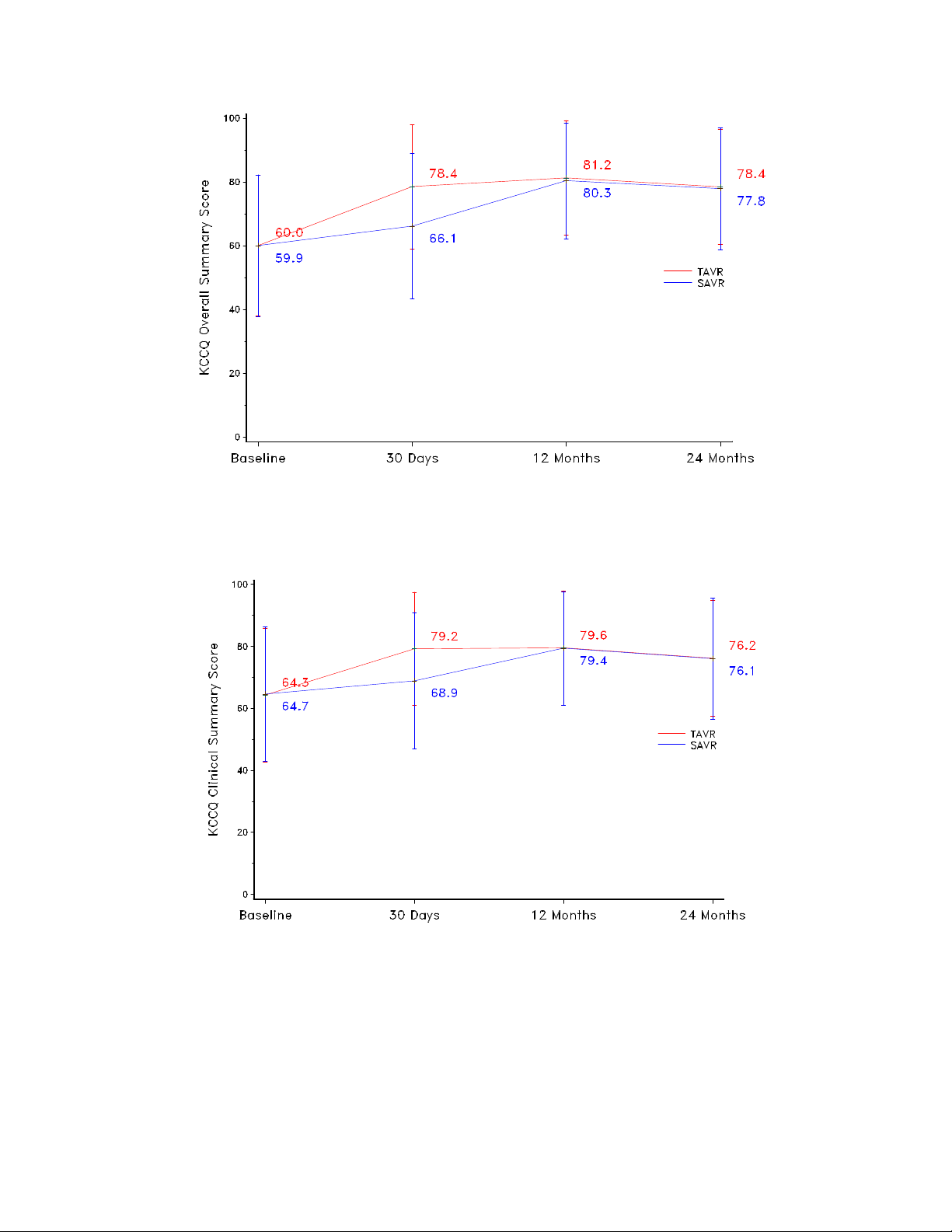
Figure 39: KCCQ Overall Summary Scores
Note: Line plot with mean and standard deviation.
Figure 40: KCCQ Clinical Summary Scores
Note: Line plot with mean and standard deviation.
The SF-36 physical and mental component summary scores for the two treatment arms are
shown in Figure 41 and Figure 42, respectively.
42
Page 45

Figure 41: SF-36 Physical Component Summary Scores
Note: Line plot with mean and standard deviation.
Figure 42: SF-36 Mental Component Summary Scores
Note: Line plot with mean and standard deviation.
The EQ-5D index scores for the two treatment arms are shown in Figure 43.
43
Page 46

2.8%
29)
3.8%
8.1%
8.7%
12.3%
13.8%
101)
2.1%
18)
1.6%
6.8%
6.9%
11.2%
11.5%
2.0%
17)
1.6%
4.8%
5.5%
7.5%
7.8%
0.0%
(0, 0)
0.0%
(0, 0)
0.0%
(0, 0)
0.1%
(1, 1)
0.0%
(0, 0)
0.1%
(1, 1)
Non-cardiovascular
0.1%
0.0%
2.1%
1.4%
4.0%
4.0%
Figure 43: EQ5D Index Scores
Note: Line plot with mean and standard deviation.
11.1.3.4 Additional safety data
Adverse events that occurred in the PMA clinical study
Procedural safety and safety during follow-up were evaluated for both TAVR and SAVR
within the SURTAVI trial. Kaplan-Meier (K-M) rates of some key CEC-adjudicated events
are presented in Table 7.
Table 7: All Adverse Events (0-24 Months) -mITT Set
Events Summary Statistics1
0-30 Days 0-12 Months 0-24 Months
TAVR SAVR TAVR SAVR TAVR SAVR
All-cause mortality or
disabling stroke
All-cause mortality
(24,
(18,
(30, 33)
(13, 13)
(66, 74)
(55, 55)
(66, 79)
(51, 51)
(87, 97)
(77, 77)
(87,
(70, 70)
Cardiovascular
Valve-related2
(17,
(13, 13)
44
(39, 39)
(41, 41)
(52, 52)
(51, 51)
Page 47

(1, 1)
(0, 0)
(16, 16)
(10, 10)
(25, 25)
(19, 19)
0.8%
(7, 7)
0.1%
(1, 1)
2.1%
(17, 19)
0.4%
(3, 3)
2.6%
(20, 22)
0.4%
(3, 3)
3.3%
29)
5.4%
5.3%
6.7%
6.3%
8.0%
1.2%
11)
2.4%
2.2%
3.4%
2.4%
4.1%
2.1%
18)
3.0%
3.1%
3.3%
4.1%
4.0%
5.7%
51)
5.9%
7.1%
7.8%
8.0%
8.4%
5.9%
55)
1.0%
6.3%
1.0%
6.3%
1.0%
Acute kidney injury - Stage
3
0.7%
(6, 6)
1.3%
(10, 10)
0.7%
(6, 6)
1.3%
(10, 10)
0.7%
(6, 6)
1.3%
(10, 10)
0.8%
(7, 7)
0.9%
(7, 7)
1.9%
(15, 15)
1.4%
(11, 11)
2.6%
(18, 18)
1.9%
(13, 13)
2.8%
26)
4.1%
8.4%
7.4%
13.2%
9.0%
28.1%
217)
6.8%
31.3%
9.0%
34.6%
10.3%
25.6%
220)
6.5%
28.5%
8.6%
31.5%
9.8%
1
Kaplan-Meier rate (# patients, # events).
Subjects with pacemaker or ICD at baseline are included. Not adjudicated by CEC.
Events Summary Statistics1
Reintervention
0-30 Days 0-12 Months 0-24 Months
TAVR SAVR TAVR SAVR TAVR SAVR
All stroke
Disabling stroke
Non-disabling stroke
Life threatening/disabling
bleeding
Major vascular
complication
MI
Aortic valve hospitalization
Permanent pacemaker
implantation3
(28,
(10,
(18,
(49,
(51,
(24,
(217,
(43, 45)
(19, 20)
(24, 25)
(47, 47)
(8, 8)
(32, 34)
(48, 48)
(44, 45)
(18, 19)
(26, 26)
(60, 66)
(54, 59)
(68, 104)
(239, 241)
(52, 55)
(26, 28)
(26, 27)
(60, 61)
(8, 8)
(55, 68)
(62, 64)
(48, 50)
(19, 20)
(30, 30)
(64, 72)
(54, 59)
(90, 134)
(253, 257)
(58, 61)
(29, 31)
(29, 30)
(63, 65)
(8, 8)
(62, 85)
(67, 70)
Permanent pacemaker
implantation4
2
Valve-related death is any death caused by structural or non-structural valve dysfunction or aortic valve
re-intervention.
3
Subjects with pacemaker or ICD at baseline are not included. Not adjudicated by CEC.
4
(220,
(51, 51)
(242, 244)
11.1.4 Additional study observations
11.1.4.1 Pre-specified analyses
The primary endpoint was examined for treatment arm differences in outcome between the
stratified randomization designation (revascularization or no revascularization) and gender.
45
(66, 68)
(256, 260)
(71, 74)
Page 48

All-cause mortality or disabling stroke stratified by need for revascularization
– mITT set
Figure 44 and Figure 45 present the all-cause mortality or disabling stroke analysis stratified
by need for coronary revascularization for the mITT set.
Figure 44: All-Cause Mortality or Disabling Stroke for Subjects with Need for
Revascularization – mITT Set
Note: The confidence intervals were calculated without multiplicity adjustment. The adjusted
confidence intervals could be wider than presented here. As such, confidence intervals are
provided to illustrate the variability only and should not be used to draw any statistical
conclusion. The trial was not powered to assess the difference between the two subgroups.
46
Page 49

Figure 45: All-Cause Mortality or Disabling Stroke for Subjects without Need for
Revascularization – mITT Set
Note: The confidence intervals were calculated without multiplicity adjustment. The adjusted
confidence intervals could be wider than presented here. As such, confidence intervals are
provided to illustrate the variability only and should not be used to draw any statistical
conclusion. The trial was not powered to assess the difference between the two subgroups.
All-cause mortality or disabling stroke analyzed by gender – mITT set
Figure 46 and Figure 47 present all-cause mortality or disabling stroke analyzed by gender
for the mITT set.
47
Page 50

Figure 46: All-Cause Mortality or Disabling Stroke at 24 Months for Male Subjects -
mITT Set
Note: The confidence intervals were calculated without multiplicity adjustment. The adjusted
confidence intervals could be wider than presented here. As such, confidence intervals are
provided to illustrate the variability only and should not be used to draw any statistical
conclusion. The trial was not powered to assess the difference between the two subgroups.
48
Page 51

Figure 47: All-Cause Mortality or Disabling Stroke at 24 Months for Female Subjects –
mITT Set
Note: The confidence intervals were calculated without multiplicity adjustment. The adjusted
confidence intervals could be wider than presented here. As such, confidence intervals are
provided to illustrate the variability only and should not be used to draw any statistical
conclusion. The trial was not powered to assess the difference between the two subgroups.
11.1.4.2 All-cause mortality by severity of aortic regurgitation
A sub-group analysis was performed to investigate the relationship between all-cause
mortality and severity of aortic regurgitation at discharge. Two sub-groups of subjects with
none/trace and mild/moderate/severe total AR as assessed at discharge were analyzed.
The results from the analysis with 2 subgroups are shown for the TAVR treatment arm in
Figure 48.
49
Page 52

Figure 48: All-Cause Mortality by Severity of Aortic Regurgitation (2 Groups) – TAVR
Implanted Set
Note: The confidence intervals were calculated without multiplicity adjustment. The adjusted
confidence intervals could be wider than presented here. As such, confidence intervals are
provided to illustrate the variability only and should not be used to draw any statistical
conclusion. The trial was not powered to assess the difference between the two subgroups.
11.1.4.3 All-cause mortality by conduction disturbance requiring a permanent pacemaker post-TAVR
An analysis was performed for implanted TAVR subjects to investigate the relationship
between all-cause mortality and permanent pacemaker implantation (PPI) through 30 days
post TAVR (Figure 49). Similar rates between subjects without a PPI and subjects with a
new PPI indicate that new-onset conduction disturbance and resultant PPI was not
significantly associated with mortality in this study.
50
Page 53

Figure 49: All-Cause Mortality by New Permanent Pacemaker – TAVR Implanted Set
Note: The confidence intervals were calculated without multiplicity adjustment. The adjusted
confidence intervals could be wider than presented here. As such, confidence intervals are
provided to illustrate the variability only and should not be used to draw any statistical
conclusion. The trial was not powered to assess the difference among the three subgroups.
All-cause mortality by patient prosthesis mismatch
The site reported aortic annular perimeters were comparable between the two treatment arms
(TAVR: 78.3 ± 7.2 mm vs. SAVR: 78.4 ± 7.1 mm). Patient prosthesis mismatch (PPM) is
defined as an indexed EOA of 0.85-0.65 cm2/m2 (moderate) and <0.65 cm2/m2 (severe) for
subjects with a BMI <30 kg/cm2, or 0.70-0.60 cm2/m2 (moderate) and <0.60 cm2/m2 (severe)
for subjects with a BMI ≥30 kg/cm2. Figure 50 and Figure 51 present the prevalence of PPM
at 12 months in the two treatment arms by valve size. The majority of SAVR patients
received a labeled valve size of ≤23 mm, and smaller valve sizes generally had more
prevalent PPM. In comparison, PPM was less prevalent in the TAVR arm.
The K-M curves for all-cause mortality by PPM grade (none, moderate, and severe) are
shown in Figure 52 and Figure 53 for the TAVR and SAVR arm, respectively.
51
Page 54

Figure 50: Prevalence of PPM at 12 Months in the TAVR Arm by Valve Size
Figure 51: Prevalence of PPM at 12 Months in the SAVR Arm by Valve Size
52
Page 55

Figure 52: All-Cause Mortality by PPM - TAVR Implanted Population
Note: The confidence intervals were calculated without multiplicity adjustment. The adjusted
confidence intervals could be wider than presented here. As such, confidence intervals are
provided to illustrate the variability only and should not be used to draw any statistical
conclusion. The trial was not powered to assess the difference among the three subgroups.
53
Page 56

Figure 53: All-Cause Mortality by PPM - SAVR Implanted Set
Note: The confidence intervals were calculated without multiplicity adjustment. The adjusted
confidence intervals could be wider than presented here. As such, confidence intervals are
provided to illustrate the variability only and should not be used to draw any statistical
conclusion. The trial was not powered to assess the difference among the three subgroups.
54
Page 57

11.2 High Risk cohort
The CoreValve™ U.S. Pivotal Trial High Risk cohort was a prospective, randomized,
unblinded, multi-center investigational study. Patients were stratified by intended access site
(iliofemoral or non-iliofemoral) prior to randomization to ensure patients were allocated to
each comparison group proportionately. Prior to randomization, patients were first evaluated
for iliofemoral access. If patients were not eligible for iliofemoral access due to their
inadequate vasculature or peripheral vascular disease, they were then considered for noniliofemoral access. Patients were then individually evaluated for subclavian or direct aortic
access.
The purpose of the study was to evaluate the safety and effectiveness of the Medtronic
CoreValve™ system in the treatment of symptomatic severe aortic stenosis in subjects
necessitating aortic valve replacement, with predicted operative mortality of ≥ 15% (and
predicted operative mortality or serious, irreversible morbidity risk of < 50%) at 30 days
(High Risk).
The High Risk cohort enrolled a total of 795 subjects with symptomatic severe aortic stenosis
(394 subjects were randomized to transcatheter aortic valve replacement [TAVR] and
401 subjects were randomized to surgical aortic valve replacement [SAVR]) at 45 activated
centers in the United States. Severe aortic stenosis was defined as an aortic valve area of
≤0.8 cm2 or aortic valve area index ≤0.5 cm2, a mean aortic valve gradient of >40 mmHg or
jet velocity >4 m/sec. The primary endpoint of the study was to demonstrate that the safety
and effectiveness of the Medtronic CoreValve™ system (TAVR), as measured by all-cause
death at 12 months, is non-inferior to surgical aortic valve replacement (SAVR) in the
treatment of symptomatic severe aortic stenosis in subjects who have a predicted high risk for
aortic valve surgery.
Of the 394 subjects randomized to TAVR, 390 received an attempted implant and comprise
the as treated (AT) TAVR population while 357 of the 401 subjects randomized to SAVR
received an attempted implant and comprise the AT SAVR population.
The following data summarize the results from the High Risk cohort (TAVR iliofemoral and
TAVR non-iliofemoral vs. SAVR iliofemoral eligible and SAVR non-iliofemoral eligible).
11.2.1 Patient population
The demographics of the study population were typical for an aortic stenosis valve
replacement study performed in the U.S., as shown in Table 8. A high proportion of the
patients had significant co-morbidities, frailties, or disabilities, and these risk factors were
generally well balanced between the study arms. The mean age for patients participating in
the trial was approximately 83 years old, and slightly greater than 50% of patients were male.
The mean STS score was approximately 7. In addition, approximately 85% of all patients
were in NYHA classes III or IV.
55
Page 58

Iliofemoral
Non-Iliofemoral
Pooled
TAVR
N=330
SAVR
N=333
TAVR
N=64
SAVR
N=68
TAVR
N=394
SAVR
N=401
P-
Values
83.6 ±
6.3
81.8 ±
8.0
82.9 ±
6.5
83.2 ±
7.1
83.5 ±
6.3
53.9%
(178/330)
53.8%
(179/333)
51.6%
(33/64)
48.5%
(33/68)
53.6%
(211/394)
52.9%
(212/401)
NYHA
Classification
13.0%
(43/330)
12.0%
(40/333)
20.3%
(13/64)
19.1%
(13/68)
14.2%
(56/394)
13.2%
(53/401)
65.2%
(215/330)
69.7%
(232/333)
67.2%
(43/64)
66.2%
(45/68)
65.5%
(258/394)
69.1%
(277/401)
21.8%
(72/330)
18.3%
(61/333)
12.5%
(8/64)
14.7%
(10/68)
20.3%
(80/394)
17.7%
(71/401)
STS Score (Risk of
Mortality, %)
Coronary Artery
Disease
75.5%
(249/330)
74.2%
(247/333)
75.0%
(48/64)
86.8%
(59/68)
75.4%
(297/394)
76.3%
(306/401)
23.3%
(77/330)
23.4%
(78/333)
37.5%
(24/64)
29.4%
(20/68)
25.6%
(101/394)
24.4%
(98/401)
Previous
Interventions
Coronary Artery
Bypass Surgery
31.2%
(103/330)
29.1%
(97/333)
21.9%
(14/64)
35.3%
(24/68)
29.7%
(117/394)
30.2%
(121/401)
Percutaneous
Intervention
Balloon
Valvuloplasty
4.5%
(15/330)
6.3%
(21/333)
12.5%
(8/64)
7.4%
(5/68)
5.8%
(23/394)
6.5%
(26/401)
Cerebral Vascular
Disease
24.7%
(81/328)
23.2%
(77/332)
29.0%
(18/62)
35.9%
(23/64)
25.4%
(99/390)
25.3%
(100/396)
13.6%
(45/330)
12.6%
(42/333)
9.4%
(6/64)
16.4%
(11/67)
12.9%
(51/394)
13.3%
(53/400)
Peripheral
Vascular Disease
37.6%
(123/327)
37.2%
(123/331)
62.5%
(40/64)
68.7%
(46/67)
41.7%
(163/391)
42.5%
(169/398)
Chronic Lung
Disease/COPD
44.5%
(147/330)
46.2%
(154/333)
45.3%
(29/64)
38.2%
(26/68)
44.7%
(176/394)
44.9%
(180/401)
13.4%
(44/329)
12.3%
(41/333)
9.4%
(6/64)
10.3%
(7/68)
12.7%
(50/393)
12.0%
(48/401)
Creatinine Level
>2 mg/dl
3.3%
(11/330)
4.5%
(15/333)
3.1%
(2/64)
5.9%
(4/68)
3.3%
(13/394)
4.7%
(19/401)
Atrial
Flutter
Table 8: High Risk Cohort Baseline Characteristics and Echocardiographic Findings
(ITT)
Demographic
Age (years) 83.4 ± 6.8
Gender (Male)
II
III
IV
7.3 ± 3.1 7.5 ± 3.1 7.2 ± 2.6 7.6 ± 3.9 7.3 ± 3.0 7.5 ± 3.2 0.2680
Previous MI
Coronary
32.1%
(106/330)
37.8%
(126/333)
42.2%
(27/64)
38.2%
(26/68)
33.8%
(133/394)
37.9%
(152/401)
0.5102
0.8464
0.6723
0.7597
0.6972
0.8828
0.2226
Prior Stroke
Home Oxygen
Fibrillation/Atrial
40.6%
(134/330)
48.8%
(162/332)
42.9%
(27/63)
56
41.2%
(28/68)
41.0%
(161/393)
47.5%
(190/400)
0.7048
0.9660
0.8984
0.8257
0.9508
0.7472
0.3021
0.0640
Page 59

Iliofemoral
Non-Iliofemoral
Pooled
TAVR
N=330
SAVR
N=333
TAVR
N=64
SAVR
N=68
TAVR
N=394
SAVR
N=401
P-
Values
Preexisting
Placement/ICD
Aorta Calcification1
10.6%
(35/330)
10.5%
(35/333)
19.0%
(12/63)
16.2%
(11/68)
12.0%
(47/393)
11.5%
(46/401)
0.0%
(0/330)
0.0%
(0/333)
1.6%
(1/63)
0.0%
(0/68)
0.3%
(1/393)
0.0%
(0/401)
Chest Wall
Deformity
1.8%
(6/330)
0.3%
(1/333)
4.7%
(3/64)
0.0%
(0/68)
2.3%
(9/394)
0.2%
(1/401)
Hostile
Mediastinum
3.9%
(13/330)
0.9%
(3/331)
3.1%
(2/64)
2.9%
(2/68)
3.8%
(15/394)
1.3%
(5/399)
Cirrhosis of the
Liver
2.4%
(8/330)
1.8%
(6/333)
3.1%
(2/64)
1.5%
(1/68)
2.5%
(10/394)
1.7%
(7/401)
4.8%
(16/330)
7.5%
(25/333)
0.0%
(0/64)
7.4%
(5/68)
4.1%
(16/394)
7.5%
(30/401)
Echocardiographic
Findings
Ejection Fraction
%)
Aortic Valve Area
(cm2)
0.72 ±
0.23
0.73 ±
0.24
0.70 ±
0.18
0.71 ±
0.22
0.72 ±
0.23
0.73 ±
0.23
Mean Gradient
mmHg)
Mitral
Moderate/Severe
1. Aorta Calcification is measured on screening CT Angiogram
Plus-minus values present the mean ± standard deviation.
Demographic
Permanent
Pacemaker
Severe
Porcelain
Wheelchair Bound
(visual estimate,
24.8%
(82/330)
58.1 ±
10.9
21.6%
(72/333)
57.5 ±
11.8
15.6%
(10/64)
57.4 ±
13.4
16.2%
(11/68)
58.3 ±
12.4
23.4%
(92/394)
58.0 ±
11.3
20.7%
(83/401)
57.7 ±
11.9
0.3669
0.8307
0.4950
0.0106
0.0218
0.4400
0.0389
0.7110
0. 5801
Across Aortic
Valve (MGV2,
Regurgitation:
48.36 ±
15.09
10.2%
(33/325)
47.69 ±
14.39
10.5%
(34/324)
47.38 ±
16.74
8.1%
(5/62)
48.08 ±
12.51
9.0%
(6/67)
48.20 ±
15.35
9.8%
(38/387)
47.75 ±
14.07
10.2%
(40/391)
The STS score predicted a 30-day mortality of 7.5% for the average surgeon at the average
hospital. The Kaplan-Meier (K-M) 30-day mortality for the As Treated SAVR arm was
4.5%. Therefore, the observed/expected ratio was 0.60 in this trial, indicating better than
average care in the SAVR arm.
11.2.2 Procedure data
As recommended in the protocol, the procedure was to occur within 30 days of
randomization. As such, time to procedure was calculated between the randomization date
and the date of the first attempted procedure. It was 13.1 ± 10.9 days for TAVR patients and
18.1 ± 14.3 days for SAVR patients.
0. 6725
0. 8486
57
Page 60

Number of Index Procedures1
323
66
389
Total Time in Cath Lab or OR
(min)
Total Procedure Time (min)
(skin to skin)
93.8% (303/323)
98.5% (65/66)
94.6% (368/389)
Valve-in-Valve Procedure
4.3% (14/323)
0.0% (0/66)
3.6% (14/389)
Converted to surgical AVR
0.3% (1/323)
1.5% (1/66)
0.5% (2/389)
Number of Valves Used
02
0.0% (0/323)
1.5% (1/67)
0.3% (1/390)
1
91.3% (295/323)
92.5% (62/67)
91.5% (357/390)
2
8.4% (27/323)
6.0% (4/67)
7.9% (31/390)
3
0.3% (1/323)
0.0% (0/67)
0.3% (1/390)
Number of Valves Implanted
0
0.0% (0/323)
1.5% (1/67)
0.3% (1/390)
1
95.0% (307/323)
98.5% (66/67)
95.6% (373/390)
2
5.0% (16/323)
0.0% (0/67)
4.1% (16/390)
3
0.0% (0/323)
0.0% (0/67)
0.0% (0/390)
Valve Size Implanted
23 mm
1.5% (5/323)
1.5% (1/66)
1.5% (6/389)
26 mm
29.4% (95/323)
40.9% (27/66)
31.4% (122/389)
29 mm
49.5% (160/323)
48.5% (32/66)
49.4% (192/389)
31 mm
19.5% (63/323)
9.1% (6/66)
17.7% (69/389)
Device Success3
86.9% (273/314)
87.3% (55/63)
87.0% (328/377)
Procedure Success4
81.5% (260/319)
82.8% (53/64)
81.7% (313/383)
1
The table includes patients with the index procedure. Index procedure (TAVR): the first procedure that the Medtronic
Procedure success is defined as device success and absence of in-hospital MACCE.
Table 9 provides a summary of the procedure data for the TAVR cohort. The overall device
success rate was 86.9% for the iliofemoral cohort and 87.3% for the non-iliofemoral cohort.
Procedure success was defined as device success and absence of in-hospital MACCE and
procedure success rates were 81.5% and 82.8% for the iliofemoral and non-iliofemoral
cohorts, respectively.
Table 9: High Risk Cohort TAVR Procedure Data - As Treated Population
General Anesthesia
TAVR IF
N=323
TAVR NIF
N=67
TAVR Pooled
N=390
209.6 ± 58.6 (323) 249.1 ± 63.4 (66) 216.3 ± 61.2 (389)
61.4 ± 33.9 (321) 55.6 ± 41.7 (65) 60.4 ± 35.3 (386)
CoreValve™ system catheter is introduced.
2
A single patient had no valves used or implanted during the procedure as the patient became hypotensive after the
TEE probe was placed and the patient was converted to SAVR.
3
Device success is defined as deployment, only 1 valve implanted, only 1 valve in correct anatomic location, EOA
>1.2 cm2 for 26, 29, and 31mm and ≥0.9 cm2 for 23 mm, mean gradient <20 mmHg, and aortic regurgitation <
moderate.
4
11.2.3 Safety and effectiveness results
11.2.3.1 Primary safety and effectiveness endpoint
The primary endpoint of all-cause mortality at 12 months included all deaths (cardiovascular
and non-cardiovascular) from any cause after a valve intervention. Figure 54 shows the
58
Page 61

Total # of Patients
390
357
# of Patients Died within 1 Year
55
67
# of Patients Censored prior to 1 Year
7
16
# of Patients Alive at 1 Year
328
274
Mortality Rate at 1 Year (K-M)
14.22%
19.12%
Standard Error at 1 Year
1.78%
2.10%
Mortality Difference (TAVR-SAVR)
-4.89%
Standard Error of Difference
2.75%
95% 1-sided UCB for Difference
-0.37%
Primary Objective – Non-Inferiority
Non-inferiority Margin
7.50%
Z-Score
-4.5019
P-Value
<0.0001
Non-Inferiority Test
Passed
Primary Objective – Superiority
Z-Score
-1.7776
P-Value
0.0377
Superiority Test
Passed
Kaplan-Meier (K-M) rates of all-cause mortality in the AT population for both treatment
arms up to 12 months follow-up. The K-M rate of all-cause mortality at 12 months was
14.22% for TAVR and 19.12% for SAVR with a difference of -4.89% (TAVR-SAVR) and
an upper 1-sided 95% confidence interval of −0.37%, which was statistically less than the
pre-specified non-inferiority margin of 7.5% (p<0.0001). Therefore, the null hypothesis that
TAVR was inferior to SAVR for the primary endpoint of all-cause mortality at 12 months
was rejected and the alternative hypothesis that TAVR was non-inferior to SAVR within a
non-inferiority margin of 7.5% was accepted. Subsequently, a pre-specified test for
superiority of TAVR over SAVR was also conducted, which demonstrated that the rate of
all-cause mortality at 12 months for TAVR was significantly less than that for SAVR at the
one-sided 0.05 level (p=0.0377).
Table 10: Primary Endpoint: All-Cause Mortality at 12 Months – As Treated Population
TAVR N=390 SAVR N=357
59
Page 62

Total # of Patients
394
401
389
353
365
326
# of Patients Died within
1 Year
Figure 54: High Risk Cohort All-Cause Mortality Kaplan-Meier Event Rate – As Treated
Population
The primary endpoint hypothesis testing was also pre-specified for the intent-to-treat (ITT),
Implanted, and Per Protocol populations, as presented in Table 11 and Figure 55 - Figure 57.
The ITT population consisted of all randomized patients. The Implanted population consisted
of all AT patients who were actually implanted with either a CoreValve device or a surgical
valve. The Per Protocol population consisted of all implanted subjects who: (1) were
implanted according to their randomization and access site stratification; (2) had at least
12 months (365 days) of follow-up or had experienced the primary endpoint (death) prior to
12 months; (3) did not cross to a different type of procedure from their first attempted
procedure types (TAVR or SAVR) before their 12 month visit; and (4) had satisfied all
inclusion/exclusion criteria. Non-inferiority of TAVR compared to SAVR was concluded for
all analysis populations (p<0.0001 for all). Subsequent superiority null hypothesis was
rejected at one-sided 0.05 level for the ITT (p=0.0365) and Implanted (p=0.042) populations,
but not for the Per Protocol population (p=0.07).
Table 11: Primary Endpoint: All-Cause Mortality at 12 Months – Pre-specified
Additional Populations
Intent-to-Treat Implanted Per Protocol
TAVR
N=394
SAVR
N=401
TAVR
N=389
SAVR
N=353
TAVR
N=365
SAVR
N=326
54 68 55 66 53 61
60
Page 63

# of Patients Censored
prior to 1 Year
# of Patients Alive at
1 Year
Mortality Rate at 1 Year
(K-M)
Standard Error at 1 Year
1.75%
2.05%
1.78%
2.11%
1.84%
2.16%
Mortality Difference
(TAVR-SAVR)
Standard Error of
Difference
95% 1-sided UCB for
Difference
Primary Objective – NonInferiority
Non-inferiority Margin
7.50%
7.50%
7.50%
Z-Score
-4.5734
-4.4443
-4.1164
P-Value
<0.0001
<0.0001
<0.0001
Non-Inferiority Test
Passed
Passed
Passed
Primary Objective –
Superiority
Z-Score
-1.7926
-1.7283
-1.4757
P-Value
0.0365
0.0420
0.0700
Superiority Test
Passed
Passed
Failed
Intent-to-Treat Implanted Per Protocol
TAVR
N=394
9 54 7 15 0 0
331 279 327 272 312 265
13.87% 18.70% 14.26% 19.03% 14.52% 18.71%
SAVR
N=401
-4.83% -4.77% -4.19%
2.70% 2.76% 2.84%
-0.40% -0.23% 0.48%
TAVR
N=389
SAVR
N=353
TAVR
N=365
SAVR
N=326
It is worth noting that although the study primary endpoint passed the pre-specified
superiority test after it passed the non-inferiority test in the As-Treated primary analysis
population, the statistical robustness of the superiority test across different analysis
populations should be interpreted based on the specific statistical parameters used.
61
Page 64

Figure 55: All-Cause Mortality Kaplan-Meier Event Rate – Intent-to-Treat Population
Figure 56: All-Cause Mortality Kaplan-Meier Event Rate – Implanted Population
62
Page 65

Total # of Patients
372
341
372
341
# of Patients Died within 1 Year
53
61
60
61
# of Patients Censored prior to 1 Year
7
15 0 0
# of Patients Alive at 1 Year
312
265
312
280
Mortality Rate at 1 Year (K-M)
14.38%
18.21%
16.13%
17.89%
Standard Error at 1 Year
1.83%
2.11%
1.91%
2.08%
Mortality Difference (TAVR-SAVR)
-3.83%
-1.76%
Figure 57: All-Cause Mortality Kaplan-Meier Event Rate – Per Protocol Population
A post hoc analysis was also performed on the primary endpoint hypothesis testing for the
Modified Per Protocol population. The Modified Per Protocol population included 22
additional subjects (7 TAVR, 15 SAVR) who were censored prior to 1 year as compared with
the Per Protocol population. In addition, a post hoc analysis was conducted on the worst-case
Modified Per Protocol population, which assumed all 7 censored TAVR subjects had died at
the censoring time and all 15 censored SAVR subjects were alive at 1 year). In both analyses,
non-inferiority was demonstrated. The results are presented in Table 12, Figure 58 and
Figure 59.
Table 12: Primary Endpoint: All-Cause Mortality at 12 Months – Ad-Hoc Additional
Populations
Modified Per Protocol
Modified Per Protocol
TAVR
N=372
SAVR
N=341
Worst Case Scenario
TAVR
N=372
SAVR
N=341
63
Page 66

Standard Error of Difference
2.79%
2.82%
95% 1-sided UCB for Difference
0.76%
2.88%
Primary Objective – Non-Inferiority
Non-inferiority Margin
7.50%
7.50%
Z-Score
-4.0572
-3.2853
P-Value
< 0.0001
0.0005
Non-Inferiority Test
Passed
Passed
Modified Per Protocol
Subjects (7 TAVR, 15 SAVR) censored before 1 year were included in the modified per protocol.
For worst case scenario the following assumptions were made for the censored subjects: the 7 TAVR subjects
were assumed to have died on date of censoring and the 15 SAVR subjects were assumed to be alive at 1 year.
Modified Per Protocol
TAVR
N=372
SAVR
N=341
Worst Case Scenario
TAVR
N=372
SAVR
N=341
Figure 58: All-Cause Mortality Kaplan-Meier Event Rate – Modified Per Protocol
Population
64
Page 67

Figure 59: All-Cause Mortality Kaplan-Meier Event Rate – Modified Per Protocol
Population – Worst Case Scenario
11.2.3.2 Key secondary safety and effectiveness endpoints
Hierarchical testing of secondary endpoints
Hypothesis testing was performed on six pre-specified secondary endpoints using a
hierarchical test procedure, as shown in Table 13. TAVR was found to be statistically noninferior to SAVR within the pre-specified non-inferiority margins in terms of changes in
mean gradient and EOA as well as in NYHA functional classification and Kansas City
Cardiomyopathy Questionnaire (KCCQ) from baseline to 12 months. However, TAVR was
not found to be statistically superior to SAVR with respect to the MACCE rate (p=0.1033),
which was a powered secondary endpoint, as discussed in more detail later. In other words,
the powered secondary endpoint of MACCE rate was not met. As a result, no hypothesis
testing was conducted on SF-12 per the pre-specified hierarchical testing protocol.
65
Page 68

Secondary Objective
TAVR
SAVR
Difference
Confidence
p-value
Test
Implanted Population
#9 / Mean gradient
95% Lower CI
39.04 ±
35.42 ±
3.62
1.49
<0.0001
Passed
#9 / EOA change
95% Lower CI
1.20 ±
0.81 ±
0.39
0.31
<0.0001
Passed
AT Population
#5 / NYHA change
95% Lower CI
1.46 ±
1.46 ±
-0.001
-0.11
<0.0001
Passed
#8 / KCCQ change
95% Lower CI
23.20 ±
21.88 ±
1.32
-2.84
0.0063
Passed
Powered Secondary
97.5% Upper CI
8.21%
10.93%
-2.73%
1.51%
0.1033
Failed
#8 / SF-12 change
95% two-sided CI
4.91 ±
-0.12 ±
5.03
(2.94, 7.13)
NA
Not
Table 13: High Risk Cohort Secondary Endpoints: Hierarchical Testing
change
(12 Month – Baseline;
mmHg)
Ha: TAVR > SAVR-15
(12 Month – Baseline;
cm2)
Ha: TAVR > SAVR-
0.375
(12 Month – Baseline)
Ha: TAVR > SAVR-
0.375
(12 Month – Baseline)
Ha: TAVR > SAVR-5
13.63
(n=290)
0.53
(n=253)
0.76
(n=305)
25.56
(n=243)
15.42
(n=222)
0.50
(n=182)
0.81
(n=232)
26.57
(n=189)
(TAVR-
SAVR)
Limit of the
Difference
Result
#1 / MACCE event rate
at 1 Month (K-M)
Ha: TAVR < SAVR
(1 Month – Baseline)
Ha: TAVR ≠ SAVR
Powered secondary hypothesis
For the AT population, the MACCE rate was 8.21% for TAVR and 10.93% for SAVR
(p = 0.1033). The null hypothesis that TAVR was equal to SAVR in the MACCE rate could
not be rejected. Of note is that the MACCE rate observed in the trial for SAVR was
considerably lower than that assumed in the power calculation (20% vs. 12.1%), which
resulted in the pre-specified sample size being too small to detect a difference between the
two study arms even if a difference exists. Therefore, this particular secondary endpoint was
underpowered for the specified hypothesis testing.
(n=390)
10.26
(n=215)
(n=357)
10.04
(n=158)
Tested
66
Page 69

All-Cause Mortality
13 (13)
3.3%
16 (16)
4.5%
55 (55)
14.2%
67 (67)
19.1%
Cardiovascular
12 (12)
3.1%
16 (16)
4.5%
40 (40)
10.4%
44 (44)
12.8%
Valve-Related1
9 (9)
2.3%
2 (2)
0.6%
21 (21)
5.6%
7 (7)
2.2%
NonCardiovascular
Reintervention
3 (3)
0.8%
0 (0)
0.0%
7 (7)
1.9%
0 (0)
0.0%
Surgical
2 (2)
0.5%
0 (0)
0.0%
3 (3)
0.8%
0 (0)
0.0%
Percutaneous
1 (1)
0.3%
0 (0)
0.0%
4 (4)
1.1%
0 (0)
0.0%
Neurological
Events
110
(133)
All Stroke
19 (20)
4.9%
22 (23)
6.2%
33 (34)
8.8%
42 (45)
12.6%
Major Stroke
15 (16)
3.9%
11 (11)
3.1%
22 (23)
5.8%
23 (23)
7.0%
Ischemic
14 (14)
3.6%
9 (9)
2.5%
19 (19)
5.0%
18 (18)
5.5%
Hemorrhagic
1 (2)
0.3%
0 (0)
0.0%
3 (4)
0.8%
3 (3)
0.9%
Minor Stroke
4 (4)
1.0%
12 (12)
3.4%
11 (11)
3.0%
20 (22)
6.0%
Ischemic
3 (3)
0.8%
11 (11)
3.1%
10 (10)
2.7%
18 (20)
5.4%
Hemorrhagic
0 (0)
0.0%
1 (1)
0.3%
0 (0)
0.0%
2 (2)
0.6%
TIA
3 (3)
0.8%
1 (1)
0.3%
6 (7)
1.6%
5 (5)
1.6%
Intracranial
Hemorrhage
All-Cause Mortality
or Major Stroke
11.2.3.3 Additional safety data
Adverse events that occurred in the PMA clinical study
Table 14, Table 15, and Table 16 provide a summary of the adverse events (AEs) that
occurred in this study for the pooled, iliofemoral and non-iliofemoral High Risk cohorts. AEs
for the AT population are summarized and K-M rates are provided.
The primary endpoint of all-cause mortality at 12 months includes all deaths (cardiovascular
and non-cardiovascular) from any cause after or during a valve intervention. The rates of allcause mortality at 12 months in the pooled AT population for both the TAVR and SAVR
treatment were 14.2% and 19.1% respectively.
Generally the rates of complications for the iliofemoral subjects were similar to the overall
rates of the pooled population since they comprised a significant majority of the overall study
cohort (323 of 390 subjects for TAVR and 300 of 357 subjects for SAVR).
Additionally, the rates of complications for the non-iliofemoral subjects were higher than the
rates for iliofemoral subjects for both TAVR and SAVR treatment arms.
Table 14: High Risk Cohort Adverse Event Summary – As Treated Population
Event
0-30 Days 0-12 Months
TAVR
N=390
# Pts
(#Event)
K-M
Rate
(%)
SAVR
N=357
# Pts
(#Event)
K-M
Rate
(%)
TAVR
N=390
# Pts
(#Event)
K-M
Rate
(%)
SAVR
N=357
# Pts
(#Event)
Rate
1 (1) 0.3% 0 (0) 0.0% 15 (15) 4.2% 23 (23) 7.3%
56 (61) 14.5% 79 (90) 22.2% 79 (101) 20.8%
31.9%
K-M
(%)
0 (0) 0.0% 0 (0) 0.0% 3 (3) 0.9% 2 (2) 0.7%
23 (29) 5.9% 24 (27) 6.7% 63 (78) 16.3% 79 (90) 22.5%
67
Page 70

CEC Adjudicated
Bleed
150
(161)
160
(186)
Life Threatening
or Disabling
106
(108)
110
(121)
Re-Classified
Bleed3
157
(170)
243
(258)
167
(195)
252
(290)
“Life Threatening
or Disabling”
125
(130)
136
(150)
109
(112)
123
(128)
114
(125)
130
(140)
Major Vascular
Complication
Acute Kidney Injury
23 (23)
6.0%
54 (54)
15.1%
23 (23)
6.0%
54 (54)
15.1%
MI
3 (3)
0.8%
3 (3)
0.8%
7 (7)
1.9%
5 (5)
1.5%
Peri-Procedural
3 (3)
0.8%
2 (2)
0.6%
3 (3)
0.8%
2 (2)
0.6%
Spontaneous
0 (0)
0.0%
1 (1)
0.3%
4 (4)
1.1%
3 (3)
0.9%
Cardiac Perforation
5 (5)
1.3%
0 (0)
0.0%
5 (5)
1.3%
0 (0)
0.0%
Cardiogenic Shock
9 (9)
2.3%
11 (11)
3.1%
9 (9)
2.3%
11 (11)
3.1%
Cardiac
Tamponade
Valve Endocarditis
0 (0)
0.0%
0 (0)
0.0%
2 (2)
0.6%
4 (4)
1.4%
Valve Thrombosis
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
Valve
Migration
96
(117)
200
(311)
229
(417)
Aortic Valve
Hospitalization
New Permanent
Implant7
Permanent
Implant8
Event
2, 6
Major Bleed
“Major Bleed”
0-30 Days 0-12 Months
TAVR
N=390
# Pts
(#Event)
K-M
Rate
(%)
38.5% NA NA
SAVR
N=357
# Pts
(#Event)
K-M
Rate
(%)
TAVR
N=390
# Pts
(#Event)
SAVR
N=357
K-M
Rate
(%)
# Pts
(#Event)
K-M
Rate
(%)
41.2% NA NA
48 (53) 12.3% NA NA 59 (65) 15.3% NA NA
27.3% NA NA
40.3%
53 (58) 13.6%
28.1%
68.1%
35.0% 64 (70) 16.6%
34.5%
28.4% NA NA
43.0%
70.8%
38.4%
29.5%
36.7%
23 (23) 5.9% 6 (6) 1.7% 24 (24) 6.2% 7 (7) 2.0%
6 (6) 1.5% 4 (4) 1.1% 7 (7) 1.8% 5 (5) 1.4%
Embolism/Device
0 (0) 0.0% 0 (0) 0.0% 0 (0) 0.0% 0 (0) 0.0%
MACCE4 30 (39) 7.7% 37 (42) 10.4% 79 (103) 20.4%
5, 6
MAE
51.3% NA NA
58.8% NA NA
15 (15) 3.9% 18 (19) 5.2% 59 (85) 16.1% 43 (59) 13.5%
Pacemaker
Pacemaker
76 (77) 25.7% 24 (24) 8.6% 84 (86) 28.6% 36 (36) 13.5%
76 (77) 19.8% 25 (25) 7.1% 85 (87) 22.3% 38 (38) 11.3%
27.3%
68
Page 71

1
All-Cause Mortality
11 (11)
3.4%
13 (13)
4.3%
43 (43)
13.4%
52 (52)
17.6%
Cardiovascular
10 (10)
3.1%
13 (13)
4.3%
32 (32)
10.1%
35 (35)
12.0%
Valve-Related1
8 (8)
2.5%
1 (1)
0.3%
18 (18)
5.7%
4 (4)
1.5%
NonCardiovascular
Reintervention
2 (2)
0.6%
0 (0)
0.0%
6 (6)
2.0%
0 (0)
0.0%
Surgical
1 (1)
0.3%
0 (0)
0.0%
2 (2)
0.7%
0 (0)
0.0%
Percutaneous
1 (1)
0.3%
0 (0)
0.0%
4 (4)
1.3%
0 (0)
0.0%
Neurological
Events
All Stroke
16 (17)
5.0%
15 (15)
5.0%
28 (29)
9.0%
30 (32)
10.8%
Event
(#Event)
Valve-related death is any death caused by prosthetic valve dysfunction, valve thrombosis, embolism, bleeding event, or
implanted valve endocarditis or related to reintervention on the operated valve.
2
For TAVR, periprocedural transfusions meeting VARC I major and life-threatening bleeding criteria were adjudicated as
events by the CEC irrespective of whether an overt bleeding complication had occurred. Since peri-procedural
transfusions meeting VARC I criteria may be considered standard of care for SAVR procedures depending on the clinical
circumstances, the same criteria were not applied and evidence of an overt bleeding complication (in addition to units
transfused) were required to adjudicate an event for SAVR only. This makes a direct comparison of the CEC adjudicated
bleeding rates in the trial inappropriate. For this reason, CEC adjudicated bleeding complications are shown for TAVR
only.
3
For the transfusion-based reclassification of bleeding events, units transfused were summed during the procedure, on
the day of the procedure and the day following the procedure. Patients who received 2-3 units of packed red blood cells or
homologous whole blood were considered to have had a “major bleeding complication” and patients receiving ≥4 units
were considered to have had a “life-threatening or disabling bleeding complication” for both TAVR and SAVR. The
nomenclature of the original adjudication was applied for consistency with this transfusion based re-classification.
4
MACCE includes all-cause death, myocardial infarction (MI), all stroke, and reintervention.
5
MAE includes all death, MI, all stroke, reintervention, cardiac perforation, cardiac tamponade, cardiogenic shock, valve
embolism/device migration, prosthetic valve dysfunction, acute kidney injury, major vascular complication, life threatening
or disabling bleed, major bleed, valve endocarditis VARC I Definitions.
6
Bleeding complications and MAE rate cells have been intentionally left blank for SAVR in this table because of differing
definitions employed for bleeding complications have made comparison of the rates to TAVR inappropriate.
7
Patients with pacemaker or ICD at baseline are excluded from the numerator and denominator. Note one (1) TAVR
patient and two (2) SAVR patients with baseline pacemaker/ICD, received new pacemaker/ICD between 30–365 days.
8
Patients with pacemaker or ICD at baseline are included in the denominator.
TAVR
N=390
# Pts
0-30 Days 0-12 Months
K-M
Rate
(%)
SAVR
N=357
# Pts
(#Event)
K-M
Rate
(%)
TAVR
N=390
# Pts
(#Event)
K-M
Rate
(%)
SAVR
N=357
# Pts
(#Event)
K-M
Rate
(%)
Table 15: High Risk Cohort Adverse Event Summary – Iliofemoral As Treated
Population
Event 0-30 Days 0-12 Months
TAVR
N=323
# Pts
(#Event)
K-M
Rate
(%)
SAVR
N=300
# Pts
(#Event)
K-M
Rate
(%)
TAVR
N=323
# Pts
(#Event)
K-M
Rate
(%)
SAVR
N=300
# Pts
(#Event)
K-M
Rate
(%)
1 (1) 0.3% 0 (0) 0.0% 11 (11) 3.7% 17 (17) 6.3%
41 (43) 12.8% 60 (67) 20.1% 62 (79) 19.7% 85 (103) 29.4%
69
Page 72

Major Stroke
12 (13)
3.7%
6 (6)
2.0%
18 (19)
5.8%
13 (13)
4.7%
Ischemic
11 (11)
3.4%
5 (5)
1.7%
15 (15)
4.8%
10 (10)
3.6%
Hemorrhagic
1 (2)
0.3%
0 (0)
0.0%
3 (4)
1.0%
2 (2)
0.7%
Minor Stroke
4 (4)
1.3%
9 (9)
3.0%
10 (10)
3.3%
17 (19)
6.2%
Ischemic
3 (3)
0.9%
8 (8)
2.7%
9 (9)
2.9%
15 (17)
5.4%
Hemorrhagic
0 (0)
0.0%
1 (1)
0.3%
0 (0)
0.0%
2 (2)
0.7%
TIA
3 (3)
0.9%
1 (1)
0.3%
6 (7)
2.0%
5 (5)
1.9%
Intracranial
Hemorrhage
CEC Adjudicated
Bleed
107
(112)
117
(136)
Life Threatening
or Disabling
Major Bleed
76 (78)
23.6%
NA
NA
80 (91)
25.0%
NA
NA
Re-Classified
Bleed3
113
(119)
204
(218)
123
(143)
210
(238)
“Life Threatening
or Disabling”
105
(110)
113
(121)
103
(108)
108
(117)
Major Vascular
Complication
Acute Kidney Injury
16 (16)
5.0%
43 (43)
14.3%
16 (16)
5.0%
43 (43)
14.3%
MI
3 (3)
0.9%
2 (2)
0.7%
7 (7)
2.3%
4 (4)
1.4%
Peri-Procedural
3 (3)
0.9%
2 (2)
0.7%
3 (3)
0.9%
2 (2)
0.7%
Spontaneous
0 (0)
0.0%
0 (0)
0.0%
4 (4)
1.4%
2 (2)
0.8%
Cardiac Perforation
4 (4)
1.2%
0 (0)
0.0%
4 (4)
1.2%
0 (0)
0.0%
Cardiogenic Shock
6 (6)
1.9%
8 (8)
2.7%
6 (6)
1.9%
8 (8)
2.7%
Cardiac
Tamponade
Valve Endocarditis
0 (0)
0.0%
0 (0)
0.0%
2 (2)
0.7%
3 (3)
1.2%
Valve Thrombosis
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
Valve
Migration
MACCE4
26 (33)
8.0%
29 (30)
9.7%
65 (85)
20.2%
76 (88)
25.6%
151
(239)
177
(331)
Aortic Valve
Hospitalization
Event 0-30 Days 0-12 Months
2, 6
TAVR
N=323
# Pts
(#Event)
K-M
Rate
(%)
SAVR
N=300
# Pts
(#Event)
K-M
Rate
(%)
TAVR
N=323
# Pts
(#Event)
K-M
Rate
(%)
SAVR
N=300
# Pts
(#Event)
K-M
Rate
(%)
0 (0) 0.0% 0 (0) 0.0% 3 (3) 1.0% 2 (2) 0.8%
33.1% NA NA
36.4% NA NA
32 (34) 9.9% NA NA 42 (45) 13.2% NA NA
35.0%
68.0%
38.3%
70.2%
35 (37) 10.8%
“Major Bleed” 80 (82) 24.9%
21 (21) 6.5% 5 (5) 1.7% 22 (22) 6.8% 6 (6) 2.0%
5 (5) 1.5% 3 (3) 1.0% 6(6) 1.9% 4 (4) 1.3%
Embolism/Device
5, 6
MAE
0 (0) 0.0% 0 (0) 0.0% 0 (0) 0.0% 0 (0) 0.0%
46.7% NA NA
8 (8) 2.5% 11 (12) 3.8% 44 (68) 14.5% 30 (43) 11.3%
35.0% 45 (48) 14.1%
34.4% 85 (95) 26.6%
54.8% NA NA
38.0%
36.2%
70
Page 73

New Permanent
Implant7
Permanent
Implant8
All-Cause Mortality
2 (2)
3.0%
3 (3)
5.3%
12 (12)
18.1%
15 (15)
27.3%
Cardiovascular
2 (2)
3.0%
3 (3)
5.3%
8 (8)
12.4%
9 (9)
16.8%
Valve-Related1
1 (1)
1.5%
1 (1)
1.8%
3 (3)
4.8%
3 (3)
6.0%
NonCardiovascular
Reintervention
1 (1)
1.5%
0 (0)
0.0%
1 (1)
1.5%
0 (0)
0.0%
Event 0-30 Days 0-12 Months
TAVR
N=323
# Pts
(#Event)
K-M
Rate
(%)
Pacemaker
Pacemaker
1
Valve-related death is any death caused by prosthetic valve dysfunction, valve thrombosis, embolism, bleeding event, or
implanted valve endocarditis or related to reintervention on the operated valve.
2
For TAVR, periprocedural transfusions meeting VARC I major and life-threatening bleeding criteria were adjudicated as
events by the CEC irrespective of whether an overt bleeding complication had occurred. Since peri-procedural transfusions
meeting VARC I criteria may be considered standard of care for SAVR procedures depending on the clinical circumstances,
the same criteria were not applied and evidence of an overt bleeding complication (in addition to units transfused) were
required to adjudicate an event for SAVR only. This makes a direct comparison of the CEC adjudicated bleeding rates in the
trial inappropriate. For this reason, CEC adjudicated bleeding complications are shown for TAVR only.
3
For the transfusion-based reclassification of bleeding events, units transfused were summed during the procedure, on the
day of the procedure and the day following the procedure. Patients who received 2-3 units of packed red blood cells or
homologous whole blood were considered to have had a “major bleeding complication” and patients receiving ≥4 units were
considered to have had a “life-threatening or disabling bleeding complication” for both TAVR and SAVR. The nomenclature
of the original adjudication was applied for consistency with this transfusion based re-classification.
4
MACCE includes all-cause death, myocardial infarction (MI), all stroke, and reintervention.
5
MAE includes all death, MI, all stroke, reintervention, cardiac perforation, cardiac tamponade, cardiogenic shock, valve
embolism/device migration, prosthetic valve dysfunction, acute kidney injury, major vascular complication, life threatening or
disabling bleed, major bleed, valve endocarditis VARC I Definitions.
6
Bleeding complications and MAE rate cells have been intentionally left blank for SAVR in this table due to differing
definitions employed for bleeding complications have made comparison of the rates to TAVR inappropriate.
7
Patients with pacemaker or ICD at baseline are not included.
8
Patients with pacemaker or ICD at baseline are included.
64 (65) 26.5% 21 (21) 9.0% 70 (72) 29.1% 33 (33) 14.9%
64 (65) 20.1% 21 (21) 7.1% 71 (73) 22.4% 34 (24) 12.1%
SAVR
N=300
# Pts
(#Event)
K-M
Rate
(%)
TAVR
N=323
# Pts
(#Event)
K-M
Rate
(%)
SAVR
N=300
# Pts
(#Event)
K-M
Rate
(%)
Table 16: High Risk Cohort Adverse Event Summary – Non-Iliofemoral As Treated
Population
Event 0-30 Days 0-12 Months
TAVR
N=67
# Pts
(#Event)
K-M
Rate
(%)
(#Event)
# Pts
SAVR
N=57
K-M
Rate
(%)
TAVR
N=67
# Pts
(#Event)
K-M
Rate
(%)
SAVR
# Pts
(#Event)
0 (0) 0.0% 0 (0) 0.0% 4 (4) 6.6% 6 (6) 12.6%
71
N=57
K-M
Rate
(%)
Page 74

Surgical
1 (1)
1.5%
0 (0)
0.0%
1 (1)
1.5%
0 (0)
0.0%
Percutaneous
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
Neurological
Events
All Stroke
3 (3)
4.5%
7 (8)
12.3%
5 (5)
7.7%
12 (13)
22.3%
Major Stroke
3 (3)
4.5%
5 (5)
8.8%
4 (4)
6.1%
10 (10)
18.7%
Ischemic
3 (3)
4.5%
4 (4)
7.0%
4 (4)
6.1%
8 (8)
15.4%
Hemorrhagic
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
1 (1)
1.9%
Minor Stroke
0 (0)
0.0%
3 (3)
5.3%
1 (1)
1.6%
3 (3)
5.3%
Ischemic
0 (0)
0.0%
3 (3)
5.3%
1 (1)
1.6%
3 (3)
5.3%
Hemorrhagic
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
TIA
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
Intracranial
Hemorrhage
CEC Adjudicated
Bleed
Life Threatening
or Disabling
Major Bleed
30 (30)
45.1%
NA
NA
30 (30)
45.1%
NA
NA
Re-Classified
Bleed3
“Life Threatening
or Disabling”
“Major Bleed”
29 (30)
43.6%
20 (20)
35.1%
29 (30)
43.6%
22 (23)
39.8%
Major Vascular
Complication
Acute Kidney Injury
7 (7)
10.6%
11 (11)
19.3%
7 (7)
10.6%
11 (11)
19.3%
MI
0 (0)
0.0%
1 (1)
1.8%
0 (0)
0.0%
1 (1)
1.8%
Peri-Procedural
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
Spontaneous
0 (0)
0.0%
1 (1)
1.8%
0 (0)
0.0%
1 (1)
1.8%
Cardiac Perforation
1 (1)
1.5%
0 (0)
0.0%
1 (1)
1.5%
0 (0)
0.0%
Cardiogenic Shock
3 (3)
4.5%
3 (3)
5.3%
3 (3)
4.5%
3 (3)
5.3%
Cardiac
Tamponade
Valve Endocarditis
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
1 (1)
2.3%
Valve Thrombosis
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
0 (0)
0.0%
Valve
Migration
MACCE4
4 (6)
6.1%
8 (12)
14.0%
14 (18)
21.4%
20 (29)
36.1%
MAE
49 (72)
73.1%
NA
NA
52 (86)
77.6%
NA
NA
Event 0-30 Days 0-12 Months
2, 6
TAVR
N=67
# Pts
(#Event)
K-M
Rate
(%)
(#Event)
# Pts
SAVR
N=57
K-M
Rate
(%)
TAVR
N=67
# Pts
(#Event)
K-M
Rate
(%)
SAVR
N=57
# Pts
(#Event)
K-M
Rate
(%)
15 (18) 22.7% 19 (23) 33.4% 17 (22) 25.8% 25 (30) 44.6%
0 (0) 0.0% 0 (0) 0.0% 0 (0) 0.0% 0 (0) 0.0%
43 (49) 64.2% NA NA 43 (50) 64.2% NA NA
Embolism/Device
16 (19) 23.9% NA NA 17 (20) 25.5% NA NA
44 (51) 65.7% 39 (40) 68.4% 44 (52) 65.7% 42 (52) 74.3%
18 (21) 26.9% 20 (20) 35.1% 19 (22) 28.5% 23 (29) 40.5%
2 (2) 3.0% 1 (1) 1.8% 2 (2) 3.0% 1 (1) 1.8%
1 (1) 1.5% 1 (1) 1.9% 1 (1) 1.5% 1 (1) 1.9%
0 (0) 0.0% 0 (0) 0.0% 0 (0) 0.0% 0 (0) 0.0%
5, 6
72
Page 75

Aortic Valve
Hospitalization
New Permanent
Implant7
Permanent
Implant8
1
Event 0-30 Days 0-12 Months
TAVR
N=67
# Pts
(#Event)
K-M
Rate
(%)
(#Event)
# Pts
SAVR
N=57
K-M
Rate
(%)
TAVR
N=67
# Pts
(#Event)
K-M
Rate
(%)
SAVR
N=57
# Pts
(#Event)
K-M
Rate
(%)
7 (7) 10.7% 7 (7) 12.9% 15 (17) 23.8% 13 (16) 25.2%
Pacemaker
Pacemaker
Valve-related death is any death caused by prosthetic valve dysfunction, valve thrombosis, embolism, bleeding event, or
implanted valve endocarditis or related to reintervention on the operated valve.
2
For TAVR, periprocedural transfusions meeting VARC I major and life-threatening bleeding criteria were adjudicated as
events by the CEC irrespective of whether an overt bleeding complication had occurred. Since peri-procedural transfusions
meeting VARC I criteria may be considered standard of care for SAVR procedures depending on the clinical circumstances,
the same criteria were not applied and evidence of an overt bleeding complication (in addition to units transfused) were
required to adjudicate an event for SAVR only. This makes a direct comparison of the CEC adjudicated bleeding rates in the
trial inappropriate. For this reason, CEC adjudicated bleeding complications are shown for TAVR only.
3
For the transfusion-based reclassification of bleeding events, units transfused were summed during the procedure, on the day
of the procedure and the day following the procedure. Patients who received 2-3 units of packed red blood cells or homologous
whole blood were considered to have had a “major bleeding complication” and patients receiving ≥4 units were considered to
have had a “life-threatening or disabling bleeding complication” for both TAVR and SAVR. The nomenclature of the original
adjudication was applied for consistency with this transfusion based re-classification.
4
MACCE includes all-cause death, myocardial infarction (MI), all stroke, and reintervention.
5
MAE includes all death, MI, all stroke, reintervention, cardiac perforation, cardiac tamponade, cardiogenic shock, valve
embolism/device migration, prosthetic valve dysfunction, acute kidney injury, major vascular complication, life threatening or
disabling bleed, major bleed, valve endocarditis VARC I Definitions.
6
Bleeding complications and MAE rate cells have been intentionally left blank for SAVR in this table due to differing definitions
employed for bleeding complications have made comparison of the rates to TAVR inappropriate.
7
Patients with pacemaker or ICD at baseline are not included.
8
Patients with pacemaker or ICD at baseline are included.
12 (12) 22.2% 3 (3) 6.3% 14 (14) 26.3% 3 (3) 6.3%
12 (12) 18.2% 4 (4) 7.1% 14 (14) 21.7% 4 (4) 7.1%
11.2.3.4 Additional effectiveness data
Improvement in NYHA functional classification was evaluated for As Treated TAVR and
SAVR patients. An evaluation of cardiac symptom severity based on NYHA classification
was conducted at several evaluation time points through the first year of follow-up
(Figure 60). Change from baseline to 12 months was evaluated for measures of forward flow
hemodynamic performance (EOA and mean gradient) for TAVR and SAVR patients
(Figure 61).
73
Page 76

Figure 60: High Risk Cohort NYHA Classification By Visit –As Treated Population
Figure 61: High Risk Cohort EOA and Mean Gradient by Visit –HR Cohort Implanted
TAVR & SAVR population
74
Page 77

Baseline
1 Month
6 Month
12 Month
KCCQ (n)
Overall Summary Score
TAVR
46.9 ± 23.4
(375)
66.2 ± 24.1
(248)
72.3 ± 22.3
(276)
72.1 ± 21.8
(252)
SAVR
46.6 ± 22.3
(327)
51.6 ± 25.4
(178)
70.6 ± 21.8
(219)
70.5 ± 22.1
(200)
Clinical Summary Score
TAVR
51.4 ± 23.3
66.8 ± 23.5
70.5 ± 21.9
69.9 ± 22.1
Figure 62 shows total aortic regurgitation (AR) severity over time in the Implanted TAVR
and SAVR arms. These data are presented per valve size as well as for all sizes combined for
both arms of the High Risk cohort.
The valve sizes had a relatively similar distribution of total AR during follow-up, although at
each evaluation a greater percentage of subjects with a 26 mm valve had no AR and a smaller
percentage had moderate or greater AR than for the 29 and 31 mm valves. All valve sizes
pooled are shown for the SAVR treatment arm. A notably smaller percentage of subjects in
the TAVR treatment arm had no AR than in the SAVR treatment arm (28.6% vs. 68.2% at
12 months) and a greater percentage of subjects in the TAVR treatment arm had moderate or
greater AR (7.1% vs. 1.3% at 12 months).
Figure 62: High Risk Cohort Total Aortic Regurgitation By Visit (Core Lab) – Implanted
Population
The Quality of Life (QoL) was evaluated using the Kansas City Cardiomyopathy
Questionnaire (KCCQ), the QualityMetric’s SF-12v2® Health Survey (SF12), and the
EuroQoL (EQ-5D), as shown in Table 17.
Table 17: High Risk Cohort Quality of Life – As Treated
75
Page 78

Baseline
1 Month
6 Month
12 Month
(375)
(248)
(276)
(252)
SAVR
50.8 ± 22.3
(327)
54.8 ± 24.5
(178)
70.3 ± 21.2
(219)
68.3 ± 22.2
(200)
SF12 (n)
Physical Component
TAVR
30.8 ± 9.2
(361)
35.9 ± 9.5
(228)
37.3 ± 10.3
(263)
37.0 ± 11.2
(237)
SAVR
31.0 ± 8.6
(309)
31.7 ± 8.5
(167)
37.6 ± 10.1
(209)
36.9 ± 9.7
(188)
Mental Component
TAVR
47.5 ± 12.1
(361)
51.1 ± 11.1
(228)
52.5 ± 10.9
(263)
52.8 ± 10.8
(237)
SAVR
48.4 ± 11.7
(309)
45.0 ± 13.1
(167)
51.1 ± 10.9
(209)
52.5 ± 10.5
(188)
EQ-5D (n)
TAVR
0.73 ± 0.20
(370)
0.78 ± 0.19
(244)
0.79 ± 0.19
(270)
0.78 ± 0.18
(248)
SAVR
0.73 ± 0.18
(326)
0.67 ± 0.25
(173)
0.80 ± 0.15
(215)
0.78 ± 0.18
(193)
Plus-minus values are mean ± standard deviation.
11.2.4 Additional study observations
Primary endpoint stratified by access route
The study was powered to demonstrate non-inferiority of TAVR compared to SAVR for the
primary endpoint for all patients (iliofemoral and non-iliofemoral) pooled. It was prespecified that the primary endpoint would be assessed for different access route subgroups
independently, but this assessment was not powered and would not be the basis for assessing
success or failure of the primary endpoint. The all-cause mortality rates are shown in
Figure 63 for the iliofemoral subgroup and Figure 64 for the non-iliofemoral subgroup.
76
Page 79

Figure 63: All-Cause Mortality – Iliofemoral As Treated Population
Figure 64: All-Cause Mortality – Non-Iliofemoral As Treated Population
77
Page 80

Gender analysis
The primary endpoint and the powered secondary endpoint of MACCE rate were examined
for gender differences as shown in Figure 65 and Figure 66.
Figure 65: All-Cause Mortality at 12 Months for Male Patients – As Treated Population
Figure 66: All-Cause Mortality at 12 Months for Female Patients – As Treated
Population
78
Page 81

Mortality stratified by STS score
An analysis was performed for TAVR patients to examine the relationship between all-cause
mortality and STS predicted risk of mortality at baseline (Figure 67). Patients were stratified
by STS score with the subgroups being STS <4, STS 4–7, STS >7–15, and STS >15.
Figure 67: High Risk Cohort All-Cause Mortality by STS – TAVR As Treated
Population
Post-implant aortic regurgitation and all-cause mortality
A post hoc subgroup analysis was performed for all TAVR patients (iliofemoral and noniliofemoral) of the Implanted population to investigate the relationship between all-cause
mortality and severity of aortic regurgitation at discharge (7 days post procedure or
discharge, whichever is first). Two subgroups of iliofemoral patients with none/trace and
greater than or equal to mild total aortic regurgitation at discharge were analyzed. The results
from the analysis are shown in Figure 68 which show that residual aortic regurgitation at
discharge appeared to be associated with long-term mortality in the TAVR patients.
However, it was also noted that there were some differences in important baseline clinical
characteristics of the patients between the two subgroups, as summarized in Table 18. As a
result, it is not clear whether there was a causal relationship between residual aortic
regurgitation and mortality. Nevertheless, the incidence of residual aortic regurgitation and
its apparent association with late-term mortality will need to be carefully monitored in postapproval follow-up.
79
Page 82

Demographics
Age (yrs)
82.7 ± 7.4
83.8 ± 6.4
Male
46.7% (92/197)
60.8% (101/166)
NYHA Class
II
16.2% (32/197)
12.7% (21/166)
III
65.0% (128/197)
66.9% (111/166)
IV
18.8% (37/197)
20.5% (34/166)
STS Score (Risk of Mortality, %)
7.3 ± 3.1
7.2 ± 2.8
Coronary Artery Disease
73.1% (144/197)
77.7% (129/166)
Previous MI
25.4% (50/197)
22.9% (38/166)
Previous Interventions
Coronary Artery Bypass
Surgery
Percutaneous Coronary
Intervention
Balloon Valvuloplasty
5.6% (11/197)
5.4% (9/166)
Cerebrovascular Disease
22.4% (44/196)
25.8% (42/163)
Prior Stroke
10.2% (20/197)
15.1% (25/166)
Peripheral Vascular Disease
44.7% (88/197)
34.1% (56/164)
Figure 68: All-Cause Mortality by Severity of Aortic Regurgitation (None/Trace vs
Mild/Moderate/Severe) – TAVR Implanted Population
Table 18: Patient Demographics and Clinical Characteristics Stratified by AR – TAVR
Implanted Population
None/Trace AR
N=197
30.5% (60/197) 28.9% (48/166)
Mild/Moderate/Severe AR
N=166
35.5% (70/197) 31.9% (53/166)
80
Page 83

Chronic Lung Disease/COPD
39.6% (78/197)
50.0% (83/166)
Home Oxygen
10.7% (21/196)
13.9% (23/166)
Creatinine Level >2 mg/dl
2.0% (4/197)
4.8% (8/166)
Atrial Fibrillation/Atrial Flutter
38.1% (75/197)
45.5% (75/165)
Preexisting Permanent
Pacemaker Placement / ICD
Aorta Calcification1
Severe
14.2% (28/197)
9.7% (16/165)
Porcelain
0.5% (1/197)
0.0% (0/165)
Chest Wall Deformity
2.5% (5/197)
2.4% (4/166)
Hostile Mediastinum
4.6% (9/197)
3.6% (6/166)
Wheelchair Bound
3.6% (7/197)
3.6% (6/166)
None/Trace AR
N=197
19.3% (38/197) 26.5% (44/166)
1. Aorta Calcification is measured on screening CT Angiogram.
Plus-minus values present the mean ± standard deviation.
Mild/Moderate/Severe AR
N=166
81
Page 84

11.3 Extreme risk cohort
The CoreValve™ U.S. Pivotal Trial Extreme Risk cohort was a prospective, nonrandomized, unblinded, multi-center investigational study. All enrolled patients were
assigned to transcatheter aortic valve replacement (TAVR) with the Medtronic CoreValve™
system. The purpose of this clinical study was to evaluate the safety and effectiveness of the
Medtronic CoreValve™ system in the treatment of symptomatic severe aortic stenosis in
patients requiring aortic valve replacement with predicted operative mortality or serious,
irreversible morbidity risk of ≥50% at 30 days (Extreme Risk).
This Extreme Risk cohort enrolled 656 patients with symptomatic severe aortic stenosis
(500 iliofemoral and 156 non-iliofemoral patients) at 41 of the 43 activated centers in the
United States with baseline characteristics described in Table 19. Severe aortic stenosis was
defined as an aortic valve area of ≤0.8 cm2 or aortic valve area index ≤0.5 cm2, a mean aortic
valve gradient of >40 mmHg or jet velocity >4 m/sec. The primary endpoint was all-cause
mortality or major stroke at 12 months. The primary analysis compared the primary endpoint
against a pre-specified performance goal.
Patients received the CoreValve™ bioprosthesis either through the iliofemoral access route
or through the non-illiofemoral (subclavian and direct aortic) access routes. An attempted
implant was performed on 489 patients via iliofemoral access and who embody the
Attempted Implantc iliofemoral cohort (n=489), which was the basis for assessment of the
primary endpoint. Of the 489 attempted implants via iliofemoral access, 486 patients were
implanted with the CoreValve™ bioprosthesis and embody the Implantedd iliofemoral cohort
(n=486), which was the basis for secondary endpoints related to hemodynamic data.
An attempted implant was performed on 150 patients via non-iliofemoral access and these
patients embody the Attempted Implant cohort. Of these 150 patients, 148 were implanted
with the CoreValve™ bioprosthesis and embody the Implanted non-iliofemoral cohort. Per
protocol, non-iliofemoral patients were not included in the primary analysis due to
anticipated heterogeneity in patient selection and outcome. Compared with patients enrolled
in the iliofemoral cohort, patients in the non-iliofemoral cohort were, generally, at a higher
risk with respect to specific critical co-morbidities.
The following data summarize the results from the Extreme Risk cohort (iliofemoral and
non-iliofemoral).
11.3.1 Patient population
The patient characteristics analyzed for the iliofemoral and non-iliofemoral enrolled cohorts
include demographics, clinical characteristics, medical history, and potentially prohibitive
anatomic factors for surgical aortic valve replacement (SAVR) and assessments for comorbidity, frailty, and disability (Table 19). The ability of a patient to obtain a functional
recovery after SAVR is largely based on the presence of significant co-morbidities, frailties,
and disabilities, with the combination of the factors having higher weight than the individual
c
The Attempted Implant population consisted of all patients with an attempted procedure, defined as when the
patient was brought into the procedure room and any of the following had occurred: anesthesia administered,
vascular line placed, TEE placed, or any monitoring line placed.
d
The Implanted population consisted of all Attempted Implant patients who were actually implanted with the
CoreValve™ bioprosthesis.
82
Page 85

II
Coronary Artery Bypass
Surgery
Percutaneous Coronary
Intervention
Peripheral Vascular Disease
Preexisting Permanent
Pacemaker Placement/ICD
Aorta Calcification1:
Severe/Porcelain
factors alone. As detailed in Table 19, a high proportion of the CoreValve™ Extreme Risk
patients had significant co-morbidities, frailties, or disabilities, which established the study
population as “Extreme Risk.” The mean age for patients participating in the trial was
approximately 83 years old, and slightly less than 50% of patients were male. The mean
Society of Thoracic Surgeons (STS) score was approximately 10. Greater than 90% of all
patients were in NYHA classes III or IV.
Additionally, coronary artery disease was present in approximately 80% of patients, and
greater than 30% of patients had previous MI. Peripheral vascular disease, COPD, and home
oxygen use were more prevalent for non-iliofemoral patients.
Table 19: Extreme Risk Cohort Baseline Characteristics and Echocardiographic
Findings (All Enrolled)
Demographic
Age (years) 83.1 ± 8.6 (500) 81.6 ± 7.7 (156)
Gender (Male)
NYHA Classification
III
IV
STS Score (Risk of Mortality, %)
Coronary Artery Disease
Previous MI 31.0% (155/500) 31.4% (49/156)
Previous Interventions
Balloon Valvuloplasty 20.4% (102/500) 22.4% (35/156)
Cerebral Vascular Disease
Iliofemoral
N=500
48.0% (240/500) 44.9% (70/156)
8.6% (43/500) 8.3% (13/156)
63.6% (318/500) 66.0% (103/156)
27.8% (139/500) 25.6% (40/156)
10.3 ± 5.5 10.5 ± 5.7
81.8% (409/500) 78.8% (123/156)
39.0% (195/500) 41.0% (64/156)
37.4% (187/500) 30.1% (47/156)
24.0% (119/496) 28.4% (44/155)
Non-Iliofemoral
N=156
Prior Stroke
Chronic Lung Disease/COPD
Home Oxygen
Creatinine Level >2 mg/dl
Atrial Fibrillation/Atrial Flutter
Severe 16.6% (83/499) 17.5% (27/154)
13.6% (68/499) 14.2% (22/155)
36.0% (179/497) 59.0% (92/156)
59.6% (298/500) 69.9% (109/156)
30.8% (154/500) 41.7% (65/156)
4.6% (23/500) 2.6% (4/156)
47.4% (236/498) 48.4% (75/155)
25.8% (129/500) 24.4% (38/156)
83
Page 86

Porcelain
Ejection Fraction (Visual
Estimate, %)
Mean Gradient across Aortic
Valve (MGV2, mmHg)
Mitral Regurgitation:
Moderate/Severe
1. Aorta Calcification is measured on screening CT Angiogram.
Plus-minus values present the mean ± standard deviation.
Total Procedure Time (min)
(skin to skin)
Emergent Operation Due to Device
or Procedure
1
Demographic
Iliofemoral
N=500
Non-Iliofemoral
N=156
5.2% (26/499) 7.8% (12/154)
Chest Wall Deformity
Hostile Mediastinum
Cirrhosis of the Liver
5.6% (28/500) 1.9% (3/156)
12.0% (60/499) 9.0% (14/156)
3.0% (15/500) 1.3% (2/156)
Wheelchair Bound 16.6% (83/500) 12.2% (19/156)
Echocardiographic Findings
53.2 ± 13.6 (498) 54.3 ± 15.3 (156)
Aortic Valve Area (cm2) 0.67 ± 0.25 (485) 0.62 ± 0.23 (153)
47.72 ± 13.53 (498) 49.67 ± 16.85 (156)
24.2% (120/496) 23.2% (36/155)
11.3.2 Procedure data
Table 20 provides a summary of the transcatheter valve implantation procedures for the
iliofemoral and non-iliofemoral cohorts, respectively. Overall device success rate was 84.6%
for the iliofemoral cohort and 88.7% for the non-iliofemoral cohort. Procedure success was
defined as device success and absence of in-hospital MACCE and procedure success rates
were 77.6% and 77.5% for the iliofemoral and non-iliofemoral cohorts, respectively.
Table 20: Extreme Risk Cohort TAVR Procedure Data (Attempted Implant)
Time to Procedure (days)
Iliofemoral
N=489
8.9 ± 12.3 (489) 10.2 ± 15.5 (150)
Non-Iliofemoral
N=150
Total Time in Cath Lab or OR (min) 214.8 ± 64.9 (486) 258.7 ± 72.5 (148)
66.1 ± 39.0 (484) 60.5 ± 46.5 (145)
General Anesthesia 94.4% (459/486)
99.3% (147/148)
Valve-in-Valve Procedure 2.5% (12/486) 0.7% (1/148)
0.0% (0/486) 0.0% (0/148)
Number of Devices Used
0
0.6% (3/489) 1.3% (2/150)
93.3% (456/489) 94.7% (142/150)
2
Valve Size Implanted
23 mm
6.1% (30/489) 4.0% (6/150)
2.5% (12/486) 6.1% (9/148)
84
Page 87

26 mm
1. Device success is defined as deployment, only 1 valve implanted, only 1 valve in correct anatomic location,
Plus-minus values present the mean ± standard deviation.
Iliofemoral
N=489
Non-Iliofemoral
N=150
35.0% (170/486) 41.2% (61/148)
29 mm
31 mm
Device Success1
58.4% (284/486) 49.3% (73/148)
4.1% (20/486) 3.4% (5/148)
84.6% (397/469) 88.7% (125/141)
Procedure Success2 77.6% (370/477) 77.5% (110/142)
EOA >1.2 cm2 for 26, 29, and 31 mm and ≥0.9 cm2 for 23 mm, mean gradient <20mmHg, and aortic regurgitation
< moderate.
2. Procedure success is defined as device success and absence of in-hospital MACCE.
11.3.3 Safety and effectiveness results
11.3.3.1 Primary safety and effectiveness endpoint
The estimated K-M rate for all-cause mortality or major stroke at 12 months for the
Attempted Implant iliofemoral cohort was 26.0% with an upper 2-sided 95% CI of 29.9%.
The upper 95% CI was lower than the pre-specified Performance Goal rate of 43% for this
primary endpoint (p<0.0001) (Figure 69).
85
Page 88

Figure 69: Extreme Risk Cohort Primary Endpoint: All-Cause Mortality or Major Stroke
Kaplan-Meier Event Rate — Iliofemoral Attempted Implant
11.3.3.2 Additional safety data
Table 21 and Table 22 provide a summary of the adverse events (AEs) that occurred in this
study for the iliofemoral and non-iliofemoral cohorts. AEs for the Attempted Implant
populations are summarized and Kaplan-Meier (K-M) rates are provided.
The rates of all-cause mortality or major stroke (the primary endpoint of the trial) were
26.0% and 39.4% at 1 year for the iliofemoral and non-iliofemoral cohorts, respectively.
Mortality was the primary driver of the primary endpoint for both the iliofemoral and noniliofemoral cohorts and cardiovascular mortality made up the majority of all deaths
experienced in both cohorts. The greater event rate of all-cause mortality or major stroke in
the non-iliofemoral cohort was expected based on the comorbidities identified in this group
of patients.
Several important periprocedural complications including acute kidney injury, myocardial
infarction, and major vascular complications generally occurred at similar rates for
iliofemoral and non-iliofemoral patients. Bleeding complications were the most frequently
observed early adverse events. Early (within 30 days) permanent pacemaker implantation
(PPI) occurred in a significant minority of patients in both cohorts.
86
Page 89

Iliofemoral N=489
Event
0-30 Days
0-12 Months
All-Cause
Major Stroke
All-Cause
Mortality
Cardiovascular
41
41
8.4%
88
88
18.3%
Valve-Related
1
12
12
2.5%
23
23
5.1%
Neurological
Events
All Stroke
20
19
4.0%
34
31
7.0%
Major Stroke
11
11
2.3%
20
19
4.3%
Ischemic
9
9
1.9%
17
16
3.6%
Hemorrhagic
2
2
0.4% 3 3
0.7%
Minor Stroke
9
9
1.9%
14
14
3.2%
Ischemic
9
9
1.9%
14
14
3.2%
Hemorrhagic
0
0
0.0% 0 0
0.0%
TIA 3 3
0.6% 5 5
1.1%
Intracranial
Hemorrhage
Bleed
191
179
36.7%
236
206
42.8%
Life
or Disabling
Major Bleed
128
121
24.9%
148
136
28.5%
Major Vascular
Complication
Acute Kidney
Injury
MI 6 6
1.2% 9 9
2.0%
PeriProcedural
Spontaneous
0
0
0.0% 3 3
0.7%
MACCE
2
72
60
12.3%
171
143
29.2%
Cardiac
Perforation
Cardiogenic
Shock
Reintervention
5
5
1.1% 9 8
1.8%
Table 21: Extreme Risk Cohort Adverse Event Summary - Iliofemoral Attempted
Implant
Mortality or
K-M
#
Events # Patients
Rate
(%)
#
Events # Patients
52 48 9.8% 139 127 26.0%
41 41 8.4% 119 119 24.3%
80 74 15.5% 141 117 25.3%
1 1 0.2% 2 2 0.4%
K-M
Rate
(%)
Threatening
Cardiogenic
Tamponade
63 62 12.7% 88 83 17.6%
44 40 8.2% 45 41 8.4%
57 57 11.8% 57 57 11.8%
6 6 1.2% 6 6 1.2%
9 9 1.8% 9 9 1.8%
13 13 2.7% 13 13 2.7%
9 9 1.9% 10 10 2.1%
87
Page 90

Iliofemoral N=489
Event
0-30 Days
0-12 Months
Surgical
0
0
0.0% 0 0
0.0%
Percutaneous
5
5
1.1% 9 8
1.8%
Valve
Endocarditis
Valve
Thrombosis
Valve Embolism/
Device Migration
MAE3
458
263
53.8%
625
307
62.8%
Aortic Valve
Hospitalization
New Permanent
Implant4
Permanent
Implant5
All-Cause
Major Stroke
All-Cause
Mortality
#
Events # Patients
K-M
Rate
(%)
#
Events # Patients
K-M
Rate
(%)
0 0 0.0% 5 5 1.3%
0 0 0.0% 0 0 0.0%
0 0 0.0% 1 1 0.2%
32 31 6.7% 124 94 21.6%
Pacemaker
Pacemaker
1
Valve-related death is any death caused by prosthetic valve dysfunction, valve thrombosis,
embolism, bleeding event, or implanted valve endocarditis or related to reintervention on the
operated valve.
2
MACCE includes all-cause death, myocardial infarction (MI), all stroke, and reintervention.
3
MAE includes all death, MI, all stroke, reintervention, cardiac perforation, cardiac
tamponade, cardiogenic shock, valve embolism/device migration, prosthetic valve
dysfunction, acute kidney injury, major vascular complication, life threatening or disabling
bleed, major bleed, valve endocarditis VARC I Definitions.
4
Patients with pacemaker or ICD at baseline are not included.
5
Patients with pacemaker or ICD at baseline are included.
105 104 29.4% 125 121 34.9%
105 104 21.6% 127 123 26.2%
Table 22: Extreme Risk Cohort Adverse Event Summary – Non-Iliofemoral Attempted
Implant
Non-Iliofemoral N=150
Event 0-30 Days 0-12 Months
Mortality or
#
Events # Patients
28
23
K-M
Rate
(%)
15.3%
#
Events # Patients
67
59
K-M
Rate
(%)
39.4%
17 17 11.3% 54 54 36.0%
88
Page 91

Cardiovascular
17
17
11.3%
42
42
28.8%
Valve-Related
4 4 2.8% 7 7
5.4%
Neurological
Events
36 32
21.8%
46 40
28.5%
All Stroke
14
13
8.8%
19
18
13.0%
Major Stroke
11
11
7.5%
13
13
9.1%
Ischemic
11
11
7.5%
12
12
8.3%
Hemorrhagic
0 0 0.0% 1 1
0.9%
Minor Stroke
3 3 2.1% 6 6
4.7%
Ischemic
3 3 2.1% 5 5
3.8%
Hemorrhagic
0 0 0.0% 1 1
0.8%
TIA 2 2
1.4% 3 3
2.3%
Intracranial
Hemorrhage
Bleed
92
87
58.3%
106
96
65.1%
Life
Disabling
Major Bleed
56
55
37.1%
63
60
41.9%
Major Vascular
Complication
Acute Kidney
Injury
MI 3 3
2.1% 3 3
2.1%
PeriProcedural
Spontaneous
1 1 0.7% 1 1
0.7%
MACCE
2
34
26
17.3%
77
62
41.4%
Cardiac
Perforation
Cardiogenic
Shock
Cardiogenic
Tamponade
Reintervention
0 0 0.0% 1 1
1.0%
Surgical
0 0 0.0% 0 0
0.0%
Percutaneous
0 0 0.0% 1 1
1.0%
Valve
Endocarditis
Valve
Thrombosis
Non-Iliofemoral N=150
Event 0-30 Days 0-12 Months
Threatening or
#
Events # Patients
1
0 0 0.0% 1 1 0.9%
36
36
K-M
Rate
(%)
24.2%
#
Events # Patients
43
43
K-M
Rate
(%)
29.4%
13 13 8.7% 14 14 9.5%
21 21 14.2% 21 21 14.2%
2 2 1.3% 2 2 1.3%
2 2 1.3% 2 2 1.3%
9 9 6.0% 9 9 6.0%
2 2 1.3% 2 2 1.3%
1 1 0.7% 2 2 1.7%
0 0 0.0% 2 1 0.8%
89
Page 92

Valve Embolism/
Device Migration
MAE3
189
104
69.3%
253
120
80.0%
Aortic Valve
Hospitalization
New Permanent
Implant4
Permanent
Implant5
Non-Iliofemoral N=150
Event 0-30 Days 0-12 Months
#
Events # Patients
K-M
Rate
(%)
#
Events # Patients
K-M
Rate
(%)
0 0 0.0% 0 0 0.0%
13 12 8.7% 37 27 21.2%
Pacemaker
Pacemaker
1
Valve-related death is any death caused by prosthetic valve dysfunction, valve thrombosis,
embolism, bleeding event, or implanted valve endocarditis or related to reintervention on the
operated valve.
2
MACCE includes all-cause death, myocardial infarction (MI), all stroke, and reintervention.
3
MAE includes all death, MI, all stroke, reintervention, cardiac perforation, cardiac tamponade,
cardiogenic shock, valve embolism/device migration, prosthetic valve dysfunction, acute kidney
injury, major vascular complication, life threatening or disabling bleed, major bleed, valve
endocarditis VARC I Definitions.
4
Patients with pacemaker or ICD at baseline are not included.
5
Patients with pacemaker or ICD at baseline are included.
24 24 22.0% 30 30 28.8%
24 24 16.4% 30 30 21.5%
Patients with unsuitable iliofemoral anatomy for placement of an 18-Fr sheath are at a higher
risk with respect to specific critical co-morbidities including peripheral vascular disease,
cerebrovascular disease, and chronic lung disease. While at a higher risk, these noniliofemoral patients with suitable axillary/subclavian or direct aortic access may be treated
with the CoreValve™ device. Given the unavailability of any viable treatment option, the
overall performance of the device and the associated benefits of treatment outweigh the risks
for this non-iliofemoral Extreme Risk patient population.
The estimated K-M rate of all-cause mortality or major stroke at 12 months for the
Attempted Implant non-iliofemoral cohort was 39.4% with an upper 95% CI of 47.2%, which
was higher than for the iliofemoral cohort (Figure 70).
Table 23 provides a summary of the K-M estimate of event free rates of key outcomes for
both the iliofemoral and non-iliofemoral cohorts. As shown in Table 23, the non-iliofemoral
cohort reported higher rates of all-cause death and all-stroke, which resulted in higher
MACCE and MAE rates compared to the iliofemoral cohort.
90
Page 93

IF
87.7
77.5
70.8
NIF
82.7
65.3
58.6
All-Cause
Death
IF
91.6
81.4
75.7
NIF
88.7
71.3
64.0
Myocardial
Infarction
IF
98.8
98.5
98.0
NIF
97.9
97.9
97.9
IF
96.0
94.8
93.0
NIF
91.2
88.0
87.0
IF
98.9
98.5
98.2
NIF
100.0
100.0
99.0
IF
46.2
40.1
37.2
NIF
30.7
24.0
20.0
Figure 70: Extreme Risk Cohort All-Cause Mortality or Major Stroke Kaplan-Meier
Event Rate Attempted Implant (IF: iliofemoral; NIF: non-iliofemoral)
Table 23: Extreme Risk Cohort Kaplan-Meier Estimate of Event-Free Rates: Results by
IF (N=489) and NIF (N=150) Cohorts
Days post Attempted Implant
Event
MACCE
All Stroke
Reintervention
MAE
Access
Site
30 days
6 months
(183 days)
12 months
(365 days)
p-value*
0.004
0.004
0.861
0.015
0.408
<0.001
*p-value from Log-Rank test comparing freedom from curves through 365 days
A post hoc analysis was conducted to compare the K-M event rates for all-cause mortality or
major stroke at 12 months between Attempted Implant iliofemoral patients in different
Society of Thoracic Surgeons (STS) risk score categories (<5%, 5–15%, >15%). The STS
91
Page 94

risk score calculates the risk of operative mortality and morbidity of adult cardiac surgery on
the basis of patient demographic and clinical variables. The Log-rank p-value for the K-M
analysis was 0.042, indicating a statistically significant difference in the event rate between
different STS score categories (Figure 71).
Figure 71: Extreme Risk Cohort Primary Endpoint: All-Cause Mortality or Major Stroke
Stratified by STS Score – Attempted Implant Iliofemoral
11.3.3.3 Additional effectiveness data
Improvement in NYHA functional classification was evaluated for Implanted iliofemoral and
non-iliofemoral patients. An evaluation of cardiac symptom severity based on NYHA
classification was conducted at several evaluation time points through the first year of
follow-up (Figure 72). Change from baseline to 12 months was evaluated for measures of
forward flow hemodynamic performance (EOA and mean gradient) for iliofemoral and noniliofemoral patients (Figure 73 and Figure 74).
92
Page 95
Figure 72: Extreme Risk Cohort NYHA Classification by Visit – Attempted Implant
Figure 73: Extreme Risk Cohort EOA and Mean Gradient by Visit – Iliofemoral
Implanted
93
Page 96

Figure 74: Extreme Risk Cohort EOA and Mean Gradient by Visit – Non-Iliofemoral
Implanted
Figure 75 shows total aortic regurgitation (AR) severity over time in the Implanted
iliofemoral population. These data are presented per valve size as well as for all sizes
combined. Considering all valve sizes, the majority of patients presented at
1, 6, and 12 months with AR severity classified as trivial or mild. Over time, the percentage
of patients with moderate or severe AR decreased to 0% at 12 months. The number of
patients with no AR increased over time to 21.3% at 12 months.
Figure 75: Extreme Risk Cohort Total Aortic Regurgitation by Visit – Iliofemoral
Implanted
94
Page 97

KCCQ (n)
Overall Summary Score
37.9 ± 22.1
(454)
62.3 ± 25.5
67.7 ± 24.2
68.8 ± 23.6
Clinical Summary Score
42.0 ± 22.4
(454)
62.3 ± 24.9
66.7 ± 23.8
66.3 ± 23.4
SF12 (n)
Physical Component
28.5 ± 8.3
(422)
34.9 ± 10.1
33.8 ± 11.3
34.3 ± 10.5
Mental Component
45.8 ± 12.3
(422)
49.8 ± 12.0
51.6 ± 11.0
51.9 ± 11.8
EQ-5D (n)
0.65 ± 0.23
(445)
0.73 ± 0.24
0.76 ± 0.20
0.73 ± 0.21
Plus-minus values are mean ± standard deviation.
Figure 76 shows total AR severity over time in the Implanted non-iliofemoral population.
Considering all valve sizes, the majority of patients presented at 1 month with AR severity
classified as mild or less. Over time, the percentage of patients with no AR increased to
39.0% at 12 months.
Figure 76: Extreme Risk Cohort Total Aortic Regurgitation by Visit – Non-Iliofemoral
Implanted
The Quality of Life (QoL) was evaluated using the Kansas City Cardiomyopathy
Questionnaire (KCCQ), the QualityMetric's SF-12v2® Health Survey (SF12), and the
EuroQoL (EQ-5D), as shown in Table 24 and Table 25.
Table 24: Extreme Risk Cohort Quality of Life – Iliofemoral Attempted Implant
Baseline 1 Month 6 Month 12 Month
(266)
(266)
(245)
(245)
(301)
(301)
(276)
(276)
(287)
(287)
(259)
(259)
95
(261)
(295)
(275)
Page 98

KCCQ (n)
Overall Summary Score
42.5 ± 22.3
(141)
51.0 ± 25.5
(74)
65.7 ± 23.8
(77)
65.1 ± 22.4
(81)
Clinical Summary Score
46.7 ± 23.0
(141)
53.7 ± 24.6
(74)
64.2 ± 23.2
(77)
65.2 ± 21.3
(81)
SF12 (n)
Physical Component
27.9 ± 8.0
(130)
32.0 ± 9.2
(66)
32.5 ± 10.7
(74)
34.0 ± 9.4
(80)
Mental Component
47.6 ± 12.0
(130)
45.1 ± 14.7
(66)
51.3 ± 11.2
(74)
49.0 ± 13.3
(80)
EQ-5D (n)
0.67 ± 0.23
(138)
0.66 ± 0.25
(72)
0.74 ± 0.19
(75)
0.73 ± 0.20
(80)
Plus-minus values are mean ± standard deviation.
Table 25: Extreme Risk Cohort Quality of Life – Non-Iliofemoral Attempted Implant
Baseline 1 Month 6 Month 12 Month
11.4 Expanded Use cohorts
The CoreValve US Expanded Use Study is a prospective, non-randomized, multi-center
investigational study designed to evaluate the safety and efficacy of the Medtronic
CoreValve™ system for the treatment of severe native calcific aortic stenosis or failure
(stenosed, insufficient, or combined) of a surgical bioprosthetic aortic valve in subjects with
significant co-morbidities in whom the risk of surgical aortic valve replacement has a
predicted operative mortality or serious, irreversible morbidity risk of ≥50% at 30 days. The
study consisted of the following six cohorts: (1) end stage renal disease (ESRD), (2) lowflow/low-gradient (LFLG), (3) severe mitral regurgitation, (4) severe tricuspid regurgitation,
(5) failed bioprosthetic surgical valve (TAV in SAV), and (6) 2 or more above conditions.
Patients received the CoreValve™ bioprosthesis either through the iliofemoral access route
or through the non-illiofemoral (subclavian and direct aortic) access routes.
The following data summarize the results from the Expanded Use end stage renal disease
(ESRD), low-flow/low-gradient (LFLG), and failed bioprosthetic surgical valve
(TAV in SAV) cohorts.
11.4.1 End stage renal disease (ESRD)
11.4.1.1 Patient population
Eligible subjects presented with ESRD requiring renal replacement therapy or creatinine
clearance of <20 cc/min but not requiring renal replacement therapy, and a mean gradient of
>40 mmHg or a jet velocity >4.0 m/sec by either resting or on dobutamine stress echo if the
LVEF <50% or simultaneous pressure recordings at cardiac catheterization by either resting
or with dobutamine stress echo. The initial aortic valve area was ≤0.8 cm² or an aortic valve
index of ≤0.5 cm²/m² on resting echocardiogram or cardiac catheterization.
A total of 105 ESRD subjects were enrolled in the Expanded Use study at 35 of the
43 activated centers in the United States. Of the 105 enrolled ESRD subjects, a total of
96
Page 99

Age (years)
75.9 ± 8.8
Gender (Male)
66.3% (69/104)
NYHA Classification
I
0.0% (0/104)
II
6.7% (7/104)
III
73.1% (76/104)
IV
20.2% (21/104)
STS Score (Risk of Mortality, %)
15.9 ± 7.8
Coronary Artery Disease
78.8% (82/104)
Previous MI
34.6% (36/104)
Previous Interventions
Coronary Artery Bypass Surgery
34.6% (36/104)
Percutaneous Coronary Intervention
44.2% (46/104)
Balloon Valvuloplasty
14.4% (15/104)
Cerebral Vascular Disease
17.5% (18/103)
Prior Stroke
14.4% (15/104)
Peripheral Vascular Disease
54.8% (57/104)
Chronic Lung Disease/COPD
70.2% (73/104)
Home Oxygen
23.1% (24/104)
Creatinine Level >2 mg/dl
99.0% (103/104)
Chronic Kidney Disease (Stage 4/5)
97.1% (101/104)
Chronic Renal Replacement Therapy
99.0% (103/104)
Atrial Fibrillation/Atrial Flutter
41.2% (42/102)
Preexisting Permanent Pacemaker
Placement/ICD
Aorta Calcification1: Severe/Porcelain
104 subjects received an attempted implant and comprise the attempted implant ESRD
cohort.
The patient characteristics analyzed include demographics, clinical characteristics, medical
history, and potentially prohibitive anatomic factors for surgical aortic valve replacement
(SAVR) and assessments for co-morbidity, frailty, and disability (Table 26). The ability of a
patient to obtain a functional recovery after SAVR is largely based on the presence of
significant co-morbidities, frailties, and disabilities, with the combination of the factors
having higher weight than the individual factors alone. As detailed in Table 26, a high
proportion of patients had significant co-morbidities, frailties, or disabilities, which
established the study population as “Extreme Risk.” The mean age for patients participating
in the trial was approximately 76 years old, and 66% of patients were male. The mean
Society of Thoracic Surgeons (STS) score was approximately 16. Greater than 90% of all
patients were in NYHA classes III or IV.
Additionally, coronary artery disease was present in approximately 80% of patients, and
greater than 50% of patients had peripheral vascular disease.
Table 26: ESRD Cohort Subject Demographics and Clinical Characteristics –
Attempted Implant
Demographic
ESRD
N=104
11.5% (12/104)
97
Page 100

Severe
6.7% (7/104)
Porcelain
1.0% (1/104)
Chest Wall Deformity
0.0% (0/104)
Hostile Mediastinum
2.0% (2/101)
Cirrhosis of the Liver
1.9% (2/103)
Wheelchair Bound
14.4% (15/104)
Echocardiographic Findings
Ejection Fraction (Visual Estimate, %)
49.7 ± 14.8
Aortic Valve Area (cm2)
0.7 ± 0.2
Mean Gradient across Aortic Valve (MGV2,
mmHg)
Mitral Regurgitation: Moderate/Severe
20.2% (21/104)
1
Aorta Calcification is measured on screening CT Angiogram.
Plus-minus values present the mean ± standard deviation.
Time to Procedure (days)
3.7 ± 5.1
Total Time in Cath Lab or OR (min)
199.4 ± 54.1
Total Procedure Time (min)
(skin to skin)
General Anesthesia
84.5% (87/103)
Valve-in-Valve Procedure
5.8% (6/103)
Emergent Operation Due to Device or
Procedure
1
91.3% (95/104)
2
7.7% (8/104)
Number of Devices Implanted
0
1.0% (1/104)
1
93.3% (97/104)
2
5.8% (6/104)
3
0.0% (0/104)
Valve Size Implanted
23 mm
0.0% (0/103)
26 mm
23.3% (24/103)
Demographic
ESRD
N=104
45.5 ± 14.5
11.4.1.2 Procedure data
Table 27 provides a summary of the transcatheter valve implantation procedure. Overall
device success rate was 85.3%. Procedure success was defined as device success and absence
of in-hospital MACCE and procedure success rates were 81.4%.
Table 27: ESRD Cohort Procedure Data - Attempted Implant
Number of Devices Used
0
ESRD
N=104
45.9 ± 23.5
0.0% (0/103)
1.0% (1/104)
98
 Loading...
Loading...