

Page 1

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
ENGLISH
DESCRIPTION:
The INTER FIX™ Threaded Fusion Device consists of a hollow, perforated, metallic cylinder and endcap. The INTER FIX™ RP Threaded Fusion Device consists of a hollow, perforated, metallic cylinder
with a single, large, outer radiused groove along the entire longitudinal axis that extends into the inside diameter of the device. Both ends of the INTER FIX™ RP implant are closed.
The INTER FIX™ and INTER FIX™ RP implants are available in diameters ranging from 12mm to 24mm and in lengths ranging from 20mm to 29mm. The endcaps of the INTER FIX™
implants are sized according to the diameter of the cylinders and are applied to the open end of the cylinders after they are filled with autogenous bone graft.
The INTER FIX™ and the INTER FIX™ RP implants are made from implant grade titanium alloy (Ti-6Al-4V) conforming to ASTM F136.
Implied warranties of merchantability and fitness for a particular purpose or use are specifically excluded. See the Medtronic Sofamor Danek Catalog or price list for further information about
warranties and limitations of liability.
INDICATIONS:
The INTER FIX™ Threaded Fusion Device and the INTER FIX™ RP Threaded Fusion Device are indicated for spinal fusion procedures in skeletally mature patients with degenerative disc
disease (DDD) at one level from L2-S1. DDD is defined as discogenic back pain with degeneration of the disc confirmed by patient history and radiographic studies. These DDD patients
may also have up to Grade I spondylolisthesis or retrolisthesis at the involved level. INTER FIX™ and INTER FIX™ RP implants are to be used with autogenous bone graft and implanted
via an open anterior approach.
CONTRAINDICATIONS:
The INTER FIX™ Threaded Fusion Device and the INTER FIX™ RP Threaded Fusion Device should not be implanted in patients with an active infection at the operative site or with an
allergy to titanium or titanium alloy.
WARNINGS:
• The INTER FIX™ Threaded Fusion Device and the INTER FIX™ RP Threaded Fusion Device should only be used by surgeons who are experienced in spinal fusion procedures and
have undergone adequate training with this device.
• A lack of adequate experience and/or training may lead to a higher incidence of adverse events such as vascular injuries, neurological events, and/or urogenital events (including
retrograde ejaculation).
PRECAUTIONS:
• The safety and effectiveness of the INTER FIX™ Threaded Fusion Device and the INTER FIX™ RP Threaded Fusion Device have not been established in patients with any of the
following conditions:
- spondylolisthesis or retrolisthesis of Grade II or greater;
- more than one level to be fused;
- revision of previous interbody fusion procedure(s);
- postoperative steroidal or nonsteroidal anti-inflammatory medication requirements;
The following contains important medical information on the INTER FIX™ Threaded Fusion Device and the INTER FIX™ RP Threaded Fusion Device.
Page 2

- gross obesity;
- ages less than 18 years or greater than 65 years;
- osteoporosis, osteopenia, and/or osteomalacia;
- pregnancy.
• The safety and effectiveness of the INTER FIX™ Threaded Fusion Device and the INTER FIX™ RP Threaded Fusion Device have only been established in anterior lumbar interbody
fusion (ALIF) procedures using autologous bone graft.
• Patients receiving the INTER FIX™ Threaded Fusion Device should have had at least six months of nonoperative treatment. Patients receiving the INTER FIX™ RP Threaded Fusion
Device should also have had at least six months of nonoperative treatment.
• Two INTER FIX™ Threaded Fusion Devices should be implanted side by side at the surgical level. One INTER FIX™ Threaded Fusion Device and one INTER FIX™ RP Threaded
Fusion Device must be implanted side by side at the surgical level. The longitudinal groove on the INTER FIX™ RP device facilitates implanting two devices in closer proximity than
that which is manageable with two INTER FIX™ devices, thus resulting in a reduced lateral profile.
• The long axis of the INTER FIX™ Threaded Fusion Devices and the INTER FIX™ RP Threaded Fusion Devices should be in the anterior-posterior direction.
• The implants and instruments must be sterilized prior to use according to the sterilization instructions provided in this package insert, unless supplied sterile and clearly labeled as such.
ADVERSE EFFECTS:
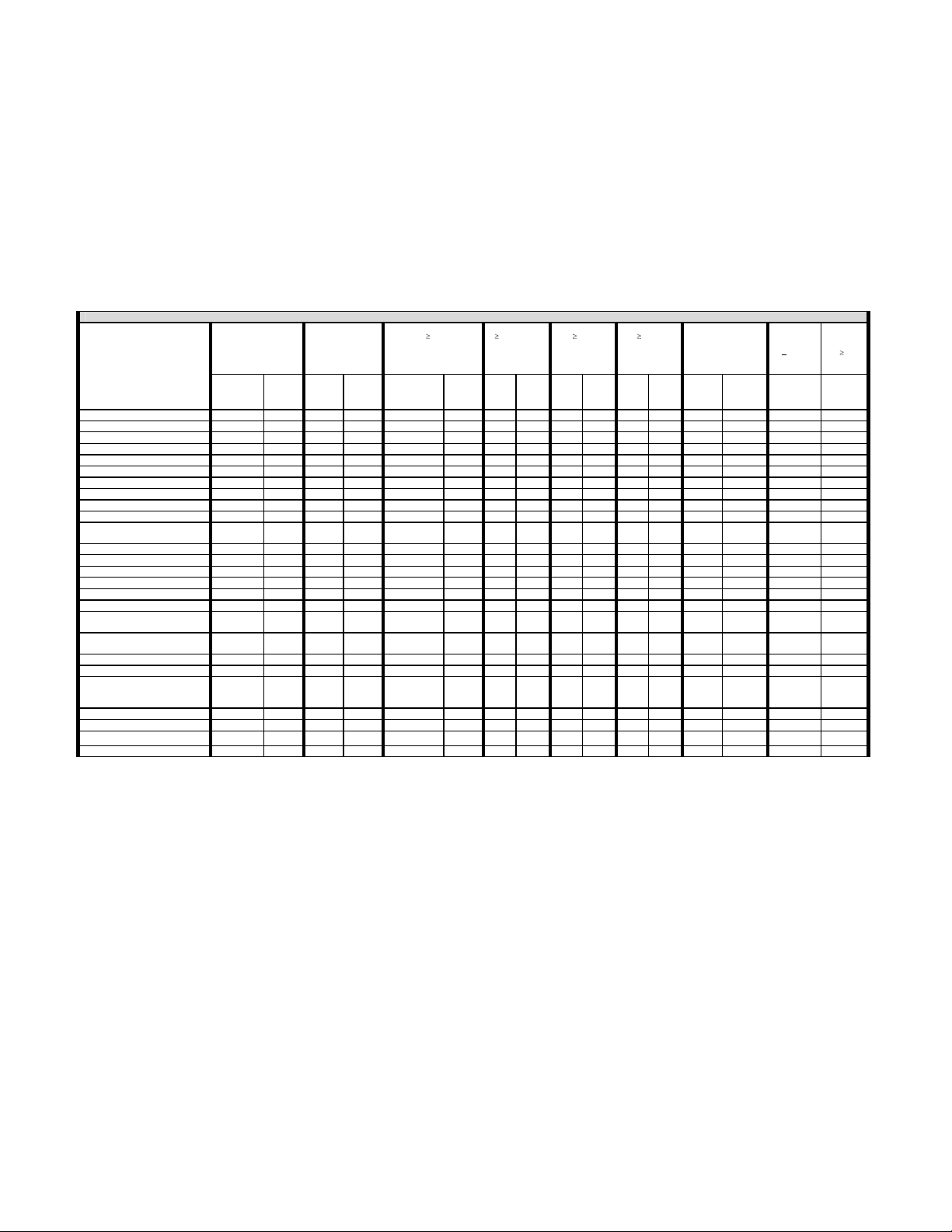
The adverse effects, as shown in Table I, were reported from the 185 INTER FIX™ device patients and 62 control patients enrolled in the multi-center clinical study of the INTER FIX™
Threaded Fusion Device. In addition, adverse effects were reported from 116 patients at 48 months and 109 patients at 72 months on patients in the post-approval study. The rates presented
are based on the total number of patients enrolled in the study at each timepoint. The adverse effects occurring in the randomized and nonrandomized INTER FIX™ device treatment groups
are combined in order to present an overall rate.
TABLE 1 – ADVERSE EVENTS
3 Month
(2-<5 Months)
[Number (%)]
INTER FIX
N=185
™
CONTROL
N=62
6 Month
(5-<9 Months)
[Number (%)]
INTER FIX
N=185
™
CONTROL
N=62
INTER FIX
12 Month
9-<19 Months)
[Number (%)]
™
CONTROL
N=185
N=62
24 Month
(19-< 30 Months)
[Number (%)]
™
INTER FIX
CONTROL
N=185
N=62
TOTAL ADVERSE
EVENTS (THROUGH
24 MONTHS)
™
INTER FIX
N=185
CONTROL
N=62
48 Month
(30 - < 60
Months)
INTER FIX
N=116
72 Month
(60
Months)
™
INTER FIX
N=109
™
Complication
Vascular
Intra-Op
Sacroiliac
Pain
N=62
Post-operative
(1 Day-<2 Months)
[Number (%)]
™
INTER FIX
CONTROL
N=185
N=62
Operative
[Number (%)]
INTER FIX
N=185
™
CONTROL
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
Neurological 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Back Pain 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
Incisional 11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
Spinal Event 1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
Urological 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Other 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Other Pain 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
Gastrointestinal 7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
Retrograde
Ejaculation
2(1.1) 3(1.6) 1(0.5) 6(3.2)
(7.4)
1
0
Respiratory 3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
Leg Pain 2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
Trauma 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
Peritoneal 3(1.6) 3(1.6) 0
Vascular
Post-Op
Bone
Fracture
Implant
Displacement
/ Loosening
Graft Site
Pain
2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
Non-Union 4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
Subsidence 2(1.1) 2(1.1) 0
Non-Union
(OUTCOME
PENDING)
1(1.6)2 0 1(1.6) 2(1.7)
Meningitis 1(1.6) 0 1(1.6)
Implant
Breakage
Cancer 1
5(8.1) 0 5(8.1)
(0.5)
1
(0.5)
2 (1.1) 1(0.8) 1(0.9)
Death 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
1 - Six (6) of 81 males enrolled in the INTER FIX™ Device group (7.4%) experienced retrograde ejaculation through the 24-month evaluation.
2 - This patient (175) has subsequently been reported as fused at approximately 52 months postoperative, and no other surgical procedure or treatment was required.
Two important adverse events in the clinical trial were intraoperative vascular and neurological injuries. A total of 15 vascular intraoperative events occurred in 14 patients in the INTER
FIX™ device group through the 24-month evaluation. These events included: 2 injuries to the vena cava, 9 injuries to the iliac vein, 1 lacerated hypogastric vein, 1 segmental vein bleeder,
1 sacral vein injury, and 1 superficial bleeder. A total of 2 vascular intraoperative injuries occurred in 2 patients in the control group during the same period. These included: 1 injury to an
iliac vein and 1 bleeding from the bone bed.
A total of 30 neurological events occurred in 28 patients in the INTER FIX™ device group through the 24-month evaluation. These events included: 1 footdrop; 3 nerve root injuries; 1 foraminal
stenosis; 1 reflex sympathetic dystrophy; 7 numbness or warmth in legs; 2 dysesthesia; 3 paresthesia; 2 radiculitis; 7 back and/or leg pain; 1 back and/or leg pain with other symptoms; 1
shooting pain in lower back; and 1 carpal tunnel syndrome. A total of 10 neurological events occurred in 8 patients in the control group during the same period. These events included: 1
radiculopathy with tingling extremities; 1 chronic back pain with radiculitis; 1 debilitating distribution symptoms; 2 back and leg pain with other symptoms; 1 denervated abductor magnus
muscle; and 4 with numbness or warmth in legs. During the post-approval study, an additional 10 neurological events were recorded in 10 patients. These events included: 1 radiculopathy;
2 cervical radiculopathies; 1 numbness and tingling in hands; 1 numbness or burning in legs; 3 back and/or leg pain; and 2 back and/or leg pain with other symptoms. In addition, the adverse
effects table presents leg and back pain adverse events and/or spinal events, such as disc space collapse, that, in some instances, had a neurological component.
Some of the adverse effects led to surgical interventions subsequent to the clinical trial surgery. These additional surgical interventions can be classified as revisions, removals, supplemental
fixations, or reoperations.1 Table II summarizes the secondary surgical interventions in the INTER FIX™ device (randomized and nonrandomized combined) and control treatment groups
through the IDE study and in the INTER FIX™ device group through the post-approval study.
1
Revision: A procedure that adjusts or in any way modifies the original implant configuration.
Removal: A procedure that removes one or more components of the original implant configuration without replacement with the same type of trial device.
Supplemental Fixation: A procedure in which additional spinal devices not approved as part of the protocol are placed.
Reoperation: Any surgical procedure that does not remove, modify or add any original implant components.
Page 3

IDE Study Post-Approval Study
Through 24 month
Revisions 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Removals 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Supplemental
Fixations
Reoperations 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
Table II - Secondary Surgical Procedures
INTER FIX™
Device
N=185
Timepoint
Control
N=62
1
48 month
Timepoint
INTER FIX™
Device
N=116
72 month
Timepoint
INTER FIX™
Device
N=109
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
1 - Because control patients were not required to follow-up after 24 months, information is not available for all of these patients through 72 months.
POTENTIAL ADVERSE EFFECTS:
The following is a list of potential adverse effects which may occur with spinal fusion surgery with the INTER FIX™ Threaded Fusion Device or the INTER FIX™ RP Threaded Fusion Device.
Some of these adverse effects may have been previously reported in the adverse effects table.
• Disassembly, bending, breakage, loosening, and/or migration of components.
• Foreign body (allergic) reaction.
• Tissue or nerve damage.
• Post-operative change in spinal curvature, loss of correction, height, and/or reduction.
• Infection.
• Dural tears.
• Neurological system compromise.
• Urological system compromise.
• Scar formation.
• Bone fracture.
• Non-union (or pseudarthrosis), delayed union, mal-union.
• Cessation of any potential growth of the operated portion of the spine. Loss of spinal mobility or function.
• Graft donor site complications.
• Damage to blood vessels and cardiovascular system compromise.
• Gastrointestinal complications.
• Reproductive system compromise.
• Damage to internal organs and connective tissue.
• Development of respiratory problems.
• Incisional complications.
• Change in mental status.
• Death.
Note: Additional surgery may be necessary to correct some of these potential adverse events.
CLINICAL RESULTS:
A multi-center equivalency2 clinical trial of the INTER FIX™ Threaded Fusion Device was conducted in the United States comparing the anterior spinal use of the INTER FIX™ device filled
with autogenous bone to femoral ring allograft filled with autogenous bone, the control, in the treatment of patients with symptomatic degenerative disc disease. Initially, the clinical trial
had a prospective, randomized, controlled design. Subsequently, the investigational plan was revised to allow patients to be entered into a non-randomized arm, i.e., patients treated with
INTER FIX™ device only.
Inclusion criteria for the clinical trial included symptomatic degenerative disc disease as noted by intractable leg and/or back pain with positive diagnostic imaging finding(s); spinal instability
as defined by greater than 4 mm of translation or greater than 5° of angulation on flexion/extension radiographs; single level symptomatic involvement from L2-S1; and no greater than Grade
1 spondylolisthesis. Specifically excluded from the study were patients who: had a previous anterior interbody fusion procedure at the involved spinal level; had osteopenia, osteoporosis,
or osteomalacia; or required bone growth stimulation.
A total of 185 patients were enrolled in the investigational INTER FIX™ device treatment group.3 A total of 62 control patients were entered into the randomized arm of the clinical trial.
A post approval requirement for the INTER FIX™ Threaded Fusion Device was to assess the long-term performance of the device by obtaining a total of 6 years of postoperative data from
a minimum of 100 patients. The long-term performance of the device was assessed on 116 patients at 4 years postoperatively and on 109 patients at 6 years postoperatively. The patient
population was obtained from participants in the IDE clinical trial. Based on criteria from the IDE study, the information obtained on these patients included a description of any additional
surgical procedures, radiographic assessment of fusion, and an assessment of pain and function.
Table III presents the clinical results from the study, describing the parameters of fusion success, pain (Oswestry) success, and neurological success at 12, 24, 48 and 72 months after the
treatment surgery. The success of this clinical trial was noted on the basis of an equivalence analysis between the INTER FIX™ device and control.
For this clinical trial, fusion was defined as the evidence of bridging trabecular bone, translation (<3mm) and angular motion (<5°) stability, and no radiolucencies surrounding more than 50%
of either implant. Also, patients having secondary surgeries due to nonunions were considered failed fusions in the calculations. Oswestry Pain/Disability success was defined as at least
a 15-point improvement in the postoperative score as compared to the preoperative score. Neurological status success was defined as a maintenance or improvement in at least 3 of the 4
neurological categories (motion, sensory, reflexes, and straight leg raise) postoperative as compared to the preoperative condition. Overall success was based on a patient demonstrating
fusion, Oswestry Pain/Disability success, and neurological status success, and no secondary surgical procedure classified as a “failure.”
2
INTER FIX™ device statistically no worse than the control.
3
Does not include 3 patients who did not receive the INTER FIX™ device treatment due to surgical adverse events.
The overall success results at 12 and 24 months postoperative can also be found in Table III.
Table III - Clinical Results from the INTER FIX™ Device Study
Fusion
Oswestry
Pain/Disability
Improvement
Patients with at
least 15 Point
Improvement from
Pre-Op
Neurological
Status
Maintenance
or
Improvement
Overall
Success
12 Month Rates 24 Month Rates
Investigational Control
96.1%
(122/127)
53.5%
(76/142)
99.3%
(150/151)
42.4%
(61/144)
(21/52)
(27/52)
(52/52)
(12/56)
Delta
40.4%
51.9%
100%
21.4%
(ǻ
-0.097 46.1%
1
)
Investigational Control
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
(71/154)
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
-0.043 49.5%
(17/57)
48 Month
Delta
(ǻ1)
Rates
Investigational Investigational
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
(53/107)
72 Month Rates
97.6% (82/84)
56.3%
(54/96)
1 - Minimal non-inferiority margin required for claiming non-inferiority of investigational to control.
All patients involved in the clinical trial of the INTER FIX™ Threaded Fusion Device, regardless of treatment group, were enrolled under the same inclusion/exclusion criteria. To substantiate
the comparability of the randomized INTER FIX™ device patients and the nonrandomized INTER FIX™ device patients, the demographic characteristics, preoperative medical conditions,
diagnostic imaging findings characteristic of degenerative disc disease, operative approach, and the location of the lumbar treated level were examined. The population was statistically
comparable except for operative approach, preoperative work status, and the preoperative diagnostic imaging findings of osteophyte formation of the vertebral endplates; scarring and/or
thickening of the annulus fibrosis, ligamentum flavum, and/or facet joint capsule; and herniated nucleus pulposus. Further statistical analyses showed that these variables were not associated
with differences in the 12-month clinical outcomes of fusion, Oswestry Pain/Disability improvement, neurological status, or overall success, with the exception of herniated nucleus pulposus.
More patients in the nonrandomized group had a finding of herniated nucleus pulposus, which was found to increase the probability of overall success.
An “intent-to-treat” analysis was performed. For this analysis, secondary surgery failures, deaths, patients lost-to-follow-up, and missing observations due to other causes resulted in missing
observations for the outcome variables and therefore were included in the denominators of the calculated rates, i.e., considered as “failures.” By treating these patients as treatment failures,
the clinical outcome rates in the intent-to-treat analysis were considerably lower than those in the actual observed clinical data. The following table (Table IV) provides the results for the
intent-to-treat analysis of INTER FIX™ device patients, but does not include the results of the control patients.
Page 4

Deaths, Secondary Surgery Failures, Lost-to-Follow-up, and Missing Observations Are Considered as Failures
12 Month Rates 24 Month
Fusion
Oswestry Pain/Disability Improvement
Patients with at least 15 Point Improvement from Pre-Op
Neurological Status Maintenance or
Improvement
Overall Success
Secondary Surgery Failures
Nonunions
Other
Deaths
1 - These patients are included in the fusion rate calculations but are otherwise considered as failures for clinical trial purposes.
2 - Patients due for follow up at that period who had secondary surgeries for reasons other than nonunions are considered failures for clinical trial purposes.
PACKAGING:
Packages for each of the components should be intact upon receipt. If a loaner or consignment system is used, all sets should be carefully checked for completeness and all components including
instruments should be carefully checked to ensure that there is no damage prior to use. Damaged packages or products should not be used, and should be returned to Medtronic Sofamor Danek.
CLEANING AND DECONTAMINATION:
If not supplied sterile, all implants and instruments must first be cleaned using established hospital methods before sterilization and introduction into a sterile surgical field. Used instruments
must be decontaminated, cleaned, and sterilized before reuse.
1
2
Table IV - Intent-to-Treat Analysis for INTER FIX™ Device
and Are Included in the Denominator of the Rates
65.9% (122/185) 67.6%
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
33.0% (61/185) 38.4% (71/185) 45.7%
5
8
0 1 0 0
(125/185)
(128/185)
Rates
5
3
48 Month
Rates
80.2%
(93/116)
(66/116)
87.9%
(102/116)
(53 /116)
4
4
72 Month
Rates
77.1%
( 84/109)
56.0%
(61 /109)
75.2%
75.2% (82/109)
(82/109)
49.5%
(54/109)
1
2
Note: Do not use cleaning solutions containing bleach or formalin since they may damage the device.
All products should be treated with care. Improper use or handling may lead to damage and possible improper functioning of the device.
STERILIZATION:
Unless marked sterile and clearly labeled as such in an unopened sterile package provided by the company, all implants and instruments used in surgery must be sterilized by the hospital
prior to use. Remove all packaging materials prior to sterilization. Only sterile products should be placed in the operative field. For a 10-6 Sterility Assurance Level, these products are
recommended to be steam sterilized by the hospital using one of the three sets of process parameters below:
METHOD CYCLE TEMPERATURE EXPOSURE TIME
Steam Pre-Vacuum 270°F (132°C) 4 Minutes
Steam Gravity 250°F (121°C) 60 Minutes
Steam* Gravity* 273°F (134°C)* 20 Minutes*
NOTE: Because of the many variables involved in sterilization, each medical facility should calibrate and verify the sterilization process (e.g. temperatures, times) used for their equipment.
*For use outside the United States, some non-U.S. Health Care Authorities recommend sterilization according to these parameters so as to minimize the potential risk of transmission of
Creutzfeldt-Jakob disease, especially for surgical instruments that could come into contact with the central nervous system.
PRODUCT COMPLAINTS:
Any health care professional (e.g., customer or user of this system of products), who has any complaints or who has experienced any dissatisfaction in the quality, identification, durability,
reliability, safety, effectiveness and/or performance of this product, should notify the distributor, Medtronic Sofamor Danek. Further, if any of the implanted INTER FIX™ Threaded Fusion
Device or INTER FIX™ RP Threaded Fusion Device components ever “malfunction,” (i.e., do not meet any of their performance specifications or otherwise do not perform as intended), or are
suspected of doing so, the distributor should be notified immediately. If any Medtronic Sofamor Danek product ever “malfunctions” and may have caused or contributed to the death or serious
injury of a patient, the distributor should be notified immediately by telephone, fax or written correspondence. When filing a complaint, please provide the component name and number, lot
number, your name and address, the nature of the complaint and notification of whether a written report from the distributor is requested.
PHYSICIAN NOTE: Although the physician is the learned intermediary between the company and the patient, the important medical information given in this document should be conveyed
to the patient.
For US Audiences Only
CAUTION: FEDERAL (USA) LAW RESTRICTS THESE DEVICES TO SALE BY OR ON THE ORDER OF A PHYSICIAN.
DEVICE RETRIEVAL EFFORTS:
Should it be necessary to remove an INTER FIX™ Threaded Fusion Device or an INTER FIX™ RP Threaded Fusion Device, please call Medtronic Sofamor Danek.
© 2006 Medtronic Sofamor Danek. All rights reserved.
Page 5

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
FRANCAIS
Ce document contient des informations médicales importantes relatives au dispositif fileté pour la fusion INTER FIX™ et au dispositif fileté pour la fusion INTER FIX™ RP.
DESCRIPTION :
Le dispositif fileté pour la fusion INTER FIX™ est composé d’un cylindre métallique, creux et perforé, ainsi que d’un bouchon. Le dispositif fileté pour la fusion INTER FIX™ RP est composé d’un cylindre
métallique, creux et perforé, qui comporte une rainure large, arrondie et extérieure tout le long de l’axe longitudinal et qui se prolonge jusqu’au diamètre intérieur du dispositif. Les deux extrémités de l’implant
INTER FIX™ RP sont fermées.
Le diamètre des implants INTER FIX™ et INTER FIX™ RP peut varier de 12mm à 24mm et la longueur de 20mm à 29mm. La taille des bouchons des implants INTER FIX™ correspond au diamètre des cylindres
et ils se placent sur l’extrémité ouverte du cylindre, après avoir été remplis avec un greffon d’os autogène.
Les implants INTER FIX™ et INTER FIX™ RP sont fabriqués en alliage de titane spécifique pour implants (Ti-6AI-4V), tel que décrit dans la norme ASTM F136.
Des garanties implicites de commercialisation et de capacité lorsqu’on l’utilise dans un but ou un usage particulier, sont expressément exclues. Pour de plus amples renseignements à propos
des garanties et des limites en ce qui concerne la responsabilité, se reporter à la liste de prix ou au catalogue de Medtronic Sofamor Danek.
INDICATIONS :
Le dispositif fileté pour la fusion INTER FIX™ et le dispositif fileté pour la fusion INTER FIX™ RP sont indiqués pour des interventions chirurgicales de fusion du rachis, chez les patients dont le squelette est
développé, et qui présentent une discopathie dégénérative (DDD) à un des niveaux rachidiens de L2 à S1. La discopathie dégénérative (DDD) est diagnostiquée par la présence de douleurs d’origine discale
dans le dos avec une dégénérescence du disque, confirmée par les antécédents du patient et par un contrôle en radiologie. Ces patients qui présentent une discopathie dégénérative (DDD) peuvent également
souffrir d’un spondylolisthésis ou d’un rétrolisthésis pouvant atteindre jusqu’à un degré I au niveau affecté du rachis. Les dispositifs INTER FIX™ et INTER FIX™ RP devront être utilisés avec un greffon d’os
autogène et implantés par le biais d’une chirurgie ouverte par voie antérieure.
CONTRE-INDICATIONS :
Ne pas implanter le dispositif fileté pour la fusion INTER FIX™, ni le dispositif fileté pour la fusion INTER FIX™ RP, chez les patients qui présentent une infection active sur la région qui doit être opérée, ni chez
ceux qui sont allergiques au titane ou à l’alliage de titane.
AVERTISSEMENTS :
• Le dispositif fileté pour la fusion INTER FIX™ et le dispositif fileté pour la fusion INTER FIX™ RP ne devront être implantés que par des chirurgiens qui ont de l’expérience dans la chirurgie de fusion du
rachis et qui ont suivi la formation appropriée à ces dispositifs.
• Si l’expérience et/ou la formation du chirurgien ne sont pas suffisantes, ceci pourrait accroître le risque que surviennent des effets indésirables tels que des lésions vasculaires, des lésions neurologiques
et/ou des problèmes de l’appareil uro-génital (y compris une éjaculation rétrograde).
PRECAUTIONS :
• La sécurité et l’efficacité du dispositif fileté pour la fusion INTER FIX™ ainsi que celles du dispositif fileté pour la fusion INTER FIX™ RP, n’ont pas été démontrées chez les patients qui correspondent à
l’un des cas suivants :
- en cas de spondylolisthésis ou de rétrolisthésis de degré II ou supérieur,
- lorsqu’il est nécessaire de fusionner plus d’un niveau,
- lorsqu’une intervention chirurgicale de fusion intersomatique précédente nécessite d’être corrigée,
- lorsqu’il sera requis d’administrer des anti-inflammatoires non-stéroïdiens ou stéroïdiens pendant l’étape postopératoire,
INFORMATIONS MÉDICALES IMPORTANTES LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
Page 6

qq pppp
- lorsque l’obésité est flagrante,
E
C
E
- lorsque l’âge du patient est inférieur à 18 ans ou supérieur à 65 ans,
- en cas d’ostéoporose, d’ostéopénie et/ou d’ostéomalacie,
- en cas de grossesse.
• La sécurité et l’efficacité du dispositif fileté pour la fusion INTER FIX™ ainsi que celles du dispositif fileté pour la fusion INTER FIX™ RP, n’ont été démontrées que pour les interventions chirurgicales de
fusion intersomatique dans le rachis lombaire par voie antérieure (ALIF), dans lesquelles un greffon d’os autologue est utilisé.
• Les patients à qui un dispositif fileté pour la fusion INTER FIX™ sera implanté, doivent avoir subi un traitement non-chirurgical au moins pendant six mois avant l’opération. Les patients à qui un dispositif
fileté pour la fusion INTER FIX™ RP sera implanté, doivent également avoir subi un traitement non-chirurgical au moins pendant six mois avant l’opération.
• Implanter les deux dispositifs filetés pour la fusion INTER FIX™ l’un à côté de l’autre au niveau qui doit être opéré. Implanter un dispositif fileté pour la fusion INTER FIX™ et un dispositif fileté pour
la fusion INTER FIX™ RP, l’un à côté de l’autre, au niveau qui doit être opéré. La rainure longitudinale du dispositif INTER FIX™ RP facilitera l’implantation la plus rapprochée possible entre les deux
dispositifs INTER FIX™, obtenant ainsi un profil latéral réduit.
• L’axe longitudinal du dispositif fileté pour la fusion INTER FIX™ ainsi que celui du dispositif fileté pour la fusion INTER FIX™ RP, doivent se trouver dans le sens antérieur-postérieur.
• Stériliser les implants et les instruments avant de les utiliser, selon les procédures de stérilisation indiquées dans cette notice, à moins qu’ils ne soient fournis stérilisés et que l’étiquette ne l’indique clairement.
EFFETS INDESIRABLES :
Les effets indésirables, présentés dans le tableau I, sont ceux qui ont été reportés lors de l’implantation du dispositif INTER FIX™ chez 185 patients, et lors du traitement chez 62 patients du
groupe de contrôle, qui ont participé à l’étude clinique multicentrique pour le dispositif fileté pour la fusion INTER FIX™. De même, sont indiqués dans ce tableau les effets indésirables qui
ont été reportés chez 116 patients pour la période de 48 mois et chez 109 patients pour la période de 72 mois, lors de l’étude post-approbation. Les pourcentages indiqués ont été calculés à
partir du nombre total de patients qui ont participé à cette étude, pour chaque intervalle de temps pré-établi. Les effets indésirables survenus chez les groupes, randomisés et non-randomisés,
traités avec le dispositif INTER FIX™, ont été regroupés afin de présenter un pourcentage global.
Complication
Vasculaire
péropératoire
Douleurs au
niveau sacroiliaque
Neurologique
Douleurs dans
le dos
Incisionnelle
Complication
dans le rachis
Urologique
Autres
Autres douleurs
Gastrointestinale
Ejaculation
rétrograde
Respiratoire
Douleurs dans
la jambe
Traumatisme
Péritonéale
Vasculaire
postopératoire
Fracture
osseuse
Déplacement /
débricolage de
l'implant
Douleurs sur le
site de la greffe
Absence de
consolidation
Effondrement
Absence de
consolidation
(RESULTAT EN
COURS)
Méningite
Rupture de
l'implant
Cancer
Décès
Péropératoires
(Nombre %)
INTER FIX™
N=185
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
5(2.7) 3(1.6) 4( 2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1( 1.6) 23(12.4) 3(4.8) 1(0.9)
6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11( 17.7) 7(6.0) 6(5.5)
11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14( 12.1) 9(8,3)
4(2.2) 1(1.6) 5(2.7) 3( 1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
7(3.8) 3(4.8) 1(0.5) 1( 0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
2(1.1) 3(1.6) 1( 0.5) 6(3.2)
3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1( 1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
3(1.6) 3(1.6) 0
2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2. 2) 10(5. 4) 2(3.2) 4(3.4) 2(1.8)
1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
1(0.5) 1(1.6) 5(8.1) 1( 0.5) 2(1.1) 6(9.7)
1(0.5) 1(1. 6) 1(1.6) 1(0.5) 2(3.2)
4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3( 2.6)
2(1.1) 2(1.1) 0
1(1.6)2 0 1( 1.6) 2(1.7)
1(1.6) 0 1(1.6)
5(8.1) 0 5(8.1)
1
1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
CONTRÔL
N=62
Postopératoires
(1 jour -<2 mois)
[Nombre (%)]
™
INTER FIX
CONTRÔLE
N=185
TABLEAU 1 – EFFETS INDÉSIRABLES
N=62
3 mois
(t
2-<5 mois)
[Nombre (%)]
INTER FIX
N=185
™
ONTRÔL
N=62
6 mois
(t5-<9 mois)
[Nombre (%)]
INTER FIX
N=185
™
CONTRÔLE
N=62
12 mois
(t
9-<19 mois)
[Nombre (%)]
INTER FIX
N=185
(0.5)
™
CONTRÔLE
N=62
1
24 mois
(t19-<30 mois)
[Nombre (%)]
™
INTER FIX
N=185
(0.5)
TOTAL
EFFETS
INDESIRABLES
(PENDANT 24
MOIS)
INTER FIX
N=185
(7.4)
™
CONTRÔLE
1
CONTRÔLE
N=62
2 (1.1) 1(0.8) 1(0.9)
48 mois
(t
(>30 - < 60
mois)
INTER FIX™
N=62
0
30<
N=116
72 mois
60
60 mois)
(t
INTER FIX™
N=109
1 - Au cours de la période de 24 mois, six (6) cas d’éjaculation rétrograde ont été observés parmi les 81 patients de sexe masculin appartenant au groupe du dispositif INTER FIX™ (7,4%).
2 - Ce patient (175) a été reporté ultérieurement puisque la fusion a été obtenue au bout de 52 mois après l’opération et ce, sans avoir eu recours à une autre intervention chirurgicale ou à tout autre traitement.
Les deux effets indésirables importants observés lors de cet essai clinique ont été les lésions vasculaires et les lésions neurologiques survenues pendant l’opération. Au total, 15 lésions vasculaires survenues
pendant l’opération ont été relevées chez 14 patients appartenant au groupe du dispositif INTER FIX™ au cours de la période de 24 mois. Ces lésions sont : 2 lésions de la veine cave, 9 lésions de la veine
iliaque, 1 lacération de la veine hypogastrique, 1 hémorragie sur un segment de la veine, 1 lésion de la veine sacrée et 1 hémorragie superficielle. En ce qui concerne le groupe de contrôle, un total de 2 lésions
vasculaires sont survenues pendant l’opération chez 2 patients au cours de la même période. Ces lésions sont : 1 lésion de la veine iliaque et 1 hémorragie sur la base de l’os.
Au total, 30 cas neurologiques ont été observés chez 28 patients appartenant au groupe du dispositif INTER FIX™ au cours de la période de 24 mois. Ces cas sont : 1 pied tombant, 3 lésions de la racine des
nerfs, 1 sténose foraminale, 1 dystrophie du réflexe sympathique, 7 engourdissements ou sensations de chaleur dans les jambes, 2 dysesthésies, 3 paresthésies, 2 radiculites, 7 douleurs dans le dos et/ou la
jambe, 1 douleur dans le dos et/ou la jambe accompagnée d’autres symptômes, 1 douleur lancinante dans le bas du dos et 1 syndrome du canal carpien.
Au total, 10 cas neurologiques ont été observés chez 8 patients appartenant au groupe de contrôle au cours de la même période. Ces cas sont : 1 radiculopathie accompagnée de fourmillements dans les extrémités,
1 douleur chronique dans le dos accompagnée de radiculite, 1 patient présentant des symptômes d’affaiblissement généralisé, 2 douleurs dans le dos et la jambe accompagnées d’autres symptômes, 1 muscle
abducteur majeur dénervé, et 4 avec des engourdissements ou des sensations de chaleur dans les jambes. Au cours de l’étude post-approbation, 10 cas neurologiques supplémentaires ont été observés chez 10
patients. Ces cas sont : 1 radiculopathie, 2 radiculopathies cervicales, 1 engourdissement et fourmillements dans les mains, 1 engourdissement ou sensations de chaleur dans les jambes, 3 douleurs dans le dos
et/ou la jambe, et 2 douleurs dans le dos et/ou la jambe accompagnées d’autres symptômes. Le tableau dans lequel sont présentés les effets indésirables, nous montre de même des effets indésirables tels que
des douleurs dans le dos et les jambes et/ou des effets indésirables en rapport avec le rachis, tel que le tassement du disque, ayant un lien neurologique dans certains cas.
Certains des effets indésirables ont entraîné une nouvelle intervention chirurgicale après l’intervention réalisée pour l’essai clinique. Ces interventions chirurgicales supplémentaires sont classifiées de la façon
suivante : correction, extraction, fixation supplémentaire ou nouvelle intervention.
FIX™ (randomisé et non-randomisé regroupés), chez ceux du groupe de contrôle pendant l’étude IDE et chez ceux du groupe du dispositif INTER FIX™ pendant l’étude post-approbation.
1
Correction : Intervention lors de laquelle l’ajustement ou toute modification de la configuration initiale de l’implant est réalisé.
Extraction: Intervention lors de laquelle un ou plusieurs composants de la configuration initiale de l’implant sont extraits, sans qu’ils ne soient remplacés par le même type de dispositif utilisé pour l’essai.
Fixation supplémentaire: Intervention lors de laquelle sont implantés des dispositifs rachidiens supplémentaires qui ne font pas partie de la configuration initialement prévue.
Nouvelle intervention: Intervention chirurgicale lors de laquelle un composant de l’implant d’origine n’est pas extrait, ni modifié, ni ajouté.
1
Le tableau II regroupe les interventions chirurgicales supplémentaires effectuées chez les patients du groupe du dispositif INTER
Page 7

Tableau II – Interventions chirurgicales supplémentaires
Correction 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Extraction 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Fixation
supplémentaire
Nouvelle
intervention
Etude IDE Etude post-approbation
Intervalle de temps
pré-établi
24 mois
Dispositif
INTER FIX™
N=185
Contrôle
N=62
Intervalle de temps
1
pré-établi
48 mois
Dispositif
INTER FIX™
N=116
Intervalle de temps
pré-établi
72 mois
Dispositif
INTER FIX™
N=109
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
1 – Aucune donnée n’est disponible pour la période de 72 mois pour les patients du groupe de contrôle, car pour ces patients, seul un suivi pendant 24 mois était prévu.
EFFETS INDÉSIRABLES POTENTIELS :
Les effets indésirables potentiels qui peuvent survenir lors d’une intervention chirurgicale de fusion du rachis, lors de laquelle un dispositif fileté pour la fusion INTER FIX™ ou un dispositif fileté pour la fusion INTER FIX™
RP est utilisé, sont énumérés dans la liste suivante. Certains de ces effets indésirables peuvent avoir été mentionnés précédemment dans le tableau des effets indésirables.
• Désassemblage, déformation, rupture, débricolage et/ou migration des composants.
• Réaction (allergique) au corps étranger.
• Lésion des tissus ou des nerfs.
• Changement postopératoire de la courbure du rachis, perte de la correction, de la hauteur, et/ou de la réduction du rachis.
• Infection.
• Déchirure durale.
• Dysfonctionnement du système neurologique.
• Dysfonctionnement du système urinaire.
• Formation de cicatrices.
• Fracture osseuse.
• Absence de consolidation osseuse (ou pseudarthrose), consolidation retardée, cal vicieux.
• Interruption de la croissance potentielle du segment opéré du rachis. Perte de la mobilité ou de la fonction du rachis.
• Complications au niveau du site donneur de greffe.
• Lésion des vaisseaux sanguins et dysfonctionnements du système cardio-vasculaire.
• Complications du système gastro-intestinal.
• Dysfonctionnement du système reproducteur.
• Lésion des organes internes et du tissu conjonctif.
• Développement de problèmes respiratoires.
• Complications liées à l’incision.
• Modification de l’état mental.
• Décès.
Observations : Une intervention chirurgicale supplémentaire peut s’avérer nécessaire pour corriger certains de ces effets indésirables potentiels.
RESULTATS CLINIQUES :
Un essai clinique comparatif2 multicentrique portant sur le dispositif fileté pour la fusion INTER FIX™ a été effectué aux Etats-Unis d’Amérique, dans le but de comparer le dispositif INTER FIX™ lorsqu’il est utilisé dans le
rachis antérieur et rempli d’os autogène, à un groupe de contrôle traité avec une allogreffe d’anneau fémoral rempli d’os autogène, lors du traitement des patients présentant une discopathie dégénérative symptomatique.
Initialement, cet essai clinique était prévu prospectif, randomisé et contrôlé. Par la suite, les plans pour cette recherche ont été changés afin de permettre aux patients d’être intégrés dans le groupe non-randomisé, c’est-à-dire
les patients traités uniquement avec le dispositif INTER FIX™.
Les critères d’inclusion pour cet essai clinique ont été les suivants : une discopathie dégénérative symptomatique diagnostiquée par une douleur réfractaire au traitement, dans la jambe et/ou dans le dos et confirmée en
radiologie ; une instabilité rachidienne déterminée par une déviation supérieure à 4mm ou par un angle supérieur à 5°, constatée par des radiographies flexion/extension ; manifestation des symptômes à un seul niveau du
rachis L2-S1 ; et un spondylolisthésis non supérieur au degré I. Ont été expressément exclus de cette étude clinique les patients qui avaient déjà subi une intervention de fusion intersomatique, par voie antérieure, au niveau
rachidien concerné ; ceux qui présentaient une ostéopénie, une ostéoporose ou une ostéomalacie ; ainsi que ceux qui nécessitaient une stimulation de la croissance osseuse.
Le nombre total de patients, appartenant au groupe du dispositif INTER FIX™, inscrits à l’étude clinique était de 185.
groupe randomisé, était de 62.
Une étude post-approbation a été requise pour le dispositif fileté pour la fusion INTER FIX™ afin d’évaluer la performance à long terme de ce dispositif, étude pour laquelle il fallait recueillir les résultats auprès d’au moins
100 patients au bout de 6 ans après l’intervention. Ainsi, la performance à long terme du dispositif a été évaluée auprès de 116 patients au bout de 4 ans après l’intervention, et auprès de 109 patients au bout de 6 ans après
l’intervention. La population était issue des patients qui ont participé à l’essai clinique IDE. En accord avec les critères de l’étude IDE, la description des interventions chirurgicales supplémentaires, le contrôle de la fusion en
radiologie, ainsi que l’évaluation de la douleur et de la fonction, faisaient partie des informations qui ont pu être obtenues pour ces patients.
Le tableau III nous montre les résultats de cette étude clinique et il met en évidence les pourcentages du succès en ce qui concerne la fusion, la douleur d’après Oswestry, et l’état neurologique, au bout de 12, 24, 48 et 72
mois après l’intervention. Le succès de cet essai clinique s’est basé sur une analyse comparative entre le dispositif INTER FIX™ et celui du contrôle.
Dans le cadre de cet essai clinique, la fusion a été définie lorsque : l’union avec l’os trabéculaire et la stabilité (déviation inférieure ou égale à 3mm et angle de mouvement inférieur à 5°) ont été obtenues, ainsi que lorsqu’il n’y
avait aucune radiotransparence autour de chaque implant supérieur à 50 %. De même, les patients qui ont dû subir une intervention chirurgicale supplémentaire en raison d’une absence de consolidation, ont été pris en compte
pour les calculs, en tant qu’échecs de fusion. En ce qui concerne l’incapacité/douleur d’après Oswestry, le succès a été considéré comme atteint lorsque le résultat postopératoire s’est amélioré d’au moins 15 points par rapport au
résultat préopératoire. Pour ce qui est de l’état neurologique, le succès a été obtenu si cet état s’est maintenu ou amélioré après l’opération, pour au moins 3 des 4 catégories neurologiques (le mouvement, les sens, les réflexes
et le levé de jambe tendue), par rapport à l’état préopératoire. Le succès total a été considéré comme atteint pour un patient si : il a été démontré que la fusion a été obtenue, si le succès en ce qui concerne l’incapacité/douleur
d’après Oswestry a été atteint, si on est parvenu au succès pour ce qui est de l’état neurologique, ainsi que si le patient n’a pas dû subir d’intervention chirurgicale supplémentaire, classifiée comme “échec”.
2
Le dispositif INTER FIX™ n’est statistiquement pas plus mauvais que celui du contrôle.
3
Les 3 patients chez lesquels le dispositif INTER FIX™ n’a pas été implanté en raison des effets indésirables liés à l’intervention, n’ont pas été repris
3
Pour l’essai clinique, le nombre total de patients inscrits dans le groupe de contrôle, appartenant au
.
Le tableau III nous montre également les pourcentages en ce qui concerne le succès total, au bout de 12 et 24 mois après l’intervention.
Fusion
Amélioration
incapacité/douleur
d'après Oswestry
Amélioration d'au
moins 15 points par
rapport au résultat
préopératoire
Amélioration ou
maintien de l'état
neurologique
Succès total
Pourcentages
12 mois
Dispositif test Con trôle
96.1%
(122/127)
53.5%
(76/142)
99.3%
(150/151)
42.4%
(61/144)
Tableau III – Résultats de l ’étude clinique pour le dispositif INTE R FIX™
Delta
1
(ǻ
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
(12/56)
-0.097 46.1%
Pourcentages
)
Dispositif test Con trôle
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
(71/154)
24 mois
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
(17/57)
Pourcentages
Delta
1
(ǻ
)
48 mois
Dispositi f test Dispositif test
94.9% (93/98) 94.4% (84/ 89)
59.5% (66/111) 68.5% (61/89)
99.0% (102/103) 97. 6% (82/84)
-0.043 49.5%
(53/107)
Pourcentages
72 mois
56.3%
(54/96)
1 - Marge de non-infériorité minimale requise pour affirmer la non-infériorité du dispositif test par rapport à celui du contrôle.
Tous les patients qui ont participé à l’essai clinique portant sur le dispositif fileté pour la fusion INTER FIX™, indépendamment du groupe, ont été sélectionnés selon les mêmes critères d’inclusion/exclusion. Afin de confirmer le
caractère comparable entre les patients du groupe randomisé du dispositif INTER FIX™ et les patients du groupe non-randomisé du dispositif INTER FIX™, ce qui suit a été contrôlé : les caractéristiques démographiques, les
conditions médicales préopératoires, le diagnostic obtenu en radiologie caractéristique d’une discopathie dégénérative, l’approche opératoire, ainsi que le positionnement dans le niveau du rachis lombaire qui devait être traité.
D’un point de vue statistique la population était comparable, sauf en ce qui concerne l’approche opératoire, la situation professionnelle du patient avant l’opération, ainsi que le diagnostic radiologique préopératoire afin de détecter
la formation d’ostéophytes sur la plaque terminale de la vertèbre, la cicatrisation et/ou l’épaississement de l’anneau fibreux, du ligament jaune et/ou de la capsule de la facette articulaire, ainsi que la hernie du nucleus pulposus.
D’autres analyses statistiques ont montré que ces variables n’étaient pas liées aux résultats cliniques obtenus sur 12 mois en ce qui concerne la fusion, l’amélioration incapacité/douleur d’après Oswestry, l’état neurologique ou
le succès total, sauf pour les hernies du nucleus pulposus. On a observé plus de patients ayant des hernies du nucleus pulposus dans le groupe non-randomisé, ce qui a fait accroître la probabilité de succès total.
Une analyse en intention de traiter a été réalisée. Pour cette analyse, les interventions chirurgicales supplémentaires classifiées comme des échecs, les décès, les patients qui n’ont pas pu être suivis, ainsi que les observations
manquantes pour toute autre raison, ont été considérés comme observations perdues pour les variables des résultats, et par conséquent, ils ont été inclus dans le dénominateur des pourcentages calculés, c’est à dire qu’ils ont
été pris en compte en tant qu’« échecs ». Comme ces patients ont été considérés en tant qu’échecs, les pourcentages pour ce qui est du résultat clinique obtenus pour l’analyse en intention de traiter, ont été considérablement
inférieurs par rapport à ceux qui ont été obtenus pour l’analyse actuelle.
Le tableau ci-dessous (Tableau IV) nous montre les résultats de l’analyse en intention de traiter pour les patients du dispositif INTER FIX™, par contre, il n’inclut pas les résultats obtenus pour les patients du groupe de contrôle.
Page 8

() y pp p p p ppgp
Les décès, les interventions chirurgicales supplémentaires classifiées comme des échecs, les patients qui n’ont pas pu être
suivis, et les observations manquantes ont été considérés en tant qu’échecs et ont été inclus dans le dénominateur des
Pourcentages
Fusion
Amélioration incapacité/douleur d'après
Oswestry
Amélioration d'au moins 15 points par rapport au
résultat préopératoire
Amélioration ou maintien de l'état
neurologique
Succès total
Echecs interventions chirurgicales
supplémentaires
Absence de consolidation
2
Autre s
Décès
CONDITIONNEMENT :
L’emballage de chaque composant devra être intact à réception. Si on a recours à un système de prêt ou de dépôt, il faudra vérifier soigneusement, avant de les utiliser, que tous les implants sont complets, et qu’aucun
composant, ni instrument, ne présente de dommage. Les produits endommagés ou ceux se trouvant dans des emballages endommagés ne devront pas être utilisés et devront être retournés à Medtronic Sofamor Danek.
NETTOYAGE ET DECONTAMINATION :
Tous les implants et instruments devront d’abord être nettoyés selon la méthode définie par l’hôpital, avant d’être stérilisés et introduits dans la zone chirurgicale stérilisée, sauf s’ils sont fournis stérilisés.
Décontaminer, nettoyer et stériliser les instruments qui ont été utilisés avant de les réutiliser.
Tableau IV - Analyse en intention de traiter pour le dispositif INTER FIX™
pourcentages
12 mois
65.9%
(122/185)
41.1%
(76/185)
81.1%
(150/185)
33.0%
(61/185)
1
5
8
0 1 0 0
Pourcentages
24 mois
67.6%
(125/185)
42.7%
(79/185)
69.2%
(128/185)
38.4% (71/185) 45.7%
Pourcentages
48 mois
80.2%
(93/116)
56.9%
(66/116)
87.9%
(102/116)
(53 /116)
5
3
4
4
Pourcentages
72 mois
77.1%
( 84/109)
56.0%
(61 /109)
75.2%
(82/109)
49.5%
(54/109)
1
2
1- Ces patients ont été inclus dans le calcul des pourcentages pour ce
qui est de la fusion, mais ils ont été considérés en tant qu’échecs par
rapport aux objectifs de l’essai clinique.
2- Les patients qui n’ont pas pu être suivis pendant cette période et qui ont
subi une intervention chirurgicale supplémentaire pour des raisons autres
qu’une absence de consolidation, ont été considérés en tant qu’échecs
par rapport aux objectifs de l’essai clinique.
Observations : Ne pas utiliser de solutions de nettoyage contenant de l’eau de javel ou de la formaline, car elles peuvent endommager l’implant.
Tous les produits devront être manipulés avec soin. Une mauvaise utilisation ou manipulation pourra endommager l’implant et l’empêcher de fonctionner correctement.
STERILISATION :
Sauf s’il est écrit “ stérilisé ” clairement sur l’étiquette de l’emballage stérilisé et intact fourni par le fabricant, tous les implants et instruments employés en chirurgie devront être stérilisés par l’hôpital avant d’être
utilisés. Avant la stérilisation, enlever tout matériel d’emballage. Seuls les produits stérilisés devront être introduits dans le bloc opératoire. Pour atteindre un niveau d’assurance de stérilité de 10-6, il est recommandé
que ces produits soient stérilisés à la vapeur par l’hôpital, selon les paramètres de l’un des trois procédés suivants :
METHODE CYCLE TEMPERATURE DUREE D’EXPOSITION
Vapeur Pré-aspiration 270°F (132°C) 4 minutes
Vapeur Sous pression 250°F (121°C) 60 minutes
Vapeur* Sous pression* 273°F (134°C)* 20 minutes*
OBSERVATIONS : Etant donné le nombre de variables qui entrent en ligne de compte dans le processus de stérilisation, chaque service médical devra juger et contrôler le processus de stérilisation (par exemple
la température, la durée) qui sera utilisé pour son matériel.
*Lorsqu’il est utilisé en dehors des Etats-Unis d’Amérique, certaines autorités médicales n’appartenant pas aux Etats-Unis recommandent que la stérilisation soit effectuée selon ces paramètres, dans le but de
minimiser le risque potentiel de transmission de la maladie de Creutzfeldt-Jakob, en particulier pour les instruments chirurgicaux qui pourraient entrer en contact avec le système nerveux central.
RECLAMATIONS CONCERNANT CE PRODUIT :
Tout professionnel de la santé (par exemple, tout client ou utilisateur de nos produits), qui a une réclamation ou un motif d’insatisfaction relatif à la qualité du produit, à son identité, à sa durabilité, à sa fiabilité,
à sa sécurité, à son efficacité et/ou à ses performances, devra le notifier à son distributeur Medtronic Sofamor Danek. De plus, si jamais l’un des composants du dispositif fileté pour la fusion INTER FIX™ ou du
dispositif fileté pour la fusion INTER FIX™ RP avait été implanté et si celui-ci « ne fonctionne pas correctement », (c’est-à-dire s’il n’accomplit pas les performances spécifiées ou s’il ne fonctionne pas comme
prévu), ou s’il était suspecté de mauvais fonctionnement, contacter le distributeur immédiatement. Si jamais un produit de Medtronic Sofamor Danek “ n’avait pas fonctionné correctement ” et avait provoqué ou
contribué au décès ou à une lésion grave d’un patient, contacter le distributeur immédiatement par téléphone, par fax ou par courrier. Pour toute réclamation, veuillez indiquer le nom et le numéro du composant,
le numéro de lot du composant, votre nom et votre adresse et la nature de votre réclamation. Veuillez spécifier si vous souhaitez un rapport écrit de la part du distributeur.
REMARQUE POUR LE MEDECIN : Bien que le médecin soit celui qui possède les connaissances, et qu’il soit l’intermédiaire entre le fabricant et le patient, les informations médicales
importantes figurant dans ce document devront être transmises au patient.
Ne s’applique qu’aux États-Unis
ATTENTION : LA LOI FEDERALE DES ETATS-UNIS D’AMERIQUE (USA) RESTREINT LA VENTE DE CES IMPLANTS PAR UN MEDECIN OU SELON LA PRESCRIPTION D’UN MEDECIN.
EXTRACTION DU DISPOSITIF :
S’il s’avérait nécessaire d’extraire un dispositif fileté pour la fusion INTER FIX™ ou un dispositif fileté pour la fusion INTER FIX™ RP, veuillez contacter Medtronic Sofamor Danek.
© 2006 Medtronic Sofamor Danek. Tous droits réservés.
Page 9

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
ESPAÑOL
Este documento contiene informaciones médicas importantes sobre el dispositivo fileteado para la fusión INTER FIX™ y, sobre el dispositivo fileteado para la fusión INTER FIX™ RP.
DESCRIPCIÓN:
El dispositivo fileteado para la fusión INTER FIX™ está compuesto de un cilindro metálico, hueco y perforado, así como de un tapón. El dispositivo fileteado para la fusión INTER FIX™ RP
está compuesto de un cilindro metálico, hueco y perforado, conteniendo una gran ranura redondeada exterior a lo largo del eje longitudinal y, la cual se prolonga hasta el diámetro interior
del dispositivo. Ambas extremidades del implante INTER FIX™ RP se encuentran cerradas.
El diámetro de los implantes INTER FIX™ e INTER FIX™ RP puede variar de 12 mm hasta 24 mm y, la longitud de 20 mm hasta 29 mm. El tamaño de los tapones de los implantes INTER
FIX™ corresponde con el diámetro de los cilindros y, se colocan en la extremidad abierta del cilindro una vez que se hayan rellenado con injerto óseo autógeno.
Los implantes INTER FIX™ e INTER FIX™ RP están fabricados con una aleación de titanio especifica para implantes (Ti-6Al-4V), según lo previsto en la norma ASTM F136.
Se excluyen expresamente, las garantías implícitas en cuanto a la comercialización y a la capacidad cuando se utilice o se intente utilizar de forma particular. Para mayor información en lo
que se refiere a las garantías y a los límites en cuanto a la responsabilidad, véase la lista de precios o el catálogo de Medtronic Sofamor Danek.
INDICACIONES:
El dispositivo fileteado para la fusión INTER FIX™ y el dispositivo fileteado para la fusión INTER FIX™ RP están indicados para las intervenciones quirúrgicas de fusión del raquis en pacientes
cuyo esqueleto se haya desarrollado y, que padezcan de discopatía degenerativa (DDD) en uno de los niveles del raquis de L2-S1. La discopatía degenerativa (DDD) se diagnostica ante
la presencia de dolor de origen discal en la espalda con degeneración del disco, confirmada por el historial médico del paciente y por control en radiología. Estos pacientes, que padecen
de discopatía degenerativa (DDD), también pueden presentar en el mismo nivel del raquis, espondilolistesis o retrolistesis pudiendo llegar hasta grado I. Utilizar con los dispositivos INTER
FIX™ e INTER FIX™ RP un injerto óseo autógeno e implantar dichos dispositivos mediante cirugía abierta por vía anterior.
CONTRAINDICACIONES:
No implantar el dispositivo fileteado par la fusión INTER FIX™, ni el dispositivo fileteado para la fusión INTER FIX ™ RP, en pacientes que presenten una infección activa en la región donde
se tenga que operar, o en pacientes que sean alérgicos al titanio o a la aleación de titanio.
ADVERTENCIAS:
• El dispositivo fileteado para la fusión INTER FIX™ y el dispositivo fileteado para la fusión INTER FIX™ RP, solamente los pueden utilizar los cirujanos que tengan una experiencia
suficiente en cirugía de fusión del raquis y, que hayan recibido la instrucción adecuada para este tipo de dispositivos.
• Si el cirujano carece de la experiencia y/o de la instrucción adecuada, esto podría aumentar el riesgo de que surjan efectos adversos como lesiones vasculares, lesiones
neurológicas, y/o problemas en el sistema urogenital (incluyendo eyaculación retrógrada).
PRECAUCIONES:
• La seguridad y la eficacia del dispositivo fileteado para la fusión INTER FIX™ así como la del dispositivo fileteado para la fusión INTER FIX™ RP, no han sido demostradas en
pacientes que se encuentren en uno de los casos siguientes:
- cuando la espondilolistesis o retrolistesis sea de grado II o superior;
- cuando sea necesario fusionar mas de un nivel;
INFORMACIONES MÉDICAS IMPORTANTES : EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX™ Y
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX™ RP
Page 10
;
- cuando sea preciso corregir una intervención quirúrgica de fusión entre los cuerpos vertebrales realizada anteriormente;
L
L
- cuando se necesite suministrar antiflamatorios esteroidales o no esteroidales durante el periodo postoperatorio;
- cuando la obesidad sea flagrante;
- cuando la edad del paciente sea inferior a 18 años o superior a 65 años;
- en caso de osteoporosis, osteopenia, y/u osteomalacia;
- en caso de embarazo.
• La seguridad y la eficacia del dispositivo fileteado para la fusión INTER FIX™ así como la del dispositivo fileteado para la fusión INTER FIX™ RP, han sido solo demostradas en caso
de intervención quirúrgica de fusión entre los cuerpos vertebrales del raquis lumbar por vía anterior (ALIF) y, en las que se utiliza un injerto óseo autólogo.
• Los pacientes a los que se les tenga que implantar un dispositivo fileteado para la fusión INTER FIX™, tienen que haber seguido un tratamiento no quirúrgico durante al menos
seis meses antes de operarlos. Los pacientes a los que se les tenga que implantar un dispositivo fileteado para la fusión INTER FIX™ RP, también tienen que haber seguido un
tratamiento no quirúrgico durante al menos seis meses antes de operarlos.
• Implantar los dos dispositivos fileteados para la fusión INTER FIX™, uno al lado del otro en el nivel que tenga que operar. Implantar un dispositivo fileteado para la fusión INTER
FIX™ y un dispositivo fileteado para la fusión INTER FIX™ RP, uno al lado del otro en nivel que se tenga que operar. La ranura longitudinal del dispositivo INTER FIX™ RP facilita la
implantación de los dos dispositivos lo más cerca posible entre dos dispositivos INTER FIX™ , de este modo se logrará un perfil lateral reducido.
• El eje longitudinal del dispositivo fileteado para la fusión INTER FIX™ así como el del dispositivo fileteado para la fusión INTER FIX™ RP, tienen que encontrarse en dirección
anterior-posterior.
• Esterilizar los implantes y los instrumentos antes de utilizarlos, según los procesos de esterilización que se indican en este folleto, excepto si se suministraran estériles y que se
indicarse claramente en la etiqueta.
EFECTOS ADVERSOS:
Los efectos adversos, como se muestra en la tabla I, son los que han surgido al implantar el dispositivo INTER FIX™ en 185 pacientes y, en el tratamiento de los 62 pacientes pertenecientes
al grupo control, los cuales han participado en el estudio clínico multicéntrico para el dispositivo fileteado para la fusión INTER FIX™. Se indican también en dicha tabla, los efectos adversos
que han surgido en 116 pacientes al cabo de 48 meses y, en 109 pacientes al cabo de 72 meses, durante el estudio post-autorización. Los porcentajes indicados han sido calculados a
partir del número total de pacientes que han participado en este estudio, para cada intervalo de tiempo preestablecido. Los efectos adversos que han surgido en los grupos, aleatorios o no
aleatorios, tratados con el dispositivo INTER FIX™, se han reunido para poder presentar de este modo una visión del porcentaje global.
Intraoperatorios
(Número %)
CONTROL
INTER
™
Complicación
Vascular intraoperatoria
Dolor a nivel sacro ilíaco
Neurológica
Dolor de espalda
Incisional
Complicación en el raquis
Urológica
Otras
Otro dolor
Gastrointestinal
Eyaculación retrógrada
Respiratoria
Dolor en la pierna
Traumatismo
Peritoneal
Vascular postoperatoria
Fractura ósea
Desplazamiento /
aflojamiento del implante
Dolor donde se ha tomado el
injerto
Ninguna consolidación
Desmoronamiento
Ninguna consolidación
(EN ESPERA DEL
RESULTADO)
Meningitis
Rotura del implante
Cáncer
Fallecimiento
1- Se han observado durante el periodo de 24 meses, seis (6) casos de eyaculación retrógrada en 81 pacientes de sexo masculino pertenecientes al grupo del dispositivo INTER FIX™
(7,4%).
2- Este paciente (175) se ha indicado posteriormente, puesto que la fusión se ha conseguido al cabo de los 52 meses después de la operación, y sin tener que recurrir a otra intervención
quirúrgica u otro tratamiento.
Se han observado dos efectos adversos importantes durante este ensayo clínico, es decir las lesiones vasculares y las lesiones neurológicas que se han producido durante la operación.
Se han presentado un total de 15 casos de lesiones vasculares durante la operación, en 14 pacientes pertenecientes al grupo del dispositivo INTER FIX™ durante el periodo de 24 meses,
siendo dichas lesiones las siguientes: 2 lesiones en la vena cava, 9 lesiones en la vena ilíaca, 1 laceración de la vena hipogástrica, 1 hemorragia en un segmento de la vena, 1 lesión en la
vena sacra y, 1 hemorragia superficial. En lo que se refiere al grupo control, se produjeron durante el mismo periodo un total de 2 lesiones vasculares durante la operación en 2 pacientes,
las cuales fueron: 1 lesión en la vena ilíaca y 1 hemorragia en la base del hueso.
Se han presentado un total de 30 casos neurológicos en 28 pacientes pertenecientes al grupo del dispositivo INTER FIX™ durante el periodo de 24 meses, siendo dichos casos los siguientes: 1
pie péndulo, 3 lesiones en la raíz de los nervios, 1 estenosis foraminal, 1 distrofia del reflejo simpático, 7 entumecimientos o calor en las piernas, 2 disestesias, 3 parestesias, 2 radiculitis, 7 dolor
en la espalda y/o en la pierna, 1 dolor en la espalda y/o en la pierna acompañados de otros síntomas, 1 arranque del dolor en la zona inferior del dorso y, 1 síndrome del túnel carpiano.
Se han presentado un total de 10 casos neurológicos en 8 pacientes pertenecientes al grupo control durante el mismo periodo, siendo dichos casos los siguientes: 1 radiculopatía con hormigueos en
las extremidades, 1 dolor crónico en la espalda acompañado de radiculitis, 1 paciente presentando síntomas de debilidad generalizada, 2 dolor en la espalda y en la pierna acompañados de otros
síntomas, 1 músculo abductor mayor desnervado y, 4 con entumecimiento o calor en las piernas. Durante el estudio post-autorización, se observaron 10 casos mas neurológicos en 10 pacientes,
siendo dichos casos los siguientes: 1 radiculopatía, 2 radiculopatías cervical, 1 entumecimiento y hormigueos en las manos, 1 entumecimiento o calor en las piernas, 3 dolor en la espalda y/o en
la pierna y, 2 dolor en la espalda y/o en la pierna acompañados de otros síntomas. La tabla que presenta los efectos adversos, nos muestra también efectos adversos como el dolor en la espalda
y en las piernas y/o los efectos adversos vinculados con el raquis, como la desaparición del espacio del disco, que en algunos casos, tiene una relación neurológica.
Ciertos efectos adversos implicaron el tener que operar otra vez después de haber realizado la cirugía correspondiente al ensayo clínico. Dichas intervenciones quirúrgicas suplementarias
pueden clasificarse del modo siguiente: corrección, extracción, fijación suplementaria o nueva operación.
en los pacientes del grupo del dispositivo INTER FIX™ (aleatorios y no aleatorios reunidos), en los del grupo control durante el estudio IDE y, en los del grupo del dispositivo INTER FIX™
durante el estudio post-autorización.
1
Corrección: Es la intervención que reajusta o de alguna manera modifica la configuración inicial del implante.
Extracción: Es la intervención que consiste en extraer uno o varios componentes de la configuración inicial del implante, sin que se sustituyan con el mismo tipo de dispositivo que el
del ensayo.
Fijación suplementaria: Es la intervención en la se implantan otros dispositivos para el raquis y los cuales no pertenecen a la configuración prevista inicialmente.
Nueva operación: Todo tipo de intervención quirúrgica en la que no se extraiga, ni se modifique, ni se añada uno de los componentes del implante inicial.
FIX
N=185
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
5(2.7) 3(1.6) 4(2.2) 1(1.6)
6(3.2) 2(3.2) 9( 4.9) 3(4.8) 5(2.7) 1(1.6)
1(0.5) 3(1.6) 2(3.2)
11(5.9) 6(9.7) 1(0.5)
1(0.5) 1(0.5) 1(1.6) 1(0.5)
1(0.5) 8(4.3) 2(3.2) 2(1.1)
2(1.1) 1(0.5) 2(3.2) 3(1.6)
4(2.2) 1(1.6) 5( 2.7)
7(3.8) 3(4.8) 1( 0.5) 1(0.5) 1(1.6)
2(1.1) 3(1.6) 1(0.5)
3(1.6) 1(1.6) 1( 0.5) 1(0.5)
2(1.1) 1(0.5)
2(1.1) 1(1.6) 5( 2.7) 5(2.7)
3(1.6)
2(1.1) 2(3.2) 1(0.5)
1(0.5) 2(1.1)
1(0.5) 1(1.6) 5(8.1)
1(0.5)
1(1.6)2
1(1.6)
5(8.1)
N=62
Postoperatorios
(1 Día -< 2
meses)
[Número (%)]
INTER
CONTROL
FIX™
N=62
N=185
TABLA I – EFECTOS ADVERSOS
[Número (%)]
INTER FIX
N=185
3 meses
( t
2-<5
meses)
™
CONTROL
N=62
6 meses
(t5-<9 meses)
[Número (%)]
INTER
™
FIX
N=185
5(2.7)
2(1.1)
3(1.6)
1(0.5)
2(1.1)
3(1.6)
1(0.5)
4(2.2)
2(1.1)
12 meses
(t
9-<19
meses)
[Número (%)]
CONTROL
INTER
CONTRO
™
N=62
1(1.6)
1(1.6)
3(4.8)
2(3.2)
2(3.2)
1
La tabla II resume las intervenciones quirúrgicas suplementarias que se realizaron,
FIX
N=185
7(3.8) 1(1.6) 4(2.2) 1(1. 6)
4(2.2) 3(4.8) 6(3.2) 4(6.5)
4(2.2) 3(4.8) 6(3.2) 6(9. 7)
2(1.1) 1(0.5) 1(1.6)
10(5.4) 1(1.6) 11(5.9) 3(4. 8)
1(1.6) 4(2.2)
7(3.8) 1(1.6) 7(3.8) 2(3.2)
9(4.9) 6(3.2) 2(3.2)
2(1.1) 2(1.1) 3(4.8)
1(0.5) 3(1.6)
2(1.1) 1(0.5)
6(3.2) 1(1. 6) 10(5.4) 1(1.6)
3(1.6) 4(2.2)
1(1.6) 1(1.6)
7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
2(1.1) 0
0 1( 1.6) 2(1.7)
0 1( 1.6)
0 5( 8.1)
1
(0.5)
1(1.6) 1(0.5) 1(0. 5) 1(1.6) 1(0.8)
24 meses
(t19-<30
[Número (%)]
INTER
N=62
FIX™
N=185
1
(0.5)
TOTAL
EFECTOS
meses)
CONTRO
ADVERSOS
(DURANTE 24
MESES)
CONTROL
INTER
N=62
™
N=62
FIX
N=185
23(12.4) 3(4.8) 1(0.9)
30(16.2) 13(21.0) 7(6.0) 3(2.8)
19(10.3) 11(17.7) 7(6.0) 6(5.5)
17(9.2) 8(12.9) 1(0.9)
27(14.6) 6(9.7) 13(11.2) 10(9.2)
16(8.6) 3(4.8) 2(1.7) 1(0.9)
22(11.9) 8(12.9) 14(12.1) 9(8,3)
27(14.6) 5(8.1) 7(6.0) 8(7.3)
13(7.0) 7(11.3) 9(7.8) 7(6.4)
6(3.2)
(7.4)
9(4.9) 1(1.6) 5(4.3) 6(5.5)
6(3.2) 0 2(1.7) 2(1.8)
28(15.1) 3(4.8) 11(9.5) 8(7.3)
3(1.6) 0
10(5.4) 2(3.2) 4(3.4) 2(1.8)
3(1.6) 0 1(0.9)
2(1.1) 6(9.7)
1(0.5) 2(3.2)
2 (1.1) 1(0. 8)
0
1
48 meses
(>30 - < 60
30-<60
(t
meses)
INTER
FIX™
N=116
72
meses
(t60
meses)
INTER
FIX
N=109
1(0.9)
™
Page 11

Estudio IDE Estudio post-autorización
Intervalo de tiempo
Corrección 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Extracción 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Fijación
Suplementaria
Nueva operación 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
Tabla II – Intervenciones quirúrgicas suplementarias
1
Intervalo de tiempo
preestablecido
48 meses
Dispositivo INTER
FIX™
N=116
preestablecido
24 meses
Dispositivo
INTER FIX™
N=185
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
Control
N=62
Intervalo de tiempo
preestablecido
72 meses
Dispositivo INTER FIX™
N=109
1- No se disponen los datos para el periodo de 72 meses para los pacientes del grupo control, puesto que para dichos pacientes solo se previó un seguimiento durante 24 meses.
POSIBLES EFECTOS ADVERSOS:
A continuación se listan los posibles efectos adversos, los cuales podrían surgir cuando se utilice en cirugía para la fusión del raquis, un dispositivo fileteado para la fusión INTER FIX™ o un
dispositivo fileteado para la fusión INTER FIX™ RP. Algunos de estos efectos adversos, ya se han indicado en la tabla de los posibles efectos adversos.
• Desensamblaje, deformación, ruptura, aflojamiento y/o migración de los componentes
• Reacción (alérgica) al cuerpo extraño.
• Lesión en los tejidos o en los nervios.
• Cambio postoperatorio de la curva del raquis, pérdida de la corrección, de la altura, y/o de la reducción del raquis.
• Infección.
• Ruptura dural.
• Disfunción del sistema neurológico.
• Disfunción del sistema urinario.
• Formación de cicatrices.
• Fractura ósea.
• Ninguna consolidación ósea (o seudoartrosis), consolidación tardía, mala consolidación.
• Interrupción del crecimiento posible del segmento del raquis operado. Pérdida de la movilidad o de la función del raquis.
• Complicaciones en la región donde se haya tomado el injerto.
• Lesión en los vasos sanguíneos y disfunción del sistema cardiovascular
• Complicaciones en el sistema gastrointestinal.
• Disfunción del sistema reproductor.
• Lesión en los órganos internos y en el tejido conectivo.
• Desarrollo de problemas respiratorios.
• Complicaciones vinculadas con la incisión.
• Cambio del estado mental.
• Fallecimiento
Observaciones: Para corregir ciertos posibles efectos adversos, podría ser necesario tener que operar de nuevo
RESULTADOS CLÍNICOS:
En los Estados Unidos de América se ha llevado a cabo un ensayo clínico comparativo2 multicéntrico para el dispositivo fileteado para la fusión INTER FIX™, con el fin de comparar el
dispositivo INTER FIX™ cuando se utiliza en el raquis anterior rellenándolo con hueso autógeno, a un grupo control tratado con aloinjerto del anillo femoral rellenado con hueso autógeno,
en el tratamiento de pacientes presentando discopatía degenerativa sintomática. Al principio, se previó un ensayo clínico prospectivo, aleatorio y controlado. Posteriormente, los planes de la
investigación cambiaron para permitir a los pacientes incorporar el grupo no aleatorio, es decir, los pacientes tratados solamente con el dispositivo INTER FIX™.
Los criterios de inclusión para este ensayo clínico fueron los siguientes: discopatía degenerativa sintomática diagnosticada por el dolor resistente al tratamiento, en la pierna y/o en la espalda
y, confirmada en radiología; inestabilidad del raquis determinada por una desviación superior a 4 mm o por un ángulo superior a 5°, en radiografías flexión/extensión; manifestación de los
síntomas en un solo nivel del raquis entre L2-S1; y en espondilolistesis no superior a grado I. Se han excluido expresamente de este estudio clínico los pacientes que ya se habían sometido
a una intervención quirúrgica de fusión entre los cuerpos vertebrales, por vía anterior, en el mismo nivel del raquis; los que presentaban osteopenia, osteoporosis u osteomalacia; así como
los que necesitaban estimulación del crecimiento óseo.
El número total de pacientes, pertenecientes al grupo del dispositivo INTER FIX™, inscritos en el estudio clínico fueron 185
en el grupo control, pertenecientes al grupo aleatorio, fueron 62.
Para el dispositivo fileteado para la fusión INTER FIX™, se requirió un estudio post-autorización con el fin de evaluar a largo plazo el resultado de dicho dispositivo, en el cual se tenia que
obtener los resultados al cabo de 6 años tras la operación, por lo menos en 100 pacientes. Por consiguiente, se evaluó el resultado del dispositivo a largo plazo en 116 pacientes al cabo
de 4 años después de operarlos y en 109 pacientes al cabo de 6 años después de operarlos. La población se obtuvo a partir de los pacientes que participaron en el ensayo clínico IDE. En
conformidad con los criterios del estudio IDE, la información que se pudo obtener para dichos pacientes incluye la descripción de las intervenciones quirúrgicas suplementarias, el control de
la fusión en radiología, así como la evaluación en cuanto al dolor y a la función.
La tabla III nos muestra los resultados de este estudio clínico et nos indica los porcentajes del éxito en cuanto a la fusión, al dolor según Oswestry y al estado neurológico, al cabo de 12, 24,
48 y 72 meses después de la operación. El éxito de este ensayo clínico se basó en un análisis comparativo entre el dispositivo INTER FIX™ y del control.
En este ensayo clínico, se definió haber conseguido la fusión cuando se obtuvo: la unión con el hueso trabecular, una estabilidad siendo la desviación inferior o igual a 3mm y el ángulo de
movimiento inferior a 5°, así como ninguna radiotransparencia alrededor de cada implante superior al 50%. Además, los pacientes que se tuvieron que someter a una intervención quirúrgica
suplementaria puesto que la consolidación no se consiguió, se han tenido en cuenta para el cálculo, como fusiones que han fracasado. En lo que se refiere a la incapacidad/dolor según Oswestry,
se ha considerado que se obtuvo éxito, cuando el resultado postoperatorio fue al menos 15 puntos más, con respecto al resultado preoperatorio. El éxito, en lo que se refiere al estado neurológico,
se obtuvo si dicho estado se ha podido mantener o se ha mejorado tras intervención, al menos en 3 de las 4 categorías neurológicas (movimiento, sentidos, reflejos y alzar la pierna estirada), con
respecto al estado preoperatorio. Se logró el éxito total en un paciente si: se ha demostrado haber conseguido la fusión, se ha obtenido éxito en lo que se refiere a la incapacidad/dolor según
Oswestry, se ha logrado éxito en cuanto al estado neurológico, así como si el paciente no se tuvo que someter a una intervención quirúrgica suplementaria, clasificada como “fracaso”.
2
El dispositivo INTER FIX™ , de un punto de vista estadístico, no es peor que el del control
3
No se han incluido 3 pacientes a quienes no se les ha implantado el dispositivo INTER FIX™ a causa de los efectos adversos vinculados con la operación.
En la Tabla III, también se puede observar el éxito total al cabo de 12 y 24 meses tras la operación.
3
. Para el ensayo clínico, el número total de pacientes inscritos
Fusión
Mejoría de la
incapacidad/dolor
según Oswestry
Mejoría de al menos
15 puntos con
respecto al resultado
preoperatorio
Mejoría o
mantenimiento del
estado neurológico
Tabla III – Resultados del estudio clínico para el dispositivo INTER FIX™
Porcentajes 12 meses Porcentajes 24 meses
Dispositivo
investigado
96.1%
(122/127)
53.5%
(76/142)
99.3%
(150/151)
Control
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
Delta
(ǻ
1
)
Dispositivo
investigado
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
Control
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
Delta
Porcentajes
(ǻ1)
48 meses
Dispositivo
investigado
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
Porcentajes
72 meses
Dispositivo
investigado
97.6% (82/84)
1 – Margen de no-inferioridad mínimo requerido para afirmar la no-inferioridad del dispositivo investigado con respecto al del control.
Todos los pacientes implicados en el ensayo clínico del dispositivo fileteado para la fusión INTER FIX™, sin tener en cuenta del grupo que se trate, se han elegido bajo los mismos criterios
de inclusión/exclusión. Para confirmar el carácter comparativo entre los pacientes pertenecientes al grupo aleatorio del dispositivo INTER FIX™ y los pacientes pertenecientes al grupo no
aleatorio del dispositivo INTER FIX™, se controló lo siguiente: las características demográficas, las condiciones médicas preoperatorias, el diagnóstico obtenido en radiología y característico
a una discopatía degenerativa, el modo de operar, y la posición en el nivel del raquis lumbar que se tenia que tratar. Desde un punto de vista estadístico la población fue comparable, excepto
en lo que se refiere al modo de operar, a la situación profesional del paciente antes de operarlo así como al diagnóstico preoperatorio radiológico para encontrar la formación de osteófitos
en la placa terminal de la vértebra; cicatrización y/o espesamiento del anillo fibroso, del ligamento amarillo y/o de la cápsula de la carilla articular, así como hernia del núcleo pulposo. Otros
análisis estadísticos demostraron que estas variables no estuvieron vinculadas con los resultados clínicos obtenidos durante 12 meses, en cuanto se refiere a la fusión, a la incapacidad/dolor
según Oswestry, al estado neurológico o al éxito total, excepto para la hernia del núcleo pulposo. Se observaron más pacientes con hernias del núcleo pulposo en el grupo no aleatorio, por
lo que la probabilidad del éxito total aumentó.
Page 12

Se ha llevado a cabo un análisis por intención de tratar. Para dicho análisis, las intervenciones quirúrgicas suplementarias clasificadas como fracasos, los fallecimientos, los pacientes que no
se han podido seguir, y las observaciones que faltaban por otras causas, se han considerado como observaciones perdidas para las variables de los resultados y, por lo tanto se han incluido
en el denominador de los porcentajes calculados, es decir se han tenido en cuenta como “fracasos”. El hecho de considerar dichos pacientes como fracasos, los porcentajes en cuanto al
resultado clínico obtenidos para el análisis por intención de tratar fueron considerablemente inferiores con respecto a los que se han obtenido para el análisis clínico actual.
La tabla siguiente (Tabla IV) nos muestra los resultados del análisis por intención de tratar obtenidos para los pacientes del dispositivo INTER FIX™, sin embargo no incluye los resultados
obtenidos para los pacientes del grupo control.
Los fallecimientos, las intervenciones quirúrgicas suplementarias clasificadas como fracasos, los pacientes que no se han
podido seguir, y las observaciones que faltaban, se han considerado como fracasos y se han incluido en el denominador de
Porcentajes 12
Fusión
Mejoría de la incapacidad/dolor
según Oswestry
Mejoría de al menos 15 puntos con respecto al
resultado preoperatorio
Mejoría o mantenimiento del estado
neurológico
Éxito total
Fracasos intervenciones quirúrgicas
suplementarias
Ninguna consolidación
2
Otros
Fallecimientos
Tabla IV – Análisis por intención de tratar para el dispositivo INTER FIX™
los porcentajes.
Meses
65.9% (122/185) 67.6%
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
33.0% (61/185) 38.4% (71/185) 45.7%
1
5
8
0 1 0 0
Porcentajes
24 Meses
(125/185)
(128/185)
5
3
Porcentajes
48 Meses
(102/116)
(53 /116)
80.2%
(93/116)
(66/116)
87.9%
4
4
Porcentajes
72 Meses
77.1%
( 84/109)
56.0%
(61 /109)
75.2%
75.2% (82/109)
(82/109)
49.5%
(54/109)
1
2
1-Estos pacientes se han incluido en el cálculo de los porcentajes para la fusión pero por otro lado se han considerado como fracasos en cuanto a los objetivos del ensayo clínico.
2- Los pacientes que no se han podido seguir durante este periodo y los cuales se han tenido que operar de nuevo, por otras razones que el hecho de no haber conseguido la consolidación,
se han considerado como fracasos en cuanto a los objetivos del ensayo clínico.
EMBALAJE:
Cuando se reciban, el embalaje de cada componente tiene que encontrase intacto. Si se recurriera a un sistema de préstamo o de depósito, controlar con mucho cuidado, y antes de utilizarlos,
que todos los componentes se encuentren completos, y además controlar minuciosamente todos los componentes e instrumentos para asegurarse que no se hayan dañado. No utilizar
productos deteriorados ni los que se encuentren en embalajes deteriorados, devuélvanse a Medtronic Sofamor Danek.
LIMPIEZA Y DESCONTAMINACIÓN:
Limpiar todos los implantes e instrumentos según el método que el hospital haya determinado, antes que se esterilicen y que se introduzcan en la zona de cirugía estéril, excepto si se
suministraran estériles. Descontaminar, limpiar y esterilizar los instrumentos que se hayan utilizado antes que se utilicen de nuevo.
Observaciones: No utilizar soluciones limpiadoras que contengan lejía o formalina ya que pueden deteriorar el implante.
Utilizar todos los productos con cuidado. Si la utilización o el manejo no se realizara correctamente, esto podría dañar el implante y/o podría impedir que funcione correctamente.
ESTERILIZACIÓN:
Excepto si fuera escrito “estéril” claramente en la etiqueta del embalaje estéril e intacto proveniente de nuestra empresa, es necesario que el hospital esterilice todos los implantes y los instrumentos,
que se utilizarán en cirugía, antes de que se empleen. Retirar el embalaje antes de esterilizarlos. Introducir en el quirófano solamente productos estériles. Para obtener un nivel de garantía de
esterilización de 10–6, se recomienda que el hospital esterilice estos productos con vapor, utilizando uno de los tres procesos de esterilización según los parámetros siguientes:
MÉTODO CICLO TEMPERATURA TIEMPO DE EXPOSICIÓN
Vapor Aspiración previa 270° F (132° C) 4 minutos
Vapor Bajo presión 250° F (121° C) 60 minutos
Vapor* Bajo presión* 273° F (134° C)* 20 minutos*
OBSERVACIONES: Puesto que en el proceso de esterilización varias variables se encuentran relacionadas, cada unidad médica tendrá que evaluar y controlar el proceso de esterilización
(por ejemplo, la temperatura, el tiempo) que utilizará para su material.
* Cuando se utilice fuera de los Estados Unidos de América, algunas autoridades sanitarias no estadounidenses recomiendan que se esterilice según estos parámetros, para minimizar el
riesgo posible de transmisión de la enfermedad de Creutzfeldt-Jakob, especialmente los instrumentos quirúrgicos que pudieran alcanzar el sistema nervioso central.
RECLAMACIONES REFERENTES A ESTE PRODUCTO:
Cualquier persona que pertenezca al equipo médico (por ejemplo, clientes o usuarios de nuestros productos), que deseara reclamar o que no esté satisfecha en lo que se refiere a la calidad,
la identidad, la durabilidad, la fiabilidad, la seguridad, la eficacia y/u otras calidades técnicas del producto, que lo notifique a su distribuidor Medtronic Sofamor Danek. Además, si uno de los
componentes implantados pertenecientes al dispositivo fileteado para la fusión INTER FIX™ o al dispositivo fileteado para la fusión INTER FIX™ RP “no funcionara correctamente”, (es decir,
que no cumpliera con las funciones indicadas o bien no funcionara según lo previsto), o si se sospechara que funciona mal, pónganse en contacto inmediatamente con su distribuidor. Si uno
de los productos de Medtronic Sofamor Danek “no hubiera funcionado correctamente” y, hubiera provocado o contribuido a que un paciente fallezca o sufra alguna lesión grave, pónganse
en contacto inmediatamente con su distribuidor, por teléfono, por fax o por correo. Para cualquier reclamación, por favor indique el nombre y el número del componente, el número del lote
del componente, su nombre y apellidos, su dirección y el tipo de reclamación. Por favor indiquen si desean un informe escrito del distribuidor.
OBSERVACIONES PARA EL MÉDICO: Aunque el médico sea el que tenga la capacitación y, además sea el intermediario entre el fabricante y el paciente, hay que remitir al paciente las
informaciones médicas importantes que se describen en este documento.
Solo aplicable en EE.UU.
ATENCIÓN: LA LEY FEDERAL DE LOS ESTADOS UNIDOS DE AMÉRICA (EE.UU.) RESTRINGE LA VENTA DE ESTOS IMPLANTES POR UN MÉDICO O SEGÚN LA PRESCRIPCIÓN
DE UN MÉDICO.
EXTRACCIÓN DEL DISPOSITIVO:
Si fuera necesario extraer un dispositivo fileteado para la fusión INTER FIX™ o un dispositivo fileteado para la fusión INTER FIX™ RP, por favor póngase en contacto con Medtronic
Sofamor Danek.
© 2006 Medtronic Sofamor Danek. Reservados todos los derechos.
Page 13

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
DEUTSCH
Diese Dokumentation enthält wichtige medizinische Informationen über die INTER FIX™ Gewinde-Instrumentation für Fusionen und die INTER FIX™ RP Gewinde-Instrumentation für
Fusionen.
BESCHREIBUNG:
Die INTER FIX™ Gewinde-Instrumentation für Fusionen besteht aus einem hohlen, perforierten, metallischen Zylinder und einer Endkappe. Die INTER FIX™ RP Gewinde-Instrumentation für
Fusionen besteht aus einem hohlen, perforierten, metallischen Zylinder mit einer breiten, äusseren gerundete Rille entlang der ganzen Längsachse, die sich bis in den inseitigen Durchmesser
der Instrumentation zieht. Beide Enden des INTER FIX™ RP Implantats sind geschlossen.
Der Durchmesser der INTER FIX™ und INTER FIX™ RP Implantate kann von 12 mm bis 24 mm varrieren und die Länge von 20 bis 29 mm. Die Endkappengrössen der INTER FIX™ Implantate
sind passend zum Durchmesser der Zylinder; sie werden auf das offene Ende des Zylinders gesetzt, nachdem der Zylinder mit autologen Knochenanlagerungen gefüllt worden ist.
Die INTER FIX™ und INTER FIX™ RP Implantate sind hergestellt aus für Implantate vorgesehener Titaniumlegierung (Ti-6Al-4V), beschrieben unter Standarden wie ASTM F136.
Die Gewährleistung der Marktängigkeit und die Eignung für einen bestimmten Zweck oder Gebrauch sind ausdrücklich ausgeschlossen. Konsultieren Sie den Medtronic Sofamor Danek
Katalog bzw. die Preisliste für weitere Informationen über die Garantie und die Haftungsgrenzen.
INDIKATIONEN:
Die INTER FIX™ Gewinde-Instrumentationen für Fusionen und INTER FIX™ RP Gewinde-Instrumentation für Fusionen sind angezeigt für spinale Fusionsoperationen bei Patienten mit
ausgewachsenem Skelett und degenerativer Diskopathie (DDD) auf einer der Wirbelebenen von L2 bis S1. Degenerative Diskopathie (DDD) ist diagnostiziert durch Rückenschmerz mit
Ursprung in den Bandscheiben, mit einer Degeneration der Bandscheibe, bestätigt durch die Krankengeschichte des Patienten und radiographische Studien. Diese Patienten, die von einer
degenerativen Diskopathie (DDD) betroffen sind, können ebenso unter Spondylolisthesis oder Retrolisthesis bis zu Grad 1 auf derselben Höhe des Rückgrats leiden. Die INTER FIX™ und
INTER FIX™ RP Instrumentationen müssen mit autologen Knochenanlagerungen gebraucht und über eine offene vordere Chirurgie implantiert werden.
KONTRAINDIKATIONEN:
Die INTER FIX™ Gewinde-Instrumentation für Fusionen und die INTER FIX™ RP Gewinde-Instrumentation für Fusionen sollten nicht bei Patienten implantiert werden, die eine aktive Infektion
an der Operationsstelle haben oder die allergisch sind gegen Titanium oder Titaniumlegierung.
WARNUNGEN:
• Die INTER FIX™ Gewinde-Instrumentation für Fusionen und die INTER FIX™ RP Gewinde-Instrumentation für Fusionen sollten nur von Chirurgen implantiert werden, die mit spinalen
Fusionsoperationen Erfahrung haben und die sich der angemessenen Ausbildung mit diesen Instrumentationen unterzogen haben.
• Wenn die Erfahrung und/oder die Ausbildung des Chirurgen ungenügend ist, kann es zu einem höheren Auftreten von nachteiligen Wirkungen führen, wie etwa vaskuläre
Verletzungen, neurologische Verletzungen und/oder Probleme des urogenitalen Systems (rückschrittliche Ejakulation eingeschlossen).
VORSICHTSMASSNAHMEN:
• Die Sicherheit und die Wirksamkeit der INTER FIX™ Gewinde-Instrumentation für Fusionen und der INTER FIX™ RP Gewinde-Instrumentation für Fusionen sind nicht bewiesen
worden bei Patienten die zu den folgenden Fällen gehören:
- im Fall von Spondylolisthesis oder Retrolisthesis von Grad II oder höher,
WICHTIGE MEDIZINISCHE INFORMATIONEN : DIE INTER FIX™ GEWINDE-INSTRUMENTATION FÜR FUSIONEN
DIE INTER FIX™ RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
Page 14

py
- wenn mehr als eine Höhe der Wirbelsäule fusionieren soll,
)
e
E
E
m
- wenn eine vorausgegangene interkörperliche Intervention korrigiert werden muss,
- wenn postoperative anti-inflammatorische Medikationsbedürfnisse mit oder ohne Steroide während der postoperativen Periode notwendig sind,
- wenn die Patienten von latenter Dickleibigkeit betroffen sind,
- wenn das Alter des Patienten unter 18 Jahren oder über 65 Jahren liegt,
- im Fall von Osteoporose, Osteopenie und/oder Osteomalazie,
- im Fall von Schwangerschaft.
• Die Sicherheit und die Wirksamkeit der INTER FIX™ Gewinde-Instrumentation für Fusionen und der INTER FIX™ RP Gewinde-Instrumentation für Fusionen sind nur festgestellt
worden in lumbalen interkörperliche Fusionsoperationen über eine vordere Annäherung (ALIF) bei denen eine autologe Knochenanlagerung gebraucht wurde.
• Patienten, bei denen eine INTER FIX™ Gewinde-Instrumentation für Fusionen implantiert wird, sollten mindestens sechs Monate lang eine nicht-operative Behandlung vor der
Operation erhalten haben. Patienten, bei denen eine INTER FIX™ RP Gewinde-Instrumentation für Fusionen implantiert wird, sollten auch mindestens sechs Monate lang eine nichtoperative Behandlung vor der Operation erhalten haben.
• Die zwei INTER FIX™ Gewinde-Instrumentationen für Fusionen sollten nebeneinander auf der Höhe der Operationsstelle implantiert werden. Eine INTER FIX™ GewindeInstrumentation für Fusionen und eine INTER FIX™ RP Gewinde-Instrumentationen für Fusionen sollten nebeneinander auf der Höhe der Operationsstelle implantiert werden. Die
Längsrille der INTER FIX™ RP Instrumentation erleichtert eine Implantation mit der höchst möglichen Nähe zwischen den zwei INTER FIX™ Instrumentationen, dadurch erhält man
ein reduziertes seitliches Profil.
• Die lange Axe der INTER FIX™ Gewinde-Instrumentation für Fusionen und diejenige der INTER FIX™ RP Gewinde-Instrumentation für Fusionen sollten von vorne nach hinten
gerichtet sein.
• Die Implantate und Instrumente müssen vor dem Gebrauch sterilisiert werden nach den Sterilisierungsanweisungen, die in dieser Verpackunsbeilage angegeben werden, ausser wenn
sie steril geliefert werden und klar als solche etikettiert sind.
NACHTEILIGE WIRKUNGEN:
Die nachteiligen Wirkungen, wie sie in der Tafel I gezeigt werden, sind bei 185 INTER FIX™ Instrumentationspatienten festgestellt worden und bei 62 Kontrollpatienten, die an der Multi-center
klinischen Studie der INTER FIX™ Gewinde-Instrumentation teilgenommen haben. Zusätzlich sind in dieser Tafel die nachteiligen Wirkungen aufgeführt, die bei 116 Patienten, die an der
Studie der postoperativen Billigung teilnahmen, nach 48 Monaten und bei 109 Patienten die teilnahmen, nach 72 Monaten auftraten. Die angegebenen Prozentsätze wurden auf der Basis
der gesamten Anzahl der Patienten berechnet, die zu jedem Zeitpunkt an dieser Studie teilgenommen haben. Die nachteilige Wirkungen, die in den randomisierten und nicht-randomisierten
INTER FIX™ Instrumentation Behandlungsgruppen auftraten sind kombiniert, um eine allgemeine Rate vorzustellen.
TAFEL 1 –NACHTEILIGE WIRKUNGEN
Intraoperativ
[(Anzahl %)]
INTER
KONT
™
FIX
ROLL
N=185
Komplikation
Vaskuläre IntraOperativ
Sacro-illiake
Schmerzen
Neurologisch 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Rückenschmerzen 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
Inzisionskomplikation 11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
Komplikation i
Rückgrat
Urologisch 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Andere 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Andere Schmerzen 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
Gastrointestinal 7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
Rückschrittliche
Ejakulation
Atmung 3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
Beinschmerzen 2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
Trauma 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
Peritoneal 3(1.6) 3(1.6) 0
Vaskuläre
postoperativ
Komplikationen
Knochenbruch 1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
Implantatwanderun
g/
Lockerung
Schmerz an der
Entnahmestelle
Keine
Konsolidierung
Einsenken 2(1.1) 2(1.1) 0
Keine
Konsolidierung
(ERGEBNISSE IN
ERWARTUNG)
Meningitis 1(1.6) 0 1(1.6)
Brechen des
Implantats
Krebs 1 (0.5) 1 (0.5) 2 (1.1) 1(0.8) 1(0.9)
Tod 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
N=62
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
2(1.1) 3(1.6) 1(0.5) 6(3.2)
2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
1(1.6)
5(8.1) 0 5(8.1)
Post-operativ
(1 Tag -<2 Monate
[Anzahl (%)]
KONTR
INTER
™
OLLE
FIX
N=185
N=62
3 Monate
(t
2-<5 Monate)
[Anzahl (%)]
INTER
FIX™
N=185
KONT
ROLL
L
N=62
6 Monate
(t5-<9 Monate)
[Anzahl (%)]
KONTR
INTER
™
OLLE
FIX
N=185
N=62
12 Monate
(t
9-<19 Monate)
[Anzahl (%)]
INTER
KONTR
FIX™
OLLE
N=185
N=62
2
0 1(1.6) 2(1.7)
24 Monate
(t19-<30 Monate)
[Anzahl (%)]
INTER
KONTR
FIX™
OLLE
N=185
N=62
GESAMTE
NACHTEILIGE
WIRKUNGEN
(ÜBER 24 MONATE)
INTER
FIX™
N=185
(7.4)
KONTRO
LLE
N=62
0
1
48 Monate
(>30 - < 60
(t
30-<60
Monate)
INTER FIX™
N=116
72 Monat
(t60
Monate)
INTER
FIX™
N=109
1- Während der Periode über 24 Monate, wurden sechs (6) Fälle von rückschrittlicher Ejakulation unter den 81 männlichen Patienten der INTER FIX™ Instrumentationsgruppe, beobachtet (7,4%).
2- Dieser Patient (175) wurde erst später aufgeführt, da bei ihm nach 52 Monaten nach der Operation, eine Fusion erzielt wurde ohne dass eine andere Operation oder Behandlung notwendig wurde.
Zwei bedeutende nachteilige Wirkungen bei der klinischen Versuchsreihe waren vaskuläre Verletzungen und neurologische Verletzungen, die während der Operation auftraten. Eine Gesamtzahl
von 15 vaskulären Verletzungen, die während der Operation auftraten, zeigte sich bei 14 Patienten der INTER FIX™ Instrumentationsgruppe während der Periode über 24 Monate. Diese
Verletzungen sind: 2 Verletzungen an der Vena Cava, 9 Verletzungen der Vena Iliaca, 1 aufgerissene Vena Hypogastrica, 1 segmentale Venenblutung, 1 sacrale Venenverletzung und 1
oberflächliche Blutung. Eine Gesamtzahl von 2 intraoperativen vaskulären Verletzungen trat bei 2 Patienten der Kontrollgruppe im Verlauf derselben Pariode auf. Diese Verletzungen sind:
1 Verletzung der Vena Iliaca und 1 Blutung am Knochenbett.
Eine Gesamtzahl von 30 neurologischen Vorkommnissen zeigte sich bei 28 Patienten der INTER FIX™ Instrumentationsgruppe während der Periode über 24 Monate. Diese Vorkommnisse
sind: 1 Ozillationsfuss, 3 Verletzungen der Nervenwurzeln, 1 foraminale Stenose, 1 Distrophie des sympathischen Reflexes, 7 mit Taubheit oder Wärmegefühl in den Beinen, 2 Dysästhesien,
3 Paraästhesien, 2 Radiculitis, 7 mit Schmerzen im Rücken und/oder im Bein, 1 mit Schmerzen im Rücken und/oder im Bein mit anderen Symptomen, 1 schiessende Pein im unteren
Rücken und 1 Karpaltunnelsyndrom.
Eine Gesamtzahl von 10 neurologischen Vorkommnissen zeigte sich bei 8 Patienten der Kontrollgruppe im Verlauf derselben Periode. Diese Vorkommnisse sind: 1 Radikulopathie mit
kribbelnden Extremitäten, 1 Patient mit chronischen Rückenschmerzen mit Radiculitis, 1 Patient mit Symptomen allgemeiner Schwäche, 2 Schmerzen im Rücken und im Bein mit anderen
Symptomen, 1 denervierter Abductor Magnus Muskel, und 4 mit Taubheit oder Wärmegefühl in den Beinen. Während der Studie der postoperativen Billigung wurden zusätzlich 10 neurologische
Vorkommnisse bei 10 Patienten aufgezeichnet. Diese Vorkommnisse sind: 1 Radikulopathie, 2 zervikalen Radikulopathien, 1 mit Taubheit und Kribbelgefühle in den Händen, 1 mit Taubheit
oder Wärmegefühl in den Beinen, 3 Schmerzen im Rücken und/oder im Bein, und 2 Schmerzen im Rücken und/oder im Bein mit anderen Symptomen. Die Tafel mit den nachteiligen
Wirkungen zeigt ausserdem nachteilige Wirkungen wie Schmerzen in Bein- und Rücken und/oder im Bereich des Rückgrats, wie z.B. eine Senkung der Bandscheiben, welche, in manchen
Fällen, einen neurologischen Ursprungs waren.
Einige dieser nachteiligen Wirkungen führten zu einer zusätzlichen chirurgischen Intervention, die auf die klinische Versuchschirurgie folgten. Diese zusätzlichen chirurgischen Interventionen
können als Korrektur, Entfernung, zusätzliche Befestigung oder neue Operation klassiert werden
INTER FIX™ Instrumentationsgruppe (randomisiert und nicht-randomisiert kombiniert), bei denjenigen der Kontrullgruppe während der IDE-Studie und bei denjenigen der INTER FIX™
Instrumentationsgruppe während der Studie der postoperativen Billigung zusammen.
1
Korrektur : Eine Operation, bei der die originale Implantatkonfiguration zurechtgerückt oder auf die eine oder andere Art modifiziert wird.
Entfernung : Eine Operation, bei der ein oder mehrere Komponenten der originalen Implantatkonfiguration entfernt werden, ohne dass sie durch dieselbe Art von Probeinstrumentation
ersetzt werden.
Zusätzliche Befestigung : Eine Operation, bei der zusätzliche spinale Instrumentationen, welche nicht als Teile der ursprünglich geplanten Konfiguration anerkannt sind, platziert werden.
Neue Operation : Alle chirurgischen Operationen, bei denen die originalen Implantatkomponenten nicht entfernt oder geändert werden, oder wo keine zusätzlichen Komponenten
1
. Die Tafel II fasst die zusätzlichen chirurgischen Interventionen bei den Patienten der
Page 15

IDE-Studie Studie postoperative Billigung
24 Monats
Korrektur 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Entfernung 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Zusätzliche Befestigung 21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
Neue Operation 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
INTER FIX™
Instrumentation
Tafel II – Zusätzliche chirurgischen Interventionen
Zeitpunkt
Kontrolle
N=185
N=62
48 Monats
1
Zeitpunkt
INTER FIX™
Instrumentation
N=116
72 Monats
Zeitpunkt
INTER FIX™
Instrumentation
N=109
eingesetzt werden.
1 - Für die Patienten der Kontrollgruppe liegen für die Periode über 72 Monate keine Daten vor, da nur eine Überwachung von 24 Monaten für sie vorgesehen war.
MÖGLICHE NACHTEILIGE WIRKUNGEN:
Die folgende Liste zählt die möglichen nachteiligen Wirkungen auf, die im Zusammenhang mit der spinalen Fusionschirurgie mit der INTER FIX™ Gewinde-Instrumentation für Fusionen oder
der INTER FIX™ RP Gewinde-Instrumentation für Fusionen erscheinen können. Einige dieser nachteiligen Wirkungen können schon auf der vorherigen Tafel der nachteiligen Wirkungen
aufgezeichnet worden sein.
• Dislokation, Verbiegung, Brechen, Lockerung und/oder Migration der Komponenten.
• Reaktion (allergisch) gegen Fremdkörper.
• Weichteil- oder Nervenbeschädigung.
• Auf die Operation folgende Veränderung der Wirbelsäulenkrümmung, Verlust der Korrektur, der Höhe und/oder der Reposition der Wirbelsäule.
• Infektion.
• Duraleinriss.
• Störung des neurologischen Systems.
• Störung des urologischen Systems.
• Narbenbildung.
• Knochenfraktur.
• Keine knöcherne Konsolidierung (oder Pseudoarthrose), verzögerte Konsolidierung, Fehlstellung.
• Stop des potentiellen Wachstums des operierten Teils der Wirbelsäule. Verlust der spinalen Mobilität oder Funktion.
• Standortkomplikationen mit dem Knochenmaterialspender.
• Verletzung der Blutgefässe und Störung des kardiovaskulären Systems.
• Komplikationen des gastrointestinalen Systems.
• Störung des reproduktiven Systems.
• Verletzung an den inneren Organen und den verbindenden Weichteilen.
• Entwicklung von Atemproblemen.
• Inzisionskomplikationen.
• Veränderung der mentalen Lage.
• Tod.
Anmerkung: Zusätzliche chirurgische Eingriffe können nötig werden, um einige von diesen möglichen nachteiligen Wirkungen zu korrigieren.
KLINISCHE ERGEBNISSE :
Eine Multi-center klinische vergleichenden2 Versuchsreihe der INTER FIX™ Gewinde-Instrumentation für Fusionen wurde in den Vereinigten Staaten von Amerika durchgeführt, die die vordere
spinale Anwendung der INTER FIX™ Instrumentation, gefüllt mit autologen Knochen, vergleicht mit einer Kontrollgruppe, behandelt mit autologe Anlagerung des femoralen Rings, gefüllt mit
autologen Knochen bei der Behandlung von Patienten mit symptomatischer degenerativer Diskopathie. Ursprünglich wurde diese klinische Versuchsreihe als prospektiv, randomisiert und
kontrolliert geplant. Später wurde der ermittelnde Plan revidiert, um den Patienten zu erlauben, in die nicht-randomisierte Gruppe einzutreten, d.h. Patienten, die nur mit der INTER FIX™
Instrumentation behandelt wurden.
Die Kriterien für die Aufnahme in die klinische Versuchsreihe waren die folgenden: symptomatische degenerative Diskopathie, diagnostiziert durch hartnäckige Bein und/oder Rückenschmerzen,
bestätigt in der Röntgenologie; spinale Instabilität, wie definiert als eine höhere Abweichung als 4mm, oder grössere als 5° Winkelung auf Flexion/Extension-Röntgenbildern; Auftreten der
Symptome auf nur einer Höhe von L2-S1; und Spondylolisthese nicht über Grad I. Speziell aus dieser klinischen Studie ausgeschlossen waren Patienten mit: einer vorausgegangenen vorderen
interkörperlichen Fusionsoperation auf der betroffenen spinalen Höhe, Osteopenie, Osteoporose oder Osteomalazie oder notwendiger Stimulation des Knochenwachstums.
Eine Gesamtheit von 185 Patienten wurden einbezogen in die ermittelnde INTER FIX™ Instrumentation Behandlungsgruppe.
randomisierten Gruppe der klinischen Versuchsreihe aufgenommen.
Über die INTER FIX™ Gewinde-Instrumentation für Fusionen wurde eine Studie der postoperativen Billigung verlangt, um die Langzeitfunktion dieser Instrumentation einzuschätzen, hierzu
wurden die Ergebnisse von mindestens 100 Patienten nach 6 Jahren nach der Operation benötigt. Die Langzeitfunktion der Instrumentation wurde bei 116 Patienten bei 4 Jahren nach der
Operation und bei 109 Patienten bei 6 Jahren nach der Operation ausgewertet. Die Patientengruppe wurde von den Teilnehmern and den IDE klinischen Versuch gebildet. Basiert auf die
Kriterien der IDE Studie, enthielt die durch diese Patienten erhaltene Information eine Beschreibung von allen zusätzlichen chirurgischen Operationen, eine radiographische Beurteilung von
Fusionen und eine Beurteilung der Schmerzen und der Funktion.
Die Tafel III zeigt die klinischen Ergebnisse dieser klinischen Studie, beschreibt die Prozentsätze des Fusionserfolgs, die Erfolge bei Schmerzen nach Oswestry, und die neurologischen
Erfolge nach 12, 24, 48 und 72 Monaten nach der chirurgischen Behandlung. Der Erfolg dieser klinischen Versuchsreihe basierte sich auf eine vergleichende Analyse zwischen der INTER
FIX™ Instrumentation und der Kontrolle.
Für diesen klinischen Versuch, war die Fusion definiert als Bestätigung: der Verbindung mit dem Trabekelknochen, der Stabilität, d.h. eine Abweichung unter oder gleich 3mm und ein
Bewegungswinkel unter 5°, sowohl als Radiotransparenzen unter 50% um jedes Implantat herum. Auch wurden die Patienten mit einer zusätzlichen Operation, zurückzuführen auf
Nicht-Konsolidierung in den Berechnungen als gescheiterte Fusionen gezählt. Was die Unfähigkeit/Schmerz nach Oswestry betrifft, so wurde der Erfolg als erreicht betrachtet, wenn
das postoperative Ergebnis sich um mindestens 15 Punkte verbessert hatte im Vergleich zum preoperativen Ergebnis. Erfolg in neurologischer Hinsicht war definiert als postoperative
Beibehaltung oder Verbesserung in wenigstens 3 der 4 neurologischen Kategorien (Bewegung, Gefühl, Reflexe und Heben des gestreckten Beins) im Vergleich zum preoperativen Zustand.
Der vollkommene Erfolg für einen Patienten war als erreicht betrachtet: wenn es bewiesen war, dass Fusion erreicht worden war, wenn der Erfolg, die Unfähigkeit/Schmerz nach Oswestry
betreffend, erreicht worden war, wenn man den neurologischen Zustand betreffend, einen Erfolg erreicht hatte, und wenn der Patient keiner zusätzlichen Operation, die als “Misserfolg”
klassifiziert war, unterzogen werden musste.
Fusion
Verbesserung von
Unfähigkeit/Schmerz
nach Oswestry
Verbesserung von
mindestens 15 Punkten
im Vergleich zum
preoperativen Ergebnis
Behalten oder
Verbesserung des
Neurologischen
Zustands
Allgemeiner Erfolg
2
INTER FIX™ Instrumentation statistisch nicht schlechter als die Kontrolle.
3
Enthält nicht 3 Patienten, die keine INTER FIX™ Instrumentation empfangen haben, da chirurgische Gegenwirkungen auftraten.
Ermittlungsmässig Kontrolle
96.1%
(122/127)
53.5%
(76/142)
99.3%
(150/151)
42.4%
(61/144)
Tafel III – E rgebnisse der klinischen Studie über die INTER FIX™ Instrumentation
12 Monats-Prozentsatz 24 Monats-Prozentsatz
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
(12/56)
Delta
1
)
(ǻ
Ermittlungsmässig Kontrolle
-0.097 46.1%
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
(71/154)
(27/52)
(24/46)
(43/45)
(17/57)
51.9%
52.2%
95.6%
29.8%
Delta
(ǻ1)
-0.043 49.5%
48 MonatsProzentsatz
Ermittlungsmässig Ermittlungsmässig
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0% (102/103) 97.6% (82/84)
(53/107)
3
Eine Gesamtheit von 62 Kontrollpatienten wurden in der
72 Monats-
Prozentsatz
56.3%
(54/96)
Die Prozentsätze über einen absoluten Erfolg bei 12 und 24 Monaten nach der Operation kann man aus der Tafel III entnehmen.
1-Verlangte minimale Abweichung, um die Nicht-Unterlegenheit der Testinstrumentation im Vergleich zu der Kontrollinstrumentation zu erklären.
Alle an dieser klinischen Versuchsreihe mit der INTER-FIX™ Gewinde-Instrumentation für Fusionen beteiligten Patienten, egal in welcher Gruppe, wurden unter denselben Einschliessungs-/
Ausschliessungskriterien einbezogen. Um die Vergleichbarkeit der randomisierten Patientsgruppe der INTER FIX™ Instrumentation und der nicht-randomisierten Patientsgruppe der INTER
FIX™ Instrumentation, wurden die demographischen Eigenschaften, preoperativen medizinischen Bedingungen, die in der Röntgenologie erhaltene Diagnose, die für eine degenerative
Diskopathie charakteristisch ist, die operative Annäherung und die Lage des lumbalen behandelten Bereichs, geprüft. Die Patientenbevölkerung war statistisch vergleichbar, ausser für die
operative Annäherung, der preoperative Arbeitsstatus des Patienten und die preoperative diagnostische Bildbestätigung von Osteophytenbildung an der vertebralen Endplatte; die Vernarbung
und/oder die Verdickung des Anulus Fibrosus, Ligamentum Flavum und/oder der Fazettengelenkkapsel, sowie Hernien des Nucleus Pulposus. Weitere statistische Analysen zeigten, dass
diese Variabeln nicht mit den klinischen Ergebnissen der erreichten Fusionen während eines Zeitraums von 12 Monaten, verbunden waren, die Verbesserung von Pein/Behinderung nach
Oswestry, des neurologischen Zustands oder den allgemeinen Erfolg, mit Ausnahme von Hernien des Nucleus Pulposus. Mehr Patienten mit Hernien des Nucleus Pulposus wurden in der
nicht-randomisierten Gruppe gefunden, was als Wahrscheinlichkeit, den allgemeinen Erfolgs zu erhöhen, notiert wurde.
Page 16

Eine “ Behandlung-vorgesehen ” Analyse wurde durchgeführt. Für diese Analyse wurden die zusätzlichen Operationen klassifiziert als Misserfolg, Tod, Patienten, die verloren-für-die-Überwachung
waren, und die aus anderen Gründen nicht beobachtet werden konnten, als fehlende Beobachtungen betrachtet für die Variablen; sie wurden in den Denominator der kalkulierten Prozentsätze
eingeschlossen, d.h. sie wurden als “Misserfolg ” betrachtet. Da diese Patienten als gescheiterte Fälle betrachtet worden sind, waren die bei der “Behandlung-vorgesehen” Analyse erhaltenen
Prozentsätzen die klinischen Ergebnisse betreffend, deutlich niedriger im Vergleich zu den bei der aktuellen Analyse erhaltenen Prozentsätzen.
Tod, zusätzliche Operationen klassifiziert als Misserfolg, Patienten verloren-für-die-Überwachung und fehlende
Beobachtungen wurden als gescheiterte Fälle betrachtet und wurden in den Denominator der Prozentsätze eingeschlossen.
12 Monats-
Fusion
Verbesserung von Unfähigkeit/Schmerz nach
Oswestry
Verbesserung von mindestens 15 Punkten im
Vergleich zum preoperativen Ergebnis
Behalten oder Verbesserung des
Neurologischen Zustands
Allgemeiner Erfolg
Gescheiterte zusätzliche Operationen
Keine Konsolidierung
2
Ander e
Tod
Die folgende Tafel (Tafel IV) liefert die Ergebnisse der “ Behandlung-vorgesehen ” Analyse für die Patienten der INTER FIX™ Instrumentation, schliesst aber die Ergebnisse der Patienten
der Kontrollgruppe nicht ein.
1 - Diese Patienten wurden in die Berechnung der Prozentsätze die Fusion betreffend, eingeschlossen, sie wurden jedoch als gescheiterte Fälle betrachtet im Verhältnis zu den Objektiven
des klinischen Versuchs.
2 - Patienten, die während dieser Zeit nicht überwacht werden konnten und die sich zusätzlichen Operationen unterziehen mussten aus anderen Gründen als Nicht-Konsolidierung, wurden
als gescheiterte Fälle betrachtet im Verhältnis zu den Objektiven des klinischen Versuchs.
VERPACKUNG:
Die Verpackung jedes dieser Komponenten sollte bei Erhalt intakt sein. Wenn ein Leih - oder Konsignationsset benutzt wird, sollte dieses sorgfältig auf Vollständigkeit geprüft werden und
alle Komponenten und die Instrumente sollten vor Gebrauch auf Anzeichen von Beschädigungen sorgfältig untersucht werden. Beschädigte Produkte oder Produkte, die sich in beschädigten
Verpackungen befinden, sollten nicht benutzt werden und zu Medtronic Sofamor Danek zurückgeschickt werden.
REINIGUNG UND DEKONTAMINATION:
Wenn sie nicht steril geliefert werden, müssen alle Implantate und Instrumente vor der Sterilisation und bevor sie in ein steriles chirurgisches Umfeld gebracht werden, zuerst gereinigt werden
nach den vom Krankenhaus gebräuchlichen Methoden. Gebrauchte Instrumente müssen dekontaminiert, gereinigt und sterilisiert werden vor dem erneuten Gebrauch.
Tafel IV - "Behandlung-vorgesehen" Analyse für die INTER FIX™ Instrumentation
Prozentsatz
65.9% (122/185) 67.6%
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
33.0% (61/185) 38.4% (71/185) 45.7%
1
5
8
0 1 0 0
24 Monats-
Prozentsatz
(125/185)
(128/185)
5
3
48 Monats-
Prozentsatz
80.2%
(93/116)
(66/116)
87.9%
(102/116)
(53 /116)
4
4
72 Monats-
Prozentsatz
77.1%
( 84/109)
56.0%
(61 /109)
75.2%
75.2% (82/109)
(82/109)
49.5%
(54/109)
1
2
Anmerkung: Keine Reinigungslösungen benutzen, die Bleichmittel oder Formol enthalten, da diese das Implantat beschädigen könnten.
Alle Produkte sollten sorgfältig behandelt werden. Ein unrichtiger Gebrauch oder unrichtige Handhabung könnte das Implantat beschädigen und verhindern, dass es richtig funktioniert.
STERILISIERUNG:
Ausser, wenn sie als “steril” beschrieben und klar als solche in einer ungeöffneten und sterilen, von der Firma gelieferten, Verpackung ausgezeichnet sind, müssen alle Implantate und Instrumente,
die bei Operationen benutzt werden, vor dem Gebrauch vom Krankenhaus sterilisiert werden. Vor der Sterilisation alles Verpackungsmaterial entfernen. Für einen 10-6 Sterilisationsgarantiebereich
wird es empfohlen, dass diese Produkte von dem Krankenhaus dampfsterilisiert werden, unter Anwendung einer der drei folgenden Parameter.
METHODE ZYKLUS TEMPERATUR DAUERZEIT
Dampf Vorabsaugung 270°F (132°C) 4 Minuten
Dampf Unter Druck 250°F (121°C) 60 Minuten
Dampf* Unter Druck* 273°F (134°C)* 20 Minuten*
ANMERKUNG: Aus Gründen der zahlreichen Variabeln, die in die Sterilisation einbezogen sind, sollten alle medizinischen Dienste den Sterilisierungsablauf, der für ihre Ausrüstungen
angewandt sind, festlegen und kontrollieren (z.B. die Temperaturen, Dauer).
* Wenn sie außerhalb der Vereinigten Staaten gebraucht wird, empfehlen einige nicht amerikanische Gesundheitsämter die Sterilisierung nach diesen Parametern, um so das potentielle Risiko der
Übertragung der Creutzfeldt-Jakob Krankheit zu minimisieren, besonders das Sterilisieren der chirurgischen Instrumente, die in Kontakt kommen könnten mit dem zentralen Nervensystem.
BEI UNZUFRIEDENHEIT MIT DEM PRODUKT:
Alle Personen, die beruflich im Gesundheitswesen beschäftigt sind, (z.B. Kunden oder Benutzer unserer Produkte), die irgend eine Klage haben, oder die unzufrieden waren während des Gebrauchs
des Produkts mit dessen Qualität, Ausführung, Haltbarkeit, Anpassung, Sicherheit, Wirksamkeit und/oder Leistung, sollten dieses ihrer Verteiler, Medtronic Sofamor Danek benachrichtigen. Weiter,
wenn einer der implantierten Komponenten der INTER FIX™ Gewinde-Instrumentation für Fusionen oder der INTER FIX™ RP Gewinde-Instrumentation für Fusionen “schlecht funktioniert”,
(z.B. liefert nicht ihre Leistungsspezifikationen oder funktioniert andererweise nicht, wie vorgesehen), oder wird verdächtigt, so zu tun, sofort mit dem Lieferanten Kontakt aufnehmen. Wenn
immer ein Medtronic Sofamor Danek Produkt “schlecht funktioniert” und möglicherweise den Tod oder schwere Verletzungen eines Patienten verursacht oder dazu beigetragen hat, sofort mit
dem Lieferanten telephonisch, per Fax oder per Post Kontakt aufnehmen. Wenn ein Klageprotokoll aufgenommen wird, bitte Name und Nummer des Komponenten, die Warenpostennr.(-n)
des Komponenten, Ihren Namen und Adresse und den Grund der Klage angeben. Bitte mitteilen, ob ein schriftlicher Bericht von der Lieferfirma gewünscht wird.
ANMERKUNG FÜR DEN ARZT: Obwohl der Arzt der fachkundige Mittelsmann zwischen Hersteller und Patient ist, müssen die wichtigen medizinischen Informationen, die in diesem Dokument
enthalten sind, an den Patienten weitergegeben werden.
Gilt nur für Leser in den USA
VORSICHT: NACH AMERIKANISCHEM BUNDESGESETZ DÜRFEN DIESE IMPLANTATE NUR DURCH EINEN ARZT ODER AUF ANWEISUNG EINES ARZTES VERKAUFT
WERDEN.
ENTFERNUNG DER INSTRUMENTATION:
Sollte es notwendig werden, eine INTER FIX™ Gewinde-Instrumentation für Fusionen oder eine INTER FIX™ RP Gewinde-Instrumentation für Fusionen zu entfernen, bitte Medtronic
Sofamor Danek anrufen.
© 2006 Medtronic Sofamor Danek. Alle Rechte vorbehalten.
Page 17

0381030 Rev. A
,
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
ITALIANO
Il presente documento contiene informazioni mediche importanti sul dispositivo filettato per la fusione INTER FIX™ e sul dispositivo filettato per la fusione INTER FIX™ RP.
DESCRIZIONE:
Il dispositivo filettato per la fusione INTER FIX™ è composto di un cilindro metallico, cavo e perforato nonchè di un tappo. Il dispositivo filettato per la fusione INTER FIX™ RP è composto di
un cilindro metallico, cavo e perforato con una scanalatura ampia, arrotondata ed esterna lungo l’intero asse longitudinale che si estende nel diametro interno del dispositivo. Le due estremità
dell’impianto INTER FIX™ RP sono chiuse.
Il diametro degli impianti INTER FIX™ ed INTER FIX™ RP può variare da 12mm a 24mm e la lunghezza da 20mm a 29mm. La dimensione dei tappi degli impianti INTER FIX™ corrisponde
al diametro dei cilindri e sono collocati all’estremità aperta del cilindro dopo che siano stati riempiti con trapianto osseo autogeno.
Gli impianti INTER FIX™ ed INTER FIX™ RP sono fabbricati con una lega di titanio speciale per impianti (Ti-6AI-4V) descritto da norme quali ASTM F136.
Ogni garanzia implicita di commerciabilità e di idoneità per uno scopo o un uso particolare è spacificatamente esclusa. Riferirsi al listino prezzi oppure al catalogo Medtronic Sofamor Danek
per maggiori informazioni sulle garanzie e le limitazioni di responsabilità.
INDICAZIONI:
Il dispositivo filettato per la fusione INTER FIX™ ed il dispositivo filettato per la fusione INTER FIX™ RP sono indicati per gli interventi chirurgici di fusione del rachide in pazienti il cui scheletro
è sviluppato, che presentano una discopatia degenerativa (DDD) ad uno dei livelli rachidi da L2 a S1. La discopatia degenerativa (DDD) è diagnosticata dalla presenza di dolori della schiena
di origine discogenica con degenerazione del disco, confermato dell’anamnesi del paziente e da un controllo radiografico. Questi pazienti che presentano una discopatia degenerativa (DDD)
possono anche avere spondilolistesi o retrolistesi che può raggiungere un grado I al livello interessato del rachide. I dispositivi INTER FIX™ ed INTER FIX™ RP devono essere utilizzati con
trapianto osseo autogeno ed impiantati mediante un intervento aperto per via anteriore.
CONTROINDICAZIONI:
Non impiantare il dispositivo filettato per la fusione INTER FIX™, né il dispositivo filettato per la fusione INTER FIX™ RP in pazienti che presentano un’infezione attiva della sede operatoria
o un’allergia al titanio o alla lega di titanio.
AVVERTENZE:
• Il dispositivo filettato per la fusione INTER FIX™ ed il dispositivo filettato per la fusione INTER FIX™ RP devono essere impiantati soltanto da chirurghi con un’esperienza nel campo
della chirurgia di fusione del rachide e che hanno seguito la formazione adatta a questi dispositivi.
• Se l’esperienza e/o la formazione del chirurgo non sono soddisfacenti, questo potrebbe aumentare il rischio che sopravvengano effetti dannosi, quali lesioni vascolari, lesioni
neurologiche e/o problemi del sistema urogenitale (ivi compreso eiaculazione retrograda).
PRECAUZIONI:
• La sicurezza e l’efficacia del dispositivo filettato per la fusione INTER FIX™ e quelle del dispositivo filettato per la fusione INTER FIX™ RP non sono state dimostrate nei pazienti che
fanno parte dei casi seguenti:
- in caso di spondilolistesi o retrolistesi di grado II o superiore,
- quando è necessario fusionare parecchi livelli,
- quando un intervento chirurgico di fusione precedente intercorporale richiede una correzione,
- quando sarà necessario amministrare antiinfiammatorie non steroidee o steroidee durante il periodo postoperatorio,
- quando l’obesità è palese
INFORMAZIONI MEDICHE IMPORTANTI : IL DISPOSITIVO FILETTATO PER LA FUSIONE INTER FIX™
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
Page 18

PRECAUZIONI:
™
O
L
O
C
L
C
L
L
• La sicurezza e l’efficacia del dispositivo filettato per la fusione INTER FIX™ e quelle del dispositivo filettato per la fusione INTER FIX™ RP non sono state dimostrate nei pazienti che
fanno parte dei casi seguenti:
- in caso di spondilolistesi o retrolistesi di grado II o superiore,
- quando è necessario fusionare parecchi livelli,
- quando un intervento chirurgico di fusione precedente intercorporale richiede una correzione,
- quando sarà necessario amministrare antiinfiammatorie non steroidee o steroidee durante il periodo postoperatorio,
- quando l’obesità è palese,
- quando l’età del paziente è inferiore a 18 anni o superiore a 65 anni,
- in caso di osteoporosi, osteopenia e/o osteomalacia,
- in caso di gravidanza.
• La sicurezza e l’efficacia del dispositivo filettato per la fusione INTER FIX™ e quelle del dispositivo filettato per la fusione INTER FIX™ RP sono state dimostrate soltanto negli
interventi chirurgici di fusione intercorporale nel rachide lombare per via anteriore (ALIF) che utilizzano un trapianto osseo autologo.
• I pazienti ai quali verrà impiantato un dispositivo filettato per la fusione INTER FIX™ dovrebbero avere almeno sei mesi di trattamento non operatorio prima dell’interveno. I pazienti ai
quali verrà impiantato un dispositivo filettato per la fusione INTER FIX™ RP dovrebbero anche avere almeno sei mesi di trattamento non operatorio prima dell’intervento.
• Impiantare due dispositivi filettati per la fusione INTER FIX™ l’uno vicino all’altro al livello dell’intervento. Impiantare un dispositivo filettato per la fusione INTER FIX™ ed un dispositivo
filettato per la fusione INTER FIX™ RP l’uno vicino all’altro al livello dell’intervento. La scanalatura longitudinale del dispositivo INTER FIX™ RP agevolerà l’installazione la più vicina
possibile di due dispositivi INTER FIX™, verrà così ottenuto un profilo laterale ridotto.
• L’asse longitudinale del dispositivo filettato per la fusione INTER FIX™ nonchè quello del dispositivo filettato per la fusione INTER FIX™ RP devono essere nella direzione anterioreposteriore.
• Gli impianti e strumenti devono essere sterilizzati prima di essere utilizzati, in conformità con le procedure di sterilizzazione fornite nel presente foglietto, se non sono forniti sterili e
chiaramente etichettati in quanto tale.
EFFETTI DANNOSI:
Gli effetti dannosi, presentati nella tabella I, sono quelli che sono stati riportati all’atto dell’impianto del dispositivo INTER FIX™ su 185 pazienti, ed all’atto del trattamento sui 62 pazienti del
gruppo di controllo, che hanno partecipato allo studo clinico per il dispositivo filettato per la fusione INTER FIX™. Inoltre, sono indicati nella presente tabella gli effetti dannosi che sono stati
riportati in 116 pazienti per il periodo di 48 mesi e in 109 pazienti per il periodo di 72 mesi, all’atto dello studio post-approvazione. Le percentuali indicate sono state calcolate a partire dal
numero totale di pazienti che hanno partecipato allo studio, per ogni intervallo di tempo pre-stabilito. Gli effetti dannosi che succedono nei gruppi, ripartiti in modo aleatorio o non aleatorio,
trattati con il dispositivo INTER FIX™, sono stati raggruppati in modo da presentare un percentuale globale.
Durante
l'intervento
(Numero %)
CONTR
Complicazione
Vascolare Intraop. 15(8.1) 2(3.2) 15 (8.1) 2(3.2)
Dolore sacroiliaco 5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
Neurologica 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Dolore dorsale 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
Al livello
dell'incisione
Complicazione nel
rachide
Urologica 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Altre 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Altri dolori 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
Gastrointestinale 7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
Eiaculazione
retrograda
Respiratoria 3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
Dolore della gamba 2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
Trauma 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
Peritoneale 3(1.6) 3(1.6) 0
Vascolare postop. 2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
Frattura ossea 1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
Spostamento /
Allentamento
dell'impianto
Dolore della sede
trapiantata
Nessun
consolidamento
Collasso 2(1.1) 2(1.1) 0
Nessun
consolidamento
(RISULTATO IN
CORSO)
Meningite 1(1.6) 0 1(1.6)
Rottura
dell'impianto
Cancro 1
Morte 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
NTER FIX
N=185
LLO
N=62
11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
2(1.1) 3(1.6) 1(0.5) 6(3.2)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
1(1.6)
5(8.1) 0 5(8.1)
Dopo
l'intervento
(1 giorno -<2
mesi)
[Numero (%)]
INTER
FIX™
N=185
CONTRO
LO
N=62
3 mesi
(t
2-<5 mesi)
[Numero (%)]
INTER
FIX™
N=185
TABELLA I – EFFETTI DANNOSI
6 mesi
(t5-<9 mesi)
[Numero (%)]
CONTR
INTER
LLO
FIX™
N=62
N=185
12 mesi
(t
9-<19 mesi)
[Numero (%)]
INTER
ONTRO
LO
N=62
2
0 1(1.6) 2(1.7)
FIX™
N=185
(0.5)
ONTRO
LO
N=62
1
24 mesi
(t19-<30 mesi)
[Numero (%)]
INTER
CONTRO
FIX™
N=185
(0.5)
TOTALE EFFETTI
DANNOSI
(DURANTE 24 MESI)
™
INTER FIX
N=185
(7.4)
1
CONTROLL
N=62
LO
N=62
2 (1.1) 1(0.8) 1(0.9)
48 mesi
(>30 - < 60
60
30<
(t
mesi)
INTER FIX™
O
N=116
0
72 mesi
(t60
mesi)
INTER FIX
N=109
1 – Durante il periodo di 24 mesi, sei (6) casi di eiaculazione retrograda sono stati osservati fra i 81 pazienti maschi appartenenti al gruppo del dispositivo INTER FIX™ (7,4%).
2 – Questo paziente (175) è stato riportato ulteriormente poichè la fusione è stata ottenuta entro 52 mesi dall’operazione e ciò, senza dover ricorrere ad un altro intervento chirurgico o ad
ogni altro trattamento.
I due effetti dannosi più seri osservati all’atto di questa prova clinica sono stati le lesioni vascolari e lesioni neurologiche successe durante l’intervento. Un totale di 15 lesioni vascolari sono stati
rilevate durante l’intervento in 14 pazienti appartenenti al gruppo del dispositivo INTER FIX™ durante il periodo di 24 mesi. Questi lesioni sono : 2 lesioni della vena cava, 9 lesioni della vena
iliaca, 1 lacerazióne della vena ipogastrica, 1 emorragia di un segmento della vena, 1 lesione della vena sacrale, ed 1 emorragia superficiale. Per quanto riguarda il gruppo di controllo, un totale
di 2 lesioni vascolari sono stati rilevate durante l’intervento in 2 pazienti durante lo stesso periodo. Questi lesioni sono : 1 lesione della vena iliaca ed un’emorragia sulla base dell’ osso.
Un totale di 30 casi neurologici sono stati osservati in 28 pazienti appartenenti al gruppo del dispositivo INTER FIX™ durante il periodo di 24 mesi. Questi casi sono: 1 piede cadente, 3 lesioni
alle radici dei nervi, 1 stenosi foraminale, 1 distrofia del riflesso simpatico, 7 intorpidimenti o calore delle gambe, 2 disestesie, 3 parestesie, 2 radicoliti, 7 dolori della schiena e/o della gamba,
1 dolore della schiena e/o della gamba con altri sintomi, 1 dolore folgorante nella parte inferiore della schiena e 1 sindrome del canale carpale.
Un totale di 10 casi neurologici sono stati osservati in 8 pazienti appartenenti al gruppo di controllo durante lo stesso periodo. Questi casi sono: 1 radicolopatia con formicolio delle estremità,
1 dolore della schiena cronico con radicoliti, 1 paziente che presenta sintomi di indebolimento generale, 2 dolori della schiena o della gamba con altri sintomi, 1 muscolo grande abduttore
denervato e 4 con intorpidimento o calore delle gambe. Durante lo studio post-approvazione, 10 casi neurologici supplementari sono stati osservati in 10 pazienti. Questi casi sono: 1
radicolopatia, 2 radicolopatie cervicali, 1 intorpidimento e formicolii nelle mani, 1 intorpidimento o calore delle gambe, 3 dolori della schiena e/o della gamba e 2 dolori della schiena e/o della
gamba con altri sintomi. La tabella degli effetti dannosi ci mostra anche effetti dannosi quali dolori della schiena e della gamba e/o effetti dannosi con un legame con il rachide, quali collasso
del disco, che, in alcuni esempi, ha un legame neurologico.
Alcuni degli effetti dannosi hanno condotto ad un intervento chirurgico supplementare dopo la chirurgia realizzata per la prova clinica. Questi interventi chirurgici supplementari possono
essere classificati in quanto correzione, rimozione, fissaggio supplementare o ri-intervento.1 La tabella II raggruppa gli interventi chirurgici supplementari effettuati nei pazienti del gruppo del
dispositivo INTER FIX™, (raggruppati in modo aleatorio o non aleatorio), in quelli del gruppo di controllo durante lo studio IDE ed in quelli del gruppo del dispositivo INTER FIX™ durante
lo studio post-approvazione.
1
Correzione : Intervento durante il quale l’aggiustamento o qualsiasi modifica della configurazione iniziale dell’impianto viene realizzato.
Rimozione : Intervento durante il quale uno o più componenti della configurazione iniziale dell’impianto sono estratti, senza essere sostituiti con lo stesso tipo di dispositivo utilizzato
per lo studio.
Fissaggio supplementare : Intervento nel quale vengono impiantati dispositivi spinali supplementari non facendo parte della configurazione inizialmente prevista.
Ri-intervento : Intervento chirurgico nel quale nessun componente dell’impianto originale viene rimosso, modificato oppure aggiunto.
™
Page 19

g
Tabella II – Interventi chirurgici supplementari
Studio IDE Studio post-approvazione
Correzione 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Rimozione 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Fissaggio
supplementare
Ri-intervento 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
Intervallo di tempo
prestabilito
Dispositivo
INTER FIX™
N=185
24 mesi
Controllo
N=62
1
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
Intervallo di tempo
prestabilito
48 mesi
Dispositivo
INTER FIX™
N=116
Intervallo di tempo
prestabilito
72 mesi
Dispositivo
INTER FIX™
N=109
1-Nessun dato è disponibile per il periodo di 72 mesi per i pazienti del gruppo di controllo, perchè per questi pazienti, era previsto soltanto un controllo durante 24 mesi.
EFFETTI DANNOSI POTENZIALI:
Segue una lista degli effetti dannosi potenziali che potrebbero succedere all’atto della chirurgia di fusione del rachide quando il dispositivo filettato per la fusione INTER FIX™ o il dispositivo
filettato per la fusione INTER FIX™ RP viene utilizzato. Alcuni di questi effetti dannosi possono essere stati previamente indicati nella tavola degli effetti dannosi.
• Disassemblaggio, piegatura, rottura, allentamento e/o migrazione dei componenti.
• Reazione (allergica) da corpo estraneo.
• Danni ai tessuti o ai nervi.
• Cambiamento postoperatorio della curvatura del rachide, perdita di correzione, altezza e/o riduzione del rachide.
• Infezione.
• Lacerazione durale.
• Disturbo del sistema neurologico.
• Disturbo del sistema urologico.
• Formazione di cicatrice.
• Frattura ossea.
• Nessun consolidamento osseo (o pseudoartrosi), consolidamento ritardato, consolidamento scorretto.
• Interruzione di crescita potenziale del segmento operato del rachide. Perdita di mobilità o di funzione del rachide.
• Complicazioni dell’area di prelievo del trapianto.
• Lesione ai vasi sanguigni e disturbo del sistema cardiovascolare.
• Complicazioni del sistema gastrointestinale.
• Disturbo dell’apparato riproduttivo.
• Lesione agli organi interni ed al tessuto connettivo.
• Sviluppo di problemi respiratori.
• Complicazioni legate all’incisione.
• Cambiamento di stato mentale.
• Morte.
Nota: Un intervento chirurgico supplementare può rivelarsi necessario per correggere alcuni di questi effetti dannosi potenziali.
RISULTATI CLINICI :
Una prova clinica comparativa2 multicentri sul dispositivo filettato per la fusione INTER FIX™ fù condotta negli Stati Uniti paragonando il dispositivo INTER FIX™ quando è utilizzato nel rachide
anteriore e riempito con un osso autogeno ad un gruppo di controllo trattato con un omotrapianto di anello femorale riempito con osso autogeno, nel trattamento di pazienti con discopatie
degenerative sintomattiche. Inizialmente, questa prova clinica fù prevista in modo da essere prospettiva, aleatoria e controllata. In seguito, il piano di ricerca fù revisato per permettere ai
pazienti di entrare nel gruppo non aleatorio, cioè pazienti trattati con il dispositivo INTER FIX™ soltanto.
I criteri di inclusione per questa prova clinica sono stati i seguenti : una discopatia degenerativa sintomatica diagnosticata da un dolore, refrattario al trattamento, nella gamba e/o nella
schiena e confermata in radiologia; un’instabilità spinale determinata da una deviazione superiore a 4 mm o da un angolo superiore a 5°, all’atto delle radiografie di flessione/estensione ;
quando i sintomi sono stati rivelati ad un livello solo del rachide da L2 a S1; e spondilolistesi non superiore al Grado 1. Furono specificatamente esclusi dallo studio i pazienti che avevano
già subito un intervento di fusione intercorporale per via anteriore allo stesso livello del rachide; i pazienti che soffrivano di osteopenia, osteoporosi o osteomalacia ; o che necessitavano
una stimulazione della crescita ossea.
Il numero totale di pazienti appartenenti al gruppo del dispositivo INTER FIX™, iscritti nello studio clinico, era di 185.
controllo, appartenente al gruppo aleatorio, era di 62.
Per il dispositivo filettato per la fusione INTER FIX™, uno studio post-approvazione è stato richiesto per valutare le prestazioni a lungo termine di detto dispositivo, per il quale bisognava raccogliere
i risultati presso almeno 100 pazienti, 6 anni dopo l’intervento. Così, le prestazioni a lungo termine del dispositivo sono state valutate presso 116 pazienti entro 4 anni dall’intervento, e presso 109
pazienti entro 6 anni dall’intervento. La popolazione proveniva dai pazienti che hanno partecipato alla prova clinica IDE. In conformità ai criteri dello studio IDE, la descrizione degli interventi chirurgici
supplementari, il controllo della fusione in radiologia, nonchè la valutazione del dolore e della funzione, facevano parte delle informazioni che hanno potuto essere ottenute per questi pazienti.
La tabella III ci mostra i risultati di questo studio clinico ed evidenzia le percentuali di successo per quanto riguarda la fusione, il dolore secondo Oswestry, e le condizioni neurologiche, entro
12, 24, 48 e 72 mesi dall’intervento. Il successo di questa prova clinica fù classificato sulla base di un’analisi comparativa tra il dispositivo INTER FIX™ ed il controllo.
Per questa prova clinica, la fusione è stata definita come : l’unione con la trabecola ossea, la stabilità, cioè una deviazione inferiore o uguale a 3 mm ed un angolo di movimento inferiore a 5°,
nonchè nessuna radiotrasparenza attorno ad ogni impianto superiore al 50%. Inoltre, i pazienti che hanno subito un intervento chirurgico supplementare a causa dell’assenza di consolidamento
sono stati considerati, per i calcoli, fallimenti di fusione. Per quanto riguarda l’incapacità/dolore secondo Oswestry, il successo è stato considerato raggiunto quando il risultato postoperatorio è
migliorato di almeno 15 punti rispetto al risultato preoperatorio. Il successo da un punto di vista neurologico è stato definito come il mantenimento o il miglioramento postoperatorio di almeno 3
fra le 4 categorie neurologiche (il movimento, i sensi, i riflessi ed il sollevamento con gamba tesa) relativamente allo stato prima dell’intervento. Il successo totale è stato considerato raggiunto
per un paziente se : è stato dimostrato che la fusione è stata ottenuta, se il successo per quanto riguarda l’incapacità / dolore secondo Oswestry è stato raggiunto, se è stato raggiunto il
successo per quanto riguarda lo stato neurologico, e se il paziente non ha dovuto subire un intervento chirurgico supplementare, classificato come un “fallimento”.
2
Il dispositivo INTER FIX™ statisticamente non peggiore del controllo.
3
I 3 pazienti nei quali il dispositivo INTER FIX™ non è stato impiantato a causa degli effetti dannosi legati all’intervento, non sono stati ripresi.
3
Per la prova clinica, il numero totale di pazienti iscritti nel gruppo di
La tabella III ci mostra anche le percentuali per quanto riguarda il successo totale, entro 12 e 24 mesi dall’intervento.
Fusione
Miglioramento
di
invalidità/dolor
e secondo
Oswestry
Pazienti con un
miglioramento
di almeno 15
punti
relativamente
alla situazione
prima
dell'intervento
Mantenimento
o
miglioramento
dello stato
neurologico
Successo
totale
Tabella III - Risultati dello studio clinico per il dispositivo INTER FIX™
Percentuali 12 mesi Percentuali 24 mesi
Dispositivo
provato Controllo
96.1%
(122/127)
53.5%
(76/142)
99.3%
(150/151)
42.4%
(61/144)
(21/52)
(27/52)
(52/52)
(12/56)
40.4%
51.9%
100%
21.4%
Delta
Dispositivo
1
(ǻ
)
provato Controllo
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
-0.097 46.1%
(71/154)
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
(17/57)
Delta
Percentuali 48
(ǻ1)
-0.043 49.5%
mesi
Dispositivo
provato
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0% (102/103) 97.6% (82/84)
(53/107)
Percentuali 72
mesi
Dispositivo
provato
56.3%
(54/96)
1- Margine di non-inferiorità richiesto per garantire la non-inferiorità del dispositivo provato rispetto a quello del controllo
Tutti i pazienti coinvolti nella prova clinica sul dispositivo filettato per la fusione INTER FIX™, senza tener conto dei gruppi, furono arruolati secondo gli stessi criteri di inclusione/esclusioni. Per
confermare la paragonabilità tra i pazienti del gruppo aleatorio del dispositivo INTER FIX™ ed i pazienti del gruppo non aleatorio del dispositivo INTER FIX™, sono stati controllati gli elementi
uenti: le caratteristiche demografiche, le condizioni mediche prima dell’intervento, la diagnosi ottenuta in radiologia caratteristica di una discopatia degenerativa, l’approccio operatorio, ed
se
Page 20

il
posizionamento al livello del rac
hide lomb
bil
ll’i
nonchè la diagnosi radiologica preoperatoria per rivelare la formazione di osteofiti sulla piastra terminale della vertebra, cicatrizzazione e/o ristringimento dell’anello fibroso, del ligamento
giallo, e/o della capsula della faccetta articolare nonchè ernie del nucleo polposo. Altre analisi statistiche hanno dimostrato che queste variabili non erano legate ai risultati clinici ottenuti su
12 mesi, per quanto riguarda la fusione, il miglioramento incapacità/dolore secondo Oswestry, lo stato neurologico o il successo totale, eccetto le ernie del nucleo polposo. Maggiori pazienti
con ernie del nucleo polposo sono stati osservati nel gruppo non aleatorio, il che aumentava la probabilità di successo totale.
Un’analisi con intento di trattare è stata realizzata. Per quest’analisi, gli interventi chirurgici supplementari classificati come fallimenti, le morti, i pazienti che non hanno potuto essere seguiti
accuratamente, nonchè mancanze di osservazioni sono considerati mancanze di osservazioni per il calcolo delle percentuali e sono stati compresi nel denominatore delle percentuali, cioè
considerati “fallimenti”. Questi pazienti essendo stati considerati fallimenti, le percentuali del risultato clinico, ottenute per l’analisi con intento di trattare, erano notevolmente inferiori a quelli
ottenuti per l’analisi attuale.
La tabella seguente (tavola IV) ci mostra i risultati di quest’analisi con intento di trattare per i pazienti del dispositivo INTER FIX™, ma non comprende i risultati ottenuti per i pazienti del
gruppo di controllo.
Morte, interventi chirurgici supplementari classificati como fallimenti, pazienti che non hanno potuto essere suguiti
accuratamente e mancanza di osservazioni sono considerati fallimenti e sono stati compresi nel denominatore delle
Percentuali 12
Fusione
Miglioramento di invalidità/dolore secondo
Oswestry
Pazienti con un miglioramento di almeno 15
punti relativamente alla situazione prima
dell'intervento
Mantenimento o miglioramento dello stato
neurologico
Successo totale
Fallimenti di interventi chirurgici
supplementari classificati
Nessun consolidamento
2
Altr i
Morte
Tabella IV – Analisi con intento di trattare per il dispositivo INTER FIX™
1
are trattato. La popolazione era statisticamente paragona
percentuali
mesi
65.9% (122/185) 67.6%
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
33.0% (61/185) 38.4% (71/185) 45.7%
5
8
0 1 0 0
Percentuali 24
mesi
(125/185)
(128/185)
5
3
Percentuali 48
mesi
80.2%
(93/116)
(66/116)
87.9%
(102/116)
(53 /116)
4
4
75.2% (82/109)
e, eccetto per l’approccio operatorio, lo stato di lavoro del paziente prima de
Percentuali
72 mesi
77.1%
( 84/109)
56.0%
(61 /109)
75.2%
(82/109)
49.5%
(54/109)
1
2
ntervento,
1 - Questi pazienti sono stati inclusi nel calcolo delle percentuali relative alla fusione ma sono stati considerati fallimenti nello scopo dello studio clinico.
2 - I pazienti che non hanno potuto essere seguiti durante questo periodo e che hanno subito un intervento chirurgico supplementare per ragioni diverse delle mancate unioni, sono stati
considerati fallimenti nello scopo dello studio clinico.
IMBALLAGGIO:
Gli imballaggi per ognuno dei componenti devono essere intatti all’atto del collaudo. Se è stato utilizzato un sistema di prestito o di spedizione, lo stato completo degli impianti deve essere
accuratamente verificato ed occorre verificare prima dell’uso che tutti i componenti e strumenti siano esenti da segni di danno. I prodotti danneggiati o i prodotti in imballaggi danneggiati non
devono essere utilizzati e devono essere rinviati alla Medtronic Sofamor Danek.
PULITURA E DECONTAMINAZIONE:
Se non sono forniti sterili, tutti gli impianti e strumenti devono prima essere puliti utilizzando le procedure definite dall’ospedale, prima della sterilizzazione e dell’introduzione in un campo
chirurgico sterile. Gli strumenti utilizzati devono essere decontaminati, puliti e sterilizzati prima di essere riutilizzati.
Nota: Non utilizzare soluzioni di pulitura che contengono candeggina o formalina perchè potrebbero danneggiare l’impianto.
Tutti i prodotti devono essere trattati con cura. Un uso o una manipolazione scorretta potranno danneggiare l’impianto ed impedirlo di funzionare correttamente.
STERILIZZAZIONE:
Tranne se viene scritto chiaramente “sterilizzato” sull’etichetta dell’imballaggio sterilizzato ed intatto fornito dal fabbricante, tutti gli impianti e strumenti utilizzati in chirurgia devono essere
sterilizzati dall’ospedale prima dell’uso. Rimuovere tutti i materiali di imballaggio prima della sterilizzazione. Soltanto i prodotti sterili devono essere collocati nel blocco operatorio. Per raggiungere
un livello di garanzia di sterilità 10-6, è raccomandato sterilizzare questi prodotti con vapore in ospedale utilizzando uno dei tre insiemi di parametri di processo sottoindicati:
METODO CICLO TEMPERATURA DURATA DI ESPOSIZIONE
Vapore Pre-aspirazione 270° F (132° C) 4 minuti
Vapore Sotto pressione 250° F (121° C) 60 minuti
Vapore* Sotto pressione* 273° F (134° C)* 20 minuti *
NOTA: A causa delle numerose variabili coinvolte nel processo di sterilizzazione, ogni servizio medico deve valutare e verificare il processo di sterilizzazione (per es. temperature, tempi)
utilizzato per la loro attrezzatura.
* Quando viene utilizzato fuori dagli Stati Uniti d’America, alcune Autorità sanitarie non statunitensi raccomandano la sterilizzazione in conformità con questi parametri, in modo da minimizzare
il rischio potenziale di trasmissione della malattia di Creutzfeldt-Jakob, specialmente per quanto riguarda gli strumenti chirurgici che potrebbero venire in contatto con il sistema nervoso
central.
LAMENTELE RELATIVE AL PRODOTTO:
Ogni Professionale Sanitario (per esempio utente o cliente dei nostri prodotti) che desideri presentare una lamentela o che non sia stato soddisfatto relativamente alla qualità, l’identità, la
durevolezza, l’affidabilità, la sicurezza, l’efficienza e/o le prestazioni del prodotto, deve notificarlo al proprio distributore Medtronic Sofamor Danek. Inoltre, nel caso in cui un componente
impiantato del dispositivo filettato per la fusione INTER FIX™ o del dispositivo filettato per la fusione INTER FIX™ RP “non funziona correttamente” (cioè se non soddisfi le specifiche di
prestazioni o se non funzioni come desiderato), o se fosse sospettato di non funzionare correttamente, contattare il distributore immediatamente. Se un qualsiasi prodotto della Medtronic
Sofamor Danek “non funziona correttamente” e può aver causato o contribuito alla morte o ad una lesione grave di un paziente, contattare il distributore immediatamente telefonicamente, per
telefax o mediante lettera. In caso di lamentela, vogliate indicare la descrizione ed il numero del componento, il numero del lotto del componento, il vostro nome ed indirizzo e la natura della
lamentela. Vogliate indicare se desiderate ricevere una relazione scritta dal distributore.
NOTA PER IL MEDICO: Benchè il medico sia colui che possiede le conoscenze e che sia l’intermediario tra il fabbricante ed il paziente, le informazioni mediche importanti date nel presente
documento devono essere trasmesse al paziente.
Solo valido per il mercato USA
ATTENZIONE: LA LEGGE FEDERALE (U.S) LIMITA LA VENDITA DI QUESTI IMPIANTI. DEVE ESSERE REALIZZATA DA UN MEDICO O DIETRO ORDINE DA UN MEDICO.
RIMOZIONE DEL DISPOSITIVO:
Se fosse necessario rimuovere un dispositivo filettato per la fusione INTER FIX™ o un dispositivo filettato per la fusione INTER FIX™ RP, pregasi contattare la Medtronic Sofamor Danek.
© 2006 Medtronic Sofamor Danek. Ogni diritto riservato.
Page 21

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
NEDERLANDS
De onderstaande tekst bevat belangrijke medische informatie over het INTER FIX™ spondylodesesysteem met schroefdraad en het INTER FIX™ RP spondylodesesysteem met
schroefdraad.
BESCHRIJVING:
Het INTER FIX™ spondylodesesysteem met schroefdraad bestaat uit een holle, geperforeerde metalen cilinder met afsluitdop. Het INTER FIX™ RP spondylodesesysteem met schroefdraad
bestaat uit een holle, geperforeerde metalen cilinder met één brede, aan de buitenzijde afgeronde groef langs de hele lengteas, die doorloopt tot in de binnendiameter van het systeem. Beide
uiteinden van het INTER FIX™ RP implantaat zijn gesloten.
De INTER FIX™ en INTER FIX™ RP implantaten zijn verkrijgbaar in diameters variërend van 12mm tot 24mm en in lengten variërend van 20mm tot 29mm. De afmetingen van de afsluitdoppen van
de INTER FIX™ stemmen overeen met de diameter van de cilinders en worden aangebracht op het open uiteinde van de cilinders nadat deze zijn gevuld met autogeen bottransplantaat.
De INTER FIX™ en de INTER FIX™ RP implantaten zijn vervaardigd van titaanlegering (Ti-6Al-4V) van medische kwaliteit overeenkomstig de norm ASTM F136.
Impliciete waarborgen betreffende de verkoopbaarheid of de geschiktheid voor een bepaald doel of gebruik worden met name uitgesloten. Zie de Medtronic Sofamor Danek Catalogus of
prijslijst voor nadere informatie over garantievoorwaarden en aansprakelijkheidsbeperkingen.
INDICATIES:
Het INTER FIX™ spondylodesesysteem met schroefdraad en het INTER FIX™ RP spondylodesesysteem met schroefdraad zijn bestemd voor spinale artrodese bij patiënten met een volgroeid
skelet die lijden aan degeneratieve discopathie (DDD) op één niveau tussen L2-S1. Degeneratieve discopathie wordt gedefinieerd als discogene rugpijn gepaard gaande met discusdegeneratie,
bevestigd door de anamnese van de patiënt en radiografisch onderzoek. Deze degeneratieve discopathie patiënten kunnen eveneens lijden aan graad 1 spondylolisthesis of retrolisthesis op
het betrokken niveau. INTER FIX™ en INTER FIX™ RP implantaten moeten worden toegepast met autogeen bottransplantaat en worden geïmplanteerd via een open ventrale benadering.
CONTRA-INDICATIES:
Het INTER FIX™ spondylodesesysteem met schroefdraad en het INTER FIX™ RP spondylodesesysteem met schroefdraad mogen niet worden geïmplanteerd bij patiënten met een actieve
infectie op de plaats van de operatie of bij patiënten die allergisch zijn voor titaan of titaanlegering.
WAARSCHUWINGEN:
• Het INTER FIX™ spondylodesesysteem met schroefdraad en het INTER FIX™ RP spondylodesesysteem met schroefdraad mogen uitsluitend worden toegepast door chirurgen met
ervaring in spondylodesetechnieken en met voldoende training in de toepassing van dit systeem.
• Een gebrek aan voldoende ervaring en/of training kan leiden tot een hoger percentage aan voorvallen zoals vasculaire beschadigingen, neurologische voorvallen en/of urogenitale
voorvallen (waaronder teruggaande ejaculatie).
INTER FIX™ SPONDYLODESESYSTEEM MET SCHROEFDRAAD
INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
BELANGRIJKE MEDISCHE INFORMATIE
Page 22

VOORZORGSMAATREGELEN:
E
C
E
• De veiligheid en doeltreffendheid van het INTER FIX™ spondylodesesysteem met schroefdraad en het INTER FIX™ RP spondylodesesysteem met schroefdraad zijn niet bewezen bij
patiënten met een van onderstaande condities:
- spondylolisthesis of retrolisthesis van Graad II of hoger;
- noodzaak tot verstijving op meer dan één niveau;
- herziening van eerder toegepaste intercorporele spondylodesemethode(n);
- postoperatieve behoefte aan steroïde of niet-steroïde anti-inflammatoire pharmaca;
- ernstige zwaarlijvigheid;
- leeftijd onder de 18 jaar of boven de 65 jaar;
- osteoporose, osteopenie en/of osteomalacie;
- zwangerschap.
• De veiligheid en doeltreffendheid van het INTER FIX™ spondylodesesysteem met schroefdraad en het INTER FIX™ RP spondylodesesysteem met schroefdraad zijn alleen bewezen
bij ventrale lumbale intercorporele spondylodesemethoden (ALIF) die gebruikmaken van autogeen bottransplantaat.
• Patiënten bij wie het INTER FIX™ spondylodesesysteem met schroefdraad wordt toegepast moeten minstens gedurende zes maanden een niet-operatieve behandeling hebben
gehad. Patiënten bij wie het INTER FIX™ RP spondylodesesysteem met schroefdraad wordt toegepast moeten minstens gedurende zes maanden een niet-operatieve behandeling
hebben gehad.
• Op het niveau van de operatie moeten twee INTER FIX™ spondylodesesystemen met schroefdraad naast elkaar worden geïmplanteerd. Op het niveau van de operatie moeten een
INTER FIX™ spondylodesesysteem met schroefdraad en een INTER FIX™ RP spondylodesesysteem met schroefdraad naast elkaar worden geïmplanteerd. Dankzij de lengtegroef
op het INTER FIX™ RP implantaat kunnen twee implantaten gemakkelijker dichter naast elkaar worden geïmplanteerd dan mogelijk is bij twee INTER FIX™ implantaten; hierdoor
wordt het zijprofiel kleiner.
• De lengteassen van de INTER FIX™ spondylodesesysteem met schroefdraad en van de INTER FIX™ RP spondylodesesysteem met schroefdraad moeten zich in de ventraal-dorsaal
richting bevinden.
• De implantaten en instrumenten moeten vóór gebruik worden gesteriliseerd, overeenkomstig de op de bijsluiter in deze verpakking gegeven sterilisatie-instructies, tenzij zij steriel
geleverd zijn en dit duidelijk op het etiket is vermeld.
NADELIGE BIJWERKINGEN:
De in Tabel I vermelde bijwerkingen werden gerapporteerd door de 185 patiënten met een INTER FIX™ systeem en de 62 controlepatiënten die hebben deelgenomen aan het in meerdere
centra uitgevoerde klinische onderzoek betreffende het INTER FIX™ spondylodesesysteem met schroefdraad. Bovendien werden bijwerkingen gerapporteerd bij 116 patiënten na 48 maanden
en bij 109 patiënten na 72 maanden van de aan het ‘post-approval’ onderzoek deelnemende patiënten. De gepresenteerde percentages zijn gebaseerd op het totaal aantal patiënten die op elk
tijdstip in het onderzoek was opgenomen. De bijwerkingen gesignaleerd in de aselecte groep en in de niet-gerandomiseerde groep van met het INTER FIX™ systeem behandelde patiënten
zijn gecombineerd teneinde een globaal percentage te geven.
Postoperatief
Operatief
Complicatie
Vasculair
Peroperatief
Sacro-iliacale
pijn
Neurologisch 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Rugpijn 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
Incisie 11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
Spinaal voorv al 1(0.5) 1(0.5) 1(1.6) 1(0.5) 3( 1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14. 6) 6(9.7) 13(11.2) 10(9.2)
Urologisch 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Overige 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Andere pijn 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
Gastrointestinaal
Teruggaande
ejaculatie
Respiratoir 3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
Pijn in de
benen
Letsel 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
Peritoneaal 3(1.6) 3(1.6) 0
Vasculair
Postoperatief
Botfractuur 1(0.5) 2(1.1)
Verplaatsing/
losraken van
implantaat
Pijn op
transplantaatlokatie
Geenverbinding
Vermindering 2(1.1) 2(1.1) 0
Geen verb inding
(RESULTAAT
HANGENDE)
Meningitis
Impla ntaatbreuk
Kanker 1
[Aantal (%)]
CONTROL
INTER FIX
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
N=185
N=62
5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
2(1.1) 3(1.6) 1(0.5) 6(3.2)
2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
1(0.5) 1(1.6) 5(8.1) 1(0.5)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
1(1.6)2 0 1(1.6) 2(1.7)
1(1.6) 0 1(1.6)
5(8.1) 0 5(8.1)
(1 Dag-<2
Maanden)
[Aantal (%)]
INTER FIX
N=185
CONTROLE
N=62
Dood 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
1 - Zes (6) van de 81 mannelijke patiënten, opgenomen in de groep met het INTER FIX™ systeem (7,4%) ondervond teruggaande ejaculatie gedurende de hele beoordelingsperiode van
24 maanden.
TABEL I - NADELIGE BIJWERKINGEN
Na 3 maanden
t2-<5 Maanden)
(
[Aantal (%)]
INTER FIX
N=185
ONTROL
N=62
Na 6 maanden
t5-<9 Maanden)
(
[Aantal (%)]
INTER FIX
N=185
CONTROLE
N=62
Na 12 maanden
t9-<19 M aanden)
(
INTER FIX
N=185
(0.5)
[Aantal (%)]
CONTROLE
Na 24 maanden
t19-< 30
(
Maanden)
[Aantal (%)]
INTER FIX
N=62
N=185
1
(0.5)
48 maanden
(>30 - < 60
60
30<
(t
maanden)
INTER FIX
N=62
N=116
0
CONTROLE
N=62
TOTAAL AANTAL
BIJWERKINGEN
(OVER 24 MAANDEN)
CONTROLE
INTER FIX
N=185
1
(7.4)
3(1.6) 0 1(0.9)
2(1.1) 6(9.7)
2 (1.1) 1(0.8) 1(0.9)
72
maanden
(t60
maanden)
INTER FIX
N=109
2 - Bij deze (175) patiënt heeft naar later is gerapporteerd verbinding plaatsgevonden ca. 52 maanden na de operatie en was geen verdere chirurgische procedure of behandeling nodig.
Twee belangrijke nadelige bijwerkingen tijdens het klinische onderzoek waren peroperatieve vasculaire en neurologische beschadigingen. In totaal vonden 15 peroperatieve vasculaire voorvallen
plaats bij 14 patiënten in de groep met het INTER FIX™ systeem tijdens de beoordelingsperiode van 24-maanden. Deze voorvallen omvatten: 2 beschadigingen van de vena cava, 9 beschadigingen
aan de vena iliaca, 1 inscheuring van de arteria hypogastrica, 1 segmentbloeder, 1 beschadiging aan de vena sacralis en 1 oppervlakkige bloeder. In totaal vonden 2 peroperatieve vasculaire
beschadigingen plaats bij 2 patiënten in de controlegroep tijdens deze zelfde periode. Deze omvatten: 1 beschadiging van een vena iliaca en 1 bloeding van het botbed.
In totaal vonden 30 neurologische voorvallen plaats bij 28 patiënten in de groep met het INTER FIX™ systeem in de beoordelingsperiode van 24 maanden. Deze voorvallen omvatten: 1 geval
waarin dorsale flexie van de voet onmogelijk werd, 3 zenuwwortelbeschadigingen, 1 foramen stenosis, 1 reflexdystrofie, 7 gevoelloosheid of branderig gevoel in de benen, 2 dysesthesie, 3
paresthesie, 2 radiculitis, 7 pijn in rug en/of benen, 1 pijn in rug en/of benen met andere symptomen, 1 stekende pijn in de onderrug en 1 carpale-tunnelsyndroom.
In totaal vonden 10 neurologische voorvallen plaats bij 8 patiënten in de controlegroep tijdens deze zelfde periode. Deze voorvallen omvatten: 1 radiculopathie met tintelende ledematen,
1 chronische rugpijn met radiculitis, 1 symptomen van uitputtende verdeling, 2 pijn in rug en benen met andere symptomen, 1 verbroken zenuwverbinding abductor magnus spier en 4 met
gevoelloosheid of een warm gevoel in de benen. Tijdens het ‘post-approval’ onderzoek werden nog 10 neurologische voorvallen genoteerd bij 10 patiënten. Deze voorvallen omvatten: 1
radiculopathie, 2 cervicale radiculopathieën, 1 gevoelloosheid en tintelende handen, 1 gevoelloosheid of branderig gevoel in de benen, 3 pijn in rug en/of benen en 2 pijn in rug en/of benen
met andere symptomen. Bovendien toont de Bijwerkingentabel verschijnselen van pijn in rug en benen en/of spinale voorvallen, zoals inzakking van de discusruimte, die in sommige gevallen
een neurologische component hadden.
Een aantal van de bijwerkingen hebben geleid tot chirurgisch ingrijpen na de klinische proefoperatie. Deze aanvullende chirurgische ingrepen kunnen worden geklasseerd als herziening,
verwijdering, aanvullende fixatie of heroperatie
samen), in de controlegroep tijdens het IDE onderzoek en in de groep met het INTER FIX™ systeem tijdens het ‘post-approval’ onderzoek.
1
Herziening: Een procedure waarbij de oorspronkelijke configuratie van het implantaat wordt aangepast of op enigerlei manier wordt gewijzigd.
Verwijdering: Een procedure waarbij één of meer onderdelen van de oorspronkelijke implantaatconfiguratie worden verwijderd zonder vervanging door hetzelfde type proefsysteem.
Aanvullende fixatie: Een procedure waarbij aanvullende spinale systemen worden geplaatst die niet als onderdeel van het protocol zijn goedgekeurd.
Heroperatie: Elke chirurgische ingreep die geen oorspronkelijke implantaatonderdelen verwijdert, wijzigt of toevoegt.
1
. Tabel II geeft een overzicht van de secundaire chirurgische ingrepen in de groepen met het INTER FIX™ systeem (aselect en niet-gerandomiseerd
Page 23

Tabel II - Secundaire chirurgische ingrepen
Herziening 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Verwijdering 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Aanvullende
fixatie
Heroperatie 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
IDE onderzoek 'Post-approval' onderzoek
Na 24 maanden Na 48 maanden Na 72 maanden
INTER FIX™
systeem
N=185
Controle
N=62
1
INTER FIX™
systeem
N=116
INTER FIX™
systeem
N=109
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
1 - Omdat van de patiënten in de controlegroep geen follow-up na 24 maanden was vereist, is over deze patiënten geen informatie beschikbaar voor de periode van 72 maanden.
POTENTIËLE BIJWERKINGEN:
Hieronder volgt een lijst van potentiële nadelige bijwerkingen die kunnen optreden bij spinale spondylodese operaties met het INTER FIX™ spondylodesesysteem met schroefdraad of het
INTER FIX™ RP spondylodesesysteem met schroefdraad. Een aantal van deze bijwerkingen kan reeds eerder zijn gerapporteerd in de Bijwerkingentabel.
• Gedemonteerd raken, buigen, breken, losraken en/of zich verplaatsen van onderdelen.
• Afstotingsverschijnselen (allergische reactie).
• Weefsel- of zenuwbeschadiging.
• Postoperatief teloorgaan van de juiste kromming, correctie, hoogte en/of reductie van de wervelkolom.
• Infectie.
• Scheuren in het harde hersenvlies.
• Stoornis van het zenuwstelsel.
• Stoornis van het urinestelsel.
• Vorming van littekenweefsel.
• Botfractuur.
• Het uitblijven van consolidatie (of pseudartrose), uitblijven van consolidatie, slechte consolidatie.
• Stilstand van potentiële groei van het geopereerde gedeelte van de wervelkolom. Verlies van mobiliteit of functie van de wervelkolom.
• Complicaties op de plaats waar het transplantaat is weggenomen.
• Beschadiging van bloedvaten en stoornis van het cardiovasculaire stelsel.
• Gastro-intestinale complicaties.
• Aandoeningen van het voortplantingssysteem.
• Schade aan inwendige organen en bindweefsel.
• Ontstaan van ademhalingsproblemen.
• Incisiecomplicaties.
• Verandering in de geestestoestand.
• Overlijden.
Opmerking: Om bepaalde bijwerkingen te corrigeren kan een nieuwe operatie nodig zijn.
KLINISCHE RESULTATEN:
In de Verenigde Staten is in meerdere centra een klinisch equivalentie2 onderzoek van het INTER FIX™ spondylodesesysteem met schroefdraad gedaan, waarbij ventrale spinale toepassing
van het met autogeen bot gevulde INTER FIX™ systeem werd vergeleken met toepassing van een met autogeen bot gevulde allogene dijbeenring, het controlesysteem, voor de behandeling van
patiënten met symptomatische degeneratieve discopathie. Aanvankelijk had het klinisch onderzoek een prospectieve, aselecte, gecontroleerde opzet. Vervolgens is het onderzoeksprogramma
zodanig herzien dat patiënten in een niet-gerandomiseerde groep konden worden opgenomen, d.w.z. patiënten die alleen met het INTER FIX™ systeem waren behandeld.
De criteria voor opname in het klinische onderzoek waren o.a. symptomatische degeneratieve discopathie tot uiting komend in hardnekkige pijn in de benen en/of de rug met duidelijke
bevinding(en) verkregen door diagnostische beeldvorming; spinale instabiliteit gedefinieerd door een translatie van meer dan 4 mm of een hoek van meer dan 5° op flexie/extensie radiografieën;
symptomatische implicatie op één niveau tussen L2-S1 en een spondylolisthesis van niet meer dan graad 1. Van dit onderzoek waren in het bijzonder uitgesloten, patiënten die eerdere ventrale
intercorporele spondylodese hadden ondergaan op het betrokken spinale niveau, patiënten met osteopenie, osteoporose of osteomalacie of die botgroeistimulatie nodig hadden gehad.
In totaal zijn 185 patiënten opgenomen in de groep voor onderzoek van de behandeling met het INTER FIX™ systeem.
van het klinische onderzoek.
Een vereiste van het ‘post-approval’ onderzoek van het INTER FIX™ spondylodesesysteem met schroefdraad was om het gedrag van het systeem op lange termijn te beoordelen door alle
postoperatieve gegevens over een periode van 6 jaar te verkrijgen van een minimum van 100 patiënten. Het gedrag van het systeem op lange termijn werd beoordeeld bij 116 patiënten 4
jaar na de operatie en bij 109 patiënten 6 jaar na de operatie. De patiëntenpopulatie werd samengesteld uit deelnemers aan het IDE klinische onderzoek. Op basis van de criteria van het
IDE onderzoek omvatte de over deze patiënten verkregen informatie een beschrijving van bijkomende chirurgische procedures, röntgenologische beoordeling van verbinding en een pijn- /
functiebeoordeling.
Tabel III presenteert de klinische resultaten van het onderzoek en beschrijft de parameters van het succes van de verstijving, het succes m.b.t. de pijn (Oswestry) en het neurologische
succes 12, 24, 48 en 72 maanden na de behandelende operatie. Het succes van dit klinische onderzoek werd genoteerd op basis van een equivalentie onderzoek tussen het INTER FIX™
systeem en het controlesysteem.
Voor dit klinische onderzoek werd spondylodese gedefinieerd als het aangetoonde overbrugging van trabeculair bot, stabiliteit van translatie (<3mm) en van hoekbeweging (<5°) en geen
radiolucente zones die meer dan 50% van elk implantaat omgeven. Ook werden patiënten die secundaire operaties ondergingen vanwege het uitblijven van verbinding voor de berekeningen
beschouwd als mislukte spondylodese. Het Oswestry pijn/onvermogen succes werd gedefinieerd als een verbetering van minstens 15 punten op de postoperatieve puntentelling vergeleken
met de preoperatieve puntentelling. Het succes in neurologische status werd gedefinieerd als postoperatief behoud of verbetering in ten minste 3 van de 4 neurologische categorieën (beweging,
gevoel, reflexen en optillen van een gestrekt been), vergeleken met de preoperatieve conditie. Globaal succes werd gebaseerd op een patiënt met aangetoonde spondylodese, Oswestry pijn/
onvermogen succes en succes in neurologische status en geen als “mislukking” geklasseerde secundaire operatie.
2
INTER FIX™ systeem statistisch niet slechter dan het controlesysteem.
3
Omvat niet de 3 patiënten die geen behandeling met het INTER FIX™ systeem kregen wegens operatieve voorvallen.
3
In totaal zijn 62 controlepatiënten opgenomen in de aselecte groep
Tabel III bevat eveneens de algemene succesresultaten 12 en 24 maanden na de operatie.
Tabel III - Klinische resultaten van het onderzoek over het INTER FIX™ systeem
Verstijving
Oswestry
pijn/onvermog
en verbetering
Patiënten met
minstens 15
punten
verbetering
t.o.v. preop
Behoud of
verbetering
van
neurologische
status
Globaal
succes
Percentages na 12
maanden
Onderzoeks-
groep
96.1%
(122/127)
53.5%
(76/142)
99.3%
(150/151)
42.4%
(61/144)
Controle
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
(12/56)
Percentages na 24
maanden
Delta
Onderzoeks-
1
(ǻ
)
groep
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
-0.097 46.1%
(71/154)
Controle
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
(17/57)
Delta
1
(ǻ
)
-0.043 49.5%
Percentages
na 48
maanden
Onderzoeks-
groep
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
(53/107)
Percentages
na 72
maanden
Onderzoeks-
groep
97.6% (82/84)
56.3%
(54/96)
1 - Minimale marge van ‘niet-inferioriteit’ vereist om ‘niet-inferioriteit’ van de onderzoeksgroep ten opzichte van de controlegroep te claimen.
Alle patiënten die betrokken waren bij het klinische onderzoek van het INTER FIX™ spondylodesesysteem met schroefdraad, ongeacht de behandelingsgroep, werden opgenomen volgens
dezelfde criteria voor opneming of uitsluiting. Om de vergelijkbaarheid van de gerandomiseerde patiënten met het INTER FIX™ systeem en de niet-gerandomiseerde patiënten met het
INTER FIX™ systeem te substantiëren werden de demografische kenmerken, de preoperatieve medische conditie, de bevindingen van diagnostische beeldvorming die kenmerkend zijn voor
degeneratieve discopathie, de operatieve benadering en de locatie van het lumbaal behandelde niveau bekeken. De populatie was statistisch vergelijkbaar, behalve wat betreft de operatieve
benadering, de preoperatieve werkstatus en de preoperatieve bevindingen door diagnostische beeldvorming van osteofytvorming op de eindplaten van de ruggenwervels; littekenvorming en/of
verdikking van de annulus fibrosis, ligamentum flavum en/of het gewrichtskapsel en hernia nuclei pulposi. Nadere statistische analyses hebben aangetoond dat deze variabelen geen verband
hielden met verschillen in de klinische spondylodese-resultaten na 12 maanden, de Oswestry verbetering in pijn/onvermogen, de neurologische status of het globale succes, met uitzondering
van hernia nuclei pulposi. In de niet-gerandomiseerde groep was bij meer patiënten hernia nuclei pulposi geconstateerd, hetgeen de kans op globaal succes bleek te verhogen.
Page 24

Er is een “bedoeling-tot-behandeling” analyse uitgevoerd. Bij deze analyse resulteerden mislukkingen in de vorm van secundaire operatie, sterfgevallen, patiënten waarvan de follow-up niet
kon worden voortgezet en door andere oorzaken ontbrekende observaties, in voor de uitkomstvariabelen ontbrekende observaties en werden daarom opgenomen onder de noemers voor
de berekende percentages, d.w.z. beschouwd als “mislukkingen”. Door deze patiënten te beschouwen als mislukkingen van de behandeling waren de klinische uitkomstpercentages in de
“bedoeling-tot-behandeling” analyse aanzienlijk lager dan in de feitelijk geobserveerde klinische gegevens. Onderstaande tabel (Tabel IV) geeft de resultaten van de “bedoeling-tot-behandeling”
analyse van patiënten met het INTER FIX™ systeem, maar omvat geen resultaten van controlepatiënten.
Sterfgevallen, mislukking in de vorm van secundaire operatie, onmogelijkheid van voortzetting van follow-up en
ontbrekende observaties worden beschouwd als mislukkingen en zijn opgenomen onder de noemer van de
Percentages na
Verstijving
Oswestry pijn/onvermogen verbetering
Patiënten met minstens 15 punten verbetering
t.o.v. preop
Behoud of verbetering van neurologische
status
Globaal succes
Secundaire operatie mislukkingen
Geen verbinding
Overige²
Sterfgevallen
1 - Deze patiënten zijn inbegrepen in de berekeningen van spondylodesepercentages maar worden overigens om klinische onderzoeksredenen beschouwd als mislukkingen.
2 - Patiënten die in die periode follow-up moesten krijgen en die secundaire operaties hebben ondergaan om andere redenen dan het uitblijven van verbinding worden om klinische
onderzoeksredenen beschouwd als mislukkingen.
VERPAKKING:
Bij ontvangst moet de verpakking van alle onderdelen intact zijn. Als een leen- of consignatiesysteem wordt gebruikt, moet zorgvuldig worden gecontroleerd of de sets compleet zijn en moeten
alle onderdelen en instrumenten vóór het gebruik worden gecontroleerd op tekenen van beschadiging. Beschadigde verpakkingen of producten mogen niet worden gebruikt en moeten aan
Medtronic Sofamor Danek worden geretourneerd.
ONTSMETTING EN REINIGING:
Niet steriel geleverde implantaten en instrumenten moeten eerst met de gevestigde ziekenhuismethodes worden gereinigd, voordat zij worden gesteriliseerd en in een steriele operatieruimte
worden gebracht. Gebruikte instrumenten moeten worden ontsmet, gereinigd en gesteriliseerd voordat ze opnieuw gebruikt worden.
N.B.! Gebruik geen reinigingsoplossingen die bleekwater of formol bevatten, deze kunnen namelijk het systeem beschadigen.
Alle producten moeten voorzichtig worden behandeld. Verkeerd gebruik of verkeerde hantering kan schade en/of eventuele onjuiste functionering van het implantaat veroorzaken.
STERILISATIE:
Tenzij de producten als steriel zijn aangemerkt en dit duidelijk wordt vermeld op het etiket op de ongeopende steriele verpakking geleverd door de onderneming, moeten alle bij operaties
gebruikte implantaten en instrumenten door het ziekenhuis worden gesteriliseerd, voordat ze worden gebruikt. Verwijder alle verpakkingmateriaal voorafgaand aan het steriliseren. Alleen
steriele producten mogen in de operatiezone worden geplaatst. Voor een gegarandeerd steriliteitsniveau van 10-6 wordt aanbevolen dat het ziekenhuis de producten met stoom steriliseert
met toepassing van een van de drie onderstaande series procesparameters:
METHODE CYCLUS TEMPERATUUR STERILISATIETIJD
Stoom Van te voren vacumeren 270° F (132° C) 4 min.
Stoom Onder druk 250° F (121° C) 60 min.
Stoom* Onder druk* 273° F (134° C)* 20 min.*
NB! Wegens de vele variabelen die aan sterilisatie te pas komen, dient elke medische instelling het voor haar apparatuur gebruikte sterilisatieproces (temperaturen, tijden enz.) te kalibreren
en te verifiëren.
*Voor gebruik buiten de Verenigde Staten, wordt door bepaalde niet-Amerikaanse gezondheidszorgautoriteiten sterilisatie volgens deze parameters aanbevolen om het potentiële risico van
overbrenging van de ziekte van Creutzfeldt-Jakob tot een minimum te beperken, vooral voor chirurgische instrumenten die in aanraking kunnen komen met het centrale zenuwstelsel.
KLACHTEN BETREFFENDE DE PRODUCTEN:
Beroepsbeoefenaren in de gezondheidszorg (bv. afnemers of gebruikers van dit productensysteem) die klachten of enige reden tot ontevredenheid hebben betreffende de kwaliteit, de
kenmerken, de duurzaamheid, de betrouwbaarheid, de veiligheid, de doelmatigheid en/ of de prestaties van het product, dienen de distributeur, Medtronic Sofamor Danek hiervan in kennis te
stellen. Verder moet de distributeur onmiddellijk worden verwittigd, indien een onderdeel van het geïmplanteerde INTER FIX™ spondylodesesysteem met schroefdraad of het INTER FIX™
RP spondylodesesysteem met schroefdraad “niet goed functioneert” (d.w.z. niet voldoet aan de specificaties betreffende de prestaties of anderszins niet op de bedoelde wijze functioneert),
of indien wordt vermoed dat dit zo is. Indien een Medtronic Sofamor Danek product ooit “slecht mocht functioneren” en dood of ernstig letsel van een patiënt hebben kunnen veroorzaken
of hiertoe hebben kunnen bijgedragen, dan moet de distributeur hiervan zo snel mogelijk per telefoon, fax of schriftelijk op de hoogte worden gebracht. Bij indiening van een klacht wordt u
verzocht de naam en het referentienummer van het of de onderdelen en het nummer of de nummers van de betreffende partij(en) te vermelden, evenals uw naam en adres en de aard van
de klacht en mede te delen of een schriftelijk rapport is gevraagd aan de distributeur.
AANTEKENING VOOR DE ARTS: Hoewel de arts de geleerde tussenpersoon is tussen het bedrijf en de patiënt, dient de in dit document vervatte belangrijke medische informatie te worden
doorgegeven aan de patiënt.
Alleen van toepassing voor de V.S.
WAARSCHUWING: VOLGENS DE AMERIKAANSE FEDERALE WETGEVING MOGEN DEZE SYSTEMEN UITSLUITEND DOOR OF OP VOORSCHRIFT VAN EEN ARTS WORDEN VERKOCHT.
POGINGEN TOT VERWIJDERING VAN HET SYSTEEM:
Mocht het nodig zijn een INTER FIX™ spondylodesesysteem met schroefdraad of een INTER FIX™ RP spondylodesesysteem met schroefdraad te verwijderen, bel dan Medtronic Sofamor Danek.
© 2006 Medtronic Sofamor Danek. Alle rechten voorbehouden.
Tabel IV - "Bedoeling-tot-behandeling" analyse voor het INTER FIX™ systeem
percentages
12 maanden
65.9% (122/185) 67.6%
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
33.0% (61/185) 38.4% (71/185) 45.7%
1
5
8
0 1 0 1
Percentages
na 24
maanden
(125/185)
(128/185)
5
3
Percentages
na 48
maanden
80.2%
(93/116)
(66/116)
87.9%
(102/116)
(53 /116)
4
4
Percentages
na 72
maanden
77.1%
( 84/109)
56.0%
(61 /109)
75.2%
75.2% (82/109)
(82/109)
49.5%
(54/109)
1
2
Page 25

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
DANSK
Der er indeholdt vigtige oplysninger om INTER FIX™ fusionsanordning med gevind og INTER FIX™ RP fusionsanordning med gevind i denne indlægsseddel.
BESKRIVELSE:
INTER FIX™ fusionsanordning med gevind består af en hul, perforeret, metalcylinder og endedæksel. INTER FIX™ RP fusionsanordning med gevind består af en hul, perforeret,
metalcylinder med en enkelt, bred, udvendig afrundet rille langs med hele længdeaksen, som går ind til anordningens indre diameter. INTER FIX™ RP implantatet er lukket i begge ender.
INTER FIX™ og INTER FIX™ RP implantater kan fås med forskellige diametre fra 12 mm til 24 mm og længder fra 20 mm til 29 mm. Endedækslernes størrelse på INTER FIX™
implantaterne svarer til cylindrenes diameter og sættes på cylindrenes åbne ender, efter de er blevet fyldt med autologt knogletransplantationsmateriale.
INTER FIX™ og INTER FIX™ RP implantater er fremstillet i titanlegering (Ti-6Al-4V) beregnet til implantater i overensstemmelse med ASTM F136.
Underforståede kommercielle garantier og egnethed for særlige formål er anvendelser er udtrykkeligt udelukket. Se Medtronic Sofamor Danek Kataloget eller prislisten, hvis De ønsker
nærmere oplysninger om garanti og ansvarsbegrænsninger.
INDIKATIONER:
INTER FIX™ fusionsanordning med gevind og INTER FIX™ RP fusionsanordning med gevind er indiceret ved kirurgiske indgreb for spinalfusion hos patienter med et fuldt udvokset
skelet med discusdegeneration (DDD) på et niveau fra L2 til S1. Discusdegeneration er defineret ved rygsmerter forbundet med discus og bekræftet af patientens sygehistorie og
røntgenundersøgelser. Patienter med discusdegeneration kan også præsentere en Grad I spondylolisthese eller retrolisthese på det involverede niveau. INTER FIX™ og INTER FIX™ RP
implantater skal bruges sammen med autologt knogletransplantationsmateriale og implanteres via en åben anterior adgang.
KONTRAINDIKATIONER:
INTER FIX™ fusionsanordning med gevind og INTER FIX™ RP fusionsanordning med gevind bør ikke implanteres hos patienter, som har en aktiv infektion på operationsstedet eller som
er allergiske overfor titan eller titanlegering.
ADVARSLER:
• INTER FIX™ fusionsanordning med gevind og INTER FIX™ RP fusionsanordning med gevind bør kun bruges af kirurger, som har erfaring med indgreb for spinalfusion og har
modtaget en passende uddannelse i brug af denne anordning.
• En mangelfuld erfaring og/eller uddannelse kan medføre en hyppigere forekomst af bivirkninger som f.eks. vaskulære læsioner, neurologiske og/eller urogenitale tildragelser (herunder
forringet ejakulation).
FORSIGTIGHEDSREGLER:
• Sikkerheden og effektiviteten af INTER FIX™ fusionsanordning med gevind og INTER FIX™ RP fusionsanordning med gevind er ikke blevet etableret hos patienter, som præsenterer
følgende forhold:
- spondylolisthese eller retrolisthese af Grad II eller derover;
INTER FIX™ FUSIONSIMPLANTAT MED GEVIND
INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND
VIGTIGE MEDICINSKE OPLYSNINGER
Page 26

- nødvendighed for fusion af mere end et niveau;
I
L
L
- eftersyn af tidligere indgreb for fusion af hvirvellegemer;
- nødvendighed for postoperative steroide eller nonsteroide antiinflammatoriske stoffer;
- kraftig fedme;
- under 18 år eller over 65 år;
- osteoporose, osteopeni, og/eller osteomalaci;
- graviditet.
• Sikkerheden og effektiviteten af INTER FIX™ fusionsanordning med gevind og INTER FIX™ RP fusionsanordning med gevind er kun blevet etableret for indgreb på den lumbale del af
forreste columna (ALIF), hvor der bruges autologt knogletransplantationsmateriale.
• Patienter, som får indopereret INTER FIX™ fusionsanordning med gevind, bør have modtaget mindst seks måneders ikke operativ behandling. Patienter, som får indopereret INTER
FIX™ RP fusionsanordning med gevind, bør have modtaget mindst seks måneders ikke operativ behandling.
• Der bør indopereres to INTER FIX™ fusionsanordninger med gevind ved siden af hinanden på det opererede niveau. En INTER FIX™ fusionsanordning med gevind og en INTER
FIX™ RP Fusionsanordning med gevind bør implanteres side om side på det opererede niveau. Rillen på langs i INTER FIX™ RP implantatet gør det lettere at implantere to
anordninger af denne type, der ligger tæt på hinanden, end to INTER FIX™ anordninger, men det resulterer dog i en reduceret lateral profil.
• Den lange akse på INTER FIX™ fusionsanordning med gevind og på INTER FIX™ RP fusionsanordning med gevind bør ligge i anterior – posterior retning.
• Implantater og instrumenter skal steriliseres før brug efter de retningslinier, der er anført i denne indlægsseddel, med mindre de leveres sterile og er klart mærket som sådanne.
BIVIRKNINGER:
Hos 185 patienter med INTER FIX™ anordning og 62 kontrolpatienter, som deltog i en klinisk multicenter undersøgelse af INTER FIX™ fusionsanordning med gevind, blev der rapporteret
om de bivirkninger, som er anført i Skema I. Herudover blev der rapporteret om bivirkninger fra 116 patienter ved 48 måneder og 109 patienter ved 72 måneder hos patienter i ”post-approval”
undersøgelsen. De viste procenter er baseret på det samlede antal patienter optaget i undersøgelsen i hver periode. De bivirkninger, der opstod i den randomiserede og den ikke randomiserede
INTER FIX™ behandlingsgruppe, er blevet lagt sammen for at give et samlet forholdstal.
SKEMA I - BIVIRKNINGER
Postoperativ
Operativ
[Antal (%)]
KONTRO
NTER FIX
Komplikation
Vaskulær
peroperativ
Sacroiliakale
smerter
Neurologisk 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Rygsmerter 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
I Ved incision 11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
Spinal
tildragelse
Urologisk 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Andet 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Andre smerter 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
Gastrointestinal 7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
Forringet
ejakulation
Åndedrætsb esvær
Smerter i ben 2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
Trauma 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
Peritoneal 3(1.6) 3(1.6) 0
Vaskulær
postoperativ
Knoglebrud 1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
Forskydning/løs
ning af
implantat
Smerter ved
donorsted for
knogletrans plantat
Ingen
konsolidation
Skridning 2(1.1) 2(1.1) 0
Ingen
konsolidation
(UVIST
RESULTAT)
Meningitis 1(1.6) 0 1(1.6)
Afbrækning af
implantat
Cancer 1
N=62
N=185
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
2(1.1) 3(1.6) 1(0.5) 6(3.2)
3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
1(1.6)
5(8.1) 0 5(8.1)
(Dag 1 - <2
[Antal (%)]
INTER
FIX
N=185
måneder)
KONTROL
N=62
3 måneder
måneder)
[Antal (%)]
INTER
FIX
N=185
2 - < 5
(t
KONTRO
N=62
6 måneder
måneder)
[Antal (%)]
INTER
FIX
N=185
(t 5 - < 9
KONTROL
N=62
Død 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
12 måneder
(t 9 - < 19
måneder)
[Antal (%)]
INTER
KONTROL
N=62
FIX
N=185
2
0 1(1.6) 2(1.7)
(0.5)
24 måneder
(t 19 - < 30
måneder)
[Antal (%)]
INTER
KONTROL
FIX
N=185
1
(0.5)
48 Måneder
(>30 - < 60
30<60
(t
Måneder)
N=62
INTER FIX
N=116
0
N=62
SAMLET ANTAL
BIVIRKNINGER
(I 24 MÅNEDER)
INTER FIX
N=185
1
(7.4)
KONTROL
2 (1.1) 1(0.8) 1(0.9)
INTER FIX
72
Måneder
60
(t
Måneder)
N=109
1 - Seks (6) ud af 81 optagne mænd i INTER FIX™ gruppen (7.4%) erfarede retrograd ejakulation i evalueringsperioden omkring 24 måneder.
2 - Disse patienter (175) er siden blevet rapporteret konsoliderede omkring 52 måneder efter indgrebet, og intet andet kirurgisk indgreb eller behandling var nødvendig.
To vigtige bivirkninger i det kliniske forsøg var peroperative vaskulære og neurologiske læsioner. Der opstod i alt 15 vaskulære peroperative hændelser hos 14 patienter i INTER FIX™
gruppen i evalueringsperioden omkring 24 måneder. Disse tildragelser omfattede: 2 læsioner på vena cava, 9 læsioner på vena iliaca, 1 dilacereret hypogastrisk vene, 1 segmentær vene
blødning, 1 sakral venelæsion og 1 superficiel blødning. Der opstod i alt 2 vaskulære peroperative læsioner hos 2 patienter i kontrolgruppen i samme periode. Disse omfattede: 1 læsion på
vena iliaca og 1 blødning fra knoglelejet.
Der opstod i alt 30 neurologiske hændelser hos 28 patienter i INTER FIX™ gruppen i evalueringsperioden omkring 24 måneder. Disse hændelser omfattede: 1 med dropfod, 3 med læsioner
på nerverødder, 1 med foraminal stenose, 1 med dystrofirefleks, 7 med følelsesløshed eller varme i benene, 2 med dysæstesi, 3 med paræstesi, 2 med radiculitis, 7 med rygsmerter og/eller
bensmerter, 1 med rygsmerter og/eller bensmerter med andre symptomer, 1 med jagende smerter i lænden, og 1 med karpaltunnelsyndrom.
Der opstod i alt 10 neurologiske hændelser hos 8 patienter i kontrolgruppen i samme periode. Disse tildragelser omfattede: 1 patient med radikulopati med prikkende ekstremiteter, 1 med
kroniske rygsmerter med radiculitis, 1 svækkede fordelingssymptomer, 2 med rygsmerter og bensmerter med andre symptomer, 1 med denerveret musculus abductor magnus, 4 med
følelsesløse eller varme ben. Under post-approval undersøgelsen blev der rapporteret endnu 10 neurologiske hændelser hos 10 patienter. Disse hændelser omfattede: 1 med radiculopati, 2
med cervikale radiculopatier, 1 med følelsesløshed eller prikken i hænderne, 1 med følelsesløshed eller brændende fornemmelse i benene, 3 med rygsmerter og/eller bensmerter, og 2 med
rygsmerter og/eller bensmerter med andre symptomer. Herudover er der i skemaet med bivirkninger anført bensmerter og rygsmerter og/eller spinale tildragelse som f.eks. sammenbrud af
hvirvlernes mellemrum, som i visse tilfælde havde en neurologisk komponent.
Visse bivirkninger førte til kirurgiske indgreb efter indgrebet, som hørte ind under det kliniske forsøg. Disse ekstra kirurgiske indgreb kan klassificeres som eftersyn, fjernelse, supplerende
fixationer eller gentagne operationer
undersøgelsen og i INTER FIX™ gruppen i post-approval undersøgelsen.
1
Eftersyn: Et indgreb som justerer eller på én eller anden måde ændrer implantatets originale konfiguration.
Fjernelse: Et indgreb som fjerner en eller flere komponenter i implantatets originale konfiguration uden genindsætning med samme type forsøgsanordning.
Supplerende Fixation: Et indgreb hvor der indopereres ekstra spinalimplantater, som ikke er godkendt som en del af protokollen.
Gentagen operation: Et kirurgisk indgreb som ikke fjerner, ændrer eller tilføjer et originalt implantat.
1
. Skema II opsummerer de sekundære kirurgiske indgreb i INTER FIX™ gruppen (både randomiseret og ikke randomiseret) og kontrolgruppen i IDE
Page 27

g
Skema II - Sekondære Kirurgiske Indgreb
IDE Undersøgelse Post-approval undersøgelse
I 24 måneders
INTER FIX™
anordning
N=185
perioden
Kontrol
N=62
1
48 måneders
perioden
INTER FIX™
anordning
N=116
72 måneders
perioden
INTER FIX™
Anordning
N=109
Eftersyn 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Fjernelser 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Supplerende
Fixationer
Gentagne
operationer
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
1 - Da kontrolpatienterne ikke krævede opfølgning efter 24 måneder, findes der ingen information vedrørende alle disse patienter i 72 måneders perioden.
MULIGE BIVIRKNINGER:
Nedenfor er anført en liste med mulige bivirkninger, som kan opstå ved indgreb for spinalfusion med INTER FIX™ fusionsanordning med gevind eller INTER FIX™ RP fusionsanordning med
gevind. Visse af disse bivirkninger er måske allerede blevet nævnt i skemaet for bivirkninger.
• Demontering, bøjning, afbrækning, løsning og/eller forskydning af komponenter.
• Allergisk reaktion med fremmedlegemer.
• Læsioner på væv eller nerver.
• Postoperativ ændring af rygkrumning, tab af korrektion, højde og/eller reposition.
• Infektion.
• Læsioner på dura mater.
• Forstyrrelser af neurologiske funktioner.
• Forstyrrelser af urologiske funktioner.
• Arvævsdannelse.
• Knoglebrud.
• Manglende sammenvoksning (eller pseudoartrose), forsinket sammenvoksning, dårlig sammenvoksning.
• Ophør af en potentiel vækst af den opererede del af hvirvelsøjlen. Tab af hvirvelsøjlens bevægelighed eller funktion.
• Komplikationer ved donorsted for knogletransplantat.
• Beskadigelse af blodkar og forstyrrelse af cardiovaskulære funktioner.
• Gastrointestinale komplikationer.
• Forstyrrelser af forplantningssystemet.
• Beskadigelse af interne organer og tilknyttede væv.
• Udvikling af åndedrætsproblemer.
• Komplikationer forbundet med incisionen.
• Ændring af mental tilstand.
• Død.
Bemærk: Yderligere kirurgiske indgreb kan være nødvendige for at korrigere visse af disse mulige bivirkninger.
KLINISKE RESULTATER:
Et multicenter ækvivalent2 klinisk forsøg med INTER FIX™ fusionsanordning med gevind er blevet udført i USA. Forsøget sammenlignede brug på forreste columna af INTER FIX™ anordning
fyldt med brug af autologt knoglemateriale med allogent anulus femoralis fyldt med autologt knoglemateriale i kontrolgruppen ved behandling af patienter med symptomatisk discusdegeneration.
I starten havde det kliniske forsøg en undersøgende, randomiseret, kontrolleret opbygning. Senere blev forskningsplanen ændret til at tillade optagelse af patienter i en ikke randomiseret
undergruppe, dvs. patienter som kun blev behandlet med INTER FIX™ anordning.
Inklusionskriterierne for det kliniske forsøg omfattede symptomatisk discusdegeneration konstateret ved intraktable smerter i ben og/eller ryg med positiv røntgendiagnose; spinal ustabilitet
defineret ved en forskydning på over 4 mm eller en vinkel på over 5° på røntgenbilleder med fremadbøjning/bagudbøjning; symptomatisk involvering af et enkelt niveau fra L2 til S1; og ikke
over Grad 1 spondylolisthese. Eksklusionkriterierne omfattede patienter som: tidligere havde gennemgået et indgreb for fusion mellem hvirvellegemer på forreste columna på det involverede
niveau; havde osteopenia, osteoporose eller knogleblødhed; eller påkrævet stimulation af knoglevækst.
Der blev optaget i alt 185 patienter i forsøgets INTER FIX™ behandlingsgruppe3. Der blev optaget i alt 62 kontrolpatienter i det kliniske forsøgs randomiserede undergruppe.
Et post-approval krav for INTER FIX™ Fusionsanordningen med gevind skulle evaluere anordningens effektivitet på lang sigt ved at indhente postoperative data fra mindst 100 patienter
over 6 år. Anordningens effektivitet på lang sigt blev evalueret hos 116 patienter 4 år efter indgrebet og hos 109 patienter 6 år efter indgrebet. Patientgruppen bestod af deltagere i IDE klinisk
forsøg. Den indhentede information om disse patienter var baseret på kriterier fra IDE forsøget og omfattede en beskrivelse af ethvert supplerende kirurgisk indgreb, en evaluering af fusion
med røntgenundersøgelse, og en evaluering af smerter og funktion.
Skema III viser de kliniske resultater fra undersøgelsen, beskriver parametrene for vellykket fusion, vellykket udfald for smerter (Oswestry), og vellykket neurologisk udfald ved 12, 24, 48 og 72
måneder efter det kirurgiske indgreb. Det kliniske forsøgs vellykkede udfald blev noteret på basis af en ækvivalens analyse mellem INTER FIX™ implantatgruppe og kontrolgruppe.
I dette kliniske forsøg var konsolidation defineret som bevis for fast forening af trabekulære knogler, stabilitet af forskydning (<3 mm) og vinkelbevægelse (<5°), og ingen opklaringer på mere
end 50% omkring hvert implantat på røntgenbilleder. Patienter, som gennemgik et sekondært indgreb på grund af manglende konsolidation, blev anset for at være mangelnde konsolidationer
i beregningerne. Et vellykket resultat for Oswestry Smerter/Invaliditet var defineret som en forbedring på mindst 15 points i den postoperative scoring i forhold til den præoperative scoring. Et
vellykket resultat for den neurologiske tilstand var defineret som en opretholdelse eller en forbedring af mindst 3 ud af 4 neurologiske kategorier (motorisk, sensorisk, reflekser, og Lasègue) i
den postoperative tilstand i forhold til den præoperative tilstand. Et samlet vellykket resultat var baseret på en påvist konsolidation, et vellykket resultat for Oswestry Smerter/Invaliditet, og et
vellykket resultat for den neurologiske tilstand, samt intet sekondært kirurgisk indgreb klassificeret som “mislykket resultat”.
2
INTER FIX™ anordning ikke statistisk dårligere end kontrol.
3
Omfatter ikke 3 patienter som ikke modtog INTER FIX™ anordningen på grund af uønskede kirurgiske hændelser.
Skema III viser også det samlede vellykkede udfald ved 12 og 24 måneder efter indgrebet.
Konsolidation
Oswestry
Smerter/
Invaliditet
Patienter med
en forbedring på
mindst 15 points
i forhold til den
præoperative
tilstand
Opretholdelse
eller
forbedring af
neurologisk
tilstand
Samlet Succes
Skema III - Kliniske resultater fra undersøgelsens INTER FIX™ gruppe
Forholdstal for 12 måneder
Testet
anordning
96.1%
(122/127)
53.5%
(76/142)
99.3%
(150/151)
42.4%
(61/144)
Kontrol
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
(12/56)
Forholdstal for 24
Delta
1
(ǻ
)
-0.097 46.1%
måneder
Testet
anordning
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
(71/154)
Kontrol
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
(17/57)
Forholdstal
Delta
(ǻ1)
-0.043 49.5%
for 48
måneder
Testet
anordning
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
(53/107)
Forholdstal for
72 måneder
Testet
anordning
97.6% (82/84)
56.3%
(54/96)
1 - Påkrævet minimal non inferiority margin for at erklære undersøgelsesresultatet non inferiority i forhold til kontrol.
Alle patienter, som deltog i det kliniske forsøg med INTER FIX™ fusionsanordning med gevind, blev optaget efter de samme inklusions/eksklusions kriterier, uafhængig af deres behandlingsgruppe.
For at dokumentere sammenligneligheden af den randomiserede INTER FIX™ patientgruppe og den ikke randomiserede INTER FIX™ patientgruppe, blev der foretaget en undersøgelse af
demografiske karakteristika, præoperative medicinske forhold, røntgendiagnostik med karakteristika for fund af discusdegeneration, operativ adgang, og placering af det behandlede lumbale
niveau. Patientgruppen var statistisk sammenlignelig med undtagelse af operativ adgang, præoperativ jobstatus, og præoperativ røntgendiagnostik med fund af dannelse af osteofyt i bruskflade
mellem discus o
corpora; arvævsdannelse og/eller fortykkelse af anulus fibrosus, ligamentum flavum, og/eller facetledkapslen; samt diskusprolaps. Yderligere statistiske analyser viste, at
Page 28

disse variabler ikke var forbundet med forskelle på de kliniske resultater efter 12 måneders udvikling med hensyn til konsolidation, forbedring af Oswestry Smerter/Invaliditet, neurologisk
tilstand, eller samlet vellykket udfald, med undtagelse af diskusprolaps. Flere patienter i den ikke randomiserede gruppe havde diskusprolaps, som viste sig, at forøge muligheden for et
samlet vellykket resultat.
Der blev gennemført en “intent-to-treat” analyse. I denne analyse blev følgende tilfælde anset for at være “mislykkede” resultater: mislykkede sekondære indgreb, dødsfald og patienter, som
var tabt for opfølgning, og manglende observationer på grund af andre årsager, hvad der resulterede i manglende observationer for de endelige variabler og som derfor indgik i det beregnede
forholdstals nævner. Når disse patienter regnes for at have givet et mislykket resultat, bliver forholdstallene for det kliniske resultat i intent-to-treat analysen betydeligt lavere end dem for de reelt
observerede kliniske data. Følgende skema (Skema IV) viser resultaterne for intent-to-treat analysen af INTER FIX™ patientgruppen men det omfatter ikke resultaterne for kontrolgruppen.
Skema IV - Intent-to-Treat Analyse for INTER FIX™ anordning
Dødsfald, mislykkede sekondære indgreb, patienter tabt for opfølgning og manglende observationer
anses for at være mislykkede behandlingsresultater og indgår i forholdstallenes nævner
Forholdstal for 12
Konsolidation
Oswestry Smerter/Invaliditet
Patienter med en forbedring på mindst 15 points i
forhold til den præopera tive tilstand
Opretholdelse eller forbedring af neurologisk
tilstand
Samlet Succes
Mislykkede sekundære indgreb
Manglende konsolidation
2
Andet
Dødsfald
1
måneder
65.9% (122/185) 67.6%
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
33.0% (61/185) 38.4% (71/185) 45.7%
5
8
0 1 0 0
Forholdstal for
24 måneder
(125/185)
(128/185)
5
3
Forholdstal for
48 måneder
(93/116)
(66/116)
(102/116)
(53 /116)
80.2%
87.9%
4
4
Forholdstal for 72
måneder
77.1%
( 84/109)
56.0%
(61 /109)
75.2%
75.2% (82/109)
(82/109)
49.5%
(54/109)
1
2
1 - Disse patienter indgår i beregningen af forholdstallet for konsolidering men anses i øvrigt for at have givet et mislykket resultat for den kliniske målsætning.
2 - Patienter hvis evalueringsdata for opfølgning skal indhentes i denne periode og som har gennemgået et sekundært indgreb af andre årsager end manglende konsolidation, anses for at
have givet et mislykket resultat for den kliniske målsætning.
EMBALLERING:
Emballagen for hvert element skal være intakt ved modtagelsen. Hvis der anvendes et system med lån eller depot, skal det før brug omhyggeligt kontrolleres at alle sæt er komplette og at alle
komponenter, herunder instrumenter, er fri for skader. Beskadigede emballager eller produkter må ikke anvendes og skal sendes tilbage til Medtronic Sofamor Danek.
RENGØRING OG DEKONTAMINERING:
Med mindre de leveres sterile, skal alle implantater og instrumenter rengøres efter gængse hospitalsmetoder før sterilisation og fremlægning i en steril operationsstue. Brugte instrumenter
skal dekontamineres, rengøres og steriliseres før genanvendelse.
Bemærk: Brug ikke rengøringsmidler, som indeholder blegemiddel eller formalin, da sådanne opløsninger kan beskadige komponenterne.
Alle produkter bør behandles med forsigtighed. En forkert anvendelse eller håndtering kan medføre beskadigelser og/eller at implantatet muligvis fungerer fejlagtigt.
STERILISATION:
Med mindre de er mærket sterile og klart etiketteret som sådanne i en uåbnet pakning leveret af firmaet, skal alle implantater og instrumenter, som bruges under indgrebet, steriliseres på
hospitalet før brug. Fjern al emballage før sterilisation. Kun sterile produkter må bringes ind i operationsstuen. For et 10-6 sterilitetssikkerhedsniveau, anbefales det at dampsterilisere disse
produkter på hospitalet ved hjælp af en af de tre fremgangsmåder anført nedenfor:
METODE CYKLUS TEMPERATUR EXPOSITIONSTID
Damp Prævakuum 270° F (132° C) 4 min.
Damp Under tryk 250° F (121°C) 60 min.
Damp* Under tryk* 273° F (134° C)* 20 min.*
BEMÆRK: På grund af de mange variabler, som indgår i sterilisation, bør hver medicinsk enhed evaluere og kontrollere sterilisationsprocessen (f.eks. temperaturer, tider), som bruges til
deres udstyr.
*For brug udenfor USA anbefaler Sundhedsmyndighederne i visse lande en sterilisation i overensstemmelse med disse parametre for at reducere den potentielle risiko for overførelse af
Creutzfeldt Jakobs sygdom, især af kirurgiske instrumenter som kan komme i kontakt med centralnervesystemet.
REKLAMATIONER:
Enhver professionel kunde eller bruger af dette produktsystem, som ønsker at reklamere eller som har en utilfredsstillende erfaring med produktets kvalitet, identitet, holdbarhed, pålidelighed,
sikkerhed, effektivitet og / eller ydeevne bør underrette distributøren, Medtronic Sofamor Danek herom. Distributøren bør ligeledes underrettes omgående, hvis en implanteret INTER FIX™
fusionsanordning med gevind eller INTER FIX™ RP fusionsanordning med gevind fungerer dårligt (det vil sige ikke lever op til den specificerede ydeevne eller på anden vis ikke fungerer efter
hensigten), eller hvis der er mistanke om fejl. Hvis det skulle ske, at et Medtronic Sofamor Danek produkt “fungerer dårligt” og kan have forårsaget eller medvirket til en patients dødsfald eller
en alvorlig læsion, skal distributøren underrettes herom så hurtigt som muligt pr. telefon, fax eller brev. Angiv venligst komponentens(ernes) navn og nummer, serienummer, Deres eget navn
og adresse, detaljer om reklamationens baggrund samt om der ønskes skriftlig redegørelse fra distributøren.
NOTE TIL LÆGEN: Skønt lægen er det uddannede mellemled mellem selskabet og patienten, skal de vigtige medicinske oplysninger indeholdt i dette dokument gives til patienten.
Gælder kun i USA
OBS: FORBUNDSLOVGIVNINGEN (USA) INDSKRÆNKER DETTE PRODUKT TIL SALG UDELUKKENDE EFTER LÆGEORDINATION.
FORSØG PÅ FJERNELSE AF IMPLANTAT:
Hvis det skulle blive nødvendigt at fjerne et INTER FIX™ Fusionsimplantat med gevind eller et INTER FIX™ RP Fusionsimplantat med gevind, kontakt venligst Medtronic Sofamor Danek.
© 2006 Medtronic Sofamor Danek. All rights reserved.
Page 29

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
SVENSKA
Följande text innehåller viktig medicinsk information om de gängade fusionsanordningarna INTER FIX™ och om de gängade fusionsanordningarna INTER FIX™ RP.
BESKRIVNING :
Den gängade fusionsanordningen INTER FIX™ består av en ihålig perforerad metallcylinder och en ändförslutning. Den gängade fusionsanordningen INTER FIX™ RP består av en ihålig perforerad
metallcylinder med en enda stor yttre rundad skåra längs hela den längsgående axeln som sträcker sig till anordningens innerdiameter. Implantatet INTER FIX™ RP:s båda ändar är slutna.
Implantaten INTER FIX™ och INTER FIX™ RP finns i diametrar mellan 12 och 24 mm, och i längder mellan 20 och 29 mm. Implantatet INTER FIX™:s ändförslutningar är anpassade till
cylindrarnas diameter och sätts på cylindrarnas öppna ände när de har fyllts med autogent bentransplantat.
Implantaten INTER FIX™ och INTER FIX™ RP är tillverkade i titanlegering avsett för implanteringsändamål (Ti-6Al-4V) enligt ASTM F136.
Underförstådda garantier för säljbarhet och användbarhet för ett särskilt ändamål eller en särskild användning utesluts uttryckligen. Se Medtronic Sofamor Danek-katalogen eller prislistan
för mer information om garantier och begränsad ansvarighet.
INDIKATIONER:
De gängade fusionsanordningarna INTER FIX™ och de gängade fusionsanordningarna INTER FIX™ RP är avsedda för spinalfusionsingrepp i patienter med fullt utvecklat skelett som lider av
degenerativ disksjukdom (DDD) vid en nivå från L2-S1. Degenerativ sjukdomen definieras som ryggsmärtor av diskursprung med degeneration av disken som bekräftas av patientens berättelse
och röntgenstudier. Dessa patienter kan också ha spondylolistes eller retrolistes upp till grad 1 vid den involverade nivån. Implantaten INTER FIX™ och INTER FIX™ RP ska användas med
autogent bentransplantat och implanteras via ett öppet anteriort ingrepp.
KONTRAINDIKATIONER:
De gängade fusionsanordningarna INTER FIX™ och de gängade fusionsanordningarna INTER FIX™ RP ska inte implanteras i patienter med en aktiv infektion i operationsområdet eller
som är allergiska mot titan eller titanlegering.
VARNING:
• De gängade fusionsanordningarna INTER FIX™ och de gängade fusionsanordningarna INTER FIX™ RP får endast användas av kirurger med erfarenhet av spinalfusionsingrepp och
som har genomgått lämplig utbildning om denna anordning.
• Brist på lämplig erfarenhet och/eller utbildning kan leda till en högre frekvens av ogynnsamma effekter som till exempel kärlskador, neurologiska besvär och/eller urogenitala besvär
(inklusive retrograd ejakulation).
FÖRSIKTIGHETSÅTGÄRDER
•De gängade fusionsanordningarna INTER FIX™:s och INTER FIX™ RP:s säkerhet och effektivitet har inte påvisats hos patienter med följande tillstånd:
- grad II eller högre spondylolistes eller retrolistes;
- mer än en nivå att fusionera;
GÄNGAD FUSIONSANORDNING INTER FIX™
GÄNGAD FUSIONSANORDNING INTER FIX™ RP
VIKTIG MEDICINSK INFORMATION
Page 30

;
- korrektion av tidigare fusionsingrepp mellan kroppar;
L
L
- krav på postoperativ steroid eller icke-steroid antiinflammatorisk medicinering;
- svår övervikt;
- under 18 år eller över 65 år;
- osteoporos, osteopeni och/eller osteomalakia;
- graviditet.
• De gängade fusionsanordningarna INTER FIX™:s och INTER FIX™ RP:s säkerhet och effektivitet har endast påvisats vid anteriora lumbala fusionsingrepp mellan kroppar (ALIF) med
hjälp av autologt bentransplantat.
• Patienter som får den gängade fusionsanordningen INETR FIX™ ska åtminstone ha genomgått sex månaders icke-operativ behandling. Patienter som får den gängade
fusionsanordningen INTER FIX™ RP ska också ha genomgått minst sex månaders icke-operativ behandling.
• Två gängade fusionsanordningar INTER FIX™ ska implanteras bredvid varandra i operationsområdet. En gängad fusionsanordning INTER FIX™ och en gängad fusionsanordning
INTER FIX™ RP måste implanteras bredvid varandra i operationsområdet. Den längsgående skåran på anordningen INTER FIX™ RP gör det lättare att implantera två anordningar
närmare varandra, jämfört med två INTER FIX™-anordningar. Det resulterar följaktligen i en reducerad lateral profil.
• De gängade fusionsanordningarna INTER FIX™:s och INTER FIX™ RP:s långa axel ska vara i anterior-posterior riktning.
• Om implantaten och instrumenten inte tillhandahålls sterila och tydligt är märkta som sådana, måste de steriliseras före användning enligt steriliseringsinstruktioner som bifogas till
denna bipacksedel.
OGYNNSAMMA EFFEKTER :
De ogynnsamma effekterna som visas i tabell I rapporterades från 185 patienter med anordningen INTER FIX™ och 62 kontrollpatienter som deltog i en klinisk “multi-center-studie”, som
genomfördes på flera centra, av den gängade fusionsanordningen INTER FIX™. Dessutom rapporterades ogynnsamma effekter från 116 patienter efter 48 månader och från 109 patienter efter
72 månader i uppföljningsstudien efter godkännande. De uppgifter som presenteras baseras på det totala antalet patienter som deltog i studien vid varje tidpunkt. De ogynnsamma effekterna
som inträffade i de randomiserade och icke-randomiserade behandlingsgrupperna med anordningen INTER FIX™ har slagits ihop för att få en totalsiffra.
TABELL I - OGYNNSAMMA EFFEKTER
Postoperativ
Operativ
Komplikation
Vaskulär intraop 15(8.1) 2(3.2) 15 (8.1) 2(3.2)
Sakroilikala smärtor 5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
Neurologiska 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Ryggsmärtor 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
Incisionala 11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
Spinal effekt 1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
Urologisk 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Andra 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Annan smärta 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
Gastrointestinal 7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
Retrograd ejakulation 2(1.1) 3(1.6) 1(0.5) 6(3.2)
Respiratorisk 3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
Bensmärtor 2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
Trauma 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
Peritoneal 3(1.6) 3(1.6) 0
Vaskulär postop 2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2)
Benfraktur 1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
Implantat
dislokation/avlossning
Smärtor i
transplantatsområdet
Utebliven frakturläk ning 4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13( 12.0) 3(2.6)
Subsidens 2(1.1) 2(1.1) 0
Utebliven frakturläk ning (I
AVVAKTAN PÅ
RESULTAT)
Meningit 1(1.6) 0 1(1.6)
Implantatbrott 5(8.1) 0 5(8.1)
Cancer 1
Död 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
[antal (%)]
KONTROL
INTER FIX
ANT=62
ant=185
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
1(1.6)
(1 dag-<2
månader)
[antal (%)]
INTER FIX
ant=185
KONTROLL
ANT=62
3 månader
2-<5 månader)
(t
[antal(%)]
INTER FIX
ant=185
KONTROL
ANT=62
6 månader
5-<9 månader)
(t
[antal (%)]
INTER FIX
ant=185
12 månader
9-<19 m ånader)
(t
KONTROLL
ANT=62
[antal (%)]
KONTROLL
INTER FIX
ANT=62
ant=185
2
0 1(1.6) 2(1.7)
(0.5)
24 månader
(t
19-< 30
månader)
[antal (%)]
KONTROLL
INTER FIX
ant=185
1
(0.5)
ANT=62
10(5.4) 2(3.2) 4(3.4) 2(1.8)
2 (1.1) 1(0.8) 1(0.9)
OGYNNSAMMA
EFFEKTER
(UNDER 24
MÅNADER)
INTER FIX
ant=185
1
(7.4)
TOTALT
48 månader
(>30 - < 60
60
30<
(t
månader)
INTER FIX
ant=116
0
KONTROLL
ANT=62
72
månader
60
(t
månader)
INTER FIX
ant=109
1 - Sex (6) av 81 manliga patienter i gruppen med INTER FIX™-anordningen (7,4 %) upplevde retrograd ejakulation under 24-månadersundersökningen.
2 - Denna patient (175) har därefter rapporterats som fusionerad ungefär 52 månader efter operationen och inga andra kirurgiska ingrepp eller behandlingar krävdes.
Vaskulära och neurologiska intraoperativa skador var två viktiga ogynnsamma effekter i den kliniska studien. Sammanlagt 15 vaskulära intraoperativa effekter inträffade hos 14 patienter i
gruppen med anordningen INTER FIX™ under 24-månadersundersökningen. Dessa effekter inkluderade: 2 skador på vena cava, 9 skador på v.iliaca, 1 sargad v.hypogastrica, 1 segmentell
venblödare, 1 sakral venskada och 1 ytblödare. Sammanlagt 2 vaskulära intraoperativa skador uppstod hos 2 patienter i kontrollgruppen under samma period. Dessa inkluderade: 1 skada
på en v.iliaca och 1 blödning från benbädden.
Sammanlagt 30 neurologiska effekter inträffade hos 28 patienter i gruppen med anordningen INTER FIX™ under 24-månadersundersökningen. Dessa effekter inkluderade: 1 droppfot, 3
nervrotsskador, 1 foraminal stenos, 1 sympatisk reflexdystrofi, 7 känsellöshet eller brinnande känsla i benen, 2 dysestesi, 3 parestesi, 2 radikulitis, 7 rygg- och/eller bensmärtor, 1 rygg- och/eller
bensmärtor med andra symtom, 1 blixtrande smärta i nedre delen av ryggen samt 1 karpaltunnelsyndrom.
Sammanlagt 10 neurologiska effekter uppstod hos 8 patienter i kontrollgruppen under samma period. Dessa effekter inkluderade: 1 radikulopati med stickningar i extremiteter, 1 kronisk
ryggsmärta med radikulitis, 1 försvagad distributionssymtom, 2 rygg- och bensmärtor med andra symtom, 1 denerverad abducerande magnus muskel och 4 känsellöshet eller värmekänsla i
benen. Under uppföljningsstudien efter godkännande registrerades ytterligare 10 neurologiska effekter hos 10 patienter. Dessa effekter inkluderade 1 radikulopati, 2 cervikala radikulopatier,
1 känsellöshet och stickningar i händerna, 1 känsellöshet eller brännande känsla i benen, 3 rygg- och/eller bensmärtor samt 2 rygg- och/eller bensmärtor med andra symtom. Tabellen med
ogynnsamma effekter framställer dessutom ogynnsamma effekter med ben- och ryggsmärtor och/eller spinaleffekter som diskhöjdkollaps som i några fall hade ett neurologiskt inslag.
Vissa av de ogynnsamma effekterna ledde till kirurgiska åtgärder efter den kliniska försökskirurgin. Dessa extra kirurgiska åtgärder kan klassificeras som revision, avlägsnande, kompletterande
fixering eller reoperation.
ngsgrupperna i IDE-studien samt gruppen med INTER FIX™-anordningen i uppföljningsstudien efter godkännande.
1
Revision: Ett ingrepp som rättar till eller på något sätt modifierar den ursprungliga implantatkonfigurationen.
Avlägsnande: Ett ingrepp som avlägsnar en eller flera komponenter i den ursprungliga implantatkonfigurationen utan att de ersätts med samma typ av försöksanordning.
Kompletterande fixering: Ett ingrepp som avser extra spinalanordningar som inte godkänns som en del av protokollet.
Reoperation: Kirurgiskt ingrep som inte avlägsnar, modifierar eller lägger till ursprungliga implantatkomponenter.
1
I tabell II sammanfattas de sekundära kirurgiska ingreppen för anordningen INTER FIX™ (kombinerad randomiserad och icke-randomiserad) och kontrollbehandli
Tabell II - Sekundära kirurgiska ingrepp
Revision 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Avlägsnande 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Kompletterande
fixering
Reoperation 42 (22.7%) 17
IDE-studie Uppföljningsstudie efter godkännande
Vid tidpunkten 24
månader
INTER FIX™
anordning
ant=185
21 (11.4%) 17
Kontroll
(27.4%)
(27.4%)
ant=62
Vid tidpunkten 48
månader
1
INTER FIX™
anordning
ant=116
7 (6.0%) 3 (2.8%)
33 (28.4%) 17 (15.6%)
Vid tidpunkten 72
månader
INTER FIX™
anordning
ant=109
Page 31

1 - Eftersom efterkontrollen efter 24 månader inte var obligatorisk för kontrollpatienterna finns information för 72 månader inte tillgänglig för alla dessa patienter.
POTENTIELLA OGYNNSAMMA EFFEKTER
I följande text listas potentiella ogynnsamma effekter som kan uppstå vid spinal fusionskirurgi med den gängade fusionsanordningen INTER FIX™ eller med den gängade fusionsanordningen
INTER FIX™ RP. Vissa av dessa ogynnsamma effekter kan tidigare ha rapporterats i tabellen med ogynnsamma effekter.
• Frigöring, krökning, brott, avlossning och/eller komponentvandring.
• Reaktion mot främmande kroppar (allergisk).
• Vävnads- eller nervskada.
• Postoperativ förändring av ryggradskrökning, förlust av korrigering, längd och/eller reduktion.
• Infektion.
• Durala revor.
• Neurologiska besvär.
• Urologiska besvär.
• Ärrbildning.
• Benfraktur.
• Utebliven frakturläkning (eller pseudartros), fördröjd läkning, felställning.
• Den opererade delen av ryggraden upphör att växa. Bortfall av rörlighet eller funktion i ryggraden.
• Komplikationer i donatorns transplantatområde.
• Skador på blodkärl och kardiovaskulära besvär.
• Gastrointestinala komplikationer.
• Reproduktionsorgansbesvär.
• Skador på invärtes organ och bindväv.
• Utveckling av respiratoriska problem.
• Incisionala komplikationer.
• Förändring av den mentala hälsan.
• Dödsfall.
Anmärkning: Ytterligare operationer kan eventuellt vara nödvändiga för att korrigera en del av dessa möjliga ogynnsamma effekter.
KLINISKA RESULTAT:
En klinisk “multi-center-ekvivalensstudie”2 av den gängade fusionsanordningen INTER FIX™ genomfördes i Förenta staterna. Anterior spinal användning av anordningen INTER FIX™ fylld
med autogent ben jämfördes med allogen femoral ring fylld med autogent ben (kontrollgrupp) vid behandling av patienter med symtomatisk degenerativ disksjukdom. Det kliniska försöket
hade inledningsvis en undersökande, randomiserad och kontrollerad utformning. Undersökningsmetoden reviderades senare för att göra det möjligt för patienter att ingå i en icke-randomiserad
grupp, dvs patienter enbart behandlade med anordningen INTER FIX™.
Kriterierna för det kliniska försöket inkluderade symtomatisk degenerativ disksjukdom som yttrade sig som svårbehandlade ben- och/eller ryggsmärtor med positivt diagnostiskt bildfynd;
spinal instabilitet som definierades som förskjutning större än 4 mm eller med en vinkel större än 5° på flexion-/extensionröntgenbilder; symtomatiskt inblandning av en nivå från L2-S1 och
inte större än spondylolistes grad 1. Uttryckligen uteslutna från studien var patienter som redan genomgått ett anteriort fusionsingrepp mellan kroppar i den involverade spinalnivån; patienter
med osteopeni, osteoporos eller osteomalakia eller med begärd stimulering av bentillväxten.
Sammanlagt 185 patienter ingick i behandlingsgruppen med anordningen INTER FIX™.
Ett krav efter godkännande för den gängade fusionsanordningen INTER FIX™ var att utvärdera anordningens långsiktiga prestationsförmåga genom att samla in sammanlagt sex års postoperativ
information från minst 100 patienter. Anordningens långsiktiga prestationsförmåga utvärderades hos 116 patienter fyra år efter operationen och hos 109 patienter sex år efter operationen.
Patientpopulationen bestod av deltagare i den kliniska IDE-studien. Baserat på kriterier från IDE-studien omfattade informationen om dessa patienter en beskrivning av eventuellt ytterligare
kirurgiska ingrepp, radiografisk analys av fusionen samt en smärt- och funktionsanalys.
I tabell III anges de kliniska resultaten från studien som beskriver parametrarna för lyckad fusion, förbättrad smärtintensitet (Oswestry) samt neurologisk status 12, 24, 48 och 72 månader efter
operationen. Den kliniska studiens framgång noterades på basis av en ekvivalensanalys mellan anordningen INTER FIX™ och kontrollgruppen.
I detta kliniska försök definierades fusion som bevis på fast förening av trabekulärt ben, stabilitet av förskjutning (<3mm) och vinkelrörelse (<5°) samt ingen uppklarning på mer än 50 %
för något av implantaten. Dessutom betraktades patienter som gick igenom sekundärkirurgi på grund av utebliven frakturläkning som misslyckade fusioner i beräkningarna. Framgång
med Oswestry smärt/svaghet definierades som åtminstone en 15-poängsförbättring av den postoperativa poängen jämfört med den preoperativa poängen. Framgång med neurologiskt
status definierades som ett postoperativt bibehållande eller en postoperativ förbättring i åtminstone tre av de fyra kategorierna (rörelse, känsel, reflexer och rak benresning) jämfört med det
preoperativa tillståndet. Lyckade generella resultat baserades på en patientdemonstreringsfusion, framgång med Oswestry smärta/svaghet och neurologiskt status. Inga sekundära kirurgiska
ingrepp klassificerades som “misslyckanden”.
2
Anordningen INTER FIX™ var statistiskt inte sämre än kontrollgruppen.
3
Omfattar inte 3 patienter som inte behandlades med anordningen INTER FIX™ på grund av kirurgiska ogynnsamma effekter.
3
Sammanlagt 62 kontrollpatienter deltog i det kliniska försökets randomiserade grupp.
Även de lyckade generella resultaten 12 och 24 månader efter operationen anges i tabell III.
Fusion
Förbättring av
Oswestry
smärta/
svaghet
Patienter med
åtminstone 15
poängsförbättring
från preop
Neurologisk
status
bibehållande
eller förbättring
Lyckade
generella resultat
Tabell III – Kliniska resultat från studien om INTER FIX™-anordningen
12 månaderstal
Prövning Kontroll
96.1%
(122/127)
53.5%
(76/142)
99.3%
(150/151)
42.4%
(61/144)
40.4%
(21/52)
51.9%
(27/52)
(52/52)
21.4%
(12/56)
Delta
(ǻ
100%
-0.097 46.1%
1
)
24 månaderstal
Prövning Kontroll
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
(71/154)
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
(17/57)
Delta
(ǻ1)
-0.043 49.5%
48
månaderstal
Prövning Prövning
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
(53/107)
72
månaderstal
97.6% (82/84)
56.3%
(54/96)
1 - Lägst icke-inferioritetsmarginal som krävs för att göra anspråk på prövningsproduktens icke-inferioritet jämfört med kontrollen.
Alla patienter som omfattades av det kliniska försöket med den gängade fusionsanordningen INTER FIX™ (utan hänsyn till behandlingsgrupp) var föremål för samma medräknings/uteslutningskriterier. Demografiska kännetecken, preoperativa medicinska tillstånd, diagnostiska bildfynd karakteriserade av degenerativ disksjukdom, kirurgisk metod och lokalisering av
den lumbala behandlade nivån undersöktes för att bekräfta jämförbarheten mellan de randomiserade patienterna med anordningen INTER FIX™ och de icke-randomiserade patienterna
med anordningen INTER FIX™. Populationen var statistiskt jämförbar bortsett från kirurgisk metod, preoperativ arbetsstatus och preoperativa diagnostiska bildfynd på osteofysbildning av
vertebrala ändplattor; ärrbildning och/eller förtjockning av annulusfibros, ligamentum flavum och/eller fasettledkapsel samt nucleus pulposus med bråckbildning. Ytterligare statistiska analyser
visade att det inte fanns något samband mellan dessa variabler och skillnaderna i de kliniska fusionsresultaten efter tolv månader, förbättring av Oswestry smärta/svaghet, neurologiskt status
eller lyckade generella resultat med undantag för nucleus pulposus med bråckbildning. Flera patienter i den icke-randomiserade gruppen uppvisade nucleus pulposus med bråckbildning som
konstaterades öka möjligheten till lyckade generella resultat.
En “intent-to-treat”-analys utfördes. Sekundära operationsmisslyckanden, dödsfall, utelämnade patienter som ska följas upp och av andra orsaker utelämnade observationer resulterade i denna
analys i att observationer utelämnades i resultatvariabeln och därför inkluderades i det beräknade talets nämnare, dvs. ansågs som “misslyckanden”. Genom att behandla dessa patienter som
behandlingsmisslyckanden blev den kliniska resultatsiffran avsevärt lägre i “intent-to-treat”-analysen än den i den faktiskt observerade kliniska datan.
Följande tabell (tabell IV) ger resultaten av “intent-to-treat”-analysen av patienterna med anordningen INTER FIX™ men inkluderar inte kontrollpatienternas resultat.
Page 32

Dödsfall, sekundära operationsmisslyckanden, utelämnade uppföljningar och utelämnade observationer, anses
12 månaderstal 24
Fusion
Förbättring av Oswestry smärta/svaghet
Patienter med åtminstone 1 5 poängsförbättring från
preop
Neurologisk status bibehållande eller
förbättring
Lyckade generella resultat
Sekundära operationsmisslyckanden
Uteblivna frakturläkningar
Andra²
Dödsfall
som misslyckanden och inkluderas i talens nämnare
65.9% (122/185) 67.6%
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
33.0% (61/185) 38.4% (71/185) 45.7%
1
5
8
0 1 0 0
månaderstal
(125/185)
(128/185)
48
månaderstal
80.2%
(93/116)
(66/116)
87.9%
(102/116)
(53 /116)
5
3
4
4
72 månaderstal
77.1%
( 84/109)
56.0%
(61 /109)
75.2%
75.2% (82/109)
(82/109)
49.5%
49.5% (54/109)
(54/109)
1
2
1 - Dessa patienter inkluderas i beräkningarna av fusionstalet men anses i andra avseenden som misslyckanden för kliniska försöksändamål.
Tabell IV – "Intent-to-treat"-analys av anordningen INTER FIX™
2 - Patienter som ska följas upp under den period och som genomgått sekundäroperationer av andra orsaker än utebliven frakturläkning anses som misslyckanden för kliniska
försöksändamål.
FÖRPACKNINGAR:
Förpackningarna för samtliga komponenter ska vara obrutna vid leverans. Om ett låne- eller deponeringssystem används bör det noga kontrolleras att uppsättningarna är kompletta och att
inga komponenter, inklusive instrument, är skadade innan användning. Skadade förpackningar eller produkter bör inte användas utan returneras till Medtronic Sofamor Danek.
RENGÖRING OCH SANERING:
Om de inte tillhandahålls sterila måste alla implantat och instrument först rengöras enligt etablerade sjukhusmetoder, före sterilisering och innan de förs in i ett sterilt operationsområde.
Använda instrument måste saneras, rengöras och steriliseras före återanvändning.
Anmärkning: Använd inte rengöringslösningar som innehåller blekmedel eller formalin eftersom de kan skada anordningen.
Alla produkter ska behandlas med försiktighet. Felaktig användning eller hantering kan leda till skada och/eller möjlig felaktig funktion av anordningen.
STERILISERING:
Såvida det inte anges att implantat och instrument i en oöppnad steril förpackning som tillhandahålls av företaget, är sterila och de är tydligt märkta som sådana, måste samtliga implantat och
instrument som används vid kirurgi steriliseras av sjukhuset före användning. Avlägsna allt förpackningsmaterial före sterilisering. Endast sterila produkter får finnas i operationsområdet. För
en sterilitetsgarantinivå på 10-6 är det rekommenderat att produkterna ångsteriliseras av sjukhuset med hjälp av en av de tre uppsättningar processparametrar nedan:
METOD CYKEL TEMPERATUR EXPONERINGSTID
Ånga Pre-vacuum 270° F (132° C) 4 min.
Ånga Atmosfärtryck 250° F (121° C) 60 min.
Ånga* Atmosfärtryck* 273° F (134° C)* 20 min.*
ANMÄRKNING: Eftersom många variabler är inblandade vid sterilisering bör varje medicinsk enhet bedöma och granska steriliseringsprocessen (t.ex. temperaturer och tider) för deras
utrustning.
*För användning utanför USA rekommenderar en del hälso- och sjukvårdsmyndigheter sterilisering enligt dessa parametrar särskilt av kirurgiska instrument som kan komma i kontakt med
det centrala nervsystemet i syfte att minimera den potentiella risken för överföring av sjukdomen Creutzfeldt-Jakob.
PRODUKTREKLAMATION:
Alla professionellt yrkesverksamma inom sjuk- och hälsovård (kunder eller användare av detta produktsystem) som har klagomål eller är otillfredsställda med produktens kvalitet, identitet,
hållbarhet, tillförlitlighet, säkerhet, effektivitet och/eller prestationsförmåga bör underrätta distributören Medtronic Sofamor Danek. Vidare, om de implanterade fusionsanordningarna INTER
FIX™:s eller INTER FIX™ RP:s komponenter någonsin ”fungerar bristfälligt” (dvs. inte uppfyller prestationsspecifikationerna eller på annat sätt inte fungerar som förväntat) eller om detta
misstänks, bör distributören omedelbart underrättas. Om någon Medtronic Sofamor Danek-produkt någonsin skulle “fungera bristfälligt” och eventuellt kan ha orsakat eller bidragit till att en
patient har avlidit eller skadats svårt bör distributören underrättas så snart som möjligt per telefon, fax eller brev. När en reklamation lämnas in bör du uppge komponentens/-ernas namn och
nummer, partinummer, namn och adress, reklamationens natur samt meddela huruvida en skriftlig rapport begärs från distributören.
ANMÄRKNING TILL LÄKARE: Även om läkaren är den förmedlande länken mellan företaget och patienten måste den viktiga medicinska informationen i detta dokument framföras till
patienten.
Gäller endasti i USA
FÖRSIKTIGHETSÅTGÄRD: FEDERAL LAG (USA) BEGRÄNSAR DESSA ANORDNINGAR TILL FÖRSÄLJNING AV ELLER ENLIGT ORDINATION AV LÄKARE.
AVLÄGSNANDE AV ANORDNINGEN :
Var god kontakta Medtronic Sofamor Danek om blir nödvändigt att avlägsna någon av de gängade fusionsanordningarna INTER FIX™ eller de gängade fusionsanordningarna INTER FIX™ RP.
© 2006 Medtronic Sofamor Danek. Eftertryck förbjudes.
Page 33

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
NORSK
Det følgende inneholder viktig medisinsk informasjon om INTER FIX™ gjenget fiksasjonsutstyr og INTER FIX™ RP gjenget fiksasjonsutstyr.
BESKRIVELSE:
INTER FIX™ gjenget fiksasjonsutstyr består av en hul, perforert metallsylinder og endedeksel. INTER FIX™ RP gjenget fiksasjonsutstyr består av en hul, perforert metallsylinder med ett enkelt
stort, avrundet radiusspor langs hele den langsgående aksen som forlenges inn i enhetens indre diameter. Begge endene av INTER FIX™ RP-implantatet er lukket.
INTER FIX™- og INTER FIX™ RP-implantatene kan fås med forskjellige diametere fra 12 til 24 mm og lengder fra 20 til 29 mm. Størrelsen på INTER FIX™-implantatenes endehetter er
tilpasset sylindrenes diameter og settes på sylindrenes åpne ende etter at de er fylt med autogent bengraft.
INTER FIX™- og INTER FIX™ RP-implantatene er laget av titanlegering til bruk ved implantering (Ti-6Al-4V) i henhold til ASTM F136.
Implisitte garantier for salgbarhet og egnethet for et bestemt formål eller bruk, utelukkes spesielt. Ytterligere informasjon om garantier og ansvarsbegrensninger finnes i Medtronic Sofamor
Danek-katalogen eller prislisten.
INDIKASJONER:
INTER FIX™ og INTER FIX™ RP gjenget fiksasjonsutstyr er indisert ved kirurgiske inngrep for sammenvoksingsstrategier i columna hos pasienter med fullt utvokst skjelett og degenerativ
skivesykdom på ett nivå fra L2-S1. Degenerativ skivesykdom er definert som ryggsmerter av diskogen opprinnelse med degenerering av skiven bekreftet av pasientens historie og
røntgenundersøkelse. Pasienter med degenerativ skivesykdom kan også presentere en Grad 1 spondylolistese eller retrolistese på det aktuelle nivået. INTER FIX™- og INTER FIX™ RPimplantatene skal brukes sammen med autogent bengraft og skal implanteres via en åpen fremre tilgang.
KONTRAINDIKASJONER:
INTER FIX™ og INTER FIX™ RP gjenget fiksasjonsutstyr må ikke implanteres hos pasienter som har en aktiv infeksjon på operasjonsstedet, eller som er allergiske overfor titan eller
titanlegering.
ADVARSLER:
• INTER FIX™ og INTER FIX™ RP gjenget fiksasjonsutstyr må bare brukes av kirurger som har erfaring med sammenvoksingsprosedyrer i columna, og som har fått egnet opplæring i
bruken av dette utstyret.
• Manglende erfaring og/eller opplæring kan føre til en hyppigere forekomst av bivirkninger, for eksempel vaskulær skade, nevrologiske og/eller urogenitale hendelser (inkludert
retrograd ejakulasjon).
INTER FIX™ GJENGET FIKSASJONSUTSTYR
INTER FIX™ RP GJENGET FIKSASJONSUTSTYR
VIKTIG MEDISINSK INFORMASJON
Page 34

FORHOLDSREGLER:
L
L
• Sikkerheten ved og effektiviteten til INTER FIX™ og INTER FIX™ RP gjenget fiksasjonsutstyr er ikke etablert hos pasienter med følgende forhold:
- spondylolistese eller retrolistese av Grad II eller over;
- nødvendighet av sammenvoksing av mer enn ett nivå;
- korrigering av tidligere inngrep som er foretatt for å oppnå sammenvoksing av virvler;
- behov for postoperative steroidale eller ikke-steroidale antiinflammatoriske midler;
- betydelig overvekt;
- pasienter som er yngre enn 18 år og eldre enn 65 år;
- osteoporose, osteopeni og/eller osteomalasi;
- graviditet.
• Sikkerheten ved og effektiviteten til INTER FIX™ og INTER FIX™ RP gjenget fiksasjonsutstyr er kun etablert for inngrep for sammenvoksing av virvellegemer i den lumbale del av
fremre columna (ALIF) der det brukes autologt bengraft.
• Pasienter som får operert inn INTER FIX™ gjenget fiksasjonsutstyr, må minst ha fått seks måneders ikke-operativ behandling. Pasienter som får operert inn INTER FIX™ RP gjenget
fiksasjonsutstyr, må minst ha fått seks måneders ikke-operativ behandling.
• Det må implanteres to INTER FIX™ gjengete fiksasjonsenheter ved siden av hverandre på det kirurgiske nivået. Ett INTER FIX™ gjenget fiksasjonsutstyr og ett INTER FIX™ RP
gjenget fiksasjonsutstyr må implanteres ved siden av hverandre på det kirurgiske nivået. Det langsgående sporet på INTER FIX™ RP-enheten gjør det lettere å implantere to enheter
nærmere hverandre enn det som er mulig med to INTER FIX™-enheter, noe som fører til en redusert sideprofil.
• Den lange aksen til INTER FIX™ og INTER FIX™ RP gjenget fiksasjonsutstyr bør ligge i retningen foran-bak.
• Implantatene og instrumentene må steriliseres før bruk i henhold til retningslinjene for sterilisering som følger med i denne dokumentasjonen, med mindre de leveres sterile og er
tydelig merket sterile.
BIVIRKNINGER:
Hos 185 pasienter som fikk implantert INTER FIX™-utstyret og 62 kontrollpasienter som deltok i den kliniske multisenterstudien av INTER FIX™ gjenget fiksasjonsutstyr ble det rapportert
om bivirkningene som er angitt i Tabell I. Dessuten ble det i etter-valideringsstudien rapportert bivirkninger fra 116 pasienter etter 48 måneder, og fra 109 pasienter etter 72 måneder.
Forholdstallene som her er angitt, er basert på det totale antallet pasienter som deltok i studien på hvert tidspunkt. Bivirkningene som forekom i den randomiserte og den ikke-randomiserte
INTER FIX™-behandlingsgruppen, er lagt sammen for å gi et samlet forholdstall.
TABELL I - BIVIRKNINGER
N=62
3 måneder
(t
måneder)
[Antall (%)]
INTER FIX
N=185
2 - <5
KONTROL
N=62
6 måneder
5 - <9 måned er)
(t
[Antall (%)]
INTER FIX
N=185
KONTROLL
Postoperativ
Operativ
Komplikasjon
Vaskulær intraoperativ 15(8.1) 2(3.2) 15 (8.1) 2(3.2)
Sakroilikale smerter 5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
Nevrologisk 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Ryggsmerte 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
Ved incisjon 11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
Hendelse i columna 1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
Urologisk 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Annet 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Andre smerter 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
Gastrointestinal 7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
Retrograd ejakulasjon 2(1.1) 3(1.6) 1(0.5) 6(3.2)
Åndedrettsbesvær 3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
Smerter i bena 2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
Trauma 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
Peritoneal 3(1.6) 3(1.6) 0
Vaskulær postoperativ 2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
Benbrudd 1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
Forskyvning/løsning av
implantat
Smerte ved donorstedet
for bengraft
Manglende
sammenvoksing
Synkning 2(1.1) 2(1.1) 0
Manglende
sammenvoksing
(RESULTATER
AVVENTES
)
Meningitt 1(1.6) 0 1(1.6)
Brekking av implantat 5(8.1) 0 5(8.1)
Cancer 1
[Antall (%)]
KONTROL
INTER FIX
N=185
N=62
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
1(1.6)
(1 dag -<2
måneder)
[Antall (%)]
INTER FIX
N=185
KONTROLL
Død 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
12 måneder
(t
9 - <19
måneder)
[Antall (%)]
KONTROLL
INTER FIX
N=62
2
0 1(1.6) 2(1.7)
N=185
N=62
(0.5)
24 måneder
(t
19 - <30
måneder)
[Antall (%)]
KONTROLL
INTER FIX
N=185
1
(0.5)
48 mnd.
(>30 - < 60
30<60
(t
mnd.)
INTER FIX
N=116
0
N=62
TOTALT ANTALL
BIVIRK-NINGER
(24 MND.)
INTER FIX
N=185
1
(7.4)
KONTROLL
N=62
2 (1.1) 1(0.8) 1(0.9)
72 mnd.
(t60
mnd.)
INTER FIX
N=109
1 - Seks (6) av 81 menn som deltok i INTER FIX™-gruppen (7,4 %), opplevde retrograd ejakulasjon i løpet av 24 månedersevalueringen.
2 - Denne pasienten (175) er senere blitt rapportert med sammenvoksing ca. 52 måneder etter inngrepet, og ingen annen kirurgisk prosedyre eller behandling var nødvendig.
To alvorlige bivirkninger i det kliniske forsøket var intraoperative vaskulære og nevrologiske skader. Det inntraff til sammen 15 vaskulære intraoperative hendelser hos 14 pasienter i INTER
FIX™-gruppen i løpet av 24 månedersevalueringen. Disse hendelsene omfattet: 2 skader på vena cava, 9 skader på vena iliacus, 1 laserert hypogastrisk vene, 1 segmentær veneblødning,
1 sakral veneskade og 1 overfladisk blødning. Det inntraff til sammen 2 vaskulære intraoperative skader hos 2 pasienter i kontrollgruppen i samme periode. Disse omfattet: 1 skade på vena
iliacus og 1 blødning fra benleiet.
Det inntraff til sammen 30 nevrologiske hendelser hos 28 pasienter i INTER FIX™-gruppen i løpet av 24 månedersevalueringen. Dette omfattet: 1 pasient med dropfoot, 3 med skader på
nerverøtter, 1 med foraminal stenose, 1 med dystrofirefleks, 7 med nummenheter eller brennende følelse i bena, 2 med dysestesi, 3 med parestesi, 2 med radikulitt, 7 med rygg- og/eller
bensmerte, 1 med rygg- og/eller bensmerte med andre symptomer, 1 med jagende smerter nederst i ryggen og 1 med karpaltunnelsyndrom.
Det inntraff til sammen 10 nevrologiske hendelser hos 8 pasienter i kontrollgruppen i samme periode. Disse hendelsene omfattet: 1 pasient med radikulopati med prikkende ekstremiteter,
1 med kroniske ryggsmerter med radikulitt, 1 med svekkede fordelingssymptomer, 2 med ryggsmerter og bensmerter med andre symptomer, 1 med denervert musculus abductor magnus
og 4 med nummenhet i bena eller varme ben. I etter-valideringsstudien ble det registrert ytterligere 10 nevrologiske hendelser hos 10 pasienter. Dette omfattet: 1 pasient med radikulopati,
2 med cervikal radikulopati, 1 med nummenhet og kribling i hendene, 1 med nummenhet eller brennende følelse i bena, 3 med rygg- og/eller bensmerte og 2 med rygg- og/eller bensmerter
med andre symptomer. I tillegg angir denne tabellen over bivirkninger bensmerter og ryggsmerter og/eller hendelser i columna, for eksempel sammenfall av skivehøyden som i noen tilfeller
hadde en nevrologisk komponent.
Noen av bivirkningene førte til kirurgiske inngrep etter inngrepet som hørte inn under det kliniske forsøket. Disse ekstra kirurgiske inngrepene kan klassifiseres som korrigering, fjerning,
supplerende fiksering eller nye operasjoner
gjennom IDE-studien og i INTER FIX™-gruppen i løpet av etter-valideringsstudien.
1
Korrigering: Et inngrep som justerer eller på én eller annen måte endrer implantatets originale konfigurasjon.
Fjerning: Et inngrep som fjerner én eller flere komponenter i implantates originale konfigurasjon uten gjeninnsetting med samme type forsøksenhet.
Supplerende fiksering: Et inngrep der det opereres inn ekstra enheter for columna, som ikke er godkjent som en del av protokollen.
Ny operasjon: Et kirurgisk inngrep som ikke fjerner, endrer eller legger til originale implantatkomponenter.
1
. Tabell II oppsummerer de sekundære kirurgiske inngrepene i INTER FIX™ (kombinert randomisert og ikke-randomisert) og kontrollbehandlingsgrupper
Page 35

Tabell II – Sekundære kirurgiske inngrep
Korrigering 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Fjerning 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Supplerende
fiksering
Ny operasjon 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
1 - Da det ikke ble krevd oppfølging fra kontrollpasientene etter 24 måneder, finnes det ikke tilgjengelig informasjon for alle disse pasientene i etter 72 måneder.
MULIGE BIVIRKNINGER:
Nedenfor finnes en liste over mulige bivirkninger som kan opptre ved kirurgiske inngrep for sammenvoksing av columna med INTER FIX™ eller INTER FIX™ RP gjenget fiksasjonsutstyr.
Noen av disse bivirkningene er kanskje allerede nevnt i tabellen over bivirkninger.
• Komponentene kan gå fra hverandre, bøye seg, brekke, løsne og/eller forskyve seg.
• Fremmedlegemereaksjon (allergisk reaksjon).
• Vev- eller nerveskade.
• Postoperativ endring i columnas normale kurvatur, tap av korrigering, høyde og/eller reponering.
• Infeksjon.
• Rifter i dura.
• Lidelser i nervesystemet.
• Urologiske lidelser.
• Arrdannelse.
• Benbrudd.
• Manglende sammenvoksing (eller pseudartrose), forsinket sammenvoksing, feil sammenvoksing.
• Opphør av mulig vekst av den opererte delen av columna. Tap av mobilitet eller funksjon i columna.
• Komplikasjoner ved donorstedet for bengraftet.
• Skade på blodkar og kardiovaskulær lidelse.
• Gastrointestinale komplikasjoner.
• Lidelser i reproduksjonsorganene.
• Skade på indre organer og bindevevet.
• Utvikling av åndedrettsproblemer.
• Komplikasjoner forbundet med incisjonen.
• Endringer i psykisk tilstand.
• Død.
INTER FIX™-
IDE-studie Etter-godkjennelsesstudie
Etter 24 mnd. Etter 48 mnd. Etter 72 mnd.
enhet
N=185
Kontroll
N=62
1
INTER FIX™- enhet
N=116
INTER FIX™- enhet
N=109
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
Merk: Ytterligere kirurgi kan være nødvendig for å korrigere noen av disse mulige bivirkningene.
KLINISKE RESULTATER:
Et multisenterekvivalent2 klinisk forsøk med INTER FIX™ gjenget fiksasjonsutstyr er utført i USA. Forsøket sammenlignet bruk på fremre deler av columna av INTER FIX™-enheten fylt med
autogent bengraft med anulus femoralis-allograft fylt med autogent ben i kontrollgruppen ved behandling av pasienter med symptomatisk degenerativ skivelidelse. I starten hadde det kliniske
forsøket en undersøkende, randomisert og kontrollert oppbygning. Senere ble forskningsplanen endret til å tillate opptak av pasienter i en ikke-randomisert undergruppe, det vil si pasienter
som kun ble behandlet med INTER FIX™-enheten.
Inklusjonskriteriene for det kliniske forsøket omfattet symptomatisk degenerativ skivelidelse, konstatert ved intraktable smerter i ben og/eller rygg med positiv røntgendiagnose; spinal ustabilitet
definert ved en forskyvning på mer enn 4 mm eller en vinkel på over 5° på røntgenbilder med fleksjon/ekstensjon; symptomatisk involvering av ett enkelt nivå fra L2-S1 og ikke over Grad 1
spondylolistese. Eksklusjonskriteriene omfattet pasienter som tidligere hadde gjennomgått et inngrep for sammenvoksing av virvellegemer i fremre deler av columna på det involverte nivået,
hadde osteopeni, osteoporose eller osteomalasi, eller krevde stimulering av benvekst.
Det ble opptatt i alt 185 pasienter i forsøkets INTER FIX™-behandlingsgruppe
Et etter-godkjennelseskrav for INTER FIX™ gjenget fiksasjonsutstyr var å evaluere utstyrets langsiktige virkninger gjennom postoperative data fra minst 100 pasienter over 6 år. Utstyrets
langsiktige ytelser ble vurdert på 116 pasienter 4 år etter operasjonen og på 109 pasienter 6 år etter operasjonen. Pasientgruppen ble hentet fra deltakere i IDEs kliniske prøve. Ut fra
kriterier fra IDE-studien omfattet informasjonene om disse pasientene en beskrivelse av eventuelle nye kirurgiske prosedyrer, radiografisk vurdering av sammenvoksing og en vurdering av
smerte og funksjon.
Tabell III viser de kliniske resultatene fra studien, med en beskrivelse av parameterne for vellykket sammenvoksing, vellykket resultat angående smerte (Oswestry) og vellykket resultat
angående nevrologisk status 12, 24, 48 og 72 måneder etter den kirurgiske behandlingen. Det vellykkede utfallet for dette kliniske forsøket ble notert på grunnlag av en ekvivalensanalyse
mellom INTER FIX™-gruppen og kontrollgruppen.
I dette kliniske forsøket ble sammenvoksing definert som bevis for fast forening av trabekulære ben, stabilitet av forskyvning (<3 mm) og vinkelbevegelse (<5 °) og ingen oppklaringer på mer
enn 50 % omkring hvert implantat på røntgenbilder. Pasienter som gjennomgikk et sekundært inngrep på grunn av manglende sammenvoksing, ble definert som manglende sammenvoksinger
i beregningene. Et vellykket resultat for Oswestry smerter/invaliditet ble definert som en forbedring på minst 15 punkt i den postoperative scoringen i forhold til den preoperative scoringen. Et
vellykket resultat for nevrologisk tilstand ble definert som en opprettholdelse eller en forbedring i minst 3 av de 4 nevrologiske kategoriene (motorisk, sensorisk, reflekser og Lasègue) i den
postoperative tilstand i forhold til den preoperative tilstanden. Et samlet vellykket resultat ble basert på en påvist sammenvoksing, et vellykket resultat for Oswestry smerter/invaliditet og et
vellykket resultat for den nevrologiske tilstanden, samt intet sekundært kirurgisk inngrep klassifisert som “mislykket”.
2
INTER FIX™-enhet ikke statistisk dårligere enn kontroll.
3
Omfatter ikke 3 pasienter som ikke fikk behandling med INTER FIX™ på grunn av uønskede kirurgiske bivirkninger.
3
. Det ble i alt opptatt 62 kontrollpasienter i det kliniske forsøkets randomiserte undergruppe.
De globale vellykkede resultatene 12 og 24 måneder etter operasjonen er også angitt i tabell III.
Sammenvoksing
Forbedring,
Oswestry
smerter/invalidite
t
Pasienter med
minst 15 punkts
forbedring i forhold
til den
preoperative
tilstanden
Opprettholdelse
eller forbedring
av nevrologisk
tilstand
Samlet vellykket
resultat
Tabell III – Kliniske resultater fra INTER FIX™-studien
Forholdstall, 12 mnd. Forholdstall, 24 mnd.
Prøving Kontroll
96.1%
(122/127)
53.5%
(76/142)
99.3%
(150/151)
42.4%
(61/144)
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
(12/56)
Delta
1
(ǻ
)
Prøving Kontroll
-0.097 46.1%
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
(71/154)
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
(17/57)
Delta
Forholdstall,
(ǻ1)
48 mnd.
Prøving
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
-0.043 49.5%
(53/107)
Forholdstall, 72
mnd.
Prøving
97.6% (82/84)
56.3%
(54/96)
Page 36

1 - Minimal margin som kreves for å kunne hevde at prøvens resultater ikke er dårligere enn kontrollen.
Alle pasienter som deltok i det kliniske forsøket med INTER FIX™ gjenget fiksasjonsutstyr, uansett behandlingsgruppe, ble opptatt etter de samme inklusjons-/eksklusjonskriteriene. For
å dokumentere sammenlignbarheten av de randomiserte INTER FIX™-pasientene og de ikke-randomiserte INTER FIX™-pasientene, ble det foretatt en undersøkelse av demografiske
karakteristikker, preoperative medisinske forhold, røntgendiagnostikk med karakteristikker for funn av degenerativ skivelidelse, operativ tilnærming og plassering av det behandlede lumbale
nivået. Pasientgruppen var statistisk sammenlignbar med unntak av operativ tilnærming, preoperative arbeidsforhold og preoperativ røntgendiagnostikk med funn av osteofyttdannelse i
de vertebrale endeplatene; arrdannelse og/eller fortykning av anulus fibrosus, ligamentum flavum og/eller fasettleddkapselen og herniert nucleus pulposus. Ytterligere statistiske analyser
viste at disse variablene ikke var forbundet med forskjeller i de kliniske resultatene etter 12 måneders utvikling med hensyn til sammenvoksing, forbedring av Oswestry smerte/invaliditet,
nevrologisk status eller samlet vellykket utfall, med unntak av herniert nucleus pulposus. Flere pasienter i den ikke-randomiserte gruppen hadde herniert nucleus pulposus, som viste seg å
øke muligheten for et samlet vellykket resultat.
Det ble gjennomført en analyse med behandlingshensikt. I denne analysen ble følgende tilfeller ansett for å være “mislykkede” resultater: mislykkede sekundære inngrep, dødsfall og
pasienter som var tapt for oppfølgning, og manglende observasjoner på grunn av andre årsaker som resulterte i manglende observasjoner for de endelige variablene og som derfor inngikk
i det beregnede forholdstalls nevnere. Ved å regne disse pasientene for å ha mislykket resultat, ble forholdstallene for det kliniske resultatet i analysen med behandlingshensikt betydelig
lavere enn dem for de reelt observerte kliniske dataene.
Følgende tabell (Tabell IV) viser resultatene for analysen med behandlingshensikt for INTER FIX™-pasientgruppen, men omfatter ikke resultatene for kontrollgruppen.
Tabell IV - analyse med behandlingshensikt for INTER FIX™-enhet
Dødsfall, mislykkede sekundære inngrep, pasienter tapt for oppfølgning og manglende observasjoner
Forholdstall, 12
Sammenvoksing
Forbedring, Oswestry smerter/invaliditet
Pasienter med minst 15 punkts forbedring i
forhold til den preoperative tilstanden
Opprettholdelse eller forbedring av
nevrologisk tilstand
Samlet vellykket resultat
Mislykkede sekundære inngrep
Manglende sammenvoksing
Annet
Dødsfall
1 - Disse pasientene inngår i beregningen av forholdstallet for sammenvoksing, men anes for øvrig for å ha gitt et mislykket resultat for den kliniske målsetningen.
2 - Pasienter hvis evalueringsdata for oppfølging skal innhentes i denne perioden, og som har gjennomgått sekundært inngrep av andre årsaker enn manglende sammenvoksing, anses for
å ha gitt et mislykket resultat for den kliniske målsetningen.
anses for å være mislykkede behandlingsresultater og inngår i forholdstallenes nevner
mnd.
65.9% (122/185) 67.6%
81.1% (150/185) 69.2%
1
2
41.1% (76/185) 42.7% (79/185) 56.9%
33.0% (61/185) 38.4% (71/185) 45.7%
5
8
0 1 0 0
Forholdstall,
24 mnd
(125/185)
(128/185)
5
3
Forholdstall,
48 mnd.
80.2%
(93/116)
(66/116)
87.9%
(102/116)
(53 /116)
4
4
Forholdstall, 72
( 84/109)
(61 /109)
75.2% (82/109)
mnd.
77.1%
56.0%
75.2%
(82/109)
49.5%
(54/109)
1
2
EMBALLASJE:
Emballasjen for hver av komponentene skal være hel når de mottas. Hvis man bruker et utlåns- eller annet leveringssystem, må det kontrolleres nøye at alle settene er fullstendige, og
alle komponenter og instrumenter må før bruk kontrolleres nøye for å sikre at ingen av dem viser tegn på skade. Skadede emballasjer eller produkter må ikke brukes og må returneres til
Medtronic Sofamor Danek.
RENGJØRING OG DEKONTAMINERING:
Hvis de ikke leveres sterile, skal alle instrumenter og implantater rengjøres ved hjelp av etablerte sykehusteknikker før sterilisering og innføring i et sterilt kirurgisk område. Brukte instrumenter
skal dekontamineres, rengjøres og steriliseres før de brukes på nytt.
Merk: Bruk ikke rengjøringsmidler som inneholder blekemidler eller formalin, slike løsninger kan skade enheten.
Alle produktene må behandles varsomt. Uriktig bruk eller håndtering kan føre til skade og/eller til at utstyret ikke fungerer riktig.
STERILISERING:
Med mindre de er tydelig merket som sterile i en uåpnet steril pakke som leveres av selskapet, må alle implantater og instrumenter som brukes i kirurgi steriliseres av sykehuset før bruk. Fjern
alt emballasjemateriale før sterilisering. Bare sterile produkter må innføres i det kirurgiske området. For et 10-6 steriliseringsgarantinivå anbefales det at disse produktene dampsteriliseres på
sykehuset ved hjelp av én av de tre prosessparametrene nedenfor :
METODE SYKLUS TEMPERATUR EKSPONERINGSTID
Damp Prevakuum 270°F (132°C) 4 min.
Damp Under trykk 250°F (121°C) 60 min.
Damp* Under trykk* 273°F (134°C)* 20 min.*
MERK: Fordi det er mange variabler involvert i sterilisering, må hver medisinske fasilitet evaluere og kontrollere steriliseringsprosessen (f.eks. temperaturer, tider) for sitt utstyr.
*For bruk utenfor USA kan noen lokale helsemyndigheter anbefale sterilisering i henhold til disse parametrene for å minimere den potensielle faren for overføring av Creutzfeldt Jakobs sykdom,
særlig av kirurgiske instrumenter som kan komme i kontakt med sentralnervesystemet.
KLAGER PÅ PRODUKTET:
Alle helsearbeidere (for eksempel kunder eller brukere av dette systemet eller disse produktene) som har klager på eller har vært misfornøyde med produktets kvalitet, identitet, varighet,
pålitelighet, sikkerhet, effektivitet og/eller ytelse, bør underrette forhandleren, Medtronic Sofamor Danek om dette. Videre, hvis noe av det implanterte INTER FIX™ eller INTER FIX™ RP
fiksasjonsutstyret ”virker dårlig” (det vil si at det ikke oppfyller ytelsesspesifikasjonene eller på annen måte ikke fungerer som beregnet), eller det er mistanke om dette, skal forhandleren
underrettes om dette umiddelbart. Hvis et Medtronic Sofamor Danek-produkt har “fungert dårlig” og har forårsaket eller medvirket til dødsfall eller alvorlig skade på en pasient, skal forhandleren
underrettes om dette så snart som mulig per telefon, faks eller skriftlig. Når en klage sendes, vennligst oppgi komponentens(enes) navn og nummer, varenummer(re), ditt navn og din adresse,
klagens art og om hvorvidt det bes om en skriftlig rapport fra forhandleren.
MERKNAD TIL LEGEN: Selv om legen er det faglige leddet mellom firmaet og pasienten, må pasienten underrettes om den viktige medisinske informasjonen i dette dokumentet.
GJELDER KUN USA
FORSIKTIG: FØDERAL LOVGIVNING I USA ANGIR AT DETTE PRODUKTET BARE KAN SELGES AV ELLER ETTER ORDINERING AV LEGE.
FORSØK PÅ Å FJERNE ENHETEN:
Hvis det er nødvendig å fjerne en INTER FIX™ eller en INTER FIX™ RP gjenget fiksasjonsenhet, ringer du Medtronic Sofamor Danek.
© 2006 Medtronic Sofamor Danek. Alle rettigheter forbeholdes.
Page 37

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
SUOMI
Alla oleva teksti sisältää tärkeitä lääketieteellisiä tietoja laitteista INTER FIX™ kierteitetty fuusiolaite ja INTER FIX™ RP kierteitetty fuusiolaite.
SELOSTUS:
INTER FIX™ kierteitetyn fuusiolaitteen rakenne on ontto, rei’itetty metallisylinteri, jonka päässä on kansi. INTER FIX™ RP kierteitettyyn fuusiolaitteeseen kuuluu ontto,
rei’itetty metallisylinteri, jossa on yksi ainoa suuri koko pituusakselin mittainen laitteen sisähalkaisijaan ulottuva ulkopuolinen uurre. Molemmat INTER FIX™ RP-istutteen
päät ovat umpinaiset.
INTER FIX™ ja INTER FIX™ RP-istutteita on saatavina halkaisijaltaan 12 mm – 24 mm ja pituudeltaan 20 mm – 29 mm. INTER FIX™ istutteiden päät ovat kooltaan
sylinterin halkaisijan mukaisia ja niitä käytetään sylintereiden avoimiin päihin autogeenisella luusiirteellä täytettyinä.
INTER FIX™ ja INTER FIX™ RP-istutteet on valmistettu istutekäyttöön tarkoitetusta titaaniseoksesta (Ti-6Al-4V), joka vastaa standardia ASTM F-136.
Oletetut kaupalliset tai soveltuvuustakuut tiettyyn tarkoitukseen tai käyttötapaan ovat poissuljetut. Katso takuita, vastuun rajoituksia tai hintoja koskevia lisätietoja Medtronic Sofamor Danekhakemistosta.
INDIKAATIOT:
INTER FIX™ kierteitetty fuusiolaite ja INTER FIX™ RP kierteitetty fuusiolaite on tarkoitettu selkärangan luufuusiota varten aikuispotilaille, joiden selkäranka on täysin
kehittynyt ja joilla on degeneratiivinen välilevytauti (DDD) väliltä L2 – S1. DDD määritellään välilevyperäisenä selkäkipuna, kun välilevyn rappeutuminen on todettu
sairaskertomuksen ja läpivalaisun avulla. Näillä degeneratiivisen välilevyn sairauspotilailla saattaa olla Asteen I nikamansiirtymä tai taakseluiskahdus. INTER FIX™ ja
INTER FIX™ RP-istutteita on käytettävä autogeenisen luusiirteen kanssa ja ne on istutettava avoimella sisäpuolisella menetelmällä.
KONTRAINDIKAATIOT:
INTER FIX™ kierteitettyä fuusiolaitetta ja INTER FIX™ RP kierteitettyä fuusiolaitetta ei saa istuttaa potilaille, joilla on aktiivi infektio leikkausalueella tai allergia titaanille
tai titaaniseoksille.
VAROITUKSIA:
• INTER FIX™ kierteitettyä fuusiolaitetta ja INTER FIX™ RP kierteitettyä fuusiolaitetta saavat käyttää vain kirurgit, joilla on kokemusta selkärangan luufuusiomenetelmistä
ja jotka ovat saaneet tätä laitetta koskevan riittävän koulutuksen.
INTER FIX™ KIERTEITETTY FUUSIOLAITE
INTER FIX™ RP KIERTEITETTY FUUSIOLAITE
TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
Page 38

• Puutteellinen kokemus la/tai koulutus voi aiheuttaa lisääntyneen määrän haitallisia sivuvaikutuksia, kuten verisuonten vauriot, neurologiset viat ja/tai virtsa-
O
L
O
L
L
L
I
I
/sukupuolihäiriöitä (kuten sisään suuntautuva siemensyöksy).
VAROTOIMET:
• INTER FIX™ kierteitetyn fuusiolaitteen ja INTER FIX™ RP kierteitetyn fuusiolaiteen turvallisuutta ja tehokkuutta koskevia tietoja ei ole käytettävissä seuraavissa
olosuhteissa:
- asteen II tai suurempi spondylolistesia tai retrolistesia;
- useampi kuin yksi fuusioitava taso;
- aiemman nikamafuusion uusinta;
- leikkauksen jälkeisen tulehduksen hoidon tarve steroidisilla tai ei-steroidisilla lääkkeillä;
- huomattava ylipainoisuus;
- alle 18 tai yli 65 vuoden ikä;
- luun haurastuma, luukato ja/tai luun pehmenemistauti;
- raskaus.
• INTER FIX™ kierteitetyn fuusiolaitteen ja INTER FIX™ RP kierteitetyn fuusiolaiteen turvallisuus ja tehokkuus on todettu ainoastaan sisäpuolisessa vyötärön
nikamafuusiossa (ALIF) autogeenista luusiirrettä käyttäen.
• Potilailla, joille laitetaan INTER FIX™ kierteitetty fuusiolaite, olisi oltava ainakin kuuden kuukauden hoito ilman leikkausta. Potilaiden, joille asetetaan INTER FIX™
RP kierteitetty fuusiolaite, on saatava vähintään puoli vuotta muita kuin leikkaushoitoja.
• Leikkaustasolle on istutettava vierekkäin kaksi INTER FIX™ kierteitettyä fuusiolaitetta. Yksi INTER FIX™ kierteitetty fuusiolaite ja yksi INTER FIX™ RP kierteitetty
fuusiolaite on istutettava leikkaustasolla vieri viereen. INTER FIX™ RP –laitteen pitkä pituussuuntainen uurre helpottaa kahden laitteen istutusta lähekkäin paremmin
kuin kahdella INTER FIX™ laitteella, koska se antaa kapeamman sivuprofiilin.
• INTER FIX™ kierteitetyn fuusiolaitteen ja INTER FIX™ RP kierteitetyn fuusiolaitteen pituusakselin on oltava sisä-ulkosuunnassa.
• Ellei osia toimiteta steriloituina ja selvästi siten merkittyinä, istutteet ja instrumentit on steriloitava ennen käyttöä tässä pakkauksessa olevien sterilointiohjeiden
mukaisesti.
HAITTAVAIKUTUKSET:
Taulukossa 1 esitetyt haittavaikutukset on saatu 185:ltä INTER FIX™ laitepotilaalta ja 62:lta kontrollipotilaalta, jotka olivat mukana INTER FIX™ kierteisen fuusiolaitteen
kliinisessä tutkimuksessa. Lisäksi on potilaiden seurantatutkimuksessa ilmoitettu haittavaikutuksia 116 potilaalla 48 kuukauden ja 109 potilaalla 72 kuukauden kuluttua.
Nämä esitetyt luvut perustuvat tutkimukseen valittujen potilaiden kokonaislukumäärään kunakin ajankohtana. Satunnaistetuissa ja ei-satunnaistetuissa INTER FIX™
laitehoidoissa olleiden ryhmien haittatapaukset on yhdistetty, jotta tapausten kokonaisesiintymä säilyisi muuttumattomana.
Leikkauksen
jälkeen
LLI
N=62
(1 pv. -< 2 kk.)
[Lukumäärä (%)]
INTER
KONTRO
FIX
N=185
LI
N=62
Leikkauksessa
[Lukumäärä (%)]
INTER
Komplikaati
o
Verisuonet
leikkauksen
aikana
Ristiselkäkipu 5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
Neurologinen 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Selkäkipu 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
Viiltohaava 11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
Selkärangan
vaurio
Urologinen 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Muu 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Muu kipu 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
Ruoansulatus
elimet
Siemensyöks
y sisäänpäin
Hengityselim
et
Säärikipu 2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
Trauma 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
Vatsakalvooire
Suonet leikk.
jälkeen
Luunmurtuma 1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
Istutteen
siirtymä/
löystymä
Siirteen
ottopaikan
kipu
Ei luutumista 4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
Oireiden
helpottumine
n
Ei luutumista
(TULOS
RATKAISEM
ATTA )
Selkäydinkalv
on tulehdus
Istutteen
murtuminen
Syöpä 1
Kuolema 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
KONTR
FIX
N=185
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
2(1.1) 3(1.6) 1(0.5) 6(3.2)
3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
3(1.6) 3(1.6) 0
2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
2(1.1) 2(1.1) 0
1(1.6)
1(1.6) 0 1(1.6)
5(8.1) 0 5(8.1)
3 kk.
(t
2-<5 kk.)
[Lukumäärä
(%)]
INTER
FIX
N=185
1 - Kuusi (6) 81:stä INTER FIX™ laitteen ryhmässä ollutta miestä (7.4%) koki sisään suuntautuvia siemensyöksyjä 24 kuukauden seurannassa.
2 - Tämä potilas (175) on ilmoitettu jälkeenpäin fuusioiduksi noin 52 kuukauden jälkeen leikkauksesta eikä mitään muuta kirurgista toimenpidettä tai hoitoa tarvittu.
Kaksi kliinisissä kokeissa tavattua huomattavaa haittatekijää olivat leikkauksen aikaiset suoni- ja hermovauriot. Yhteensä sattui 15 leikkauksen aikaista suonivauriota 14 potilaalla INTER FIX™
laiteryhmässä 24 kuukauden seurannan aikana. Näihin vaurioihin kuului: 2 onttolaskimolle (vena cava) 9 suoliluulaskimolle (iliacus), 1 repeytynyt alavatsan laskimo (hypogastric) 1 vuotava
segmentalislaskimo, 1 vaurioitunut ristiselän laskimo ja 1 pintavuoto. Yhteensä 2 leikkauksen aikaista suonivauriota tapahtui saman jakson aikana kahdella potilaalla verrokkiryhmässä. Nämä
olivat: 1 suoliluulaskimon vaurio ja 1 verenvuoto luualustasta.
Yhteensä 30 neurologisia tapahtumia ilmeni 28 potilaalla INTER FIX™ laiteryhmässä 24 kuukauden seurannan aikana. Nämä olivat: 1 jalan kärjen halvaus (footdrop), 3 hermojuuren vauriota,
1 foraminaaliahtautuma, 1 refleksisympaattinen dystrofia, 7 säärien tunnottomuutta tai kumotusta, 2 tuntohäiriötä, 3 harhatuntoa, 2 hermojuuren tulehdusta, 7 selkä ja/tai säärikipua, 1 selkä
ja/tai säärikipu lisäoireilla, 1 alaselän vihlova kipu ja 1 rannetunnelin oireyhtymä.
TAULUKKO I - HAITTAVAIKUTUKSET
6 kk.
5-<9 kk.)
(t
[Lukumäärä
(%)]
INTER
KONTR
LLI
N=62
FIX
N=185
KONTRO
LI
N=62
kuukautta
INTER FIX
72 kk
(t
60
)
N=109
12 kk.
9-<19 kk.)
(t
[Lukumäärä
(%)]
INTER
KONTRO
(0.5)
LI
N=62
1
FIX
N=185
2
0 1(1.6) 2(1.7)
24 kk.
19-< 30 kk.)
(t
[Lukumäärä (%)]
INTER
KONTRO
(0.5)
LI
N=62
2 (1.1) 1(0.8) 1(0.9)
FIX
N=185
HAITTA-VAIKUTUK-
SET YHTEENSÄ
(24 KK. AJALTA)
KONTROLL
NTER FIX
N=185
1
(7.4)
N=62
0
48 kk
(>30 - < 60
(t
30d60
kuukautta)
INTER FIX
N=116
Page 39

Yhteensä 10 neurologista häiriötä tavattiin verrokkiryhmässä samana kautena 8 potilaalla. Nämä olivat: 1 hermojuuren tulehdus ja kihelmöinti raajojen päissä, 1 krooninen selkäkipu ja
hermojuuren tulehdus, 1 heikentävä jakosyndroma, 2 selkä- ja säärikipua ja lisäoireita, 1 pääloitontajalihaksen (abductor magnus) hermokato ja 4 säärien tunnottomuutta tai kuumotuksen
tuntoa. Seurannan jälkiarvioinnissa löydettiin 10 muuta neurologista tapausta 10 potilaalla. Nämä olivat: 1 hermojuuren vaurio, 2 kaulan hermojuuren vauriota, 1 käsien tunnottomuus ja
kihelmöinti, 1 säärien tunnottomuus tai kumotus, 3 selkä ja/tai säärikipua ja 2 selkä ja/tai säärikipua lisäoireilla. Lisäksi haittavaikutustaulukossa nähdään säären ja selän kiputapaukset, kuten
välilevyn tilan luhistuminen, joihin joissakin tapauksissa liittyy neurologinen tekijä.
Muutamat haittavaikutukset ovat antaneet aiheen kliinisen leikkauksen jälkikäsittelyyn. Nämä jatkoleikkaukset voidaan luokitella tarkistuksina, poistoina, lisäkiinnityksinä tai lisäleikkauksina.
Taulukossa II on yhteenveto kirurgisista lisäleikkauksista INTER FIX™ laitetta käytettäessä (satunnaistetut ja satunnaistamattomat yhdessä) ja verrokkihoidettujen ryhmä IDE-tutkimuksen
ja INTER FIX™ laiteryhmissä seurannan jälkiarvioinnissa.
1
Muutos: Toimenpide, jolla säädetään tai jotenkin muutetaan alkuperäisen istutteen rakennetta.
Poisto: Toimenpide, jolla poistetaan yksi tai useampi alkuperäisen rakenteen komponentti vaihtamatta sitä samantyyppiseen laitteeseen.
Lisäkiinnitys: Toimenpide, jossa selkärangan laitteisiin lisätään niihin hyväksymättömiä osia.
Jatkoleikkaus: Jokainen kirurginen toimenpide, jolla ei poisteta, muuteta tai lisätä mitään alkuperäisiä komponentteja.
1
Taulukko II - Jatkoleikkaustoimenpiteet
Muutos 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Poistot 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Lisäkiinnitykset 21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
Jatkoleikkaus 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
1 - Koska verrokkipotilaita ei pyydetty seurantaan 24 kuukauden jälkeen, tiedot puuttuvat kaikilta näistä potilaista loppukaudelta 72 kuukauteen saakka.
MAHDOLLISET HAITTAVAIKUTUKSET:
Alla on luettelo mahdollisista haittavaikutuksista selkänikamafuusioleikkauksesta, kun käytetään INTER FIX™ kierteitettyä fuusiolaitetta tai INTER FIX™ RP kierteitettyä fuusiolaitetta. Eräät
niistä sisältyvät ehkä jo edellä annettuun haittavaikutustaulukkoon.
• Hajoaminen, taipuminen, murtuminen, höltyminen ja/tai komponenttien siirtyminen.
• Vieraan esineen (allerginen) vastareaktio.
• Kudos- tai hermovaurio.
• Leikkauksen jälkeinen selkärangan käyristymä, korjautuman menetys, pituuskasvu ja/tai lyheneminen.
• Infektio.
• Kovakalvon repeämät.
• Neurologisen systeemin vaaralle altistus.
• Urologisen systeemin vaaralle altistus.
• Arven muodostus.
• Luunmurtuma.
• Yhdistymättömyys (eli pseudartroosi), viivästynyt yhdistyminen, huono yhdistyminen.
• Selkärangan leikatun osan mahdollisen kasvun pysähtyminen. Selkärangan liikkuvuuden tai toiminnan menetys.
• Siirteen ottopaikan komplikaatiot.
• Verisuonivauriot ja sydän-verenkiertosysteemin vaaralle altistus.
• Ruoansulatuselinten komplikaatiot.
• Suvunjatkamishäiriöt.
• Sisäelinten ja sidekudosten vauriot.
• Hengityselinten häiriöt.
• Aukileikkauskomplikaatiot.
• Mielentilan muutos.
• Kuolema.
IDE-tutkimus Seurannan jälkiarviointi
INTER FIX™
Laite
N=185
24 kuukauden
jakso
Kontrolli
N=62
48 kuukauden
1
INTER FIX™ Laite
jakso
N=116
72 kuukauden jakso
INTER FIX™
Laite
N=109
Huom.: Tarvitaan mahdollisesti uusi leikkaus eräiden tällaisten haittavaikutusten korjaamiseksi.
KLIINISET TUTKIMUKSET:
Yhdysvalloissa suoritettiin kliininen monikeskuksinen yhdenvertaisuus2 kokeilu INTER FIX™ kierteisellä fuusiolaitteella, jossa verrattiin autogeenisellä luusiirteellä täytettyä INTER FIX™ laitetta
etupuolisessa selkärankakäytössä samoin autogeenisella siirteellä täytettyyn reisiluurenkaaseen kontrollina hoidettaesa potilaita, joilla oli symptomaattinen degeneratiivinen välilevytauti.
Aluksi kliininen tutkimus oli aiottu suoritettavaksi käyttäen satunnaiskontrollimenettelyä. Tutkimussuunnitelmaa muutettiin myöhemmin siten, että potilaat voitiin ottaa ei-satunnaishaaraan,
eli heitä hoidettiin vain INTER FIX™ laitteella.
Kliiniseen tutkimuksen valintaperusteena oli symptomaattinen kuvauksella todettu degeneratiivinen välilevyn tauti, joka ilmeni paikallistamattomana kipuna selässä tai raajassa; selkärangan
epästabilisuus, joka määriteltiin taivutus-/venytys-läpivalaisukuvissa näkyvänä yli 4 mm siirtymänä tai yli 5° kallistumana; yksitasoinen symptomaattinen osallistuminen välillä L2 – S1; eikä astetta
I suurempaa spondylolistesiaa. Tutkimuksen ulkopuolelle suljettiin erikoisesti potilaat, joilla oli kyseisellä alueella aikaisemmin suoritettu nikaman fuusio; joilla oli luukato, luun huokoistuminen
tai luun pehmenemistauti (osteomalakia) tai jotka vaativat luunkasvustimulointia.
Yhteensä 185 tutkittavaa potilasta valittiin INTER FIX™ laitteen hoitoryhmään
INTER FIX™ kierteitetyn fuusiolaitteen jälkihyväksynnän vaatimuksena oli todeta laitteen pitkäaikainen toimivuus kokoamalla aina 6 vuoden leikkauksen jälkeiset tiedot vähintäänkin 100
potilaasta. Laitteen pitkäaikainen toimivuus todettiin 116 potilaalla 4 vuoden ajalta leikkauksesta ja 109 potilaasta 6 vuoden ajalta. Tämä potilasjoukko saatiin kokoon kliinisiin IDE-kokeisiin
osallistujista. IDE-tutkimuksen kriteerioiden perusteella näistä potilaista saadut tiedot sisälsivät mahdollisia lisäleikkauksia koskevat selostukset, fuusion läpivalaisut ja kipua ja toimintakykyä
koskevat tiedot.
Taulukossa III esitetään tutkimuksen kliiniset tulokset, joihin liittyvät fuusion onnistumisparametrit, kivun poiston (Oswestry) ja neurologinen onnistuminen 12, 24, 48 ja 72 kuukauden jälkeen
leikkaushoidosta. Tämän kliinisen tutkimuksen menestys todettiin samanarvoisuusanalyysilla INTER FIX™ laitteen ja kontrollin kesken.
Fuusio määriteltiin tässä kliinisessä tutkimuksessa todettuna taberkuliluun liittymisenä, siirtymän (<3 mm) ja kulmaliikkeen (<5°) stabilisuutena ja röntgensäteiden läpäisykyvyn yli 50%
puutteena kummankin istutteen ympärillä. Samoin potilaat, jotka tarvitsivat jatkoleikkauksen, katsottiin laskelmissa epäonnistuneiksi fuusioiksi. Oswestry-kivun-/vammaisuuden suhteen
tapahtunut onnistuminen määriteltiin vähintään 15 pisteen paranemisena leikkausta edeltävään tasoon nähden leikkauksen jälkeisessä mittauksessa. Neurologisen tilan onnistuminen
määriteltiin parempana tuloksena leikkauksen jälkeen ainakin kolmessa neljästä neurologista kategoriasta (liike, aistinta, refleksit ja säären suoraan nosto) leikkausta edeltäneeseen tilaan
verrattuna. Yleisonnistuminen perustui potilaalla nähtävään fuusioon, onistuneeseen Oswestry-kivun/vammaisuuteen ja neurologisen tilan paranemiseen ja, että mitään jatkoleikkausta ei
ole luokiteltu “epäonnistumiseksi”.
2
INTER FIX™ laite ei tilastollisesti kontrollia huonompi.
3
potilasta on jätetty pois, joita ei hoidettu INTER FIX™ laitteella kirurgisten komplikaatioiden vuoksi.
3
. Yhteensä 62 kontrolliryhmän potilasta valittiin kliinisen tutkimuksen satunnaistettuun haaraan.
Tulosten yleismenestys 12 ja 24 kuukauden jälkeen leikkauksesta nähdään myös taulukosta III.
Taulukko III –INTER FIX™ laitteen kliiniset tutkimustulokset
Fuusio
Oswestry-kipu/
vammaisuus
positiivinen
kehitys
t 15 pisteen lisäys
leikkausta
edeltäneeseen
Neurologinen tila
ennallaan tai
parempi
12 kk. suhdeluvut 24 kk. suhdeluvut
Tutkimusaihe Kontrolli
96.1%
(122/127)
53.5%
(76/142)
99.3%
(150/151)
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
Delta
1
(ǻ
)
Tutkimusaihe Kontrolli
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
Delta
(ǻ1)
48 kk.
suhdeluvut
Tutkimusaihe Tutkimusaihe
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
72 kk.
suhdeluvut
97.6% (82/84)
Onnistumisen
yleissuhdeluku
42.4%
(61/144)
21.4%
(12/56)
-0.097 46.1%
(71/154)
29.8%
(17/57)
1 - Tarvitaan « ei-alemman » vaatimukselle « ei-alempi » marginaali tutkimusen tarkkailua varten
-0.043 49.5%
(53/107)
56.3%
(54/96)
Page 40

Kaikki potilaat valittiin INTER FIX™ kierteistetyn fuusiolaitteen kliinisen tutkimuksen molempiin ryhmiin samoin hyväksyntä-/hylkäysperustein. Jotta satunnaistetut ja satunnaistamattomat
INTER FIX™ laitteen potilasryhmät voitaisiin pitää vertailukelpoisina, otettiin seuraavat tekijät huomioon ja tutkittiin: demograafiset ominaisuudet, leikkausta edeltävät terveystilat, kuvauksilla
saadut degeneratiivista välilevyn tautia osoittavat diagnoosit , leikkaustapa ja sijainti käsitellyssä vyötärössä. Otanta oli, leikkaustapaa lukuun ottamatta, tilastollisesti vertailukelpoinen.
Leikkausta edeltävien valmistelujen ja kuvausten perusteella saadut diagnostiset tulokset luupiikkimuodostuksesta (osteofyytti) selkärangan päätelevyissä ja/tai syykehän (anulus fibrosis),
keltasiteen (ligamentum flavum) ja/tai nivelkapselin paksuun-tuminen; ja välilevyn tyrä. Lisäksi tilastolliset analyysit osoittivat, että näillä muuttujilla ei ollut riippuvuutta 12 kuukauden kliinisistä
fuusiotuloksien eroavuuksista. Oswestry-kivun-/vammaisuuden helpottuminen, neurologinen tila, yleinen paraneminen paitsi välilevyn tyrän tapauksessa. Useammalta satunnaistamattomalta
potilaalta löydettiin välilevyn tyrä, jonka havaittiin lisäävän yleistä onnistumisen todennäköisyyttä.
Suoritettiin “hoitosuunnitelman” tulosten analyysi. Tässä analyysissä aiheutuivat puuttuvat havainnot seuraavista seikoista: epäonnistuneet jatkoleikkaukset, kuolintapaukset, potilaan katoaminen
seurannassa ja muista syistä saamatta jääneet havainnot. Tästä syystä nämä lukumäärät sisällytettiin laskettujen suhdelukujen nimittäjiin eli niitä pidettiin “epäonnistumisina”. Koska nämä
potilaat laskettiin lisänä hoidon epäonnistumisiin, hoitosuunnitelman analyysin kliiniset tulokset olivat huomattavasti alempia kuin todellisissa kliinisissä havainnoissa.
Seuraava taulukko (Taulukko IV) sisältää INTER FIX™ laitteen potilaiden hoitosuunnitelman tulosten analyysin, mutta ei kontrolliryhmän tuloksia.
Taulukko IV - INTER FIX™ laitteen hoitosuunnitelman tulosten analyysi
Kuolleet, epäonnistuneet jatkoleikkaukset, puuttuva seuranta ja puuttuvat havainnot katsotaan
epäonnistumisiksi ja nämä luvut sisältyvät suhdelukujen nimittäjiin
12 kk.
Fuusio
Oswestry-kipu/vammaisuus positiivinen
kehitys
t 15 pisteen lisäys leikkausta edeltäneeseen
Neurologinen tila ennallaan tai parempi
Onnistumisen yleissuhdeluku
Jatkoleikkauksen epäonnistumiset
Puuttuvat luutumiset
2
Muut
Kuolintapaukset
1 - Nämä potilaat on otettu mukaan fuusion suhdeluvun laskentaan, mutta katsotaan kliinisessä tutkimuksessa muuten epäonnistuneiksi.
2 - Tässä kaudessa jatkoseurantaan kuuluvat potilaat, joilla oli jatkoleikkauksia muista syistä kuin luutumisen puutteesta, katsotaan kliinisessä tutkimuksessa epäonnistuneiksi.
PAKKAUS:
Jokaisen komponentin pakkauksen tulee olla ehjä vastaanotettaessa. Jos käytössä on lainaus- tai tallessapitojärjestelmä, kaikki sarjat on tarkastettava huolellisesti sen varmistamiseksi, että ne
sisältävät kaikki komponentit ja niissä mahdollisten olevien vikojen varalta ennen käyttöä. Vahingoittuneita pakkauksia ei saa käyttää, ja ne on palautettava Medtronic Sofamor Danek:ille.
PUHDISTUS:
Ellei komponentteja toimiteta steriloituina ja kaikki istutteet ja instrumentit on ensin puhdistettava normaalilla sairaalamenetelmällä ennen sterilointia ja steriiliin kirurgiseen alueeseen vientiä.
Käytetyt instrumentit on aina puhdistettava ja steriloitava ennen uudelleen käyttöä.
Huom.: Älä käytä valkaisuaineita tai formaliinia sisältäviä puhdistusliuoksia, koska ne voivat vahingoittaa laitetta.
Kaikkia tuotteita on käsiteltävä varoen. Sopimaton käyttö tai käsittely voi vahingoittaa laitetta ja/tai haitata sen toimintaa.
STERILOINTI:
Ellei avaamatonta, valmistajalta saatua pakkausta ole selvästi merkitty steriiliksi, kaikki kirurgisissa toimenpiteissä käytettävät istutteet ja instrumentit täytyy steriloida sairaalassa ennen
käyttöä. Poista kaikki pakkausmateriaali ennen sterilointia. Vain steriilejä välineitä saa viedä leikkaussaliin. Nämä laitteet suositellaan taattua steriilisyysastetta 10-6 varten höyrysteriloitaviksi
sairaalassa käyttäen jotakin kolmesta alla mainitusta prosessiparametriä:
MENETELMÄ JAKSO LÄMPÖTILA ALTISTUS-Aika
Höyry Esityhjiö 270° F (132° C) 4 min.
Höyry Paineistettu 250° F (121° C) 60 min.
Höyry* Paineistettu* 273° F (134° C)* 20 min.*
HUOM.: Koska sterilointiin liittyy useita muuttujia, jokaisen lääkintälaitoksen on kalibroitava ja tarkistettava välineillensä käytettävä sterilointiprosessinsa (esim. lämpötilat, ajat).
*Muutamat muiden maiden terveydenhoitoviranomaiset suosittelevat Yhdysvaltain ulkopuolista käyttöä varten sterilointia näitä parametreja käyttäen, jotta Creutzfeldt-Jakob-taudin tarttumisvaara
minimoitaisiin, varsinkin mahdollisesti keskushermoston kanssa kosketuksiin joutuvista kirurgisista instrumenteista.
TUOTTEITA KOSKEVAT VALITUKSET:
Jokaisen terveysalan ammattilaisen (esim. tämän systeemin tuotteiden asiakkaan tai käyttäjän), jolla on jotakin valituksen aihetta tai joka on pettynyt tuotteen laadun, identiteetin, kestävyyden,
luotettavuuden, turvallisuuden, tehokkuuden ja / tai suorituksen suhteen, tulee ilmoittaa siitä MEDTRONIC SOFAMOR DANEK:ille. Jos jokin istutettu INTER FIX™ kierteitetty fuusiolaite tai
INTER FIX™ RP kierteitetty fuusiolaite tai sen osa “toimii huonosti”, (eli ei vastaa mitään määriteltyjä toimintaominaisuuksia tai muuten ei toimi odotetulla tavalla) tai on aihetta arvella niin,
laitteen myyjälle on asiasta ilmoitettava viipymättä. Jos jokin Medtronic Sofamor Danek-tuote on “toiminut huonosti” ja mahdollisesti aiheuttanut tai ollut osallisena kuolemaan tai vakavaan
vammaan, jälleenmyyjälle tulisi ilmoittaa asiasta mahdollisimman pian puhelimitse, telekopiolla tai kirjallisesti postitse. Ilmoita valituksessa komponenttien nimet ja numerot, pakkauksen
numero, nimi, osoite, valituksen aihe ja merkintä siitä, vaaditaanko jälleenmyyjän kirjallinen lausunto.
LÄÄKÄRIÄ KOSKEVA HUOMAUTUS: Vaikka lääkäri onkin oppia saanut välikäsi yhtiön ja potilaan välillä, tähän julkaisuun sisältyvät tärkeät lääketieteelliset seikat on annettava potilaan tietoon.
Koskee vain USA:ta
HUOMIOON OTETTAVAA: LIITTOVALTION (USA) LAKI RAJOITTAA NÄIDEN LAITTEIDEN MYYNNIN AINOASTAAN LÄÄKÄRILLE TAI LÄÄKÄRIN MÄÄRÄYSTÄ VASTAAN.
LAITTEEN POISOTTOYRITYKSET:
Mikäli osoittautuu välttämättömäksi poistaa INTER FIX™ kierteitetty fuusiolaite tai INTER FIX™ RP kierteitetty fuusiolaite, ota yhteys Medtronic Sofamor Danek:iin.
© 2006 Medtronic Sofamor Danek. Kaikki oikeudet pidätetään.
Implisitte garantier for salgbarhet og egnethet for et bestemt formål eller bruk, utelukkes spesielt. Ytterligere informasjon om garantier og ansvarsbegrensninger finnes i Medtronic Sofamor
Danek-katalogen eller prislisten.
1
suhdeluvut
65.9% (122/185) 67.6%
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
33.0% (61/185) 38.4% (71/185) 45.7%
5
8
0 1 0 0
24 kk.
suhdeluvut
(125/185)
(128/185)
5
3
48 kk.
suhdeluvut
80.2%
(93/116)
(66/116)
87.9%
(102/116)
(53 /116)
4
4
77.1%
( 84/109)
56.0%
(61 /109)
75.2%
75.2% (82/109)
(82/109)
49.5%
(54/109)
72 kk.
suhdeluvut
1
2
Page 41

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
ΕΛΛΗΝΙΚΑ
Το ακόλουθο κείμενο περιέχει σημαντικές ιατρικές πληροφορίες για την ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ και την ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ RP.
ΠΕΡΙΓΡΑΦΗ:
Η ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ αποτελείται από ένα κοίλο, διάτρητο, μεταλλικό κύλινδρο και τερματικό πώμα. Η ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ RP αποτελείται από ένα
κοίλο, διάτρητο, μεταλλικό κύλινδρο που φέρει κατά μήκος ολόκληρου του επιμήκους άξονα ένα μόνο, μεγάλο, εξωτερικό αύλακα με στρογγυλοκομμένες τις ακμές, ο οποίος επεκτείνεται και στην εσωτερική διάμετρο
της συσκευής. Και τα δύο τέρματα του εμφυτεύματος INTER FIX™ RP είναι κλειστά.
Τα εμφυτεύματα INTER FIX™ και INTER FIX™ RP διατίθενται σε διαμέτρους που κυμαίνονται από 12 mm μέχρι 24 mm και σε μήκη που κυμαίνονται από 20 mm μέχρι 29 mm. Τα τερματικά πώματα των εμφυτευμάτων
συγχώνευσης INTER FIX™ έχουν μέγεθος ανάλογο με τη διάμετρο των κυλίνδρων και τοποθετούνται στο ανοιχτό άκρο των κυλίνδρων αφού αυτοί πληρωθούν με αυτογενές οστικ
Τα εμφυτεύματα INTER FIX™ και INTER FIX™ RP είναι κατασκευασμένα από κράμα αλουμινίου ποιότητας εμφυτεύματος (Ti-6AI-4V) σύμφωνα με το πρότυπο ASTM F 136.
Ειδικά αποκλείεται κάθε σιωπηρή εγγύηση εμπορευσιμότητας και καταλληλότητας για συγκεκριμένο σκοπό ή χρήση. Βλέπε τον κατάλογο της Medtronic Sofamor Danek ή τον τιμοκατάλογο για περισσότερες πληροφορίες
σε σχέση με εγγυήσεις και περιορισμούς ευθύνης.
ΕΝΔΕΙΞΕΙΣ:
Η ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ και η ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ RP ενδείκνυνται για διαδικασίες σπονδυλικής συγχώνευσης σε ασθενείς με ώριμο σκελετό και με
εκφυλιστική δισκοπάθεια (DDD) σε ένα επίπεδο από τα L2-S1. Η DDD ορίζεται ως δισκογενής πόνος της πλάτης με εκφυλισμό των δίσκου επιβεβαιούμενο από το ιστορικό του ασθενούς και ακτινογραφικώς. Οι εν
λόγω ασθενείς DDD μπορεί επίσης να εμφανίζουν σπονδυλολίσθηση ή παλινολίσθηση μέχρι και βαθμού Ι στο σχετικό επίπεδο. Τα εμφυτεύματα INTER FIX™ και INTER FIX™ RP πρέπει να χρησιμοποιούνται με
αυτογενές οστικό μόσχευμα και να εμφυτεύονται με ανοιχτή πρόσθια προσέγγιση.
ΑΝΤΕΝΔΕΙΞΕΙΣ:
Η ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ και η ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ RP δεν πρέπει να εμφυτεύονται σε ασθενείς με ενεργή μόλυνση στο σημείο της χειρουργικής επέμβασης
ή με αλλεργία στο τιτάνιο ή στα κράματα τιτανίου.
ΠΡΟΕΙΔΟΠΟΙΗΣΕΙΣ:
• Η ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ και η ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ RP πρέπει να χρησιμοποιούνται μόνο από χειρούργους με πείρα στις διαδικασίες σπονδυλικής
συγχώνευσης και με επαρκή κατάρτιση στη συσκευή αυτή.
• Ενδεχόμενη έλλειψη επαρκούς πείρας και/ή κατάρτισης μπορεί να οδηγήσει σε αυξημένο ποσοστό δυσμενών περιστατικών, όπως είναι οι αγγειακές κακώσεις, τα νευρολογικά επεισόδια και/ή τα επεισόδια του
ουρογεννητικού συστήματος (συμπεριλαμβανομένης της ανάδρομης εκσπερμάτωσης).
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP
Σημαντικές ιατρικές πληροφορίες
ό μόσχευμα.
Page 42

ΠΡΟΦΥΛΑΞΕΙΣ:
φαιρ
Ȉ
™
™
Dz
Ȉ
™
™
• Η ασφάλεια και αποτελεσματικότητα της ελικοτομημένης συσκευής συγχώνευσης INTER FIX™ και της ελικοτομημένης συσκευής συγχώνευσης INTER FIX™ RP δεν έχουν αποδειχθεί για ασθενείς με
οποιαδήποτε από τις εξής καταστάσεις:
- σπονδυλολίσθηση ή παλινολίσθηση βαθμού ΙΙ ή υψηλότερου,
- ανάγκη συγχώνευσης επιπέδων περισσότερων από ένα,
- αναθεώρηση προηγούμενης διαδικασίας ή προηγούμενων διαδικασιών συγχώνευσης μεταξύ σπονδυλικών σωμάτων,
- μετεγχειρητικές απαιτήσεις στεροειδών ή μη στεροειδών αντιφλεγμονικών φαρμάκων,
- χονδροειδής παχυσαρκία,
- ηλικία κάτω των 18 ή άνω των 65 ετών,
- οστεοπόρωση, οστεοπενία και/ή οστεομαλάκυνση,
- εγκυμοσύνη.
• Η ασφάλεια και αποτελεσματικότητα της ελικοτομημένης συσκευής συγχώνευσης INTER FIX™ και της ελικοτομημένης συσκευής INTER FIX™ RP έχουν αποδειχθεί μόνο για τις διαδικασίες πρόσθιας οσφυϊκής
συγχώνευσης μεταξύ σπονδυλικών σωμάτων (ALIF) με χρήση
• Οι ασθενείς που δέχονται την ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ πρέπει να έχουν ήδη υποστεί μη χειρουργική θεραπεία επί τουλάχιστον έξι μήνες. Οι ασθενείς που δέχονται την
ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ RP πρέπει να έχουν ήδη υποστεί μη χειρουργική θεραπεία επί τουλάχιστον έξι μήνες.
• Δύο ελικοτομημένες συσκευής συγχώνευσης INTER FIX™ πρέπει να εμφυτεύονται η μια δίπλα στην άλλη σε επίπεδο χειρουργικής επέμβασης. Μια ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ και μια
ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ RP πρέπει να εμφυτεύονται η μια δίπλα στην άλλη στο επίπεδο χειρουργικής επέμβασης. Ο διαμήκης αύλακας της συσκευής INTER FIX™ RP διευκολύνει
την εμφύτευση δύο συσκευών σε μεγαλύτερη εγγύτητα από εκείνη που μπορεί να επιτευχθεί με δύο συσκευές INTER FIX™, εξασφαλίζοντας έτσι περιορισμένη κατά πλάτος διατομή.
αυτογενούς οστικού μοσχεύματος.
• Ο μακρός άξονας των ελικοτομημένων συσκευών συγχώνευσης INTER FIX™ και των ελικοτομημένων συσκευών συγχώνευσης INTER FIX™ RP πρέπει να βρίσκονται σε πρόσθια-οπίσθια κατεύθυνση.
• Τα εμφυτεύματα και τα όργανα πρέπει να αποστειρώνονται πριν χρησιμοποιηθούν σύμφωνα με τις οδηγίες αποστείρωσης που παρέχονται στο παρόν ένθετο συσκευασίας, εκτός αν έχουν διατεθεί
αποστειρωμένα και σαφώς επισημασμένα ως αποστειρωμένα.
ΔΥΣΜΕΝΕΙΣ ΕΠΙΠΤΩΣΕΙΣ:
Οι δυσμενείς επιπτώσεις που εμφανίζονται στον πίνακα Ι αναφέρθηκαν με βάση τους 185 ασθενείς με συσκευή INTER FIX™ και τους 62 ασθενείς ελέγχου που εγγράφηκαν για την κλινική μελέτη πολλαπλών κέντρων
τη σχετική με την ελικοτομημένη συσκευή συγχώνευσης INTER FIX™. Επιπλέον, αναφέρθηκαν δυσμενή περιστατικά από 116 ασθενείς στους 48 μήνες και 109 ασθενείς στους 72 μήνες σε ασθενείς της μετεγκριτικής
μελέτης. Τα αναφερόμενα ποσοστά βασίζονται στο συνολικό αριθμό ασθενών που εγγράφηκαν στη μελέτη σε κάθε χρονικό σημείο αναφοράς. Οι δυσμενείς επιπτώσεις οι εμφανιζόμενες στις τυχαίες και μη τυχαίες
ομάδες θεραπείας με συσκευή INTER FIX™ έχουν συνδυασθεί ώστε να παρουσιάζεται ένα ενιαίο ποσοστό.
ȆǿȃǹȀǹȈ ǿ – ǻȊȈȂǼȃǼǿȈ ǼȆ ǿȆȉȍȈǼǿȈ
ǼȖȤİȚȡȘIJȚțȫȢ
[ĮȡȚșμȩȢ (%)]
ȆİȡȚʌȜȠțȒ
ǻȚİȖȤİ ȚȡȚIJȚț ȫȢ
ıİ ĮȖȖİȓȠ
ǿİȡȠ-İȚȜİĮțȩȢ
ʌȩȞȠȢ
ȃİȣȡȠȜȠȖȚțȒ
ȆȩȞȠȢ ıIJȘȞ
ʌȜȐIJȘ
ǼȞIJȠμȒȢ
ǼʌİȚıȩįȚȠ
ıʌȠȞįȣȜȚțȒȢ
ıIJȒȜȘȢ
ȅȣȡȠȜȠȖȚțȒ
DZȜȜȠȣ İȓįȠȣȢ
DZȜȜȠȢ ʌȩȞȠȢ
īĮıIJȡȠİȞIJİȡȚțȒ
ǹȞȐįȡȠμȘ
İțıʌİȡμȐIJȦıȘ
ǹȞĮʌȞİȣıIJȚțȒ
ȆȩȞȠȢ țȐIJȦ
ȐțȡȦȞ
ȉȡĮȪμĮ
ȆİȡȚIJȠȞȚțȒ
ȂİIJİȖȤİȚȡȘIJȚțȒ
ıİ ĮȖȖİȓȠ
ĬȡĮȪıȘ ȠıIJȠȪ
ȂİIJȐșİıȘ /
ȤĮȜȐȡȦıȘ
İμijȣIJİȪμĮIJȠȢ
ȆȩȞȠȢ ıȘμİȓȠȣ
μȠıȤİȪμĮIJȠȢ
ȂȘ ȑȞȦıȘ
ȀĮșȓȗȘıȘ
ȂȘ ȑȞȦıȘ
(ȉȅ
ǹȆȅȉǼȁǼȈȂǹ
ǹȃǹȂǼȃǼȉǹǿ)
ȂȘȞȚȖȖ ȓIJȚįĮ
ĬȡĮȪıȘ
İμijȣIJİȪμĮIJȠȢ
țĮȡțȓȞȠȢ
ĬȐȞĮIJȠȢ
INTER FIX™
N=185
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
2(1.1) 3(1.6) 1(0.5) 6(3.2)
3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
3(1.6) 3(1.6) 0
2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
2(1.1) 2(1.1) 0
1(1.6)2 0 1(1.6) 2(1.7)
1(1.6) 0 1(1.6)
5(8.1) 0 5(8.1)
1
1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
1 – Έξι (6) στους 81 εγγεγραμμένους στην ομάδα συσκευών INTER FIX™ άνδρες (7,4%) παρουσίασαν ανάδρομη εκσπερμάτωση κατά
DzȁǼīȋȅ
N=62
ȂİIJİȖȤİȚȡȚIJȚțȫȢ
(1 ȘμȑȡĮ-<2
μȒȞİȢ)
[ĮȡȚșμȩȢ (%)]
DzȁǼīȋȅȈ
NTER FIX
N=185
N=62
3 μȒȞİȢ
[ĮȡȚșμȩȢ
NTER FIX
N=185
2-<5
(t
μȒȞİȢ)
(%)]
ȁǼīȋȅ
N=62
6 μȒȞİȢ
(t
5-<9 μȒȞİȢ)
[ĮȡȚșμȩȢ (%)]
NTER FIX
DzȁǼīȋȅȈ
N=185
N=62
12 μȒȞİȢ
(t9-<19
μȒȞİȢ)
[ĮȡȚșμȩȢ (%)]
INTER
DzȁǼīȋȅȈ
FIX™
N=62
N=185
(0.5)
24 μȒȞİȢ
(t
19-< 30
μȒȞİȢ)
[ĮȡȚșμȩȢ (%)]
DzȁǼīȋȅȈ
NTER FIX
N=185
N=62
1
(0.5)
2 (1.1) 1(0.8) 1(0.9)
τη διάρκεια της 24μηνης αξιολόγησης.
ȈȊȃȅȁǿȀǹ
ǻȊȈȂǼȃǾ
ȆǼȇǿȈȉǹȉǿȀǹ
(ȈǼ 24 ȂǾȃǼȈ)
™
INTER FIX
N=185
1
(7.4)
48 ȂǾȃȅ
(>30 - < 60
(t
30<60
DzȁǼīȋȅȈ
N=62
ȂȒȞİȢ)
INTER FIX™
N=116
0
72
ȂǾȃȅ
(t
60
ȂȒȞİȢ)
INTER FIX
N=109
™
2 – Ο ασθενής αυτός (175) αναφέρθηκε στη συνέχεια ως υποβληθείς σε σπονδυλοδεσία πριν από 52 μήνες περίπου, και δεν απαιτήθηκε καμία άλλη χειρουργική διαδικασία ή αγωγή.
Δυο σημαντικά δυσμενή περιστατικά της κλινικής δοκιμής ήταν οι διεγχειρητικές αγγειακές και νευρολογικές κακώσεις. Συνολικά 15 διεγχειρητικά αγγειακά περιστατικά έλαβαν χώρα σε 14 ασθενείς της ομάδας συσκευών
INTER FIX™ κατά την 24μηνη αξιολόγηση. Στα περιστατικά αυτά συγκαταλέγονταν: 2 κακώσεις στην κοίλη φλέβα, 9 κακώσεις στην ιλιακή φλέβα, 1 διάρρηξη υπογάστριας φλέβας, 1 αιμορραγία τμηματικής φλέβας, 1
κάκωση φλέβας περί το ιερόν οστούν και 1 αιμορραγία επιφανειακού αιμοφόρου αγγείου. Συνολικά αναφέρθηκαν 2 διεγχειρητικές αγγειακές κακώσεις σε 2 ασθενείς της ομάδας ελέγχου κατά την ίδια χρονική
Στις κακώσεις αυτές περιλαμβάνονταν: 1 κάκωση σε ιλιακή φλέβα και 1 αιμορραγία από την κοίτη του οστού.
Κατά τη διάρκεια της 24μηνης αξιολόγησης της ομάδας συσκευών INTER FIX™ σημειώθηκε συνολικός αριθμός 30 νευρολογικών περιστατικών σε 28 ασθενείς. Στα περιστατικά αυτά συγκαταλέγονταν: 1 πτώση
άκρου ποδός, 3 κακώσεις ρίζας νεύρου, 1 στένωση τρήματος, 1 δυστροφία αντανακλαστικού συμπαθητικού νεύρου, 7 περιπτώσεις μουδιάσματος
περιπτώσεις παραισθησίας, 2 περιπτώσεις ριζοπάθειας, 7 περιπτώσεις πόνου στην πλάτη και/ή στο κάτω άκρο, 1 περίπτωση πόνου στην πλάτη και/ή στο κάτω άκρο με άλλα συμπτώματα, 1 περίπτωση διαπεραστικού
πόνου στην κάτω πλάτη και 1 σύνδρομο καρπιαίου σωλήνα.
Στην ομάδα ελέγχου σημειώθηκαν συνολικά 10 νευρολογικά περιστατικά σε
1 χρόνιο πόνο πλάτης με ριζίτιδα, 1 περίπτωση συμπτωμάτων εξουθενωτικής κατανομής, 2 περιπτώσεις πόνου στην πλάτη και τα κάτω άκρα συνοδευόμενου από άλλα συμπτώματα, 1 απονεύρωση του μείζονος
απαγωγού μυός και 4 περιπτώσεις μουδιάσματος ή αίσθησης θερμότητας στα κάτω άκρα. Κατά την μετεγκριτική μελέτη, σημειώθηκαν 10 επιπλέον νευρολογικά περιστατικά σε 10 ασθενείς. Στα περιστατικά αυτά
συγκαταλέγονταν: 1 περίπτωση ριζοπάθειας, 2 αυχενικής ριζοπάθειας, 1 περίπτωση μουδιάσματος και κνησμού στα χέρια, 1 περίπτωση μουδιάσματος ή αίσθησης θερμότητας στα κάτω άκρα, 3 περιπτώσεις πόνου
στην πλάτη και/ή στο κάτω άκρο και 2 περιπτώσεις πόνου στην πλάτη και/ή στο κάτω άκρο με άλλα συμπτώματα. Επιπλέον, ο πίνακας δυσμενών συνεπειών παρουσιάζει περιστατικά πόνου στα κάτω άκρα και την
πλάτη και/ή περιστατικά της σπονδυλικής στήλης, όπως είναι η κατάρρευση μεσοδιαστήματος δίσκων, τα οποία, σε ορισμένες περιπτώσεις, είχαν και νευρολογική διάσταση.
Μερικά από τα δυσμενή περιστατικά οδήγησαν σε χειρουργικές επεμβάσεις μετά
έσεις, επιπρόσθετες στερεώσεις ή επανεγχειρήσεις.1 Ο Πίνακας ΙΙΙ συνοψίζει τις δευτερογενείς χειρουργικές επεμβάσεις στις θεραπευτικές ομάδες συσκευής INTER FIX™ (τυχαία και μη τυχαία ομάδα μαζί) και
α
8 ασθενείς κατά την ίδια χρονική περίοδο. Στα περιστατικά αυτά συγκαταλέγονταν: 1 περίπτωση ριζιτικής πάθησης με κνησμό στα άκρα,
την κλινική δοκιμαστική επέμβαση. Οι πρόσθετες αυτές χειρουργικές επεμβάσεις μπορούν να χαρακτηρισθούν ως αναθεωρήσεις,
ή αισθήματος καύσης στα κάτω άκρα, 2 περιπτώσεις δυσαισθησίας, 3
περίοδο.
Page 43

ελέγχου, μέσω της μελέτης IDE και στην ομάδα συσκευής INTER FIX™ κατά τη διάρκεια της μετεγκριτικής μελέτης.
1
Αναθεώρηση: Διαδικασία που αναπροσαρμόζει ή με οποιοδήποτε τρόπο τροποποιεί την αρχική διαμόρφωση του εμφυτεύματος.
Αφαιρέσεις : Διαδικασία αφαίρεσης μιας ή περισσότερων συνιστωσών της αρχικής διαμόρφωσης του εμφυτεύματος χωρίς αντικατάσταση με τον ίδιο τύπο δοκιμαστικής συσκευής.
Συμπληρωματική στερέωση: Διαδικασία κατά την οποία τοποθετούνται πρόσθετες συσκευές σπονδυλικής στήλης, η οποίες δεν έχουν εγκριθεί ως μέρος το
Επανεγχείριση: Οποιαδήποτε χειρουργική
ȂİȜȑIJȘ IDE ȂİIJİȖțȡȚIJȚțȒ ȂİȜȑIJȘ
ȀĮIJȐ IJȘ įȚȐȡțİȚĮ IJȠȣ
ǹȞĮșİȦȡȒıİȚȢ 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
ǹijĮȚȡȑıİȚȢ 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
ȈȣμʌȜȘȡȦμĮIJȚțȑȢ
ıIJİȡİȫıİȚȢ
ǼʌĮȞİȖȤİȚȡȒıİȚȢ 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
ȆȓȞĮțĮȢ ǿǿ – ǻȚĮįȚț ĮıȓİȢ įİȣIJİȡȠȖİȞȠȪȢ ȤİȚȡȠȣȡȖȚțȒȢ
24μȒȞȠȣ
ȈȣıțİȣȒ
INTER FIX™
N=185
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
DzȜİȖȤȠȢ
N=62
1
ȀĮIJȐ IJȘ įȚȐȡțİȚĮ
IJȠȣ 48μȒȞȠȣ
ȈȣıțİȣȒ
INTER FIX™
N=185
ȀĮIJȐ IJȘ įȚȐȡțİȚĮ IJȠȣ
72μȒȞȠȣ
DzȜİȖȤȠȢ
N=109
1
υ πρωτοκόλλου.
1 - Εξαιτίας του ότι οι ασθενείς ελέγχου δεν υποχρεούνταν σε παρακολούθηση μετά τους 24 μήνες, δεν υπάρχουν διαθέσιμες πληροφορίες για όλους αυτούς τους ασθενείς για το διάστημα τω ν 72 μηνών.
Δυνάμει δυσμενείς συνέπειες:
Παρατίθεται κατωτέρω ο κατάλογος δυνάμει δυσμενών συνεπειών που μπορούν να σημειωθούν σε συνάρτηση με τη χειρουργική σπονδυλικής συγχώνευσης με ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ ή
με ελικοτομημένη συσκευή συγχώνευσης INTER FIX™ RP. Ορισμένες από τις εν λόγω δυσμενείς συνέπειες ενδέχεται να έχουν αναφερθεί προηγουμένως στον πίνακα δυσμενών συνεπειών.
• Αποσυναρμολόγηση, κάμψη, θραύση, χαλάρω ση και/ή μετακίνηση συνιστωσών.
ντίδραση ξένου σώματος (αλλεργική).
• Α
• Ζημιά σε ιστό ή νεύρο.
• Μετεγχειρητική μετατροπή της σπονδυλικής καμπυλότητας, απώλεια ορθότητας ή ύψους και/ή βράχυνση.
• Μόλυνση.
• Ρήξη της σκληρής μήνιγγας.
• Διαταραχές νευρολογικού συστήματος.
• Διαταραχές ουρολογικού συστήματος.
• Διαμόρφωση ουλής.
• Θραύση οστού.
• Μή ένωση (ή ψευδάρθρωση), καθυστέρηση ένωσης, κακή ένωση.
• Παύση κά
• Περιπλοκές σημείου προέλευσης μοσχεύματος.
• Ζημιά σε αιμοφόρα αγγεία και διαταραχές καρδιαγγειακού συστήματος.
• Γαστροεντερικές περιπλοκές.
• Ανωμαλίες αναπαραγωγικού συστήματος.
• Ζημιά σε εσωτερικά όργανα και συνδετικό ιστό.
• Ανάπτυξη αναπνευστικών προβλημάτων.
• Περιπλοκές χειρουργικής εντομής.
• Αλλαγή ψυχικής κατάστασης.
• Θάνατος.
θε δυνάμει ανάπτυξης του χειρουργημένου τμήματος της σπονδυλικής στήλης. Απώλεια κινητικότητας ή της λειτουργίας της σπονδυλικής στήλης.
Σημείωση: Για τη διόρθωση μερικών από τις παραπάνω πιθανές ανεπιθύμητες ενέργειες ενδέχεται να απαιτηθούν πρόσθετες χειρουργικές επεμβάσεις.
ΚΛΙΝΙΚΑ ΑΠΟΤΕΛΕΣΜΑΤΑ:
Πραγματοποιήθηκε στις ΗΠΑ σε πολλαπλά κέντρα κλινική δοκιμή ισοδυναμίας2 της ελικοτομημένης συσκευής συγχώνευσης INTER FIX™, προς σύγκριση της χρήσης στην εμπρόσθια σπονδυλική στήλη της συσκευής
INTER FIX™ γεμισμένης με αυτογενές οστό με αλλογενές μόσχευμα μηριαίου δακτυλίου γεμισμένου με αυτογενές οστό (αυτή ήταν η βάση ελέγχου) για τη θεραπεία ασθενών με συμπτωματική εκφυλιστική δισκοπάθεια.
Αρχικά, η κλινική αυτή δοκιμή είχε σχέδιο πιθανολογικό, τυχαίο και ελεγχόμενο. Στη συνέχεια, το ερευνητικό σχέδιο αναθεωρήθηκε, ώστε να καταστεί δυνατή η συμπερίληψη ασθενών σε μη τυχαίο σκέλος, δηλ. ασθενών
που υποβλήθηκαν σε θεραπεία μόνο με συσκευή INTER FIX™.
Στα κριτήρια συμπερίληψης για την κλινική δοκιμή συγκαταλέγονταν τα εξής: συμπτωματική εκφυλιστική δισκοπάθεια χαρακτηριζόμενη από δυσθεράπευτο πόνο κάτω άκρων και/ή πλάτης με θετικές διαγνωστικές
διαπιστ
ώσεις εικόνας; αστάθεια της σπονδυλικής στήλης οριζόμενη βάσει μετάθεσης μεγαλύτερης από 4 mm ή από γωνία άνω των 5° σε ακτινογραφίες κάμψης/ έκτασης; συμπτωματική εμπλοκή ενός επιπέδου από
τα L2-S1 και σπονδυλολίσθηση όχι μεγαλύτερη από βαθμό Ι. Εξαιρέθηκαν ειδικά από τη μελέτη ασθενείς που είχαν υποστεί προηγούμενα πρόσθια συγχώνευση διασπονδυλικών σωμάτων στο σχετικό επίπεδο της
σπονδυλικής στήλης ή υπέφεραν από οστεοπενία, οστεοπόρωση ή οστεομαλάκυνση ή χρειάζονταν διέγερση ανάπτυξης των οστών.
Στη διερευνητική ομάδα θεραπείας με συσκευή INTER FIX™ καταγράφηκαν συνολικά 185 ασθενείς.
Μια από τις μετεγκριτικές απαιτήσεις για την Ελικοτομημένη Συσκευή Συγχώνευσης INTER FIX™ ήταν η αξιολόγηση της μακροχρόνιας απόδοσης της συσκευής συλλέγοντας συνολικά μετεγχειρητικά δεδομένα 6 ετών
για τουλάχιστον 100 ασθενείς. Η μακροχρόνια απόδοση της συσκευής αξιολογήθηκε σε 116 ασθενείς στα 4 χρόνια μετά την επέμβαση και σε 109 ασθενείς στ
συνελέγη από συμμετέχοντες στην κλινική μελέτη IDE. Με βάση κριτήρια της μελέτης IDE, οι πληροφορίες που συγκεντρώθηκαν ως προς τους ασθενείς αυτούς περιελάμβαναν την περιγραφή κάθε επιπλέον χειρουργικής
διαδικασίας, ακτινογραφική αξιολόγηση της σπονδυλοδεσίας και αξιολόγηση του πόνου και της λειτουργίας.
Ο Πίνακας ΙΙΙ αναπ
48 και 72 μήνες μετά την θεραπευτική χειρουργική επέμβαση. Η επιτυχία αυτής της κλινικής δοκιμής βαθμολογήθηκε βάσει ανάλυσης ισοδυναμίας μεταξύ συσκευής INTER FIX™ και ελέγχου.
Για την κλινική αυτή δοκιμή, η συγχώνευση ορίσθηκε ως η ύπ
από τα δύο σχετικά εμφυτεύματα να περιβάλλεται από ακτινοδιαπερατότητες κα τά περισσότερο από 50%. Επίσης, στους υπολογισμούς, οι περιπτώσεις των ασθενών που υπέστησαν δευτερογενείς εγχειρητικές
επεμβάσεις λόγω μη ενώσεων θεωρήθηκαν αποτυχημένες συγχωνεύσεις. Η επιτυχία ως προς την εμφάνιση πόνου/ εξ
την προεγχειρητική κατά τουλάχιστον 15 μονάδες. Η επιτυχία νευρολογικής κατάστασης ορίσθηκε ως διατήρηση ή βελτίωση σε τουλάχιστον 3 από τις 4 νευρολογικές κατηγορίες (κίνηση, αίσθηση, αντανακλαστικά και
άρση ευθυτενούς κάτω άκρου) στο μετεγχειρητικό στάδιο σε σύγκριση με την προεγχειρητική κα
και επιτυχία νευρολογικής κατάστασης, χωρίς δευτερογενή χειρουργική διαδικασία χαρακτηριζόμενη ως “αποτυχία”.
2
Στατιστικώς τα αποτελέσματα δεν υπήρξαν χειρότερα για τη συσκευή INTER FIX™ σε σύγκριση με τη βάση ελέγχου.
3
Περιλαμβάνονται 3 ασθενείς που δεν υπέστησαν τη θεραπεία με συσκευή INTER FIX™ λόγω δυσμενών χειρουργικών περιστατικών.
αριστά τα κλινικά αποτελέσματα της μελέτης, περιγράφοντας τις παραμέτρους της επιτυχούς σπονδυλοδεσίας, την επίτευξη μείωσης πόνου (Oswestry) και νευρολογικής αποκατάστασης σε 12, 24,
αρξη στοιχείων που τεκμαίρουν σταθερότητα όσον αφορά τη γεφύρωση οστικής δοκίδας, τη μετάθεση (<3 mm) και τη γωνιακή κίνηση (<5°), χωρίς οποιοδήποτε
3
Στο τυχαίο σκέλος της κλινικής δοκιμής περιλήφθηκαν συνολικά 62 ασθενείς ελέγχου.
α 6 χρόνια μετά την επέμβαση. Ο πληθυσμός ασθενών
ασθένισης Oswestry ορίσθηκε ως η βελτίωση της μετεγχειρητικής βαθμολογίας σε σύγκριση με
τάσταση. Η γενική επιτυχία βασίστηκε σε ασθενή που εμφανίζει συγχώνευση, επιτυχία πόνου/ εξασθένισης Oswestry
Στον Πίνακα ΙΙΙ μπορείτε επίσης να βρείτε τα συνολικά αποτελέσματα επιτυχίας 12 και 24 μήνες μετά την εγχείρηση.
ȈȣȖȤȫȞİȣıȘ
ǺİȜIJȓȦıȘ ʌȩȞȠȣ/
İȟĮıșȑȞȚıȘȢ
Oswestry
ǹıșİȞİȓȢ μİ
ȕİȜIJȓȦıȘ
IJȠȣȜȐȤȚıIJȠȞ 15
μȠȞȐįȦȞ Įʌȩ IJȘȞ
ʌȡȠİȖȤİȚȡȘIJȚțȒ
țĮIJȐıIJĮıȘ
ǻȚĮIJȒȡȘıȘ Ȓ
ȕİȜIJȓȦıȘ
ȞİȣȡȠȜȠȖȚțȒȢ
țĮIJȐıIJĮıȘȢ
īİȞȚțȒ İʌȚIJȣȤȓĮ
ȆȓȞĮțĮȢ III – ȀȜȚȞȚțȐ ǹʌȠIJ İȜȑ ıμĮIJĮ IJȘȢ ȂİȜȑIJȘȢ ȈȣıțİȣȒȢ INTER FIX™
ȆȠıȠıIJȐ 12 μȘȞȫȞ ȆȠıȠıIJȐ 24 μȘȞȫȞ
ǻȚȐIJĮȟȘ
įȠțȚμȒȢ
96.1%
(122/127)
53.5%
(76/142)
99.3%
(150/151)
42.4%
(61/144)
DzȜİȖȤȠȢ
ǻȑȜIJĮ
(ǻ
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
-0.097 46.1%
(12/56)
ǻȚȐIJĮȟȘ
1
)
įȠțȚμȒȢ
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
(71/154)
DzȜİȖȤȠȢ
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
(17/57)
ȆȠıȠıIJȐ 48
ǻȑȜIJĮ
(ǻ1)
-0.043 49.5%
μȒȞȠȣ
ǻȚȐIJĮȟȘ
įȠțȚμȒȢ
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
(53/107)
ȆȠıȠıIJȐ 72
μȒȞȠȣ
ǻȚȐIJĮȟȘ
įȠțȚμȒȢ
97.6% (82/84)
56.3%
(54/96)
1 - Απαιτείται ελάχιστο περιθώριο μη-κατωτερότητας για να δηλωθεί η μη-κατωτερότητα της διάταξης δοκιμής σε σχέση με τη διάταξη ελέγχου.
Page 44

Το σύνολο των ασθενών που περιλήφθηκαν στην κλινική δοκιμή της ελικοτομημένης συσκευής συγχώνευσης INTER FIX™, ασχέτως ομάδας θεραπείας, επιλέχθηκαν βάσει των ίδιων κριτηρίων συμπερίληψης/ αποκλεισμού.
Προς τεκμηρίωση της συγκρισιμότητας των τυχαίων ασθενών με συσκευή INTER FIX™ και των μη τυχαίων ασθενών με συσκευή INTER FIX™, εξετάσθηκαν τα δημογραφικά χαρακτηριστικά, οι προεγχειρητικές ιατρικές
καταστάσεις, οι διαγνωστικές διαπιστώσεις εικόνας που είναι χαρακτηριστικές της εκφυλιστικής δισκοπάθειας, η εγχειρητική προσέγγιση και η θέση του θεραπευόμενου οσφυϊκού επιπέδου. Ο πληθυσμός υπήρξε στατιστικά
συγκρίσιμος με την εξαίρεση της εγχειρητικής προσέγγισης, της προεγχειρητικής προετοιμασίας και των προεγχειρητικών διαγνωστικών διαπιστώσεων εικόνας διαμόρφωσης οστεοφύτου των τερματικών σπονδυλικών πλακών;
της ουλοποίησης και/ή χόνδρυνσης του ινώδους δακτυλίου,
οι μεταβλητές αυτές δεν συνδέονταν με διαφορές όσον αφορά τα 12μηνα κλινικά αποτελέσματα συγχώνευσης, βελτίωσης πόνου/ εξασθένισης Oswestry, νευρολογικής κατάστασης ή γενικής επιτυχίας, με εξαίρεση την κήλη
πολφώδους πυρήνα. Μεγαλύτερος υπήρξε
Πραγματοποιήθηκε ανάλυση “θεραπευτικής πρόθεσης”. Για την ανάλυση αυτή, οι αποτυχίες δευτερογενούς χειρουργικής οι θάνατοι, οι περιπτώσεις ασθενών που διέφυγαν την παρακολούθηση και η απουσία
παρατηρήσεων λόγω άλλων αιτίων
θεωρήθηκαν “αποτυχίες”. Η αντιμετώπιση αυτών των ασθενών ως θεραπευτικών αποτυχιών οδήγησε, όσον αφορά την ανάλυση θεραπευτικής πρόθεσης, σε ποσοστά κλινικών αποτελεσμάτων σημαντικώς χαμηλότερα
από εκείνα των πραγματικών παρατηρημένων κλινικών δεδομένων. Ο ακόλουθος
αλλά δεν περιλαμβάνει τα αποτελέσματα των ασθενών ελέγχου.
ȅȚ șȐȞĮIJȠȚ, ĮʌȠIJȣȤȓİȢ įİȣIJİȡȠȖİȞȠȪȢ ȤİȚȡȠȣȡȖȚțȒȢ, įȚĮijȣȖȑȢ Įʌȩ ʌĮȡĮțȠȜȠȪșȘıȘ țĮȚ Ș ĮʌȠȣıȓĮ ʌĮȡĮIJȘȡȒıİȦȞ
ȆȠıȠıIJȐ 12
ȈȣȖȤȫȞİȣıȘ
ǺİȜIJȓȦıȘ ʌȩȞȠȣ/ İȟĮıșȑȞȚıȘȢ Oswestry
ǹıșİȞİȓȢ μİ ȕİȜIJȓȦıȘ IJȠȣȜȐȤȚıIJȠȞ 15 μȠȞȐįȦȞ
Įʌȩ IJȘȞ ʌȡȠİȖȤİȚȡȘIJȚțȒ țĮIJȐıIJĮıȘ
ǻȚĮIJȒȡȘıȘ Ȓ ȕİȜIJȓȦıȘ ȞİȣȡȠȜȠȖȚțȒȢ
țĮIJȐıIJĮıȘȢ
īİȞȚțȒ İʌȚIJȣȤȓĮ
ǹʌȠIJȣȤȓİȢ įİȣIJİȡȠȖİȞȠȪȢ ȤİȚȡȠȣȡȖȚțȒȢ
1
ȂȘ İȞȫıİȚȢ
ȁȠȚʌȑȢ ʌİȡȚʌIJȫıİȚȢ
ĬȐȞĮIJȠȚ
ȆȓȞĮțĮȢ ǿV – ǹȞȐȜȣıȘ șİȡĮʌİȣIJȚțȒȢ ʌȡȩșİıȘȢ ȖȚĮ ıȣıțİȣȒ ǿNTER FIX™
șİȦȡȠȪȞIJĮȚ ĮʌȠIJȣȤȓİȢ țĮȚ ʌİȡȚȜĮμȕȐȞȠȞIJĮȚ ıIJȠȞ ʌĮȡĮȞȠμĮıIJȒ IJȠȣ ʌȠıȠıIJȠȪ
2
ο αριθμός των ασθενών της μη τυχαίας ομάδας στους οποίους διαπιστώθηκε κήλη πολφώδους πυρήνα, πράγμα που θεωρήθηκε αυξητικό της πιθανότητας γενικής επιτυχίας.
οδήγησαν σε απουσία παρατηρήσεων όσον αφορά τις μεταβλητές αποτελέσματος και συνεπώς συμπεριλήφθηκαν στους παρανομαστές των ποσοστών που υπολογίστηκαν, δηλ.
του ωχρού συνδέσμου και/ή της κάψας οπίσθιας σπονδυλικής άρθρωσης καθώς και της κήλης πολφώδους πυρήνα. Περαιτέρω στατιστικές αναλύσεις έδειξαν ότι
πίνακας (πίνακας ΙV) παρουσιάζει τα αποτελέσματα της ανάλυσης θεραπευτικής πρόθεσης για τους ασθενείς συσκευής INTER FIX™
μȘȞȫȞ
65.9% (122/185) 67.6%
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
33.0% (61/185) 38.4% (71/185) 45.7%
5
8
0 1 0 0
ȆȠıȠıIJȐ 24
μȘȞȫȞ
(125/185)
(128/185)
5
3
ȆȠıȠıIJȐ 48
μȒȞȠȣ
80.2%
(93/116)
(66/116)
87.9%
(102/116)
(53 /116)
4
4
ȆȠıȠıIJȐ 72 μȒȞȠȣ
77.1%
( 84/109)
56.0%
(61 /109)
75.2%
75.2% (82/109)
(82/109)
49.5%
(54/109)
1
2
1- Αυτοί οι ασθενείς περιλαμβάνονται στους υπολογισμούς ποσοστών συγχώνευσης αλλά κατά τα άλλα θεωρούνται αποτυχίες για τους σκοπούς της κλινικής δοκιμής.
2- Οι ασθενείς για τους οποίους είχε έλθει
κλινικής δοκιμής.
ΣΥΣΚΕΥΑΣΙΑ:
Οι συσκευασία κάθε μιάς από τις συνιστώσες πρέπει να είναι άθικτη κατά την παραλαβή. Αν χρησιμοποιείται σύστημα δανεισμού ή παρακαταθήκης, πριν από τη χρήση απαιτείται προσεκτικός έλεγχος όλων των σετ για
πληρότητα και όλων των συνιστωσών για ίχνη φθοράς. Οι συσκευασίες ή τα προϊόντα που έχουν υποστεί ζημιά δεν πρέπει να χρησιμοποιούνται αλλά να επιστρέφονται στη Medtronic Sofamor Danek.
ΑΠΟΛΥΜΑΝΣΗ ΚΑΙ ΚΑΘΑΡΙΣΜΟΣ:
Όλα τα εμφυτεύματα και όργανα, αν δεν παρέχονται αποστειρωμένα, πρέπει πρώτα να καθαριστούν με χρήση καθιερωμένων νοσοκομειακών μεθόδων, πριν αποστειρωθούν και εισαχθούν σε αποστειρωμένη χειρουργική
περιοχή. Τα χρησιμοποιούμενα πρέπει να απολυμαίνονται, καθαρίζονται και αποστειρώνονται πριν ξαναχρησιμοποιηθούν.
Σημείωση: Μην χρησιμοποιείτε διαλύματα καθαρισμού που περιέχουν λευκαντικό ή φορμαλίνη γιατί ενδέχεται να προξενήσουν ζημιά
η στιγμή παρακολούθησης εκείνη την περίοδο και που υπέστησαν δευτερογενή χειρουργική για λόγους άλλους από μη ένωση θεωρούνται αποτυχίες για τους σκοπούς της
στη συσκευή.
Πρέπει να μεταχειρίζεσθε όλα τα προϊόντα με προσοχή. Ακατάλληλη χρήση ή χειρισμός μπορούν να επιφέρουν βλάβη και πιθανή ακατάλληλη λειτουργία της συσκευής.
ΑΠΟΣΤΕΙΡΩΣΗ:
Εκτός αν επισημαίνονται ως αποστειρωμένα και φέρουν σαφή αναγραφή αυτού στην προερχόμενη από την εταιρεία κλειστή αποστειρωμένη συσκευασία τους, όλα τα εμφυτεύματα και όργανα που χρησιμοποιούνται στη
χειρουργική πρέπει να αποστειρώνονται στο νοσοκομείο πριν τη χρήση. Αφαιρείτε πλήρως το υλικό συσκευασίας πριν την αποστείρωση. Χρησιμοποιείτε μόνο αποστειρωμένα προϊόντα στο χειρουργείο. Για Εγγυημένο
Βαθμό Αποστείρωσης 10-6 συνιστάται αποστείρωση με ατμό των προϊόντων αυτών στο νοσοκομείο, με χρήση ενός από τους τρεις ακόλουθους συνδυασμούς παραμέτρων επεξεργασίας.
ΜΕΘΟΔΟΣ ΚΥΚΛΟΣ ΘΕΡΜΟΚΡΑΣΙΑ ΧΡΟΝΟΣ ΕΚΘΕΣΗΣ
Ατμός Προκαταρκτική αποστείρωση σε κενό 270° F (132°C) 4 min
Ατμός υπό πίεση 250° F (121°C) 60 min
Ατμός* υπό πίεση* 273° F (134°C)* 20 min *
ΣΗΜ. : Λόγω των πολυάριθμων παραμέτρων που εμπλέκονται στην αποστείρωση, κάθε ιατρική εγκατάσταση θα
χρησιμοποιούνται για τον εξοπλισμό της.
* Για τη χρήση εκτός των ΗΠΑ, ορισμένες υγειονομικές αρχές χωρών εκτός ΗΠΑ συνιστούν αποστείρωση βάσει αυτών των παραμέτρων ώστε να περιορίζεται στο ελάχιστο ο δυνάμει κίνδυνος μετάδοσης της νόσου
Κρόυτσφελντ-Γιάκομπ
ΠΑΡΑΠΟΝΑ ΓΙΑ ΤΑ ΠΡΟΪΟΝΤΑ
Κάθε επαγγελματικός χρήστης μέσων ιατρικής περίθαλψης (π.χ. πελάτης ή χρήστης αυτού του συστήματος προϊόντων) ο οποίος έχει οποιοδήποτε παράπονο ή εμπειρίες που τον άφησαν μη ικανοποιημένο από την
ποιότητα, ταυτότητα, αντοχή, αξιοπιστία, ασφάλεια, αποτελεσματικότητα και/ή απόδοση του προϊόντος, καλείται να πληροφορήσει σχετικά τον διανομέα ή τη Medtronic Sofamoe Danek. Επιπλέον, αν ποτέ οποιεσδήποτε
από τις εμφυτευμένες συνιστώσες της ελικοτομημένης συσκευής συγχώνευσης INTER FIX™ και της ελικοτομημένης συσκευής συγχώνευσης INTER FIX™ RP “δυσλειτουργήσουν” (δηλαδή δεν ικανοποιούν οποιεσδήποτε
από τις προδιαγραφές απόδοσής τους ή κατά άλλο τρόπο δεν λειτουργούν όπως προβλέπεται) ή αν υπάρχουν υπόνοιες ότι συμβαίνει αυτό, πρέπει να ενημερώνεται αμέσως ο υπεύθυνος διανομής. Αν ποτέ ένα
προϊόν της Medtronic Sofamor Danek “δυσλειτουργεί” και ενδέχεται να έχει προκαλέσει θάνατο ή σοβαρή κάκωση ασθενούς ή να έχει συμβάλει σε αυτά, ο διανομέας πρέπει να ενημερώνονται το συντομότερο δυνατό
τηλεφωνικά, με τηλεομοιοτυπία ή γραπτή αλληλογραφία. Όταν υποβάλλετε παράπονο, παρακαλείσθε να αναφέρετε το όνομα της συνιστώσας ή των συνιστωσών, τον αριθμό εξαρτήματος, τον ή τους
τους, το όνομα και τη διεύθυνσή σας, τη φύση του παραπόνου και γνωστοποίηση του κατά πόσον ζητείται γραπτή έκθεση για τον υπεύθυνο διανομής.
ΣΗΜΕΙΩΣΗ ΓΙΑ ΤΟΝ ΙΑΤΡΟ: Αν και ο γιατρός αποτελεί σπουδασμένο μεσάζοντα ανάμεσα στην εταιρεία και τον ασθενή, οι σημαντικές ιατρικές πληροφορίες που αναφέρει αυτό το
στον ασθενή.
, ιδίως όσον αφορά στα χειρουργικά όργανα που ενδέχεται να έρθουν σε επαφή με το κεντρικό νευρικό σύστημα.
πρέπει να εκτιμά και να ελέγχει τις διαδικασίες αποστείρωσης (π.χ. θερμοκρασία, διάρκεια) που
αριθμούς μερίδας
κείμενο πρέπει να γνωστοποιούνται
Mόνο για πελάτες εντός των Η.Π.Α.
ΠΡΟΣΟΧΗ: Η ΟΜΟΣΠΟΝΔΙΑΚΗ ΝΟΜΟΘΕΣΙΑ (ΤΩΝ ΗΠΑ) ΕΠΙΤΡΕΠΕΙ ΤΗΝ ΠΩΛΗΣΗ ΑΥΤ Ω Ν ΤΩΝ ΣΥΣΚΕΥΩΝ ΜΟΝΟ ΑΠΟ ΓΙΑΤΡΟ Η ΒΑΣΕΙ ΣΥΝΤΑΓΗΣ ΓΙΑΤΡΟΥ.
ΠΡΟΣΠΑΘΕΙΕΣ ΑΝΑΚΤΗΣΗΣ ΣΥΣΚΕΥΗΣ:
Αν καταστεί αναγκαία η αφαίρεση μιας ελικοτομημένης συσκευής συγχώνευσης INTER FIX™ ή ελικοτομημένης συσκευής συγχώνευσης INTER FIX™ RP, παρακαλείσθε να τηλεφωνήσετε στη Medtronic Sofamor
Danek.
© 2006 Medtronic Sofamor Danek. Με επιφύλαξη κάθε δικαιώματος.
Page 45

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
PORTUGUÊS
Este documento contem informações médicas importantes relativas ao dispositivo para fusão roscado INTER FIX™ e ao dispositivo para fusão roscado INTER FIX™ RP.
DESCRIÇÃO:
O dispositivo para fusão roscado INTER FIX™ consiste num cilindro metálico, oco e perfurado assim como numa bujão. O dispositivo para fusão roscado INTER FIX™ RP consiste num
cilindro metálico, oco e perfurado, e comporta uma grande estria arredondada e externa, todo ao longo do eixo longitudinal que prolonga-se até ao diâmetro interior do dispositivo. Ambas
as extremidades do implante INTER FIX™ RP são fechadas.
O diâmetro dos implantes INTER FIX™ e INTER FIX™ RP pode variar de 12mm a 24mm e o comprimento de 20mm a 29mm. O tamanho dos bujões dos implantes INTER FIX™ corresponde
ao diâmetro dos cilindros e colócão-se na extremidade aberta do cilindro depois este terem sido enchido com enxerto ósseo autogéneo.
Os implantes INTER FIX™ e INTER FIX™ RP são fabricados em liga de titânio especial para implantes (Ti-6Al-4V) descrito pela norma ASTM F136.
Garantias implícitas de comercialização ou de aptidão para um objecto ou uma utilização particular são expressamente excluídas. Para mais informações, consultar a lista de preços ou o
catálogo da Medtronic Sofamor Danek acerca das garantias e dos limites de responsabilidades.
INDICAÇÕES:
O dispositivo para fusão roscado INTER FIX™ e o dispositivo para fusão roscado INTER FIX™ RP são indicados para as intervenções cirúrgicas de fusão da coluna em pacientes cujo
esqueleto está desenvolvido e que apresentam uma discopatia degenerativa (DDD) num dos níveis da coluna do L2 ao S1. A discopatia degenerativa (DDD) é diagnosticada pela presença
de dor no dorso de origem discal com degenerescência do disco, confirmada pelos antecedentes do paciente e por controlo radiológico. Estes pacientes que apresentam uma discopatia
degenerativa (DDD) também podem ter espondilolístese ou retrolístese até ao grau I no nível implicado da coluna. Os dispositivos INTER FIX™ e INTER FIX™ RP devem ser utilizados com
enxerto ósseo autogéneo e implantados por meio duma cirurgia aberta por via anterior.
CONTRA-INDICAÇÕES:
Não implantar o dispositivo para fusão roscado INTER FIX™ nem o dispositivo para fusão roscado INTER FIX™ RP em pacientes que apresentam uma infecção activa na região a operar,
nem naqueles alérgicos ao titânio ou á liga de titânio.
ADVERTÊNCIAS:
• O dispositivo para fusão roscado INTER FIX™ e o dispositivo para fusão roscado INTER FIX™ RP devem unicamente ser implantados por cirurgiões com experiência na cirurgia de
fusão da coluna e terem sido formados com estes dispositivos.
• Se a experiência e/ou a formação do cirurgião não são suficientes, isto poderia aumentar o risco que surjão efeitos indesejáveis tais como : lesões vasculares, lesões neurológicas
e/ou problemas no aparelho urogenital (incluindo ejaculação retrógrada).
O DISPOSITIVO PARA FUSÃO ROSCADO INTER FIX™
O DISPOSITIVO PARA FUSÃO ROSCADO INTER FIX™ RP
INFORMAÇÕES MÉDICAS IMPORTANTES
Page 46

PRECAUÇÕES:
™
O
L
O
L
L
L
O
• A segurança e a eficiência do dispositivo para fusão roscado INTER FIX™ e do dispositivo para fusão roscado INTER FIX™ RP não foi demonstradas nos pacientes que encontramse num dos casos seguintes:
- no caso de espondilolístese ou de retrolístese é de grau II ou superior,
- quando é necessário fusionar mais dum nível,
- quando é preciso corrigir uma intervenção cirúrgica de fusão entre os corpos vertebrais previamente realizada,
- quando é necessário administrar anti-inflamatórios esteróides ou não-esteróides, durante o período pós-operatório,
- quando a obesidade é flagrante,
- quando a idade do paciente é inferior a 18 anos ou superior a 65 anos,
- em caso de osteoporose, osteopenia, e/ou osteomalacia,
- no caso de gravidez.
• A segurança e a eficiência do dispositivo para fusão roscado INTER FIX™ e do dispositivo para fusão roscado INTER FIX™ RP foram demonstradas unicamente para as
intervenções cirúrgicas de fusão entre os corpos vertebrais na coluna lombar por via anterior (ALIF), utilizando enxerto ósseo autólogo.
• Os pacientes nos quais um dispositivo para fusão roscado INTER FIX™ será implantado, devem ter seguido um tratamento não-cirúrgico durante pelo menos os seis meses
precedendo a intervenção. Os pacientes nos quais um dispositivo para fusão roscado INTER FIX™ RP será implantado, devem também ter seguido um tratamento não-cirúrgico
durante pelo menos os seis meses precedendo a intervenção.
• Implantar os dois dispositivos para fusão roscados INTER FIX™ um ao lado do outro no nível a operar. Implantar um dispositivo para fusão roscado INTER FIX™ e um dispositivo
para fusão roscado INTER FIX™ RP um ao lado do outro no nível a operar. A estria longitudinal do dispositivo INTER FIX™ RP facilitará a implantação a mais próxima possível entre
os dois dispositivos INTER FIX™, obtendo por conseguinte um perfil lateral reduzido.
• O eixo longitudinal do dispositivo para fusão roscado INTER FIX™ assim como aquele do dispositivo para fusão roscado INTER FIX™ RP devem-se encontrar na direcção anteriorposterior.
• Os implantes e instrumentos devem ser esterilizados antes de ser utilizados conforme aos procedimentos de esterilização indicados neste folheto, excepto se fornecidos estéreis e
que a etiqueta o indica claramente.
EFEITOS INDESEJÁVEIS :
Os efeitos indesejáveis, como indicado no quadro I, são aqueles reportados durante a implantação do dispositivo INTER FIX™ em 185 pacientes e no tratamento dos 62 pacientes do grupo
de controlo, que participaram ao estudo clínico multicentro para o dispositivo para fusão roscado INTER FIX™. Além de mais, neste quadro estão indicados os efeitos indesejáveis que foram
reportados em 116 pacientes para o periodo de 48 meses, e em 109 pacientes para o periodo de 72 meses, durante o estudo pós-aprovação. As porcentagens indicadas foram calculadas
a partir do número total de pacientes que participaram a este estudo, para cada interval de tempo préestabelecido. Os efeitos indesejáveis que sucederam nos grupos, aleatorizados e não
aleatorizados, tratados com o dispositivo INTER FIX™, foram reunidos para poder apresentar uma porcentagem global.
QUADRO I - EFEITOS INDESEJÁVEIS
Operatórios
(Número %)
NTER FIX
Complicação
Vascular operatória
Dor sacroilíaca
Neurológica
Dor no dorso
Devida à incisão
Complicação na
coluna
Urológica
Outras
Outras dores
Gastrointestinal
Ejaculação
retrógrada
Respiratória
Dor na perna
Traumatismo
Peritoneal
Vascular pósoperatória
Fractura óssea
Deslocação/
Desconjuntamento
do implante
Dor onde se colheu
o enxerto
Não consolidação
Subsidência
Não consolidação
(À ESPERA DO
RESULTADO)
Meningite
Quebra do Implante
Cancro
Falecimento
1 – Durante o periodo de 24 meses, seis (6) casos de ejculação retrógrada foram observados perante os 81 pacientes de sexo masculino pertencendo ao grupo do dispositivo INTER
FIX™ (7,4%).
2 – Este paciente (175) foi reportado ulteriormente visto que a fusão foi obtida ao cabo de 52 meses após a operação, e isto sem que outra intervenção cirúrgica ou qualquer outro tratamento
tenham sido necessários.
Os dois efeitos indesejáveis importantes observados durante este ensaio clínico, foram lesões vasculares e lesões neurológicas que surgiram durante a operação. No total, 15 lesões vasculares
que surgiram durante a operação foram assinaladas em 14 pacientes pertencendo ao grupo do dispositivo INTER FIX™, durante o periodo de 24 meses. Estas lesões são: 2 lesões da veia
cava, 9 lesões da veia ilíaca, 1 laceração da veia hipogástrica, 1 hemorragia num segmento da veia, 1 lesão da veia sagrada, 1 hemorragia superficial. Relativamente ao grupo de controlo,
um total de 2 lesões vasculares surjiram durante a operação em 2 pacientes durante o mesmo periodo. Estas lesões são: 1 lesão da veia ilíaca, e 1 hemorragia da base de osso.
No total, 30 casos neurológicos foram observados em 28 pacientes pertencendo ao grupo do dispositivo INTER FIX™, durante o periodo de 24 meses. Estes casos são: 1 pé pendente, 3
lesões das raízes dos nervos, 1 estenose foraminal, 1 distrofia do reflexo simpático, 7 endormecimentos ou ardor nas pernas, 2 disestesias, 3 parestesias, 2 radiculites, 7 dores no dorso e/ou
na perna, 1 dor no dorso e/ou na perna acompanhada de outros sintomas, 1 dor lancinante na parte de baixo das costas e 1 síndroma do canal carpiano.
No total, 10 casos neurológicos foram observados em 8 pacientes pertencendo ao grupo de controlo durante o mesmo periodo. Estes casos são: 1 radiculopatia acompanhada de formigueiro
nas extremidades, 1 dor crónica no dorso acompanhada de radiculites, 1 paciente apresentando sintomas de enfraquecimento geral, 2 dores no dorso e na perna acompanhadas de outros
sintomas, 1 músculo grande abdutor desnervado, e 4 com endormecimento ou ardor nas pernas. Durante o estudo pós-aprovação, 10 casos neurológicos suplementares foram observados
em 10 pacientes. Estes casos são: 1 radiculopatia, 2 radiculopatias cervicais, 1 endormecimento e formigueiro nas mãos, 1 endormecimento ou ardor nas pernas, 3 dor no dorso e/ou na
perna, e 2 dores no dorso e/ou na perna acompanhadas de outros sintomas. O quadro que apresenta os efeitos indesejáveis mostra-nos também efeitos indesejáveis, tais que dores no dorso
e nas pernas e/ou efeitos indesejáveis ligados à coluna, tais que o colapso do espaço discal, que, em certas circunstâncias, tem uma relação neurológica.
Alguns dos efeitos indesejáveis implicaram uma intervenção cirúrgica suplementar após à intervenção realizada para o ensaio clínico. Estas intervenções cirúrgicas suplementares são
classificadas da maneira seguinte: correcção, extracção, fixação suplementar, ou reoperação.
do grupo do dispositivo INTER FIX™ (aleatorizados e não aleatorizados reunidos), naqueles do grupo de controlo durante o estudo IDE e naqueles do grupo do dispositivo INTER FIX™
durante o estudo pós-aprovação.
CONTR
N=185
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
LO
N=62
5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
2(1.1) 3(1.6) 1(0.5) 6(3.2)
3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
3(1.6) 3(1.6) 0
2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
2(1.1) 2(1.1) 0
1(1.6)2 0 1(1.6) 2(1.7)
1(1.6) 0 1(1.6)
5(8.1) 0 5(8.1)
1
1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
Pós-
Operatórios
(1 Dia - <2
meses)
[Número (%)]
INTER
CONTRO
FIX™
N=185
O
N=62
3 meses
(t
meses)
[Número
(%)]
INTER
FIX™
N=185
2-<5
CONTR
LO
N=62
6 meses
5-<9
(t
meses)
[Número (%)]
ONTRO
INTER
FIX™
N=185
N=62
12 meses
9-<19
(t
meses)
[Número (%)]
ONTRO
INTER
O
FIX™
N=185
N=62
(0.5)
1
O quadro II resume as intervenções cirúrgicas suplementares efectuadas nos pacientes
24 meses
19-<30
(t
meses)
[Número (%)]
INTER
FIX™
N=185
(0.5)
CONTRO
O
1
TOTAL EFEITOS
INDESEJÁVEIS
(DURANTE 24
MESES)
™
N=185
(7.4)
CONTROL
N=62
0
1
INTER FIX
O
N=62
2 (1.1) 1(0.8) 1(0.9)
48
meses
(>30 - < 60)
(t
30-<60
Meses)
INTER FIX
N=116
™
72
meses
(t
60
Meses)
INTER FIX
N=109
™
Page 47

1
p
Correcção: Intervenção durante a qual o ajustamento ou qualquer modificação da configuração inicial do implante esta realizado.
Extracção: Intervenção durante a qual um ou mais componentes da configuração inicial do implante sao extraídos, sem substituição com o mesmo tipo de dispositivo utilizado para o ensaio.
Fixação Suplementar: Intervenção durante a qual são implantados dispositivos para a coluna adicionais que não fazem parte da configuração inicialmente prevista.
Reoperação: Intervenção cirúrgica durante a qual um componente do implante de origem não é extraído, modificado ou adicionado.
Estudo IDE Estudo pós-aprovação
Intervalo de tempo
Correcção 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Extracção 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Fixação
Suplementar
Reoperação 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
Quadro II - Intervenções cirúrgicas suplementares
préestabelecido
24 meses
Dispositivo
INTER FIX™
N=185
Controlo
N=62
Intervalo de tempo
préestabelecido
1
INTER FIX™
48 meses
Dispositivo
N=116
Intervalo de tempo
préestabelecido
72 meses
Dispositivo
INTER FIX™
N=109
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
1- Nenhum dado está disponível relativamente ao periodo de 72 meses para os pacientes do grupo de controlo, porque para estes pacientes, só estava previsto um seguimento durante 24 meses
EFEITOS INDESEJÁVEIS POSSÍVEIS:
O seguinte é uma lista dos efeitos indesejáveis possíveis que podem ocorrer durante a intervenção cirúrgica de fusão da coluna utilizando um dispositivo para fusão roscado INTER FIX™
ou um dispositivo para fusão roscado INTER FIX™ RP. Alguns destes efeitos indesejáveis podem ter sido referidos anteriormente no quadro dos efeitos indesejáveis.
• Desmontagem, torção, rotura, desconjuntamento, e/ou migração dos componentes.
• Reacção (alérgica) ao corpo estranho.
• Lesão dos tecidos ou dos nervos.
• Mudança pós-operatória da curvatura da coluna, perca de correcção, altura, e/ou redução da coluna.
• Infecção.
• Rotura dural.
• Disfunção do sistema neurológico.
• Disfunção do sistema urinário.
• Formação de cicatriz.
• Fractura óssea.
• Não consolidação óssea (ou pseudoartrose), consolidação tardia, má consolidação.
• Interrupção de qualquer desenvolvimento potencial do segmento da coluna operado. Perca da mobilidade ou da função da coluna.
• Complicações na região onde se colheu o enxerto.
• Lesão dos vasos sanguíneos, e disfunção do sistema cardiovascular.
• Complicações do sistema gastrointestinal.
• Disfunção do sistema de reprodução.
• Lesão dos órgãos internos e do tecido conjuntivo.
• Desenvolvimento de problemas respiratórios.
• Complicações ligadas à incisão.
• Mudança do estado mental.
• Falecimento.
Observações: Uma cirurgia suplementar pode vir a ser necessária para corrigir alguns destes efeitos indesejáveis possíveis.
RESULTADOS CLÍNICOS:
Um ensaio clínico comparativo2 multicentro sobre o dispositivo para fusão roscado INTER FIX™ foi efectuado nos Estados Unidos de América com o fim de comparar o dispositivo INTER
FIX™ quando se utiliza na coluna anterior e que é enchido com osso autogéneo, a um grupo de controlo tratado com um autoenxerto do anel femoral enchido de osso autogéneo, isto no
tratamento dos pacientes que apresentam uma discopatia degenerativa sintomática. Inicialmente, este ensaio clínico foi previsto prospectivo, aleatorizado e controlado. Subsequentemente,
os planos desta pesquisa foram mudados para permitir aos pacientes de entrarem no grupo não aleatorizado, isto é, pacientes tratados com o dispositivo INTER FIX™ unicamente.
Os critérios de inclusão para este ensaio clínico eram os seguintes: discopatia degenerativa sintomática diagnosticada por dor resistente ao tratamento na perna e/ou no dorso e confirmada
em radiologia; instabilidade da coluna determinada por um desvio superior a 4mm ou por um ângulo superior a 5°, nas radiografias flexão/extensão; manifestação dos sintomas a um só
nível da coluna entre a L2 - S1; e espondilolístese não superior ao grau I. Foram expressamente excluídos deste estudo clínico os pacientes que tiveram uma intervenção cirúrgica de fusão
entre os corpos vertebrais, por via anterior, no mesmo nível da coluna; os pacientes que apresentam osteopenia, osteoporose, ou osteomalacia; assim como os pacientes que precisavam
duma estimulação do desenvolvimento ósseo.
O número total de pacientes, pertencendo ao grupo do dispositivo INTER FIX™, inscritos no estudo clínico era de 185.
de controlo, pertencendo ao grupo aleatorizado, era de 62.
Para o dispositivo para a fusão roscado INTER FIX™, um estudo pós-aprovação foi requerido com o fim de avaliar a longo prazo os resultados deste dispositivo, para o qual era necessário
obter os resultados ao cabo de 6 anos após a intervenção, em pelo menos 100 pacientes. Assim, avaliou-se o resultado do dispositivo a longo prazo em 116 pacientes ao cabo de 4 anos
após terem sido operados, e em 109 pacientes ao cabo de 6 anos após terem sido operados. A população fazia parte dos pacientes que participaram ao ensaio clínico IDE. Em conformidade
com os critérios do estudo IDE, a descrição das intervervenções cirúrgicas suplementares, o controlo radiológico da fusão, assim como a avaliação da dor e da função, faziam parte das
informações que poderam ser obtidas para estes pacientes.
O quadro III mostra-os os resultados deste estudo clínico e põe em evidência as porcentagens do êxito relativamente à fusão, à dor segundo Oswestry e o estado neurológico, ao cabo de 12,
24, 48 e 72 meses após a intervenção. O êxito deste ensaio clínico estava baseado sobre uma análise comparativa entre o dispositivo INTER FIX™ e àquele de controlo.
Neste ensaio clínico, a fusão foi definida quando se obteve: a união com o osso trabecular, uma estabilidade quando o desvio é inferior ou igual a 3mm e o ângulo de movimento inferior a 5°,
assim como nenhuma radiotransparência à volta de cada implante superior a 50%. Além de mais, os pacientes que sofreram uma intervenção cirúrgica suplementar consequente a ausência
de consolidação, foram considerados, nos cálculos, como fusões sem êxito. Relativamente à incapacidade/dor segundo Oswestry, considerou-se que o êxito estava atingido quando o resultado
pós-operatório melhorou-se de pelo menos 15 pontos em relação ao resultado pré-operatório. O êxito, relativamente ao estado neurológico, foi obtido se este estado pôde-se manter ou se
foi melhorado após a operação, de pelo menos 3 das 4 categorias neurológicas (o movimento, os sentidos, os reflexos, e o levantamento da perna recta), em comparação com o estado
pré-operatório. Considerou-se que o êxito estava atingido para um paciente se: foi demonstrado que a fusão foi atingida, se obteve-se êxito no que diz respeito à incapacidade/dor segundo
Oswestry, se obteve-se êxito relativamente ao estado neurológico, assim como se o paciente não teve de sofrer uma intervenção cirúrgica suplementar, classificada como “falência”.
2
Estatisticamente o dispositivo INTER FIX™ não é pior de que àquele de controlo.
3
Os 3 pacientes nos quais o dispositivo INTER FIX™ não foi implantado em razão dos efeitos indesejáveis relacinados com a intervenção, não foram tomados en conta.
3
Para o ensaio clínico, o número total de pacientes inscritos no grupo
O quadro III também mostra-nos as porcentagens relativas ao êxito total, ao cabo de 12 e 24 meses, após a intervenção.
Fusão
Melhoramento da
incapacidade/dor
segundo
Oswestry
Melhoramento de
pelo menos 15
pontos em relação
ao resultado préoperatório
Manutenção ou
melhoramento
do estado
neurológico
Êxito Total
Porcentagem 12 meses Porcentagem 24 meses
Dispositivo de
(122/127)
(76/142)
(150/151)
(61/144)
Quadro III – Resultados do estudo clínico para o dispositivo INTER FIX™
Delta
Dispositivo de
1
(ǻ
)
teste Controlo
96.1%
53.5%
99.3%
42.4%
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
(12/56)
teste Controlo
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
-0.097 46.1%
(71/154)
Delta
(ǻ1)
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
-0.043 49.5%
(17/57)
Porcentagem 48
meses
Dispositivo de
teste
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0% (102/103) 97.6% (82/84)
(53/107)
Porcentagem 72
meses
Dispositivo de
teste
56.3%
(54/96)
1 - Margem de não inferioridade minima requerida para afirmar a não inferioridade do dispositivo de teste em relação àquele de controlo
Todos os pacientes implicados no ensaio clínico sobre o dispositivo para a fusão roscado INTER FIX™, não tomando em conta o grupo, foram seleccionados segundo os mesmos critérios de
inclusão/exclusão. Para confirmar o carácter comparativo entre os pacientes do grupo aleatorizado do dispositivo INTER FIX™ e os pacientes do grupo não aleatorizado do dispositivo INTER
FIX™, examinou-se o seguinte: as características demográficas, as condições médicas pré-operatórias, o diagnóstico obtido em radiologia característico duma discopatia degenerativa, a
maneira de o
erar, e à colocação do nível da coluna lombar que deve ser tratado. A população era estatisticamente comparável, excepto no que diz respeito à maneira de operar, à actividade
Page 48

de trabalho do paciente antes da operação, ao diagnóstico radiologico pré-operatório de formação de osteófitos na placa terminal da vertebra; fibrose e/ou espessamento do anulus fibroso,
do ligamento amarelo, ou da cápsula da faceta articular; e hérnia do núcleo pulposo. Outras análises estatísticas mostraram que estas variáveis não estavam relacionadas com os resultados
clínicos da fusão num periodo de 12 meses, a melhoria invalidez/dor segundo Oswestry, o estado neurológico ou o êxito total, excepto para as hérnias do núcleo pulposo. Foram observados
mais pacientes tendo hérnias do núcleo pulposo no grupo não aleatorizado, o que aumentou a probabilidade de êxito total.
Foi realizada uma análise com intenção de tratar. Para esta análise, as intervenções cirurgias suplementares classificadas como falências, os falecimentos, os pacientes que não puderam ter
sido seguidos, e as observações que faltavam por outras razões, foram considerados como observações perdidas para as variáveis dos resultados, e por isso foram incluídos no denominador
das porcentagens calculadas, isto é, considerados como “falências”. Como estes pacientes foram considerados como falências, as porcentagens relativas ao resultado clínico obtidas para
a análise com intenção de tratar, eram consideravelmente inferiores em relação àqueles obtidos para a análise actual.
O quadro que segue (Quadro IV) mostra-nos os resultados da análise com intenção de tratar para os pacientes do dispositivo INTER FIX™, mas não inclui os resultados obtidos para os
pacientes do grupo de controlo.
Falecimentos, intervenções cirúrgicas suplementares classificadas como falências, os pacientes que não puderam ter sido
seguidos, e as observações que faltavam, foram considerados como falências e estão incluídos no denominador das
Porcentagem
Fusão
Melhoramento da incapacidade/dor segundo
Oswestry
Melhoramento de pelo menos 15 pontos em
relação ao resultado pré-operatório
Manutenção ou melhoramento do estado
neurológico
Êxito Total
Falências das Intervenções cirúrgicas
suplementares
Ausências de consolidação
2
Outros
Falecimentos
Quadro IV - Análise com intenção de tratar para o dispositivo INTER FIX™
porcentagens
12 meses
65.9%
(122/185)
41.1%
(76/185)
81.1%
(150/185)
33.0%
(61/185)
1
5
8
0 1 0 0
Porcentagem
24 meses
67.6%
(125/185)
42.7% (79/185) 56.9%
69.2%
(128/185)
38.4% (71/185) 45.7%
5
3
Porcentagem
48 meses
80.2%
(93/116)
(66/116)
87.9%
(102/116)
(53 /116)
4
4
Porcentagem
72 meses
77.1%
( 84/109)
56.0%
(61 /109)
75.2%
(82/109)
49.5%
(54/109)
1
2
1 - Estes pacientes foram incluídos no cálculo das porcentagens relativas à fusão mas foram considerados como falências em relação aos objectivos do ensaio clínico.
2 - Os pacientes que não poderam ser seguidos durante este período e que sofreram uma intervenção cirúrgica suplementar por razões outras de que ausência de consolidação, foram
considerados como falências em relação aos objectivos do ensaio clínico.
EMBALAGEM:
As embalagens de cada um dos componentes devem estar intactas no momento da recepção. Se um sistema de empréstimo ou de depósito é utilizado, deve-se cuidadosamente verificar
que todos os implantes estão completos e que todos os componentes e os instrumentos devem ser minuciosamente controlados para prevenir de qualquer dano antes da utilização. Os
produtos danificados ou aqueles ainda numa embalagem danificada não devem ser utilizados, e devem ser devolvidos à Medtronic Sofamor Danek.
LIMPEZA E DESCONTAMINAÇÃO:
Todos os instrumentos e implantes devem em primeiro lugar ser limpos segundo o método definido pelo hospital antes da esterilização e da introdução destes na zona operatória estéril,
excepto se fornecidos estéreis. Os instrumentos que foram utilizados devem ser descontaminados, limpos, e esterilizados antes de serem reutilizados.
Observação: Não utilizar soluções de limpeza contendo lixívia ou formalina porque estas podem danificar o implante.
Todos os produtos devem ser tratados com cuidado. A utilização ou o manejamento incorrectos podem danificar o implante e impedir que este funcione correctamente.
ESTERILIZAÇÃO:
Excepto se estivesse claramente escrito como “estéril” na etiqueta da embalagem esterilizada e intacta fornecida pelo fabricante, todos os implantes e instrumentos utilizados em cirurgia
devem ser esterilizados pelo hospital antes da utilização. Retirar todo o material das embalagens antes de esterilizá-lo. Unicamente produtos estéreis devem ser empregados no bloco
operatório. Para obter um nível de segurança de esterilização de 10-6, é recomendado de esterilizar estes produtos com vapor pelo hospital, utilizando um dos três conjuntos de parâmetros
processuais seguintes:
MÉTODO CICLO TEMPERATURA TEMPO DE EXPOSIÇÃO
Vapor Vácuo Prévio 270° F (132° C) 4 minutos
Vapor Em pressão 250° F (121° C) 60 minutos
Vapor* Em pressão* 273° F (134° C)* 20 minutos*
OBSERVAÇÃO: Por causa das numerosas variáveis implicadas no processo de esterilização, cada departamento médico deve avaliar e verificar o processo de esterilização (por exemplo:
temperaturas, tempos), utilizado por seus maquinismos.
*Quando é utilizado fora dos Estados Unidos de América, certas Autoridades de Saúde que não pertencem aos Estados Unidos recomendam a esterilização conforme a estes parâmetros
de maneira a reduzir ao mínimo o risco potencial de transmissão da doença de Creutzfeldt-Jakob, especialmente para os instrumentos cirúrgicos que poderiam vir a estar em contacto com
o sistema nervoso central.
RECLAMAÇÕES SOBRE O PRODUTO:
Qualquer Profissional de Saúde (por ex: cliente ou utilizador de nossos produtos), que tenha qualquer reclamação ou que esteja insatisfeito com a qualidade, a identidade, a durabilidade,
a fiabilidade, a segurança, a eficiência e/ou as outras qualidades técnicas do produto, deve notificá-lo a seu distribuidor Medtronic Sofamor Danek. Além de mais, se qualquer componente
pertencendo ao dispositivo para fusão roscado INTER FIX™ ou ao dispositivo para fusão roscado INTER FIX™ RP foi implantado e “não funciona correctamente” (isto é: não reúne nenhuma
das especificações anunciadas ou que por outro lado não funcionasse como previsto), ou está suspeitado de tal, contactar o distribuidor imediatamente. Se qualquer produto da Medtronic
Sofamor Danek “não tivesse funcionado correctamente” e que tiver causado ou contribuído ao falecimento ou a graves lesões para o paciente, contactar o distribuidor imediatamente por
telefone, fax ou correspondência escrita. Quando registar uma reclamação, faça o favor fornecer o nome e o número do componente, o número de lote do componente, seu nome e apelidos
e sua direcção e a natureza da reclamação. É favor indicar se algum relatório escrito é pedido da parte do distribuidor.
OBSERVAÇÃO PARA O MÉDICO: Embora o médico seja o que possui o conhecimentos e que seja o intermediário entre o fabricante e o doente, as informações médicas importantes
fornecidas neste folheto devem ser comunicadas ao doente.
Apenas aplicável aos EUA
ATENÇÃO: AS LEIS FEDERAIS (E.U.A.) RESTRINGEM ESTES IMPLANTES A SEREM COMERCIALIZADOS UNICAMENTE POR UM MÉDICO OU SOBRE A PRESCRIÇÃO DUM
MÉDICO.
EXTRACÇÃO DO DISPOSITIVO:
Se a extracção dum dispositivo para a fusão roscado INTER FIX™ ou dum dispositivo para a fusão roscado INTER FIX™ RP fosse necessária, é favor contactar a Medtronic Sofamor Danek.
©
2006 Medtronic Sofamor Danek. Todos direitos reservados.
Page 49

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
ČESKY
Následující text obsahuje důležité medicínské informace o šroubovitém fúzním zařízení INTER FIX™ a šroubovitém fúzním zařízení INTER FIX™ RP.
POPIS:
Šroubovité fúzní zařízení INTER FIX™ obsahuje dutý, perforovaný, kovový válec a koncový uzávěr. Šroubovité fúzní zařízení INTER FIX™ RP obsahuje dutý, perforovaný, kovový válec s
jedním velkým vnějším zakulaceným žlábkem podél celé podélné osy, který dosahuje do vnitřního průměru z. Oba konce implantátu INTER FIX™ RP jsou uzavřeny.
Implantáty INTER FIX™ a INTER FIX™ RP jsou k dispozici v poloměrech od 12 do 24 mm a délkách od 20 do 29 mm. Koncové uzávěry implantátů INTER FIX™ mají velikost podle
průměru válců a nasazují se do otevřených konců válců poté, co se naplní autogenním kostním štěpem.
Implantáty INTER FIX™ a INTER FIX™ RP se vyrábí z titanové slitiny (Ti-6Al-4V) vhodné pro implantáty, která je v souladu s ASTM F136.
Výslovně jsou vyloučeny nepřímé záruky uplatnitelnosti na trhu a vhodnosti ke konkrétnímu účelu či použití. Další informace o zárukách a omezení odpovědnosti najdete v ceníku nebo
katalogu Medtronic Sofamor Danek.
INDIKACE:
Šroubovité fúzní zařízení INTER FIX™ a šroubovité fúzní zařízení INTER FIX™ RP se indikují pro postupy při spinální fúzi u pacientů s dospělou kostrou s degenerativním onemocněním
plotének (DDD) na jedné úrovni od L2-S1. Degenerativní onemocnění plotének je definováno jako bolest zad diskogenního původu s degenerací meziobratlové ploténky potvrzené anamnézou
pacienta a radiologickým nálezem Tito pacienti s degenerativním onemocnění plotének mohou dokonce trpět až spondylolistézou nebo retrolistézou prvního stupně na dotčené úrovni.
Implantáty INTER FIX™ a INTER FIX™ RP se mají používat s autogenním kostním štěpem a mají být implantovány otevřeným anteriorním přístupem.
KONTRAINDIKACE:
Šroubovité fúzní zařízení INTER FIX™ a šroubovité fúzní zařízení INTER FIX™ RP se nesmí implantovat u pacientů s aktivní infekcí v místě operačního zákroku nebo při alergii na titan
či titanovou slitinu.
UPOZORNĚNÍ:
• Šroubovité fúzní zařízení INTER FIX™ a šroubovité fúzní zařízení INTER FIX™ RP smí používat pouze chirurgové, kteří mají zkušenosti s postupy při spinální fúzi a absolvovali
odpovídající školení s tímto zařízením.
• Nedostatek odpovídajících zkušeností a/nebo školení může vést k vyšší míře výskytu nežádoucích příhod, jako jsou vaskulární poranění, neurologické příhody a/nebo urogenitální
příhody (včetně retrográdní ejakulace).
ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™
ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE
Page 50

BEZPEČNOSTNÍ OPATŘENÍ:
A
A
• Bezpečnost a efektivita šroubovitého fúzního zařízení INTER FIX™ a šroubovitého fúzního zařízení INTER FIX™ RP nebyla zjišťována u pacientů s jakýmkoliv z následujících stavů:
- spondylolistéza nebo retrolistéza stupně II nebo vyšší;
- fúzi se má podrobit více než jedna úroveň;
- revize postupu(ů) předchozí meziobratlové fúze;
- pooperační požadavky na steroidní či nesteroidní protizánětlivou medikaci;
- vysoká obezita;
- věk do 18 let nebo nad 65 let;
- osteoporóza, osteopenie a/nebo osteomalacie;
- těhotenství.
• Bezpečnost a efektivita šroubovitého fúzního zařízení INTER FIX™ a šroubovitého fúzního zařízení INTER FIX™ RP byla zjišťována pouze u postupů anteriorní lumbální
meziobratlové fúze (ALIF) využívajících autologního kostního štěpu.
• Pacienti přijímající šroubovité fúzní zařízení INTER FIX™ by měli absolvovat přinejmenším šest měsíců neoperační léčby. Pacienti přijímající šroubovité fúzní zařízení INTER FIX™
RP by měli absolvovat přinejmenším šest měsíců neoperační léčby.
• Dvě šroubovitá fúzní zařízení INTER FIX™ je třeba implantovat vedle sebe na úrovni chirurgického výkonu. Jedno šroubovité fúzní zařízení INTER FIX™ a jedno šroubovité fúzní
zařízení INTER FIX™ RP se musí implantovat vedle sebe na úrovni chirurgického výkonu. Podélný žlábek na za
blízkosti než je vzdálenost, kterou lze zvládnout se dvěma zařízeními INTER FIX™, což způsobuje snížení laterálního profilu.
řízení INTER FIX™ RP usnadňuje implantaci dvou zařízení ve větší
• Podélná osa šroubovitých fúzních zařízení INTER FIX™ a šroubovitých fúzních zařízení INTER FIX™ RP musí být v anteriorně posteriorním směru.
• Implantáty a instrumenty musí být před použitím sterilizovány podle sterilizačních pokynů uvedených v tomto příbalovém letáku, pokud již nejsou dodávány sterilní a takto i jasně
označeny.
NEŽÁDOUCÍ ÚČINKY:
Nežádoucí účinky, jak jsou uvedeny v tabulce I, byly hlášeny u 185 pacientů se zařízením INTER FIX™ a 62 kontrolních pacientů zařazených do multicentrické klinické studie šroubovitého
fúzního zařízení INTER FIX™. Dále byly hlášeny nežádoucí účinky u 116 pacientů během 48 měsíců a u 109 pacientů během 72 měsíců u pacientů ve studii prováděné po schválení.
Uvedené poměry jsou založeny na celkovém počtu pacientů ve studii v každém časovém bodě. Nežádoucí účinky vyskytující se u skupin léčených zařízením INTER FIX™ s randomizací a
bez randomizace jsou spojeny, aby bylo možné prezentovat celkovou míru.
TABULKA I - NEŽÁDOUCÍ ÚÈINKY
PooperaÉní
(1 den -<2
Operativní
Komplikace
Vaskulární
intraoperaÉní
Sakroiliakální bolest 5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
Neurologické 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Bolest v zádech 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
Incisionální 11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
Spinální pûíhoda 1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
Urologické 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Jiná 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Jiná bolest 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
Gastroin testinální
Retrográdní
ejakulace
RespiraÉní 3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
Bolest nohou 2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
Trauma 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
Peritoneální 3(1.6) 3(1.6) 0
Vaskulární
pooperaÉní
Kostní fraktura 1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
VytlaÉení/uvolnÕní
implantátu
Bolest v místÕ
štÕpu
Nezhojení 4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
Sesednutí 2(1.1) 2(1.1) 0
Nezhojení (ÈEKÁ
NA POTVRZENÍ)
Meningitida 1(1.6) 0 1(1.6)
Zlomení implantátu 5(8.1) 0 5(8.1)
Rakovina 1
[PoÉet (%)]
KONTROL
INTER FIX
N=62
N=185
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
2(1.1) 3(1.6) 1(0.5) 6(3.2)
2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
1(1.6)2 0 1(1.6) 2(1.7)
[PoÉet (%)]
INTER FIX
N=185
mÕsíce)
KONTROLA
N=62
3 mÕsícĉ
(t
2 -<5 mÕsícĉ)
[PoÉet (%)]
INTER FIX
N=185
ONTROL
N=62
6 mÕsícĉ
(t
(5 -<9 mÕsícĉ)
[PoÉet (%)]
INTER FIX
N=185
KONTROLA
N=62
Úmrtí 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
12 mÕsícĉ
(t
9 -<19 mÕsícĉ)
[PoÉet (%)]
INTER FIX
N=185
(0.5)
(t
KONTROLA
INTER FIX
N=62
1
24 mÕsícĉ
19 -<30 mÕsícĉ)
[PoÉet (%)]
KONTROLA
N=185
(0.5)
72
mÕsícĉ
(t
60
mÕsícĉ)
INTER FIX
N=109
N=62
NEŽÁDOUCÍ
PúÍHODY CELKEM
(BÔHEM 24 MÔSÍ CĈ)
KONTROLA
INTER FIX
N=185
1
(7.4)
48 mÕsícĉ
(>30 - < 60
(t
30<
60
mÕsícĉ)
INTER FIX
N=62
N=116
0
2 (1.1) 1(0.8) 1(0.9)
1 - Retrográdní ejakulace se vyskytla v šesti (6) případech z 81 mužů začleněných do skupiny se zařízením INTER FIX™ (7,4 %) během hodnocení po 24 měsíců.
2 - Tento pacient (175) byl následně hlášen jako pacient s fúzí přibližně za 52 měsíců po operaci a nebylo zapotřebí žádného chirurgického postupu či léčby.
Dvěma nejzávažnějšími nežádoucími příhodami byla intraoperační vaskulární a neurologická poranění. U 14 pacientů
intraoperačních příhod během hodnocení po 24 měsíců. Tyto příhody zahrnují: 2 poranění vena cava, 9 poranění iliákální žíly, 1 lacerovaná žíla v hypogastriu, 1 segmentální poranění žíly
při chirurgickém výkonu, 1 poranění sakrální žíly a 1 superficiální poranění při chirurgickém výkonu. U 2 pacientů v kontrolní skupině se ve stejné době vyskytly 2 vaskulární intraoperačních
poranění. Ta zahrnovala: 1 poranění iliakální žíly a 1 krvácení z kostního lůžka.
U 28 pacientů ve skupině se zařízením INTER FIX™ se během hodnocení po 24 měsíců celkem vyskytlo 30 neurologických příhod. Tyto příhody zahrnují: 1 plantárně ohnutá noha způsobená
paralýzou svalů, 3 poranění kořenu nervu, 1 foraminální stenóza, 1 reflexní sympatická dystrofie, 7 pocitů necitlivosti či tepla v nohou, 2 dysthesie, 3 parestézie, 2 radikulitidy, 7 bolestí zad
a/nebo nohou, 1 bolest zad a/nebo nohy s dalšími symptomy, 1 vystřelování bolesti do dolních zad a 1 syndrom karpálního tunelu.
U 8 pacientů v kontrolní skupině se ve stejné době celkem vyskytlo 10 neurologických příhod. Tyto příhody zahrnují: 1 radikulopatie s mravenč
radikulitidou, 1 invalidizující distribuční symptomy, 2 bolesti zda a nohou s dalšími příznaky, 1 denervace musculus abductor magnus a 4 krát necitlivost nebo horkost v nohou. V období po
schválení studie bylo zaznamenáno dalších 10 neurologických příhod u 10 pacientů. Tyto příhody zahrnují: 1 radikulopatie, 2 cervikální radikulopatie, 1 pocit necitlivosti a brnění v rukou, 1
pocit necitlivosti nebo pálení v nohou, 3 bolesti zad a/nebo nohou a 2 bolesti zad a/nebo nohou s dalšími příznaky. Navíc tabulka nežádoucích účinků uvádí nežádoucí příhody, jako jsou
bolest v nohou a zádech a/nebo spinální nežádoucí příhody, například kolaps ploténkového prostoru, který má v některých případech neurologickou komponentu.
Některé nežádoucí účinky vedly k chirurgickým zákrokům po chirurgickém výkonu v rámci klinického hodnocení. Tyto dodatečné chirurgické zákroky lze klasifikovat jako revize, odstranění,
doplňkové fixace nebo reoperace
v kontrolní léčebné skupině na základě studie IDE a ve skupině se zařízením INTER FIX™ na základě studie po schválení.
1
Revize: Proces, který upravuje nebo jakýmkoliv způsobem mění originální konfiguraci implantátu.
Odstranění: Proces, který odstraňuje jeden či více dílů originální konfigurace implantátu bez náhrady stejným typem hodnoceného zařízení.
Doplňková fixace: Postup, při němž se použijí doplňková spinální zařízení neschválená jako součást protokolu.
Reoperace: Jakýkoliv chirurgický proces, který neodstraňuje, neupravuje ani nepřidává žádné originální díly implantátu.
1
. Tabulka II uvádí souhrn sekundárních chirurgických zákroků ve skupině se zařízením INTER FIX™ (kombinovaná randomizovaná a nerandomizovaná) a
ve skupině se zařízením INTER FIX™ se celkem vyskytlo 15 vaskulárních
ením v končetinách, 1 chronická bolest zad s
Page 51

Studie IDE Studie po schválení
Tabulka II - Sekundární chirurgické postupy
BÕhem stanovené doby 24
mÕsícĉ
Zaûízení
INTER FIX™
N=185
Kontrola1
N=62
BÕhem stanovené
doby 48 mÕsícĉ
Zaûízení
INTER FIX™
N=116
BÕhem stanovené
doby 72 mÕsícĉ
Zaûízení
INTER FIX™
N=109
Revize 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
OdstranÕní 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Doplïkové
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
fixace
Reoperace 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
1 - Protože nebylo u kontrolních pacientů vyžadováno sledování po 24 měsících, informace o všech těchto pacientech nejsou pro celé období 72 měsíců k dispozici.
MOŽNÉ NEŽÁDOUCÍ ÚČINKY:
Následuje seznam potenciálních nežádoucích účinků, které se mohou vyskytnout při spinálním fúzním chirurgickém zákroku při použití šroubovitého fúzního zařízení INTER FIX™ a šroubovitého
fúzního zařízení INTER FIX™ RP. Některé z těchto nežádoucích účinků již mohly být dříve uvedeny v tabulce nežádoucích účinků.
• Rozpojení, ohnutí, rozlomení, uvolnění a/nebo migrace dílů.
• Reakce (alergická) na cizí těleso.
• Poškození tkání či nervů.
• Pooperační změny zakřivení páteře, ztráta korekce, výšky a/nebo redukce.
• Infekce.
• Trhliny dura mater.
• Narušení neurologického systému.
• Narušení urologického systému.
• Jizvení.
• Kostní fraktura.
• Bez srůstu (nebo pseudoartróza), opoždění srůstu, nesprávný srůst.
• Útlum jakéhokoliv potenciálního růstu operované části páteře. Ztráta spinální mobility či funkce.
• Komplikace v dárcovském místě kostního štěpu.
• Poškození krevních cév a narušení kardiovaskulárního systému.
• Gastrointestinální komplikace.
• Narušení reprodukčního systému.
• Poškození vnitřních orgánů a pojivové tkáně.
• Rozvoj respiračních problémů.
• Komplikace řezné rány.
• Změna duševního stavu.
• Úmrtí.
Poznámka: Pro léčbu n
KLINICKÉ VÝSLEDKY:
Multicentrická klinická studie ekvivalentnosti2 šroubovitého fúzního zařízení INTER FIX™ byla prováděna ve Spojených státech, kdy se porovnávalo anteriorní spinální použití zařízení INTER
FIX™ naplněných autogenní kostí s alotransplantátem anulus femoralis naplněného autogenní kostí, kontrolním postupem, při léčbě pacientů trpících symptomatickým degenerativním
onemocněním plotének. Zpočátku mělo klinické hodnocení prospektivní, randomizovaný, kontrolovaný design. Následně byl plán hodnocení revidován, aby umožňoval zařazení pacientů
do nerandomizovaného ramene, tj. pacientů léčených pouze zařízením INTER FIX™.
Kritéria pro zařazení do klinického hodnocení zahrnovala symptomatické degenerativní onemocnění plotének, které se projevovalo bolestí nohou a/nebo zad z nezjištěné příčiny s pozitivním
nálezem(y) z diagnostického zobrazovacího vyšetření; spinální nestabilitou, definovanou jako translace větší než 4 mm nebo angulace větší než 5° na rentgenových snímcích flexe/extenze;
jednoúrovňovým symptomatickým zapojením od L2-S1 a spondylolistézou nejvýše stupně 1. Z této studie byli konkrétně vyloučeni pacienti, kteří: podstoupili předchozí proces meziobratlové
fúze na dotčené spinální úrovni, trpěli osteopenií, osteoporózou nebo osteomalacií nebo u nich byla nutná stimulace růstu kostí.
ěkterých z těchto možných nežádoucích příhod může být zapotřebí dodatečného chirurgického zákroku.
Celkem bylo zařazeno do léčebné skupiny se zařízením INTER FIX™ 185 pacientů.3 Do randomizovaného ramene klinického hodnocení bylo zařazeno celkem 62 kontrolních pacientů.
Požadavek kladený na šroubovité fúzní zařízení INTER FIX™ spočíval v hodnocení dlouhodobého chování zařízení po získání pooperačních údajů za celkem 6 let minimálně u 100 pacientů.
Dlouhodobé chování zařízení bylo hodnoceno u 116 pacientů během 4 let po operaci a u 109 pacientů během 6 let po operaci. Populace pacientů byla získána od účastníků klinického
hodnocení IDE. Na základě kritérií ze studie IDE zahrnovaly informace získané o těchto pacientech popis jakýchkoli dodatečných chirurgických výkonů, radiografického hodnocení fúze a
hodnocení bolesti a funkce.
Tabulka III uvádí klinické výsledky ze studie a popisuje parametry úspěšnosti fúze, úspěšnost odstranění bolesti (Oswestry) a neurologickou úspěšnost během 12, 24, 48 a 72 měsíců po
chirurgickém výkonu. Úspěch tohoto klinického hodnocení byl hodnocen na základě ekvivalenční analýzy mezi zařízením INTER FIX™ a kontrolním zařízením.
Pro tuto klinickou studii byla fúze definována jako důkaz o přemostění trabekulární kosti, translace (<3mm)a stabilita (<5°) angulárního pohybu a nulová radiolucence v okolí více než 50 %
jakéhokoliv implantátu. Pacienti se sekundárními chirurgickými zákroky kvůli nespojení byli rovněž ve výpočtech považováni za neúspěšné fúze. Úspěšnost při snížení bolesti/postižení dle
Oswestry skóre byla defi
jako zachování nebo zlepšení v nejméně 3 ze 4 neurologických kategoriích (motorika, sensorické podněty, reflexy a zvýšení rovné nohy) po operaci ve srovnání s předoperačním stavem.
Celková úspěšnost byla založena na prokázání fúze u pacienta, úspěšnosti Oswestry skóre bolesti/postižení a úspěšnosti léčby neurologického stavu, přičemž žádný sekundární chirurgický
výkon nebyl klasifikován jako “selhání“.
2
Zařízení INTER FIX ™ nebylo statisticky horší než zařízení kontrolní.
3
Nezahrnuje 3 pacienty, kteří nepodstoupili léčbu zařízením INTER FIX™ díky nežádoucím chirurgických příhodám.
nována jako zlepšení nejméně o 15 bodů u pooperačního skóre v porovnání s předoperačním hodnocením. Úspěšnost léčby neurologického stavu byla definována
Výsledky celkové úspěšnosti za 12 a 24 měsíců po operaci lze rovněž nalézt v Tabulce III.
Fúze
Zlepšení
Oswestry skóre
pro
bolest/postižení
Pacienti se
zlepšením
nejménÕ o 15
bodĉ vĉÉi stavu
pûed operací
Zachování Éi
zlepšení
neurologického
stavu
Celkový úspÕch 42.4%
Tabulka III - Klinické výsledky ze studie se zaûízením INTER FIX™
PomÕry pro 12 mÕsícĉ PomÕry pro 24 mÕsícĉ
Testované
zaûízení Kontrola
96.1%
(122/127)
53.5%
(76/142)
99.3%
(150/151)
(61/144)
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
(12/56)
Delta
Testované
1
)
(ǻ
zaûízení
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
-0.097 46.1%
(71/154)
Kontrola
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
(17/57)
Delta
PomÕry pro 48
(ǻ1)
mÕsícĉ
Testované
zaûízení
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
-0.043 49.5%
(53/107)
PomÕry pro 72
mÕsícĉ
Testované
zaûízení
97.6% (82/84)
56.3%
(54/96)
1 - Minimální rozpětí noninferiority požadované pro nárokování noninferiority studijních výsledků vůči výsledkům kontrolním.
Všichni pacienti zapojení do klinického hodnocení šroubovitého fúzního zařízení INTER FIX™, bez ohledu na léčebnou skupinu, byly zařazeni za stejných zařazovacích/vyřazovacích
kritérií. Pro odůvodnění srovnatelnosti randomizovaných pacientů se zařízením INTER FIX™ a nerandomizovaných pacientů se zařízením INTER FIX™ byly zkoumány demografické
charakteristiky, předoperační zdravotní stav, nálezy diagnostických zobrazovacích metod, charakteristika degenerativního onemocnění plotének, operační přístup a místo léčené lumbální
úrovně. Populace byla statisticky srovnatelná s výjimkou operativního přístupu, odborného stavu před operačním výkonem a předoperačních nálezů diagnostických zobrazovacích metod
Page 52

tvorby osteofytů vertebrálních koncových plotének; zjizvení a/nebo ztluštění anulus fibrosis, ligamentum flavum a/nebo hladkých ploch kloubního pouzdra a herniace nucleus pulposus.
Další statistické analýzy ukázaly, že tyto proměnné nebyly spojeny s rozdíly v 12 měsíčních klinických výsledcích fúze, zlepšení bolestí/postižení dle Oswestry skóre, neurologickém stavu
nebo celkovém úspěchu s výjimkou herniace nucleus pulposus. Více pacientů v nerandomizované skupině mělo nález v podobě herniace nucleus pulposus, u něhož se zjistilo, že zvyšuje
pravděpodobnost celkového úspěchu.
Byla provedena analýza léčebných záměrů. V této analýze selhání sekundárních chirurgických výkonů, úmrtí, pacienti ztracení při sledování a chybějící pozorování z jiných důvodů měla za
následek chybějící pozorování v případě výsledných proměnných, a proto byla zahrnuta do jmenovatelů vypočítaných poměrů, tj., považovaných za „selhání“. Tím, že se s těmito pacienty
zacházelo jako s případy neúspěšné léčby, byly míry klinických výsledků v analýze léčebných záměrů podstatně nižší než u skutečně pozorovaných klinických údajů.
Následující tabulka (Tabulka IV) uvádí výsledky analýzy léčebných záměrů pacientů se zařízením INTER FIX™, ale nezahrnuje výsledky kontrolních pacientů.
Úmrtí, sekundární chirurgická selhání, ztráta pûi sledování a chybÕjící pozorování se považují za selhání a jsou
PomÕry pro 12
Fúze 65.9% (122/185) 67.6%
Zlepšení Oswestry skóre pro bolest/postižení
Pacienti se zlepšením nejménÕ o 15 bodĉ vĉÉi stavu
pûed operací
Zachování Éi zlepšení neurologického stavu 81.1% (150/185) 69.2%
Celkový úspÕch 33.0% (61/185) 38.4% (71/185) 45.7%
Sekundární chirurgická selhání
Nespojení
Ostatní
Úmrtí 0 1 0 0
1 -
1
2
Tito pacienti jsou zahrnuti do výpočtu poměru fúze, ale jsou jinak považováni za selhání pro účely klinického hodnocení.
Tabulka IV - analýza léÉebných zámÕrĉ pro zaûízení INTER FIX™
zahrnuty ve jmenovateli pomÕrĉ
mÕsícĉ
41.1% (76/185) 42.7% (79/185) 56.9%
5
8
PomÕry pro 24
mÕsícĉ
(125/185)
(128/185)
5
3
PomÕry pro 48
mÕsícĉ
80.2% (93/116) 77.1%
(66/116)
87.9%
(102/116)
(53 /116)
4
4
PomÕry pro 72
mÕsícĉ
( 84/109)
56.0%
(61 /109)
75.2%
(82/109)
49.5%
(54/109)
1
2
2 - Pacienti určení pro sledování v této době, kteří podstoupili sekundární chirurgické výkony z jiných důvodů než nespojení, jsou považování za selhání pro účely klinického hodnocení.
BALENÍ:
Obal každého dílu musí být při převzetí neporušený. Jestliže se používá nakládací či zásilkový systém, je nutné pečlivě zkontrolovat úplnost všech sad a všechny součásti včetně
nástrojů musí být podrobeny pečlivé prohlídce, zda-li před použitím nenesou známky poškození. Poškozená balení či produkty se nesmí používat a musí se vrátit společnosti Medtronic
Sofamor Danek.
ČIŠTĚNÍ A DEKONTAMINACE:
Pokud nebudou dodávány jako sterilní, všechny implantáty a instrumenty musí být nejprve očištěny zavedenými nemocničními metodami před sterilizací a uvedeny do sterilního chirurgického
pole. Před opětovným použitím se musí použité nástroje dekontaminovat, vyčistit a vysterilizovat.
Poznámka: Nepoužívejte čisticí roztoky obsahující bělicí činidlo nebo formalín, protože mohou zařízení poškodit.
Se všemi výrobky se musí zacházet opatrně. Nesprávné použití či manipulace mohou vést k poškození či případné nesprávné funkci zařízení.
STERILIZACE:
Pokud není zařízení označeno jako sterilní a toto jasně uvedeno na neotevřeném sterilním obalu dodaném společností, všechny implantáty a nástroje použité při chirurgickém výkonu musí
být před použitím v nemocnici sterilizovány. Před sterilizací odstraňte veškerý obalový materiál. V operační poli se smí používat pouze sterilní výrobky. Pro zajištění úrovně sterility 10-6 se
doporučuje v nemocnici sterilizovat tyto výrobky parou s použitím jedné ze tří skupin dále uvedených parametrů.
METODA CYKLUS TEPLOTA DOBA EXPOZICE
Parou Předsterilizační vakuum 270°F (132°C) 4 minuty
Parou Gravitační 250°F (121°C) 60 minut
Parou* Gravitační* 273°F (134°C)* 20 minut*
POZNÁMKA: Vzhledem k mnoha proměnným faktorům při sterilizaci musí každé zdravotnické zařízení kalibrovat a ověřit sterilizační postupy (např. teplotu, dobu) použité pro svá zařízení.
* Při použití v jiných zemích než v USA některé zdravotnické orgány doporučují sterilizaci podle těchto parametrů, aby se minimalizovalo riziko přenosu Creutzfeldt-Jakobovy nemoci, zejména
u chirurgických nástrojů, které by mohly přijít do styku s centrálním nervovým systémem.
REKLAMACE VÝROBKU:
Jakýkoliv zdravotnický pracovník (např. zákazník nebo uživatel tohoto systému produktů), který má jakoukoliv reklamaci nebo je nespokojen s kvalitou, identitou, trvanlivostí, spolehlivostí,
bezpečností, účinností nebo chováním výrobku, by o tom měl informovat distributora nebo společnost Medtronic Sofamor Danek. Pokud se dále někdy objeví „vadná funkce“ jakéhokoliv dílu
implantovaného šroubovitého fúzního zařízení INTER FIX™ nebo INTER FIX™ RP (tj. tento nesplní jakékoliv své výkonové parametry nebo jinak nevyhovuje účelu, k němuž je určen) nebo
existuje-li podezření na „vadnou funkci“, je nutné to okamžitě oznámit distributorovi. Jestliže někdy dojde u jakéhokoliv výrobku Medtronic Sofamor Danek k „vadné funkci“ a ta může způsobit
či přispět k úmrtí či závažnému poranění pacienta, distributor o tom musí být co nejdříve informován telefonicky, faxem či písemně. Při uplatňování reklamace, prosím, uveďte název a číslo
součástky(tek), číslo šarže(í), své jméno a adresu a povahu reklamace a oznamte, zda-li požadujete písemnou zprávu pro distributora.
POZNÁMKA PRO LÉKAŘE: Ačkoliv je lékař poučeným prostředníkem mezi společností a pacientem, důležité lékařské informace uvedené v tomto dokumentu by měly rovněž být sděleny
pacientovi.
Pouze pro uživatele z USA
UPOZORNĚNÍ: FEDERÁLNÍ ZÁKONY (USA) OMEZUJÍ PRODEJ TĚCHTO ZAŘÍZENÍ POUZE LÉKAŘŮM A NA JEJICH OBJEDNÁVKU.
Pokud by bylo nutné odstranit šroubovité fúzní zařízení INTER FIX™ a šroubovité fúzní zařízení INTER FIX™ RP, obraťte se, prosím, na Medtronic Sofamor Danek.
©
2006 Medtronic Sofamor Danek. Všechna práva vyhrazena.
ODSTRANĚNÍ ZAŘÍZENÍ:
Page 53

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
INTER FIX™ KEERMESTATUD FUSIOONISEADE
EESTI
Järgnevas leidub olulist meditsiinilist informatsiooni INTER FIX™ keermestatud fusiooniseadme ja INTER FIX™ RP keermestatud fusiooniseadme kohta.
KIRJELDUS:
INTER FIX™ keermestatud fusiooniseade koosneb õõnsast, perforeeritud metallsilindrist ja otsakattest. INTER FIX™ RP keermestatud fusiooniseade koosneb õõnsast, perforeeritud
metallsilindrist, millel on üksik, lai, välisümardusega soon kogu pikisuunalisel teljel, mis ulatub seadme siseläbimõõdu sisse. INTER FIX™ RP implantaadi mõlemad otsad on suletud.
INTER FIX™ ja INTER FIX™ RP implantaadid on saadaval erinevate läbimõõtudega vahemikus 12 mm kuni 24 mm ja pikkustega vahemikus 20 mm kuni 29 mm. INTER FIX™ implantaatide
otsakatete mõõdud sõltuvad silindrite diameetritest ning need paigutatakse silindrite avatud otstele peale seda, kui need on täidetud autogeense luusiirikuga.
INTER FIX™ ja INTER FIX™ RP implantaadid on valmistatud implanteeritavast titaanisulamist (Ti-6AI-4V), mis vastab standardile ASTM F136.
Konkreetselt on välistatud kaudsed garantiid turustatavusele ja konkreetseks otstarbeks või kasutusalaks sobivuse kohta. Vt. Medtronic Sofamor Danek kataloogist või hinnakirjast
lisainformatsiooni garantiide ja vastutuse piirangute kohta.
NÄIDUSTUSED:
INTER FIX™ keermestatud fusiooniseade ja INTER FIX™ RP keermestatud fusiooniseade on näidustatud lülisamba fusiooni protseduurideks täiskasvanud luustikuga patsientidel, kellel on
degeneratiivne lülidevahelise ketta haigus (DDD) ühel tasandil vahemikus L2-S1. Degeneratiivset lülidevahelise ketta haigust on defineeritud kui ketta degeneratsiooniga diskogeense päritoluga
seljavalu, mida kinnitab haiguslugu ja radiograafilised uuringud. Nendel degeneratiivse lülidevahelise ketta haigusega patsientidel võib esineda ka kuni 1. astme spondülolisteesi või retrolisteesi
antud tasandil. INTER FIX™ ja INTER FIX™ RP implantaate tuleb kasutada koos autogeense luusiirikuga ja implanteerida avatud eespoolse lähenemisega operatsioonil.
VASTUNÄIDUSTUSED:
INTER FIX™ keermestatud fusiooniseadet ja INTER FIX™ RP keermestatud fusiooniseadet ei tohiks implanteerida patsientidele, kellel esineb operatsioonikohal aktiivset infektsiooni või
kellel on allergia titaani või titaanisulamite suhtes.
HOIATUSED:
• INTER FIX™ keermestatud fusiooniseadet ja INTER FIX™ RP keermestatud fusiooniseadet tohivad kasutada vaid kirurgid, kellel on kogemusi lülisamba fusiooni protseduuridega ning
kes on saanud selle seadme kasutamise kohta piisava väljaõppe.
• Piisavate kogemuste ja/või väljaõppe puudumine võib tuua kaasa suurema kõrvaltoimete, näiteks vaskulaarsete vigastuste, neuroloogiliste komplikatsioonide ja/või urogenitaalsete
komplikatsioonide (sealhulgas retrograadne ejakulatsioon) esinemissageduse.
INTER FIX™ RP KEERMESTATUD FUSIOONISEADE
OLULINE MEDITSIINILINE INFORMATSIOON
Page 54

HOIATUSED:
L
• INTER FIX™ keermestatud fusiooniseadme ja INTER FIX™ RP keermestatud fusiooniseadme ohutust ja efektiivsust ei ole tõestatud patsientidel, kes vastavad mingile järgnevatest
tingimustest:
- või kõrgema astme spondülolistees või retrolistees;
- fusioon tuleb teostada enam kui ühel tasandil;
- eelneva(te) lülidevahelise fusiooni protseduuri(de) korrigeerimine;
- postoperatiivse steroidse või mittesteroidse põletikuvastase medikamentoosse ravi vajadus;
- haiguslik rasvumine;
- vanus alla 18 või üle 65 aasta;
- osteoporoos, osteopeenia ja/või osteomalaatsia;
- rasedus.
• INTER FIX™ keermestatud fusiooniseadme ja INTER FIX™ RP keermestatud fusiooniseadme ohutust ja efektiivsust on tõestatud ainult eespoolse lumbaalse lülikehadevahelise
fusiooni protseduuridel (ALIF) koos autoloogse luusiiriku kasutamisega.
• Patsiendid, kelle paigaldatakse INTER FIX™ keermestatud fusiooniseade, peaksid olema saanud vähemalt kuus kuud mittekirurgilist ravi. Patsiendid, kelle paigaldatakse INTER
FIX™ RP keermestatud fusiooniseade, peaksid samuti olema saanud vähemalt kuus kuud mittekirurgilist ravi.
• Operatsioonitasandile tuleks kõrvuti implanteerida kaks INTER FIX™ keermestatud fusiooniseadet. Operatsioonitasandile tuleks kõrvuti implanteerida üks INTER FIX™ keermestatud
fusiooniseade ja üks INTER FIX™ RP keermestatud fusiooniseade. INTER FIX™ RP seadmel asuv pikisuunaline soon hõlbustab kahe seadme enam lähestikku implanteerimist kui on
võimalik kahe INTER FIX™ seadme korral, mille tulemusel saab saavutada väiksema lateraalse profiili.
• INTER FIX™ keermestatud fusiooniseadmete ja INTER FIX™ RP keermestatud fusiooniseadmete pikitelg peaks olema suunatud eespoolt tahapoole.
• Implantaate ja instrumente tuleb enne kasutamist steriliseerida vastavalt sellel pakendi infolehel toodud steriliseerimise juhistele, välja arvatud juhul, kui need on tarnitud steriilselt ja
nende etikettidel on sellekohane selge märgistus.
KÕRVALTOIMED:
Tabelis I näidatud kõrvaltoimetest teatati 185 INTER FIX™ seadmega patsiendil ja 62 kontrollgrupi patsiendil, kes osalesid INTER FIX™ keermestatud fusiooniseadme mitme keskusega
kliinilises uuringus. Kinnitamise järgse uuringu patsientidel teatati lisaks kõrvaltoimeid 116 patsiendil 48 kuu möödudes ja 109 patsiendil 72 kuu möödudes. Näidatud arvud põhinevad
uuringus osalevate patsientide koguarvul iga ajavahemiku kohta. Üldise esinemissageduse väljendamiseks on kombineeritud randomiseeritud ja randomiseerimata INTER FIX™ seadme
ravigruppides esinenud kõrvaltoimeid.
TABEL I - KÕRVALTOIMED
3 kuud
2-<5 kuud)
(t
[arv (%)]
INTER FIX™
N=185
KONTROL
GRUPP
N=62
GRUPP
N=62
Postoperatiivne
(1 päev-<2 kuud)
[arv (%)]
INTER FIX™
N=185
KONTROLL
GRUPP
N=62
Operatiivne
[arv (%]
INTER FIX™
Komplikatsioon
Vaskulaarne intraop. 15(8.1) 2(3.2) 15 (8.1) 2(3.2)
Sakroiliakaalne valu 5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
Neuroloogiline 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Seljavalu 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
Sisselõikega seotud 11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
Spinaalne 1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
Uroloogiline 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Muud 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Muud valud 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
Gastrointestinaalne 7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
Retrograadne
ejakulatsioon
Respiratoorne 3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
Jalavalu 2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
Trauma 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
Peritoneaalne 3(1.6) 3(1.6) 0
Vaskulaarne postop. 2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
Luu fraktuur 1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
Implantaadi nihkumine /
lõdvenemine
Luusiiriku võtmise koha
valud
Mitteühinemine 4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
Vajumine 2(1.1) 2(1.1) 0
Mitteühinemine
(TULEMUSE OOTEL)
Meningiit 1(1.6) 0 1(1.6)
Implantaadi purunemine 5(8.1) 0 5(8.1)
Vähk 1
KONTROLL
N=185
2(1.1) 3(1.6) 1(0.5) 6(3.2)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
1(1.6)2 0 1(1.6) 2(1.7)
Surm 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
1 - 81-st meespatsiendist, kes osalesid INTER FIX™ seadme grupis (7,4%) kogesid kuus (6) retrograadset ejakulatsiooni 24-kuulise hindamisperioodi jooksul.
2 - Selle patsiendi (175) kohta teatati hiljem fusiooni teke ligikaudu 52 kuul postoperatiivselt, mingeid muid kirurgilisi protseduure ega ravi vaja ei olnud.
(t
INTER FIX
N=185
6 kuud
5-<9 kuud)
[arv (%)]
™
KONTROLL
GRUPP
N=62
(t
INTER FIX
N=185
(0.5)
12 kuud
9-<19 kuud)
[arv (%)]
™
KONTROLL
GRUPP
N=62
24 kuud
19-< 30 kuud)
(t
[arv (%)]
™
INTER FIX
KONTROLL
N=185
GRUPP
N=62
1
(0.5)
KÕRVALTOIMEID
KOKKU
(24 KUU JOOKSUL)
™
N=185
(7.4)
KONTROLL
GRUPP
1
INTER FIX
48 kuud
(>30 - < 60
30-<60
(t
kuud)
INTER FIX™
N=62
N=116
0
72 kuud
60
(t
kuud)
INTER FIX™
N=109
2 (1.1) 1(0.8) 1(0.9)
Kliinilise uuringu kaks kõige tähtsamat kõrvaltoimet olid intraoperatiivsed vaskulaarsed ja neuroloogilised vigastused. INTER FIX™ seadme grupis esines 24-kuulise hindamisperioodi jooksul
kokku 15 vaskulaarset intraoperatiivset komplikatsiooni 14 patsiendil. Nende komplikatsioonide hulka kuulusid: 2 õõnesveeni vigastust, 9 niudeveeni vigastust, 1 sisemise niudeveeni rebestus,
1 segmentaarne veeni veritsus, 1 ristluuveeni vigastus ja 1 pindmine veritsus. Kokku esines samal ajavahemikul kontrollgrupis 2 vaskulaarset intraoperatiivset vigastust 2 patsiendil. Nende
hulka kuulusid: 1 niudeveeni vigastus ja 1 veritsus luuliselt aluselt.
24-kuulise hinnanguperioodi kestel esines INTER FIX™ seadme grupis kokku 30 neuroloogilist komplikatsiooni 28 patsiendil. Nende komplikatsioonide hulka kuulusid: 1 võimetus jalga
ettepoole tõsta; 3 närvijuure vigastust; 1 lülidevahemulgu stenoos; 1 sümpaatiline refleksdüstroofia; 7 jalgade tuimuse või soojuse tunnet; 2 düsteesiat, 3 paresteesiat; 2 radikuliiti; 7 selja ja/või
jala valu; 1 selja ja/või jala valu koos muude sümptomitega; 1 kiirguv valu selja alaosas ja 1 karpaaltunneli sündroom.
Sama perioodi vältel esines kontrollgrupis kokku 10 neuroloogilist komplikatsiooni 8 patsiendil. Nende komplikatsioonide hulka kuulusid: 1 radikulopaatia pakitsustundega jäsemetes; 1 krooniline
seljavalu koos radikuliidiga; 1 üldise nõrgenemise sümptomite juhtum; 2 selja- ja jalavalu koos muude sümptomitega; 1 denerveeritud suure reie lähendajalihase ja 4 jalgade tuimuse või
soojustundega juhtu. Kinnitamise järgse uuringu käigus registreeriti veel 10 neuroloogilist komplikatsiooni 10 patsiendil. Nende komplikatsioonide hulka kuulusid: 1 radikulopaatia, 2 tservikaalset
radikulopaatiat, 1 käte tuimuse ja pakitsuse tunne, 1 jalgade tuimuse või põletustunne, 3 selja ja/või jala valu ning 2 selja ja/või jala valu koos muude sümptomitega. . Lisaks on kõrvaltoimete
tabelis toodud jalgade või seljavalu juhtumid ja/või spinaalsed komplikatsioonid, näiteks lülivaheketta kollaps, milles esines mõningatel juhtudel neuroloogiline komponent.
Mõningate kõrvaltoimete tulemusel oli vajalik kirurgiline sekkumine kliinilise uuringu operatsiooni järgselt. Neid täiendavaid kirurgilisi sekkumisi võib klassifitseerida kui korrigeerimisi, eemaldamisi,
täiendavaid fikseerimisi või kordusoperatsioone
kombineeritult) ja kontrollravi gruppides IDE uuringu vältel ja INTER FIX™ seadme grupis kinnitamise järgse uuringu vältel.
1
Korrigeerimine: Protseduur, milles reguleeritakse või mistahes viisil muudetakse algset implantaadi konfiguratsiooni.
1
. Tabelis II on kokkuvõtlikult esitatud sekundaarsed kirurgilised sekkumised INTER FIX™ seadme (randomiseeritud ja randomiseerimata
Eemaldamine: Protseduur, milles eemaldatakse üks või enam algse implantaadi konfiguratsiooni komponentidest ilma sama tüüpi uuritava seadmega asendamiseta.
Täiendav fikseerimine: Protseduur, milles paigaldatakse täiendavaid seadmeid, mida ei olnud kinnitatud antud protokolli osaks.
Kordusoperatsioon: Mistahes kirurgiline protseduur, milles ei eemaldata, muudeta ega lisata ühtegi algse implantaadi komponenti.
Page 55

Tabel II – Sekundaarsed kirurgilised protseduurid
INTER FIX™
IDE uuring Kinnitamise järgne uuring
seade
N=185
Kuni 24 kuu
ajapunkt
Kontroll-
grupp
N=62
1
48 kuu
ajapunkt
INTER FIX™
seade
N=116
72 kuu
ajapunkt
INTER FIX™
seade
N=109
Korrigeerimised 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Eemaldamised 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Täiendavad
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
fikseerimised
Kordusoperatsioonid 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
1
- Kuna kontrollgrupi patsientidele ei olnud järelkontroll 24 kuu möödudes nõutav, ei leidu kõigi patsientide kohta andmeid 72 kuulise perioodi vältel.
VÕIMALIKUD KÕRVALTOIMED:
Järgnevalt on loetletud võimalikud kõrvaltoimed, mida võib esineda seoses lülisamba fusiooni operatsiooniga INTER FIX™ keermestatud fusiooniseadme või INTER FIX™ RP keermestatud
fusiooniseadme kasutamisel. Mõnda nendest kõrvaltoimetest võib olla eelnevalt nimetatud kõrvaltoimete tabelis.
• Komponentide osadeks lahti tulemine, paindumine, purunemine, lõdvenemine ja/või nihkumine.
• Võõrkehareaktsioon (allergiline).
• Kudede või närvikahjustus.
• Postoperatiivsed muutused lülisamba kõveruses, korrektsiooni, pikkuse ja/või reduktsiooni kaotus.
• Infektsioon.
• Kõvakelme rebendid.
• Närvisüsteemi häired.
• Urogenitaalsüsteemi häired.
• Armkoe teke.
• Luu fraktuur.
• Mitteühinemine (või pseudartroos), viivitunud ühinemine, väärühinemine.
• Opereeritud selgroo osa potentsiaalse kasvu peatumine. Spinaalse liikuvuse või funktsiooni kaotus.
• Luusiiriku võtmise koha komplikatsioonid.
• Veresoonte kahjustus ja kardiovaskulaarsüsteemi häired.
• Gastrointestinaalsed komplikatsioonid.
• Reproduktiivsüsteemi häired.
• Siseorganite ja sidekudede kahjustused.
• Respiratoorsete probleemide teke.
• Sisselõike koha komplikatsioonid.
• Vaimse seisundi muutus.
• Surm.
Märkus: Täiendav operatsioon võib olla vajalik mõnede nende võimalike kõrvaltoimete korrigeerimiseks.
KLIINILISED TULEMUSED:
USA-s viidi läbi INTER FIX™ keermestatud fusiooniseadme mitme keskusega ekvivalentsuse kliiniline uuring, milles võrreldi sümptomaatilise degeneratiivse lülidevahelise ketta haigusega
patsientide ravis autogeense luuga täidetud INTER FIX™ seadme eespoolset spinaalset kasutamist kontrollseadmega, autogeense luuga täidetud femoraalrõnga allotransplantaadiga.
Algselt oli kliinilise uuringu vormiks prospektiivne, randomiseeritud ja kontrollitud uuring. Hiljem muudeti uuringuplaani, võimaldamaks patsientidel lülituda randomiseerimata grupiga, s.t ainult
INTER FIX™ seadmega ravitud patsiendigrupiga.
Kliinilisse uuringusse kaasamise kriteeriumide hulka kuulus sümptomaatiline degeneratiivne lülivaheketta haigus, mis väljendus ravile allumatus jala ja/või selja valus koos positiivse piltdiagnostika
leiuga (leidudega); lülisamba ebastabiilsus, mida määratleti kui enam kui 4 mm translatsiooni või enam kui 5-kraadist nurka fleksiooni/ekstensiooni radiograafidel; ühetasandiline sümptomaatiline
haaratus tasandil L2-S1 ning kuni 1. astme spondülolistees. Uuringust olid spetsiifiliselt väljaarvatud need patsiendid, kellele oli eelnevalt teostatud eespoolne lülikehadevaheline fusiooni
protseduur antud spinaalsel tasandil; kellel oli osteopeenia, osteoporoos või osteomalaatsia või kes vajasid luu kasvu stimuleerimist.
Uuritava INTER FIX™ seadme ravigruppi kaasati kokku 185 patsienti. Kliinilise uuringu randomiseeritud kontrollgruppi kaasati kokku 62 patsienti.
Üks INTER FIX™ keermestatud fusiooniseadme kinnitamise järgne nõue oli hinnata seadme pikaajalist talitlust, hankides vähemalt 100 patsiendi kohta postoperatiivseid andmeid kokku 6
aasta vältel. Seadme pikaajalist talitlust hinnati 116 patsiendil 4 aastal postoperatiivselt ja 109 patsiendil 6 aastal postoperatiivselt. Patsientide populatsioon valiti IDE kliinilises uuringus
osalejate hulgast. IDE uuringu kriteeriumide põhjal sisaldasid nende patsientide andmed kõigi täiendavate kirurgiliste protseduuride kirjeldust, fusiooni radiograafilist hinnangut ning valu ja
funktsiooni hinnangut.
Tabelis III esitatakse uuringu kliinilisi tulemusi, kirjeldades fusiooni edukuse näitajaid, valu (Oswestry) edukust ja neuroloogilist edukust 12, 24, 48 ja 72 kuul peale ravioperatsiooni. Selle
kliinilise uuringu õnnestumist määrati INTER FIX™ seadme ja kontrollseadme vahelise ekvivalentsuse analüüsi alusel.
Selles kliinilises uuringus defineeriti fusiooni kui sildava trabekulaarse luu tõendatust, translatsiooni (<3 mm) ja nurkliikuvuse (<5°) stabiilsust ning radioloogilise läbipaistvuse puudumist
enam kui 50% ulatuses mõlema implantaadi ümber. Samuti loeti mitteühinemisest tulenevate sekundaarsete operatsioonidega patsiente arvutustes ebaõnnestunud fusioonideks. Oswestry
valu/puude skaalal loeti õnnestumiseks vähemalt 15-punktilist paranemist postoperatiivses skooris, võrreldes preoperatiivse skooriga. Õnnestumist neuroloogilises seisundis defineeriti kui
samana säilimist või paranemist vähemalt 3-s neljast neuroloogilisest kategooriast (liikuvus, sensoorsus, refleksid ja sirge jala tõste) postoperatiivselt, võrreldes preoperatiivse seisundiga. Üldise
õnnestumise aluseks oli patsiendil ilmne fusiooni teke, Oswestry valu/puude skaala alusel õnnestumine ja õnnestumine neuroloogilises seisundis ning ebaõnnestumisena klassifitseeritava
sekundaarse kirurgilise protseduuri puudumine.
2
INTER FIX™ seade ei ole statistiliselt kontrollseadmest halvem.
3
Selle hulka ei kuulu 3 patsienti, kellele ei teostatud INTER FIX™ seadmega ravi tulenevalt kirurgilistest kõrvaltoimetest
Üldise õnnestumise tulemused 12 ja 24 kuul postoperatiivselt leiduvad samuti tabelis III.
Tabel III - INTER FIX™ seadme uuringu kliinilised tulemused
Fusioon 96.1%
Oswestry
valu/puude
skaalal
paranemine
Patsiendid
vähemalt 15punktilise
paranemisega,
võrreldes
preoperatiivse
skooriga
Neuroloogilise
seisundi
püsimine või
paranemine
Üldine
õnnestumine
12 kuu suhtarvud 24 kuu suhtarvud
Uuritav Kontrollgrupp
(122/127)
53.5%
(76/142)
99.3%
(150/151)
42.4%
(61/144)
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
(12/56)
Delta
1
)
('
Uuritav Kontrollgrupp
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
-0.097 46.1%
(71/154)
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
(17/57)
1 - Vähim võrdväärsuse hälve, mis on nõutav, et saaks väita uuritava võrdväärsust kontrollseadmega.
Delta
('1)
-0.043 49.5%
48 kuu
suhtarvud
Uuritav
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0% (102/103) 97.6% (82/84)
(53/107)
72 kuu
suhtarvud
Uuritav
56.3%
(54/96)
Page 56

Kõik INTER FIX™ keermestatud fusiooniseadme kliinilises uuringus osalenud patsiendid kaasati uuringusse sõltumata ravigrupist samade sisse-/väljaarvamise kriteeriumide alusel.
Randomiseeritud INTER FIX™ seadmega patsientide ja randomiseerimata INTER FIX™ seadmega patsientide võrreldavuse kindlustamiseks vaadeldi demograafilisi näitajaid, preoperatiivseid
meditsiinilisi seisundeid, degeneratiivsele lülivaheketta haigusele iseloomulikke piltdiagnostika leide, kirurgilist lähenemisviisi ja ravitud lumbaalpiirkonna asukohta. Antud populatsioon oli
statistiliselt võrreldav, välja arvatud kirurgilise lähenemisviisi, preoperatiivse tööalase staatuse ja lülikehade pindadel osteofüütide moodustumise preoperatiivsete piltdiagnostika leidude;
fibroosvõru, kollasideme ja/või liigesjätke kihnu armistumise ja/või paksenemise ning säsituuma väljasopistuse osas. Edasistest statistilistest analüüsidest nähtus, et need muutujad ei olnud
seotud erinevustega fusiooni kliinilistes tulemustes 12 kuu möödumisel, paranemises Oswestry valu/puude skaalal, neuroloogilises seisundis ega üldises õnnestumises, välja arvatud säsituuma
väljasopistumise korral. Randomiseerimata grupis esines enamatel patsientidel säsituuma väljasopistumist ning leiti, et see suurendab üldise õnnestumise tõenäosust.
Viidi läbi “raviks ettenähtute” analüüs. Selle analüüsi seisukohalt põhjustasid sekundaarse operatsiooni ebaõnnestumised, surmajuhtumid, järelkontrolli mitteilmunud patsiendid ja muudel
põhjustel puuduvad vaatlustulemused tulemuste muutujate määramiseks puuduvaid vaatlustulemusi ning seetõttu kaasati need arvutatud suhtarvude nimetajatesse, s.t loeti “ebaõnnestumisteks”.
Lugedes neid patsiente ravi ebaõnnestumisteks, saadi raviks ettenähtute analüüsis oluliselt madalamad kliinilised tulemusmäärad kui tegelikes kliinilistes vaatlusandmetes.
Järgnevas tabelis (Tabel IV) on antud INTER FIX™ seadme patsientide osas raviks ettenähtute analüüsi tulemused, kuid selles ei sisaldu kontrollpatsientide tulemusi.
Surmajuhtumeid, sekundaarse operatsiooni ebaõnnestumisi, järelkontrolli mitteilmunuid ja puuduvaid
12 kuu
Fusioon 65.9% (122/185) 67.6%
Oswestry valu/puude skaalal paranemine
Patsiendid vähemalt 15-punktilise
paranemisega, võrreldes preoperatiivse
skooriga
Neuroloogilise seisundi püsimine või
paranemine
Üldine õnnestumine 33.0% (61/185) 38.4% (71/185) 45.7%
Sekundaarse operatsiooni ebaõnnestumised
Mitteühinemised
Muud
Surmajuhtumid 0 1 0 0
vaatlustulemusi loeti ebaõnnestumisteks ning need sisalduvad suhtarvude nimetajates.
1
2
Tabel IV - INTER FIX™ seadme raviks ettenähtute analüüs
suhtarvud
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
5
8
24 kuu
suhtarvud
(125/185)
(128/185)
5
3
48 kuu
suhtarvud
80.2% (93/116) 77.1%
(66/116)
87.9%
(102/116)
(53 /116)
4
4
72 kuu
suhtarvud
( 84/109)
56.0%
(61 /109)
75.2%
(82/109)
49.5%
(54/109)
1
2
1 - Need patsiendid on kaasatud fusiooni määra arvutustesse, kuid muidu loetakse kliinilise uuringu seisukohalt ebaõnnestumisteks.
2 - Antud perioodil järelkontrolliks ettenähtud patsiente, kellele oli tehtud sekundaarseid operatsioone muudel põhjustel peale mitteühinemiste, loeti kliinilise uuringu seisukohalt
ebaõnnestumisteks.
PAKEND:
Iga komponendi pakendid peaksid kättesaamisel olema puutumata. Kui kasutatakse laenu- või konsignatsioonisüsteemi, siis tuleks enne kasutamist kõigi komplektide komplektsust hoolikalt
kontrollida ja hoolikalt tuleks kontrollida kõiki komponente, sealhulgas instrumente, tagamaks, et nad ei ole rikutud. Kahjustatud pakendeid ega tooteid ei tohiks kasutada ja need tuleks
tagastada Medtronic Sofamor Danek-ile.
PUHASTAMINE JA DEKONTAMINATSIOON:
Kõik implantaadid ja instrumendid, kui nad ei ole tarnitud steriilselt, tuleb enne steriliseerimist ja steriilsele operatsioonialale viimist esmalt puhastada kehtestatud haiglametoodika alusel.
Kasutatud instrumentidelt tuleb enne uuesti kasutamist mustus eemaldada, need tuleb puhastada ja steriliseerida.
Märkus: Mitte kasutada pleegitusaineid või formaliini sisaldavaid puhastuslahuseid, kuna need võivad seadet kahjustada.
Kõiki tooteid tuleb käsitseda hoolikalt. Mittekorrektne kasutus või käsitsemine võib tuua kaasa kahjustusi ja seadme võimaliku ebakorrektse funktsioneerimise.
STERILISEERIMINE:
Juhul kui nad ei ole steriilsuse markeeringuga ja sellekohaselt sildistatud firma poolt tarnitud avamata steriilses pakendis, siis kõiki operatsioonil kasutatavaid implantaate ja instrumente tuleb
haigla poolt enne kasutamist steriliseerida. Enne steriliseerimist eemaldada kõik pakkematerjalid. Operatsioonialale tohib viia ainult steriilseid tooteid. 10-6 SAL steriilsustaseme tagamiseks
on neid tooteid soovitatav haigla poolt auruga steriliseerida, kasutades ühte kolmest järgnevast protsessi parameetrite komplektist:
MEETOD TSÜKKEL TEMPERATUUR TÖÖTLUSPERIOOD
Aur Eel-vaakum 270°F (132°C) 4 minutit
Aur Raskusjõul 250°F (121°C) 60 minutit
Aur * Raskusjõul * 273°F (134°C)* 20 minutit *
MÄRKUS: Tulenevalt steriliseerimisega seotud paljudest muutujatest peaks iga meditsiiniasutus kalibreerima ja kinnitama steriliseerimisprotsessi (s.t. temperatuurid, ajaperioodid), mida
nende seadmetel kasutatakse.
*Väljaspool USA-d kasutamiseks soovitavad mõningad mitte-USA tervishoiuametkonnad steriliseerimist vastavalt nendele parameetritele, et minimeerida Creutzfeldt-Jakobi haiguse ülekandmise
potentsiaalset riski, eriti kirurgiliste instrumentide puhul, mis võivad sattuda kontakti kesknärvisüsteemiga.
KAEBUSED TOOTE KOHTA:
Kõik tervishoiu alal töötajad (näiteks selle tootesüsteemi kliendid või kasutajad), kellel on mingeid kaebusi või kes on olnud rahulolematud toote kvaliteedi, identiteedi, vastupidavuse, töökindluse,
ohutuse, tõhususe ja/või talitlusega, peaksid sellest teatama turustajale või Medtronic Sofamor Danekile. Peale selle, kui mingil implanteeritud INTER FIX™ keermestatud fusiooniseadmel või
INTER FIX™ RP keermestatud fusiooniseadmel esineb “talitlushäireid” (s.t ei vasta mingile oma talitluse spetsifikatsioonile või muul viisil ei talitle nagu ette nähtud) või kui on selline kahtlus,
siis tuleb sellest kohe teatada turustajale. Juhul kui mingil Medtronic Sofamor Danek tootel esineb kunagi “talitlushäireid” ja see võib olla põhjustanud patsiendi surma või tõsise vigastamise,
või aidanud sellele kaasa, siis tuleks sellest turustajale otsekohe teatada telefoni, faksi või kirja teel. Kaebuse esitamisel tuleks lisada komponendi (komponentide) nimetus, number, partii
number (numbrid), esitaja nimi ja aadress, kaebuse sisu ning märge sellest, kas soovitakse kirjalikku aruannet turustajale.
ARSTI MÄRKUS: Kuigi arst on õpetatud vahemees firma ja patsiendi vahel, tuleks selles dokumendis toodud oluline meditsiiniline informatsioon patsiendile edastada.
Ainult USA tarijaile
ETTEVAATUST: RIIKLIK (USA) SEADUSANDLUS PIIRAB NENDE SEADMETE MÜÜKI, MIS VÕIB TOIMUDA AINULT ARSTI POOLT VÕI ARSTI KORRALDUSEL.
SEADME EEMALDAMINE:
Juhul kui peaks tekkima vajadus INTER FIX™ keermestatud fusiooniseadet või INTER FIX™ RP keermestatud fusiooniseadet eemaldada, võtke ühendust Medtronic Sofamor Danek-iga.
©
2006 Medtronic Sofamor Danek. Kõik õigused reservee.
Page 57

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
.
MAGYAR
Az alábbi dokumentum fontos orvosi információkat tartalmaz az INTER FIX™ menetes fúziós eszközre és az INTER FIX™ RP menetes fúziós eszközre vonatkozóan.
LEÍRÁS:
Az INTER FIX™ menetes fúziós eszköz egy üreges, perforált fémhengerből és egy zárósapkából tevődik össze. Az INTER FIX™ RP menetes fúziós eszköz pedig egy üreges, perforált
fémhengerből áll, melynek egész hosszanti tengelye mentén egy nagyméretű, külső körkörös bemetszés helyezkedik el, mely az eszköz belső átmérőjébe is benyúli. Az INTER FIX™ RP
implantátum mindkét vége zárt.
A rendelkezésre álló INTER FIX™ és az INTER FIX™ RP implantátumok átmérője 12 mm-től 24 mm-ig, hosszúsága 20 mm-től 29 mm-ig terjed. Az INTER FIX™ implantátumok zárósapkáit
a hengerek átmérői szerint méretezték, ezeket a hengerek nyitott végére kell ráhelyezni, miután utóbbiakat autogén csontgrafttal feltöltötték.
Az INTER FIX™ és az INTER FIX™ RP eszközök implantátum osztályú titánötvözetből készültek (Ti-6Al-4V), mely az ASTM F136 szabványnak megfelel.
Az értékesíthetőségre és egy bizonyos célra vagy alkalmazásra való megfelelőségre vonatkozó hallgatólagos szavatosságot kifejezetten kizárjuk. A szavatossággal és a felelősség korlátozásával
kapcsolatos további információt lásd az Medtronic Sofamor Danek katalógusban vagy árlistában.
JAVALLATOK:
Az INTER FIX™ menetes fúziós eszközt, valamint az INTER FIX™ RP menetes fúziós eszközt degeneratív porckorongbetegségben (DDD) szenvedő érett csontvázú betegeken végzett
gerincfúziós eljárások során használják egy adott szinten az L2-S1-től kezdődően. A degeneratív porckorongbetegség meghatározás szerint discogén eredetű hátfájás, mely a porckorong
degenerációjával jár, amint az a kórtörténet és a röntgenfelvételek által megerősítést nyer. E degeneratív porckorongbetegségben szenvedő betegek rendelkezhetnek I. fokú spondylolisthesissel
vagy retrolisthesissel a szóban forgó szinten. Az INTER FIX™ és az INTER FIX™ RP implantátumokat autogén csontgrafttal kell használni és nyitott anterior eljárással kell beültetni.
ELLENJAVALLATOK:
Az INTER FIX™ menetes fúziós eszközt és az INTER FIX™ RP menetes fúziós eszközt nem szabad olyan betegek testébe beültetni, akiknél a műtét helyén aktív fertőzés lépett fel, vagy
akik titánra illetve titánötvözetre allergiásak.
FIGYELMEZTETÉSEK:
• Az INTER FIX™ menetes fúziós eszközt és az INTER FIX™ RP menetes fúziós eszközt csak olyan sebészek használhatják, akik a gerincfúziós eljárások terén tapasztaltak, és az
eszköz használatával kapcsolatban megfelelő gyakorlattal rendelkeznek.
• A megfelelő tapasztalat és/vagy gyakorlat hiánya a káros események, mint például érsérülések, neurológiai és/vagy urogenitalis esetek (retrográd ejakulációt beleértve) gyakoribb
előfordulásához vezethet.
INTER FIX™ MENETES FÚZIÓS ESZKÖZ
INTER FIX™ RP MENETES FÚZIÓS ESZKÖZ
FONTOS ORVOSI INFORMÁCIÓK
Page 58

ÓVINTÉZKEDÉSEK:
L
L
• Az INTER FIX™ menetes fúziós eszköz, valamint az INTER FIX™ RP menetes fúziós eszköz biztonságosságát és hatékonyságát még nem határozták meg az alábbi betegségekben
szenvedő betegek esetén:
- II. vagy magasabb fokú spondylolisthesis vagy retrolisthesis;
- több, mint egy szinten végrehajtandó fúzió;
- korábbi csigolyatest közötti fúziós eljárás(ok) revíziója;
- posztoperatív szteroidos vagy nem szteroidos gyulladáscsökkentő gyógyszer szükségessége;
- nagyfokú obesitas;
- 18 évnél fiatalabb vagy 65 évnél idősebb kor;
- osteoporosis, osteopenia, és/vagy osteomalacia;
- terhesség.
• Az INTER FIX™ menetes fúziós eszköz és az INTER FIX™ RP menetes fúziós eszköz biztonságosságát és hatékonyságát csak autológ csontgraft alkalmazásával történő anterior
lumbalis csigolyatest közötti fúziós (ALIF) eljárásokra vonatkozóan állapították meg.
• Azoknak a betegeknek, akikbe az INTER FIX™ menetes fúziós eszközt ültetik be, korábban legalább hat hónapos nonoperatív kezelésben, míg azoknak a betegeknek, akikbe az
INTER FIX™ RP menetes fúziós eszközt ültetik be, korábban úgyszintén legalább hat hónapos nonoperatív kezelésben kellett részesülniük.
• A műtét szintjén két INTER FIX™ menetes fúziós eszközt kell egymás mellé beültetni. Egy INTER FIX™ menetes fúziós eszközt és egy INTER FIX™ RP menetes fúziós eszközt kell
egymás mellett beültetni a műtét szintjén. Az INTER FIX™ RP eszközön található hosszanti bemetszés lehetővé teszi, hogy a két eszközt egymáshoz közelebb ültessék be, mint ami
két INTER FIX™ eszköz esetén megvalósítható lenne, mely a laterális profil csökkenését eredményezi.
• Az INTER FIX™ menetes fúziós eszközök, valamint az INTER FIX™ RP menetes fúziós eszközök hosszanti tengelyének anterior-posterior irányban kell elhelyezkednie.
• Használat előtt az implantátumokat és a műszereket sterilizálni kell a jelen csomaghoz mellékelt utasítások szerint, kivéve, ha steril állapotban kerültek szállításra, és ez a címkén
egyértelműen szerepel.
KÁROS HATÁSOK:
A káros hatásokat, amint az I. táblázatban szerepelnek, 185, INTER FIX™ eszközzel kezelt és 62 kontroll betegen végzett többközpontos klinikai vizsgálat során állapították meg, melyet az
INTER FIX™ menetes fúziós eszközre vonatkozóan végeztek. Továbbá, a jóváhagyást követő vizsgálatban részt vevő betegek körében káros hatásról számoltak meg 116 beteg esetén a 48
hónapos és 109 beteg esetén a 72 hónapos csoportban. Az itt bemutatott arányszámok mindegyik időszakban a vizsgálatban résztvevő betegek teljes számán alapulnak . Az INTER FIX™
eszközzel kezelt, randomizált és nem randomizált csoportban előforduló káros hatásokat kombinálták, s ezzel az összarányszám kerül bemutatásra.
I. TÁBLÁZAT – KÁROS HATÁSOK
Posztoperatív
N=62
(1 nap-<2 hónap)
[Szám (% )]
KONTROLL
INTER FIX
N=185
N=62
Operatív
Szövódmény
Vascularis
intraoperatív
Sacroiliacalis
fájdalom
Neurológiai 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Hátfájdalom 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
Incisionalis 11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
Spinalis
szövódmény
Urológiai 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Egyéb 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Egyéb fájdalom 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
Gastrointestinalis 7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
Retrográd
ejakuláció
Respiratorikus 3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
Lábfájdalom 2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
Trauma 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
Peritonealis 3(1.6) 3(1.6) 0
Vascularis postop. 2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
Csonttörés 1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
Az implantátum
elmozdulása/kilazul
ása
A graft helyének
fájdalma
Az összeforradás
meghiúsulása
Besüllyedés 2(1.1) 2(1.1) 0
Az összeforradás
meghiúsulása (AZ
EREDMÉNY
MEGERòSÍTÉSRE
VÁR)
Meningitis 1(1.6) 0 1(1.6)
Az implantátum
eltörése
Rák 1
[Szám (% )]
KONTROL
INTER FIX
N=185
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
2(1.1) 3(1.6) 1(0.5) 6(3.2)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
1(1.6)
5(8.1) 0 5(8.1)
Halál 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
3 hónap
(
t2-<5 hónap)
[Szám (% )]
INTER FIX
N=185
KONTROL
N=62
(
t5-<9 hónap)
[Szám (% )]
INTER FIX
N=185
6 hónap
KONTROLL
12 hónap
(
t9-<19 hónap)
[Szám (% )]
KONTROLL
INTER FIX
N=62
2
0 1(1.6) 2(1.7)
N=185
N=62
(0.5)
24 hónap
(
t19-< 30 hónap)
[Szám (% )]
KONTROLL
INTER FIX
N=185
1
(0.5)
ÖSSZES KÁROS
HATÁS
(24 HÓNAP ALATT)
KONTROLL
INTER FIX
N=62
N=185
(7.4)
N=62
1
2 (1.1) 1(0.8) 1(0.9)
48 hónap
(t
30<60
hónap)
INTER FIX
N=116
72 hónap
(t60
hónap)
INTER FIX
N=109
0
1 - Az INTER FIX™ eszközzel kezelt csoportban részt vevő 81 férfibetegből hat (6) esetében (7,4%) fordult elő retrográd ejakuláció a 24-hónapos kiértékelés alatt.
2 - E beteg (175) esetén a fúzió megtörténtéről utólagosan, körülbelül 52 hónappal a műtét után számoltak be, s nem volt szükség egyéb műtéti beavatkozásra vagy kezelésre.
A klinikai vizsgálat során az alábbi két káros esemény fordult elő jelentős mértékben: intraoperatív vascularis és neurológiai sérülések. Összesen 15 vascularis intraoperatív esemény fordulat
elő az INTER FIX™ eszközcsoporttal kezelt 14 beteg körében a 24 hónapos kiértékelés alatt. Ezek az alábbi eseményeket foglalták magukban: 2 véna cava sérülés, 9 iliacus véna sérülés,
1 hypogastricus véna laceratio, 1 szegmentális vénás vérzés, 1 sacralis vénasérülés és 1 felületi vérzés. A kontroll csoportban ugyanazon időszak alatt összesen 2 vascularis intraoperatív
sérülés fordult elő 2 beteg esetén. Ezek az alábbiakat foglalták magukban: 1 iliacus véna sérülés és 1 vérzés a csontágyból.
Összesen 30 neurológiai esemény fordult elő 28, az INTER FIX™ eszköz-csoportban levő beteg esetében a 24-hónapos kiértékelés során. Ezek az alábbiakat foglalták magukban: 1 lábfejlelógás, 3 ideggyökér-sérülés, 1 foraminalis stenosis, 1 sympathicus reflex dystrophia, 7 lábzsibbadás vagy meleg érzés, 2 dysaesthesia, 3 paraesthesia, 2 radiculitis, 7 hát- és/vagy lábfájás,
1 hát és/vagy lábfájás egyéb tünetekkel, 1 nyilalló fájdalom a hát alsó részén és 1 carpalis alagút szindróma.
A kontroll csoportban ugyanazon időszak alatt összesen 10 neurológiai esemény fordult elő 8 beteg esetén. Ezek az alábbiakat foglalták magukban: 1 radiculopathia bizsergő végtagokkal,
1 krónikus hátfájdalom radiculitis mellett, 1 disztribúció által kiváltott debilitáló tünetek, 2 hát- és lábfájdalom egyéb tünettel együtt, 1 denervált abductor magnus izom, 4 lábzsibbadás vagy
melegség. A jóváhagyást követő vizsgálat során további 10 neurológiai eseményt jegyeztek fel 10 beteg esetén. Ezek az alábbiakat foglalták magukban: 1 radiculopathia, 2 cervicalis
radiculopathia, 1 kézzsibbadás és bizsergés, 1 lábzsibbadás vagy melegség, 3 hát- és/vagy lábfájdalom, valamint 2 hát- és/vagy lábfájdalom egyéb tünetekkel. Továbbá, a káros hatásokat
bemutató táblázat olyan, a lábbal, háttal és/vagy gerinccel kapcsolatos eseteket mutat be, mint például a porckorong által kitöltött tér hirtelen csökkenése, melyeknek bizonyos esetekben
neurológiai komponensük is volt.
Bizonyos káros hatások a klinikai vizsgálati műtétet követően sebészeti beavatkozáshoz vezettek. E további sebészeti beavatkozások a következő csoportokba sorolhatók: revízió, eltávolítás,
pótlólagos rögzítés vagy reoperáció
csoportban az IDE vizsgálat során, valamint az INTER FIX™ eszközzel kezelt csoportban a jóváhagyást követő vizsgálat során.
1
Revízió: Olyan eljárás, melynek során az eredeti implantátum konfigurációját beállítják vagy bármilyen módon megváltoztatják.
1
. A II. táblázat összegzi a másodlagos sebészeti beavatkozásokat az INTER FIX™ eszközzel (randomizált és nem randomizált együtt) kezelt és a kontroll
Eltávolítás: Olyan eljárás, melynek során az eredeti implantátum konfigurációjának egy vagy több alkotóelemét eltávolítják, anélkül, hogy ugyanazon típusú próbaeszközzel pótolnák.
Pótlólagos rögzítés: Olyan eljárás, melynek során további, a protokoll részeként jóvá nem hagyott gerincterápiás eszközöket helyeznek be.
Reoperáció: Olyan műtéti eljárás, melynek során semmilyen eredeti implantátum-alkotórészt nem távolítanak el, nem módosítanak és nem adnak hozzá.
Page 59

INTER FIX™
IDE vizsgálat Jóváhagyást követó vizsgálat
24-hónapos 48-hónapos 72-hónapos
eszköz
N=185
Kontroll1
N=62
INTER FIX™
eszköz
N=116
INTER FIX™
eszköz
N=109
Revízió 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Eltávolítás 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
II. táblázat – Másodlagos sebészeti eljárások
Pótlólagos
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
rögzítés
Reoperáció 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
1 - Mivel a kontrollbetegeknek nem kellett utólagos vizsgálatokon részt venniük 24 hónap után, ezen betegek mindegyikére nem áll rendelkezésre 72 hónapos információ.
LEHETSÉGES KÁROS HATÁSOK:
Az alábbi lista azokat a lehetséges káros hatásokat tartalmazza, melyek az INTER FIX™ menetes fúziós eszközzel vagy az INTER FIX™ RP menetes fúziós eszközzel végzett gerincfúziós
műtétek során fordulnak el. A káros hatások közül néhány már korábban is előfordulhatott az őket tartalmazó táblázatban.
• Az alkotórészek szétválása, meghajlása, törése, kilazulása és/vagy vándorlása.
• Idegen test (allergiás) reakció.
• Szöveti vagy idegi károsodás.
• A gerinc görbületének posztoperatív változása, a korrekció, magasság és/vagy redukció elvesztése.
• Fertőzés.
• Duralis repedés.
• A neurológiai rendszer károsodása.
• Az urológiai rendszer károsodása.
• Hegképződés.
• Csonttörés.
• Az összeforradás elmaradása (vagy pseudoarthrosis), késleltetett vagy helytelen összeforradás.
• A gerinc műtött szakaszán a potenciális növekedés megszakadása. A gerinc mobiltásának vagy funkciójának elvesztése.
• A graft donor hely komplikációi.
• A véredények károsodása vagy a cardiovascularis rendszer egyéb károsodása.
• Gastronintestinalis szövődmények.
• A reproduktív rendszer romlása.
• A belső szervek és az összekötő szövet károsodása.
• Légzési problémák kialakulása.
• Incisionalis szövődmények.
• A mentális állapot változása.
• Halál.
Megjegyzés: Ezen lehetséges káros hatások korrekciója érdekében további műtétre lehet szükség.
KLINIKAI EREDMÉNYEK:
Egy többközpontos ekvivalencia1 klinikai vizsgálatot végeztek az INTER FIX™ menetes fúziós eszközzel az Egyesült Államokban, melynek során összehasonlították a gerincen anterior
módon használt, autogén csonttal feltöltött INTER FIX™ eszközt az autogén csonttal feltöltött femoralis gyűrű allografttal, melyet kontrollként használtak, szimptomatikus degeneratív
porckorongbetegségben szenvedő betegek esetében. Kezdetben a klinikai vizsgálatot prospektív, randomizált, ellenőrzött módon tervezték. Ezt követően, a vizsgálatot módosították, olyan
módon, hogy lehetővé tegyék a betegek számára, hogy a vizsgálat nem randomizált ágába belépjenek, azaz csak az INTER FIX™ eszközzel kezelt betegek alkalmazásával.
A klinikai vizsgálatban történő részvétel kritériumai közé tartoztak az alábbi betegségek: szimptomatikus degeneratív porckorongbetegség, amint az a nehezen mozgatható láb és/vagy hátfájdalom
révén pozitív diagnosztikus képalkotó eljárások eredményével (eredményeivel) kimutathatóvá vált; a gerinc instabilitása, melyet a hajlításkor/nyújtáskor készített röntgenfelvételen 4 mm-nél
nagyobb mértékű eltolódással vagy 5°-nál nagyobb mértékű elhajlással határoztak meg; egy szintű tüneti folyamat a L2-S1-től kezdve; 1. fokúnál nem magasabb szintű spondylolisthesis. A
vizsgálatból kimondottan kizárták azokat a betegeket, akiknél korábban anterior csigolyatest közötti fúziós eljárást hajtottak végre a szóban forgó gerincszinten; osteopeniában, osteoporosisban,
vagy osteomalaciában szenvedtek; vagy a csontnövekedés stimulálására volt szükségük.
Összesen 185 beteg vett részt az INTER FIX™ eszközzel kezelt vizsgálati csoportban3. Összesen 62 kontroll beteg vett részt a klinikai vizsgálat randomizált ágában.
Az INTER FIX™ menetes fúziós eszközre vonatkozó, jóváhagyást követő vizsgálat célja az volt, hogy kiértékelje az eszköz hosszú távú teljesítményét, azáltal, hogy minimum 100 betegből
összesen 6 éven át posztoperatív adatokat gyűjtenek. Az eszköz hosszú távú teljesítményét 116 betegen értékelték 4 éves posztoperatív és 109 betegen 6 éves posztoperatív időszakon
át. A betegpopulációt az IDE klinikai vizsgálat résztvevői alkották. Az IDE vizsgálatból származó kritériumok alapján, e betegekről kapott információ magában foglalta bármely egyéb műtéti
eljárás leírását, a fúzió radiográfiás felmérését, valamint a fájdalom és a működés kiértékelését.
A III. táblázat bemutatja a vizsgálat klinikai eredményeit, leírván a sikeres fúzió, fájdalom- (Oswestry) és neurológiai kezelés paramétereit a műtétet követő 12, 24, 48 és 72 hónappal. E klinikai
vizsgálat sikerességét az INTER FIX™ eszköz és a kontrol közötti ekvivalencia-elemzés alapján állapították meg.
E klinikai vizsgálat esetében, a fúziót úgy definiálták, mint a nyilvánvalóan látható áthidaló trabecularis csontot, az eltolódás (<3mm) vagy az elhajlás (<5°) stabilitását, illetve olyan radiolucencia
hiányát, mely bármelyik implantátum több, mint 50%-át körülveszi. Emellett, az összeforrás meghiúsulása miatt másodlagos műtéten részt vevő betegek esetét a számításokban sikertelen
fúziónak tekintették. Az Oswestry fájdalom/rokkantság kezelésének sikerét úgy határozták meg, mint a postoperatív értékelés legalább 15 pontos javulását a preoperatív értékeléshez képest.
A neurológiai állapot sikerét úgy definiálták, mint a 4 neurológiai kategória (mozgás, érzékelés, reflexek és a kiegyenesített láb felemelése) közül 3 postoperatív fennmaradását vagy javulását
a preoperatív állapothoz viszonyítva. Az általános sikert olyan betegre alapozták, akinél a fúzió, az Oswestry fájdalom/rokkantság kezelése és a neurógiai állapot sikert mutatott és semmilyen
másodlagos sebészeti eljárást nem osztályoztak „kudarcként”.
2
Az INTER FIX™ eszköz statisztikailag nem volt rosszabb, mint a kontroll.
3
Nem tartalmaz 3 olyan beteget, akiket a káros sebészeti hatások miatt nem kezeltek az INTER FIX™ eszközzel.
A III. táblázat tartalmazza a műtétet követő 12 és 24 hónapos időszakokra vonatkozó összesített sikerességi mutatókat is.
III. táblázat - Az INTER FIX™ eszköz vizsgálatából származó klinikai eredmények
Fúzió 96.1%
Oswestry
fájdalom/
rokkantság
javulása A
pre-operatív
állapothoz
képest legalább
15 pontos
javulást mutató
betegek
Neurológiai
állapot
fennmaradása
vagy javulása
Általános siker 42.4%
12 hónapos arányszámok 24 hónapos arányszámok
Vizsgálati Kontroll
(122/127)
53.5%
(76/142)
99.3%
(150/151)
(61/144)
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
(12/56)
Delta
1
(ǻ
)
Vizsgálati Kontroll
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
-0.097 46.1%
(71/154)
(27/52)
(24/46)
(43/45)
(17/57)
Delta
(ǻ1)
51.9%
52.2%
95.6%
29.8%
-0.043 49.5%
48 hónapos
arányszámok
Vizsgálati
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
(53/107)
72 hónapos
arányszámok
Vizsgálati
97.6% (82/84)
56.3%
(54/96)
1 - Minimális eltérés, amely szükséges annak kinyilvánításához, hogy a tesztelt eszközök a kontroll eszközökhöz képest nem rosszabb minőségűek.
Az INTER FIX™ menetes fúziós eszköz klinikai vizsgálatában részt vett összes beteget, a kezelt csoporttól függetlenül ugyanazon felvételi/kizárási kritériumok alapján sorolták be. A
randomizált, INTER FIX™ eszközzel kezelt betegek és a nem randomizált, INTER FIX™ eszközzel kezelt betegek összehasonlításának bizonyítása érdekében megvizsgálták a demográfiai
jellemzőket, a betegek preoperatív egészségi állapotát, a degeneratív porckorongbetegségre jellemző diagnosztikai képalkotó eredményeket, a műtéti módszert, valamint lumbalis kezelés
szintjét. A populáció statisztikailag összehasonlítható volt, kivéve a műtéti módszert, a beteg mûtét elõtti szakmai tevékenységét, valamint a csigolya véglemezen osteophyta képződésről
kapott preoperatív diagnosztikai képalkotási eredményeket; az annulus fibrosis, a ligamentum flavum és/vagy a kisízületi tokok forradását és/vagy megvastagodását; valamint a nucleus
Page 60

pulposus sérvét. A további statisztikai elemzések kimutatták, hogy ezek a változók nem álltak kapcsolatban a fúzióra, Oswestry fájdalom/rokkantság javulására, neurológiai állapotra vagy
összességében a terápia sikerére vonatkozó 12 hónapos klinikai eredmények különbségeivel, kivéve a nucleus pulposus sérvét. A nem randomizált csoportban több betegnek volt nucleus
pulposus sérv eredménye, melyről úgy találták, hogy összességében megnöveli a siker valószínűségét.
Végrehajtottak egy kezelési szándék szerinti elemzést is. Ezen elemzés céljára a másodlagos műtét sikertelenségét, a halált, az utólagos vizsgálatból kimaradt betegeket és az egyéb okokból
hiányzó megfigyeléseket az eredményváltozók szempontjából hiányzó megfigyelésnek tekintették, ennélfogva a kiszámított arányszámok nevezőibe beszámították, azaz „kudarcnak” tekintették
őket. Azáltal, hogy ezeket a betegeket is a sikertelen kezelés kategóriájába számították, a kezelési szándék szerinti elemzés klinikai eredményeire vonatkozó eredmények arányszámai
lényegesen alacsonyabbak voltak, mint a ténylegesen megfigyelt klinikai adatok esetén.
A következő táblázat (IV. táblázat) bemutatja az INTER FIX™ eszközzel kezelt betegek kezelési szándék szerinti elemzésének eredményeit, de nem tartalmazza a kontrollbetegek adatait.
A halál, a másodlagos mċtét sikertelensége, az utólagos vizsgálatból történó kimaradás és a hiányzó
12 hónapos
Fúzió 65.9% (122/185) 67.6%
Oswestry fájdalom/rokkantság javulása
A pre-operatív áll apothoz képest legalább 15 pontos
javulást mutató betegek
Neurológiai állapot fennmaradása vagy
javulása
Általános siker 33.0% (61/185) 38.4% (71/185) 45.7%
Másodlagos mċtét kudarca
Sikertelen összeforradás
2
Egyéb
Halál 0 1 0 0
IV. táblázat – Kezelési szándék szerinti elemzés az INTER FIX™ eszköz esetén
megfigyelések sikertelenségnek számítanak és az arányszámok nevezóibe beleszámítanak
arányszámok
41.1% (76/185) 42.7% (79/185) 56.9%
1
81.1% (150/185) 69.2%
5
8
24 hónapos
arányszámok
(125/185)
(128/185)
5
3
48 hónapos
arányszámok
80.2% (93/116) 77.1%
(66/116)
87.9%
(102/116)
(53 /116)
4
4
72 hónapos
arányszámok
( 84/109)
56.0%
(61 /109)
75.2%
(82/109)
49.5%
(54/109)
1
2
1 - E betegek szerepelnek a fúziós arányszámok számításában, de a klinikai vizsgálat szempontjából kudarcnak tekintendők.
2 - Azok a betegek, akiknek a szóban forgó időszakban utólagos kezelésen kellett részt venniük, és másodlagos műtétet hajtottak végre rajtuk a sikertelen összeforradástól eltérő okokból,
a klinikai vizsgálat szempontjából kudarcnak tekintendők.
CSOMAGOLÁS:
Szállításkor az összes alkotórész csomagolásának érintetlennek kell lennie. Kölcsönkapott eszközök, vagy konszignációs rendszer használata esetén az összes készletet figyelmesen meg
kell vizsgálni, hogy teljes-e, és az összes alkotórészt a műszereket beleértve figyelmesen meg kell vizsgálni annak biztosítása érdekében, hogy nem szenvedtek károsodást használat előtt.
A megrongálódott csomagokat vagy termékeket ne használja, hanem küldje vissza őket a Medtronic Sofamor Danek részére.
TISZTÍTÁS ÉS FERTŐTLENÍTÉS:
Amennyiben nem steril módon szállítják, az összes implantátumot és műszert először a bevált kórházi módszerekkel meg kell tisztítani, mielőtt sterilizálnák és steril sebészeti területre vinnék
őket. A használt műszereket újrafelhasználás előtt szennyezés-mentesíteni, tisztítani és sterilizálni kell.
Megjegyzés: Ne használjon olyan tisztító oldatot, amelyek fehérítőszert vagy formalint tartalmaznak, mivel ezek megrongálhatják az eszközt.
Az összes termékkel körültekintően kell bánni. A helytelen használat vagy kezelés az eszköz károsodását és esetleg nem megfelelő működését eredményezheti.
STERILIZÁLÁS:
Amennyiben a steril megjelölés nem szerepel egyértelműen egy gyárilag szállított felbontatlan steril csomag címkéjén, az operáció során használt összes implantátumot és műszert a kórházi
személyzetnek sterilizálnia kell használat előtt. Sterilizálás előtt távolítson el minden csomagolóanyagot. Csak steril termékeket helyezzen a műtét területére. 10-6 sterilitási szint (SAL)
eléréséhez ezen termékeket a kórházi személyzetnek gőzzel ajánlatos sterilizálnia az alábbi három eljárási paramétersorozat egyikének alkalmazásával.
MÓDSZER CIKLUS HŐMÉRSÉKLET STERILIZÁLÁS IDEJE
Gőz Elővákuum 270°F (132°C) 4 perc
Gőz Gravitáció 250°F (121°C) 60 perc
Gőz* Gravitáció* 273°F (134°C)* 20 perc*
MEGJEGYZÉS: Mivel a sterilizálás során számos változó játszik szerepet, minden egyes egészségügyi intézménynek kalibrálnia és hitelesítenie kell a saját készülékeik esetében használt
sterilizáló eljárást (pl. hőmérséklet, idő).
*Az Egyesült Államokon kívüli használat esetén, bizonyos nem amerikai egészségügyi hatóságok a sterilizálást ezen paraméterek alapján ajánlják, hogy a Creutzfeldt-Jakob betegség terjedésének
potenciális kockázata a minimumra csökkenthető legyen, különösen olyan sebészeti műszerek esetén, melyek a központi idegrendszerrel érintkezésbe kerülhetnek.
A TERMÉKKEL KAPCSOLATOS PANASZOK:
Bármely egészségügyi szakember (pl. e termékrendszerek vásárlója vagy felhasználója), aki nincs megelégedve vagy bármilyen panasza van a termék minőségét, mibenlétét, tartósságát,
megbízhatóságát, biztonságosságát, hatékonyságát és/vagy teljesítményét illetően, forduljon a Medtronic Sofamor Danek-hez vagy a cég disztribútorához. Továbbá, ha a beültetett INTER
FIX™ menetes fúziós eszköz vagy INTER FIX™ RP menetes fúziós eszköz bármelyik alkotórésze hibásan működik (azaz nem tesz eleget bármelyik teljesítményspecifikációjának vagy
egyébként nem a rendeltetésének megfelelően működik), vagy a hibás működés gyanúja felmerül, azonnal értesíteni kell a disztribútort. Amennyiben bármely Medtronic Sofamor Danek
termék valamikor is hibásan működik, és fennáll a lehetőség, hogy egy beteg halálát vagy súlyos sérülését okozta, vagy ahhoz hozzájárult, a disztribútort azonnal értesíteni kell telefonon,
telefaxon vagy írásban. Panasz benyújtásakor, kérjük adja meg az alkotórész(ek) nevét és számát, a tételszámot (tételszámokat), a nevét és címét, a panasz mibenlétét, valamint azt, hogy
kér-e írásos jelentést a disztribútortól.
FIGYELMEZTETÉS AZ ORVOS SZÁMÁRA: Bár a gyártó vállalat és a beteg között az orvos játssza a képzett közvetítő szerepét, a jelen dokumentumban szereplő fontos orvosi információkat
át kell adni a beteg számára.
Csak egyesült államokbeli felhasználóknak
FIGYELEM! AZ USA SZÖVETSÉGI TÖRVÉNYEINEK ÉRTELMÉBEN A JELEN ESZKÖZÖK CSAK ORVOSOK ÁLTAL VAGY ORVOSI RENDELVÉNYRE ÉRTÉKESÍTHETŐK.
AZ ESZKÖZ ELTÁVOLÍTÁSA:
Amennyiben szükséges lenne egy INTER FIX™ menetes fúziós eszköz vagy egy INTER FIX™ RP menetes fúziós eszköz eltávolítása, kérjük hívja a Medtronic Sofamor Danek-et.
©
2006 Medtronic Sofamor Danek. Minden jog fenntartva
Page 61

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
LATVISKI
Šeit ir sniegta svarīga medicīniska informācija par INTER FIX™ vītņoto spondilosindēzes ierīci un par INTER FIX™ RP vītņoto spondilosindēzes ierīci.
APRAKSTS:
INTER FIX™ vītņotā spondilosindēzes ierīce sastāv no doba, perforēta metāla cilindra un gala vāciņa. INTER FIX™ RP vītņotā spondilosindēzes ierīce ir dobs, perforēts metāla cilindrs ar
vienu lielu noapaļotu izgriezumu pa cilindra garenasi visā cilindra garumā. Abi INTER FIX™ RP implanta gali ir slēgti.
INTER FIX™ un INTER FIX™ RP implanti ir pieejami ar diametru no 12 mm līdz 24 mm un garumu no 20 mm līdz 29 mm. INTER FIX™ implantu gala vāciņi ir ar cilindram atbilstošu diametru
un tiek uzlikti cilindra vaļējam galam pēc tam, kad cilindrā ir iepildīts autogēnais pārstādāmais kauls.
INTER FIX™ un INTER FIX™ RP implanti ir izgatavoti no implantu kvalitātes titāna sakausējuma (Ti-6Al-4V), kas atbilst ASTM F136 prasībām.
Netieši izteiktas garantijas attiecībā uz komerciālo kvalitāti vai derīgumu noteiktām vajadzībām ir speciāli atrunātas. Papildu informāciju par garantijām un atbildības ierobežojumiem varat
uzzināt Medtronic Sofamor Danek katalogā vai cenrādī.
INDIKĀCIJAS:
INTER FIX™ vītņotā spondilosindēzes ierīce un INTER FIX™ RP vītņotā spondilosindēzes ierīce ir indicēta mugurkaula spondilosindēzes procedūrām pacientiem ar nobriedušu skeletu, kam
ir deģeneratīvā disku slimība (DDD) vienā līmenī no L2 līdz S1. Deģeneratīvā disku slimība tiek definēta ar muguras sāpēm, kas ceļas no diskiem, kur disku deģenerācija ir apstiprināta ar
slimības vēsturi un radiogrāfiskiem pētījumiem. Šiem deģeneratīvās disku slimības pacientiem slimības skartajā līmenī var būt arī I pakāpes spondilolistēze vai retrolistēze. INTER FIX™ un
INTER FIX™ RP implanti ir jālieto ar pārstādītu autogēno kaulu un jāimplantē, izmantojot atklātu anterioro piekļuvi.
KONTRINDIKĀCIJAS:
INTER FIX™ vītņoto spondilosindēzes ierīci un INTER FIX™ RP vītņoto spondilosindēzes ierīci nedrīkst implantēt pacientiem ar aktīvu infekciju operācijas vietā vai ar alerģiju pret titānu
vai titāna sakausējumu.
BRĪDINĀJUMI:
• INTER FIX™ vītņoto spondilosindēzes ierīci un INTER FIX™ RP vītņoto spondilosindēzes ierīci drīkst lietot tikai tādi ārsti, kam ir pieredze muguras spondilosindēzes procedūru
veikšanā, un kas ir izgājuši pienācīgu apmācību darbam ar šo ierīci.
• Pienācīgas pieredzes un/vai apmācības trūkums var palielināt nelabvēlīgo parādību skaitu, piemēram, asinsvadu bojājumus, neiroloģiskas parādības un/vai uroģenitālas parādības
(ieskaitot sauso ejakulāciju).
INTER FIX™ VĪTŅOTĀ SPONDILOSINDĒZES IERĪCE
INTER FIX™ RP VĪTŅOTĀ SPONDILOSINDĒZES IERĪCE
SVARĪGA MEDICĪNISKĀ INFORMĀCIJA
Page 62

PIESARDZĪBA:
E
E
• INTER FIX™ vītņotās spondilosindēzes ierīces un INTER FIX™ RP vītņotās spondilosindēzes ierīces drošība un efektivitāte nav noskaidrota pie pacientiem ar kādu no šiem
stāvokļiem:
- II vai augstākas pakāpes spondilolistēze vai retrolistēze;
- nepieciešamība veikt spondilosindēzi vairāk kā vienā līmenī;
- iepriekšējās(o) spondilosindēzes procedūras(u) revīzija;
- nepieciešamība pēc operācijas lietot steroīdos vai nesteroīdos pretiekaisuma līdzekļus;
- stipri izteikta aptaukošanās;
- vecums, kas mazāks par 18 gadiem vai lielāks par 65 gadiem;
- osteoporoze, osteopēnija un/vai osteomalācija;
- grūtniecība.
• INTER FIX™ vītņotās spondilosindēzes ierīces un INTER FIX™ RP vītņotās spondilosindēzes ierīces drošība un efektivitāte ir noskaidrota tikai pie anteriorām lumbārās
spondilosindēzes (ALIF) procedūrām, izmantojot autogēnā kaula pārstādīšanu.
• Pacientiem, kam implantē INTER FIX™ vītņoto spondilosindēzes ierīci, ir jābūt vismaz sešu mēnešu ilgam bezoperācijas periodam. Pacientiem, kam implantē INTER FIX™ RP
vītņoto spondilosindēzes ierīci, arī ir jābūt vismaz sešu mēnešu ilgam bezoperācijas periodam.
• Operācijas līmenī ir jāimplantē blakus divas INTER FIX™ vīt
spondilosindēzes ierīce un viena INTER FIX™ RP vītņotā spondilosindēzes ierīce. Gareniskais izgriezums INTER FIX™ RP vītņotajā spondilosindēzes ierīcē artvieglo divu ierīču
implantēšanu ciešākā tuvumā, nekā to ir iespējams izdarīt ar divām INTER FIX™ vītņotajām spondilosindēzes ierīcēm, tā iegūstot samazinātu laterālo profilu.
• INTER FIX™ vītņotās spondilosindēzes ierīces un INTER FIX™ RP vītņotās spondilosindēzes ierīces garenasij ir jābūt vērstai anteriori-posteriorā virzienā.
ņotās spondilosindēzes ierīces. Operējamā līmenī blakus vienu otrai ir jāimplantē viena INTER FIX™ vītņotā
• Pirms lietošanas implanti un instrumenti ir jāsterilizē atbilstoši sterilizācijas instrukcijām, kas ievietotas šajā iepakojumā, ja vien tie nav piegādāti sterili un kā tādi skidri apzīmēti.
NELABVĒLĪGĀS SEKAS:
1. tabulā parādītās nelabvēlīgās sekas ir apkopotas no 185 pacienta ar INTER FIX™ ierīci un 62 kontroles pacientiem, kas tika iesaistīti INTER FIX™ vītņotās spondilosindēzes ierīces
multicentriskā klīniskajā pētījumā. Pētījumā, kas tika veikts pēc reģistrācijas apliecības saņemšanas, nelabvēlīgas sekas tika novērotas 116 pacientiem pēc 48 mēnešiem un 109 pacientiem
pēc 72 mēnešiem. Parādītie dati ir attiecināti uz kopējo pacientu skaitu, kas katrā laika brīdī bija iesaistīti pētījumā. Lai iegūtuu vispārinātu rādītāju, ir apvienotas nelabvēlīgās sekas, kas tika
novērotas randomizētajā un nerandomizētajā grupā, kurās pacientiem implantēja INTER FIX™ ierīci.
PÏc operÁcijas
(1 diena < 2
OperÁcijÁ
[Skaits (%)]
KONTROL
KomplikÁcija
VaskulÁras, operÁcijÁ 15(8.1) 2(3.2) 15 (8.1) 2(3.2)
SÁpes krustu-ieguría rajonÁ 5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
NeiroloÙiskas 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Muguras sÁpes 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
Iegriezumi 11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
Mugurkaula komplikÁcijas 1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
UroloÙiskÁs 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Citas 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Citas sÁpes 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
GastrointestinÁlas 7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
SausÁ ejakulÁcija 2(1.1) 3(1.6) 1(0.5) 6(3.2)
RespiratorÁs 3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
SÁpes kÁjÁs 2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
Trauma 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
PeritoneÁlas 3(1.6) 3(1.6) 0
VaskulÁras, pÏc operÁcijas 2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
Kaula fraktćra 1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
Implanta
pÁrvietošanÁs/atslÁbinÁšanÁs
SÁpes pÁrstÁdÛtÁ kaula
íemšanas vietÁ
Nesaaugšana 4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
NosÏšanÁs 2(1.1) 2(1.1) 0
Nesaaugš ana
(GALAREZULTÀTS NEZINÀMS)
MeningÛts 1(1.6) 0 1(1.6)
Implanta lćzums 5(8.1) 0 5(8.1)
VÏzis 1
INTER FIX
N=185
N=62
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
1(1.6)2 0 1(1.6) 2(1.7)
[Skaits (%)]
INTER FIX
N=185
mÏneši)
KONTROLE
1. TABULA - NELABVÎLÚGÀS SEKAS
N=62
3 mÏnesis
(t
2 - < 5 mÏneši
[Skaits (%)]
INTER FIX
N=185
KONTROL
N=62
6 mÏnesis
(t5 - < 9 mÏneši
[Skaits (%)]
INTER FIX
N=185
KONTROLE
N=62
(t
INTER FIX
N=185
NÁve 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
12 mÏnesis
9 - < 19 mÏneši
[Skaits (%)]
KONTROLE
N=62
(0.5)
24 mÏnesis
19 - < 30
(t
mÏneši
[Skaits (%)]
KONTROLE
INTER FIX
N=185
1
(0.5)
72.
mÏnesis
60
(t
60
mÏneši)
INTER FIX
N=109
N=62
NELABVÎLÚGAS
SEKAS KOPÀ
(24 MÎNEŠOS)
INTER FIX
N=185
1
(7.4)
48. mÏnesis
(>30 - < 60
30<
(t
KONTROLE
N=62
mÏneši
INTER FIX
N=116
0
2 (1.1) 1(0.8) 1(0.9)
1 - Sešiem (6) no 81 vīrieša, kas bija iesaistīti INTER FIX™ ierīces grupā (7,4%), 24 mēnešu novērtēšanas periodā novēroja sauso ejakulāciju.
2 - Vēlāk tika ziņots, ka šim pacientam (175.) apmēram 52 mēnešus pēc operācijas bija notikusi spondilosindēze, un nebija nepieciešama cita ķirurģiskā operācija vai ārstēšana.
Klīniskajā pētījumā tika novērotas divas svarīgas nelabvēlīgas sekas: vaskulā
grupā 14 pacientiem kopā novēroja 15 vaskulāras parādības, kas bija radušās operācijas laikā. Šīs parādības ietvēra: 2 dobās vēnas ievainojumi, 9 iegurņa vēnas ievainojumi, 1 plēsta
hipogastrālā vēna, 1 segmentāla vēnas asiņošana, 1 sakrālās vēnas ievainojums un 1 virspusēja asiņošana. Kontroles grupā tajā pašā laikā periodā 2 pacientiem kopā novēroja 2 vaskulārus
operācijas laikā iegūtus ievainojumus. Tie ietvē
24 mēnešu novērtēšanas periodā INTER FIX™ ierīces pacientu grupā 28 pacientiem kopā novēroja 30 operācijas laikā iegūtas neiroloģiskas parādības. Šīs parādības ietvēra: 1 pēdas
krišanu, 3 nervu saknīšu ievainojumus, 1 foraminālo stenozi, 1 refleksa simpātisko distrofiju, 7 kāju nejutīguma vai dedzināšanas gadījumus, 2 disestēzijas gadījumus, 3 parestēzijas gadījumus,
2 radikulīta gadījumus, 7 muguras un/vai kājas sāpju gadījumus, 1 gadījumu, kad muguras un/vai kāju sāpes novēroja kopā ar citiem simptomiem, 1 asas sāpes muguras lejasdaļā un 1
karpāl
ā tuneļa sindromu.
Ta jā pašā laika periodā kontroles grupā 10 pacientiem kopā novēroja 8 neiroloģiskās parādības. Šīs parādības ietvēra: 1 radikulopātiju ar ekstremitāšu tirpšanu, 1 hroniskas muguras sāpes
ar radikulītu, 1 novājinošus izplatīšanās simptomus, 2 muguras un kāju sāpes ar citiem simptomiem, 1 denervētu abductor magnus muskuli un 4 kāju tirpšanas vai silšanas gadījumus.
Pētījumā, kas tika veikts pēc reģistrācijas, 10 pacientiem tika papildus novērotas 10 neiroloģiskas parādības. Šīs parādības ietvēra: 1 radikulopātiju, 2 cervikālās radikulopātijas, 1 roku
nejūtības un tirpšanas gadī
citiem simptomiem. Bez tam nevēlamo seku tabula ietver kāju un muguras sāpju nevēlamās sekas un/vai spinālās parādības, piemēram, starpdisku telpas sabrukumu, kas dažos gadījumos
ietver neiroloģisko komponentu.
Dažām no nevēlamajām sekām bija nepieciešama ķirurģiska iejaukšanās pēc klīniskājā pētījumā veiktās operācojas. Šīs papildu ķirurģiskās iejaukšanās var klasificēt kā
papildu fiksācijas vai reoperācijas
pētījumā IDE, kā arī par INTER FIX™ ierīces grupu pētījumā pēc reģistrācijas.
1
Revīzija: Procedūra, kurā sakārto vai citādi izmaina sākotnējo implanta konfigurāciju.
jumu, 1 kāju nejūtības vai dedzināšanas gadījumu, 3 muguras un/vai kājas sāpju gadījumus un 2 gadījumus, kad muguras un/vai kāju sāpes novēroja kopā ar
1
. 2. tabulā apkopoti sekundārās ķirurģiskās iejaukšanās dati par INTER FIX™ ierīces (kopā randomizētie un nerandomizētie pacienti) un kontroles grupu
ra: 1 iegurņa vēnas ievainojums un 1 asiņošana no kaula pamatnes.
rie un neiroloģiskie ievainojumi operācijas gaitā. 24 mēnešu novērtēšanas periodā INTER FIX™ ierīces pacientu
revīzijas, izņemšanas,
Izņemšana: Procedūra, kurā izņem vienu vai vairākus oriģinālā implanta konfigurācijas komponentus bez to aizvietošanas ar tā paša tipa pētījumā lietoto ierīci.
Papildu fiksācija: Procedūra, kurā tiek ievietotas papildu spinālās ierīces, kas nav iekļautas protokolā.
Reoperācija: Jebkura ķirurģiskā operācija, kurā neizņem, neizmaina vai nepieliek klāt jebkuru oriģinālā implanta komponentu.
Page 63

PÏtÛjums IDE PÏtÛjums pÏc reÙistrÁcijas
24 mÏnešos 48. mÏnesis
INTER FIX™
2. tabula -SekundÁrÁs áirurÙiskÁs procedćras
ierÛce
N=185
Kontrole1
N=62
INTER FIX™
ierÛce
N=116
72. mÏnesis
INTER FIX™
ierÛce
N=109
RevÛzijas 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Izíemšanas 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Papildu
fiksÁcijas
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
ReoperÁcijas 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
1 - Apsekošanas pētījumā pēc 24. mēneša nebija vajadzīgi kontroles pacienti, tāpēc līdz 72. mēnesim par šiem pacientiem nav pieejama informācija.
IESPĒJAMĀS NELABVĒLĪGĀS SEKAS:
Šajā sarakstā uzskaitītas iespējamās nelabvēlīgās sekas, kas var sekot muguras spondilosindēzes operācijām ar INTER FIX™ vītņoto spondilosindēzes ierīci vai INTER FIX™ RP vītņoto
spondilosindēzes ierīci. Dažas no šīm nelabvēlīgām sekām var jau būt ietvertas nelabvēlīgo seku tabulā.
• Komponentu atvienošanās, saliekšanās, salūšana, atslābināšanās un/vai pārvietošanās.
• (Alerģiska) reakcija uz svešķermeņiem.
• Audu vai nervu bojājums.
• Pēcoperācijas izmaiņas muguras izliekumā, korekcijas zudums, skriemeļu augstuma samazināšanās un/vai redukcija.
• Infekcija.
• Muguras smadzeņu cietā apvalka plīsumi.
• Neiroloģiskās sistēmas bojājumi.
• Uroloģiskās sistēmas bojājumi.
• Rētu veidošanās.
• Kaula fraktūra.
• Nesaaugšana (vai pseidartroze), novēlota saaugšana, nepareiza saaugšana.
• Mugurkaula operētās daļas iespējamās attīstības apstāšanās. Mugurkaula kustīguma vai funkciju zudums.
• Pārstādāmā kaula donora vietas komplikācijas.
• Asinsvadu bojājumi un cita veida sirds un asinsvadu sistēmas traucējumi.
• Gastrointestinālas komplikācijas.
• Reproduktīvās sistēmas bojājumi.
• Iekšējo orgānu un saistaudu bojājumi.
• Respiratoro traucējumu izveidošanās.
• Griezumu komplikācijas.
• Garīgā stāvokļa izmaiņas.
• Nāve.
Piezīme: Dažu iepējamo nelabvēlīgo seku korekcijai var būt nepieciešams veikt papildu ķirurģisko operāciju.
KLĪNISKIE REZULTĀTI:
Multicentrisks INTER FIX™ vītņotās spondilosindēzes ierīces ekvivalences2 klīniskais pētījums tika veikts Savienotajās Valstīs, kurā tika salīdzināts ar autogēno kaulu pildītas INTER FIX™
ierīces anteriorais spinālais pielietojums salīdzinājumā ar allokaula (augšstilba kaula) gredzena, kas pildīts ar autogēno kaulu pielietojumu un kontroli pacientu ārstēšanā ar simptomātisku
deģeneratīvo disku slimību. Sākotnēji kīniskajam pētījumam bija perspektīva, randomizēta, kontrolēta pētījuma plāns. Pēc tam pētījuma plāns tika pārskatīts, atļaujot iesaistīt pacientus
nerandomizētajā grupā, t.i., pie pacientiem, kurus ārstēja tikai ar INTER FIX™ ierīci.
Kritēriji iekļaušanai klīniskajā pētījumā ietvēra simptomātisku deģeneratīvu disku slimību, kas konstatējama pēc nevaldāmas kājas un/vai muguras sāpēm ar pozitīviem rezultātiem attēlveidošanas
diagnostikā; muguras nestabilitāti, kas definēta ar nobīdi, lielāku par 4 mm vai nošķiebumu, lielāku par 5° fleksijas/ekstensijas radiogrammās; vienlīmeņa simptomātiskus sarežģījumus no L2
līdz S1; spondilolistēzi, kas nepārsniedza 1. pakāpi. No pētījuma specifiski tika izslēgti pacienti, kuriem: bija agrāk veikta anterioras spondilosindēzes procedūra tajā pašā mugurkaula līmenī;
bija osteopēnija, osteoporoze vai osteomalācija; vai kuriem bija nepieciešama kaulu augšanas stimulācija.
Ar INTER FIX™ ierīci ārstējamo pacientu pētījuma grupā kopā tika iekļauts 185 pacients.
INTER FIX™ vītņotās spondilosindēzes ierīces pētījumā pēc reģistrācijas bija nepieciešams novērtēt ierīces uzvedību ilgstošā periodā, iegūstot datus par 6 gadiem pēc operācijas minimums
100 pacientiem. Ierīces uzvedība ilgstošā periodā tika novērtēta 116 pacientiem 4 gadus pēc operācijas un 109 pacientiem 6 gadus pēc operācijas. Pacientu populācija tika ņemta no
IDE klīniskā pētījuma dalībniekiem. Pamatojoties uz pētījuma IDE kritē
radiogrāfisko novērtējumu, kā arī sāpju un funkciju novērtējumu.
3. tabulā parādīti pētījuma klīniskie rezultāti, aprakstot veiksmīgas spondilosindēzes parametrus, sāpju mazināšanas (pēc Oswestry skalas) panākumus un neiroloģiskos panākumus 12, 24, 48
un 72 mēnešus pēc ārstnieciskās ķirurģiskās operācijas. Šī klīniskā pētījuma panākumi tika vērtēti, balstoties uz ekvivalences analīzi starp INTER FIX™ ier
Šajā klīniskajā pētījumā spondilosindēze tika definēta kā trabekulārā kaula saaugšana, nobīdes (<3mm) un leņķiskā kustīguma (<5°) stabilizēšanās un ne vairāk par 50% rentgenstaros
caurspīdīgas zonas ap katru implantu. Bez tam, aprēķinos tika uzskatīts, ka pacientiem, kuriem bija nepieciešama otrreizēja operācija sakarā ar nesaaugšanu, spondilosindēze nav izdevusies.
Panākumi pēc Oswestry sāpju/invaliditātes rādītājiem tika definēti kā uzlabojums rādītājos pēc operācijas vismaz par 15 punktiem, salīdzinot ar rādītājiem pirms operācijas. Panā
neiroloģiskā stāvokļa uzlabojumā tika definēts kā stāvokļa saglabāšanās vai uzlabošanās vismaz 3 no 4 neiroloģiskajām kategorijām (kustības, sajūtas, refleksi un izstieptas kājas pacelšana)
pēc operācijas, salīdzinot ar stāvokli pirms operācijas. Vispārējie panākumi tika pamatoti ar spondilosindēzes panākumiem, panākumiem Oswestry sāpju/imvaliditātes rādītājos un panākumiem
neiroloģiskā stāvokļa uzlabojumā, kā arī sekundārām ķirurģiskām procedūrām bez nevienas neveiksmes.
2
INTER FIX™ ierīce statistiski ne sliktāka par kontroli.
3
Neietver 3 pacientus, kam netika izmantota ārstēšana ar INTER FIX™ ierīci sakarā ar nelabvēlīgām ķirurģiskās iejaukšanās sekām.
rijiem, par šiem pacientiem iegūtā informācija ietvēra visu papildu ķirurģisko procedūru aprakstus, spondilosindēzes
3
Klīniskā pētījuma randomizētajā grupā kopumā bija iekļauti 62 kontroles pacienti.
īces grupu un kontroles grupu.
kums
3. tabulā var iepazīties arī ar kopējo panākumu rezultātiem 12 un 24 mēnešus pēc operācijas.
SpondilosindÏze 96.1%
Uzlabojums
Oswestry
sÁpju/invaliditÁtes
rÁdÛtÁjos Pacienti
ar vismaz 15
punktu
uzlabojumu,
salÛdzinot ar
pirmsoperÁcijas
rÁdÛtÁjiem
NeiroloÙiskÁ
stÁvokåa
saglabÁšanÁs vai
uzlabošanÁs
VispÁrÏjie
panÁkumi
12 mÏneša rÁdÛtÁji 24 mÏneša rÁdÛtÁji
PÏtÛjuma Kontrole
(122/127)
53.5%
(76/142)
99.3%
(150/151)
42.4%
(61/144)
3. tabulaI – INTER FIX™ ierÛces pÏtÛjuma klÛniskie rezultÁti
Delta
1
)
(ǻ
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
-0.097 46.1%
(12/56)
PÏtÛjuma Kontrole
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
(71/154)
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
(17/57)
48 mÏneša
Delta
1
)
(ǻ
-0.043 49.5%
rÁdÛtÁji
PÏtÛjuma
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
(53/107)
72 mÏneša
rÁdÛtÁji
PÏtÛjuma
97.6% (82/84)
56.3%
(54/96)
1 - Minimālā robeža, ko nepieciešams pārsniegt, lai apgalvotu, ka pētījuma rezultāti nav sliktāki par kontroli.
Page 64

Visi pacienti, kas bija iekļauti INTER FIX™ vītņotās spondilosindēzes ierīces klīniskajā pētījumā, neatkarīgi no ārstēšanas grupas, bija iekļauti pēc vieniem un tiem pašiem iekļaušanas/
izslēgšanas kritērijiem. Lai pamatotu rezultātu salīdzināmību starp randomizēto INTER FIX™ ierīces pacientu grupu un nerandomizēto INTER FIX™ ierīces pacientu grupu, tika pētīti pacientu
demogrāfiskie raksturojumi, pirmsoperācijas medicīniskais stāvoklis, diagnostiskās attēlveidošanas konstatējumi, kas raksturo deģeneratīvo disku slimību, operācijas pieeja un ārstētās
lumbārās joslas līmeņa lokalizācija. Popul
attēlveidošanas konstatējumus par osteofītu veidošanos uz skriemeļu galaplāksnēm, annulus fibrosus, ligamentum flavum un/vai fasešu locītavas kapsulas rētojumu un/vai sabiezinājumu, kā
arī nucleus pulposus plīsumu. Tālākās statistiskās analīzes parādīja, ka šīe mainīgie nav saistāmi ar atšķirībām spondilosindēzes iznākumā pēc 12 mēnešiem, Oswestry sāpju/invaliditātes
rādītāja uzlabojumā, neirolo
izrādījās, palielināja vispārējo panākumu varbūtību.
Tika veikta arī visu sākotnēji iesaistīto pacientu rezultātu analīze. Šai analīzei neveiksmes sekundārajās ķirurģiskajās operācijās, nāves gadījumi, no novērojumu loka izkritušie pacienti un citu
iemeslu dēļ neveiktie novērojumi, kas nedeva novērojumus rezultātu mainīgajiem, aprēķinos tika iekļauti sauc
ārstēšanas gadījumus, klīnisko rezultātu rādījumi sākotnēji iesaistīto pacientu analīzē bija ievērojami zemāki par faktiski novērotajiem klīniskajiem datiem.
ģiskajā stāvoklī vai vispārējos panākumos, izņemot nucleus pulposus plīsumu. Nerandomizētajā grupā biežāk tika konstatēts nucleus pulposus plīsums, kas, kā
ācija bija statistiski salīdzināma, izņemot operācijas pieejas aspektu, pirmsoperācijas profesionālos apstākļus un pirmsoperācijas diagnostiskās
ējā, t.i., tika uzskatīti par “neveiksmēm”. Aplūkojot šos pacientus kā nesekmīgas
Šajā tabulā (4. tabula) ir dota visu sākotnēji ārstēšanā ar INTER FIX™ ierīci iesaistīto pacientu rezultātu analīze, bet tajā nav ietverti kontroles pacientu rezultāti.
NÁves gadÛjumi, neveiksmÛga otrreizÏjÁ operÁcija, izkrišana no novÏrojumu loka un trćkstoši novÏrojumi tika uzskatÛti par
12 mÏneša
SpondilosindÏze 65.9% (122/185) 67.6%
Uzlabojums Oswestry sÁpju/invaliditÁtes
rÁdÛtÁjos
Pacienti ar vismaz 15 punktu uzlabojumu,
salÛdzinot ar pirmsoperÁcijas rÁdÛtÁjiem
NeiroloÙiskÁ stÁvokåa saglabÁšanÁs vai
uzlabošanÁs
VispÁrÏjie panÁkumi 33.0% (61/185) 38.4% (71/185) 45.7%
OtrreizÏjÁs operÁcijas neveiksmes
Nesaaugšana
2
Citi
NÁves gadÛjumi 0 1 0 0
1 - Šie pacienti ir iekļauti spondilosindēzes rādītāju aprēķinos, bet citādi tiek uzskatīti par neveiksmēm klīniskā pētījuma vajadzī
2 - Pacienti ar šajā periodā paredzētu atkārtotu apsekošanu, kam bija orreizēja operācija no nesaaugšanas atšķirīgu iemeslu dēļ, no klīniskā pētījuma viedokļa tika pieskaitīti pie neveiksmīgajiem
gadījumiem.
IEPAKOJUMS:
Visu komponentu iepakojumam pie saņemšanas ir jābūt neskartam. Ja sistēma ir saņemta kā aizdevums vai konosaments, ir jāpārbauda visu komplektu pilnība; visi komponenti, ieskaitot
instrumentus, ir rūpīgi jāpārbauda pirms lietošanas, vai tiem nav defekti. Bojātus iepakojumus vai izstrādājumus nedrīkst izmantot, tie ir jānosūta atpakaļ Medtronic Sofamor Danek.
TĪRĪŠANA UN DEKONTAMINĀCIJA:
Ja implanti un instrumenti nav piegādāti sterili, tos pirms sterilizēšanas un ienešanas sterilajā operācijas zonā ir jānotīra, izmantojot slimnīcā apstiprinātos paņēmienus. Izmantotie instrumenti
ir jādekontaminē, jāntīra un pirms atkārtotas lietošanas jāsterilizē.
4. tabula - Visu sÁkotnÏji ÁrstÏšanai ar INTER FIX™ ierÛci iesaistÛto pacientu rezultÁtu analÛze
neveiksmÏm un iekåauti rÁdÛtÁju aprÏáinos saucÏjÁ
rÁdÛtÁji
41.1% (76/185) 42.7% (79/185) 56.9%
1
81.1% (150/185) 69.2%
5
8
24 mÏneša
rÁdÛtÁji
(125/185)
(128/185)
48 mÏneša
rÁdÛtÁji
80.2% (93/116) 77.1%
(66/116)
87.9%
(102/116)
(53 /116)
5
3
4
4
72 mÏneša
rÁdÛtÁji
( 84/109)
56.0%
(61 /109)
75.2%
(82/109)
49.5%
(54/109)
1
2
bām.
Piezīme: Nelietojiet tādus tīrīšanas šķīdumus, kas satur balinātāju vai formalīnu, jo tie var sabojāt šo ierīci.
Ar visiem izstrādājumiem ir jāapietas uzmanīgi. Nepareiza pielietošana vai apiešanās ar ierīci var izsaukt bojājumus un ierīces nepareizu funkcionēšanu.
STERILIZĀCIJA:
Ja nav norādījuma par sterilitāti un skaidra uzraksta uz etiķetes neatvērtam sterilam iepakojumam, kas saņemts no kompānijas, visi implanti un instrumenti, kas tiek izmantoti ķirurģiskā operācijā,
ir obligāti jāsterilizē slimnīcā pirms to izmantošanas. Pirms sterilizācijas noņemiet visus iesaiņojuma materiālus. Operācijas laukā drīkst ienest tikai sterilus izstrādājumus. Lai panāktu 10-6
drošības līmeni, ieteicams šos izstrādājumus slimnīcā sterilizēt ar tvaiku pēc viena no trim tālāk norādītajiem režīmiem:
PAŅĒMIENS CIKLS TEMPERATŪRA APSTRĀDES ILGUMS
Tvaiks Ar vakuumēšanu 270° F (132°C) 4 minūtes
Tvaiks Parastais 250° F (121°C) 60 minūtes
Tvaiks* Parastais* 273° F (134°C)* 20 minūtes*
PIEZĪME: Sakarā ar to, ka sterilizācijā ir jāņem vērā daudzi mainīgie faktori, katrai medicīnas iestādei ir jākalibrē un jāpārbauda sterilizācijas process (piem., temperatūra, ilgums) attiecīgajām
iekārtām.
*Izmantojot ārpus ASV, dažas ar ASV nesaistītas veselības aizsardzības iestādes iesaka sterilizēt pēc šiem parametriem, lai līdz minimumam samazinātu iespējamo Kreucfelda-Jakoba
slimības izplatīšanas risku, sevišķi attiecībā uz ķirurģiskajiem instrumentiem, kas var nonākt kontaktā ar centrālo nervu sistēmu.
SŪDZĪBAS PAR IZSTRĀDĀJUMU:
Veselības aprūpes profesionāļiem (piem., klientiem vai šīs sistēmas produktu lietotājiem), kuriem ir jebkādas sūdzības vai kuri nav apmierināti ar izstrādājuma kvalitāti, identiskumu, izturību,
uzticamību, drošību, efektivitāti un/vai darbību, ir jāvēršas pie izplatītāja vai Medtronic Sofamor Danek. Papildus tam, ja kāda implantētā INTER FIX™ vītņotās spondilosindēzes ierīces vai
INTER FIX™ RP vītņotās spondilosindēzes ierīces komponents ‘slikti darbojas’ (t.i., neatbilst kādam no uzrādītajiem darba parametriem vai citā veidā neatbilst paredzētajam uzdevumam)
vai rada aizdomas, ka varētu slikti darboties, ir nekavējoties jāziņo izplatītājam. Ja kāds Medtronic Sofamor Danek produkts jebkad ir “slikti darbojies” un iespējams izsaucis tieši vai netieši
pacienta nāvi vai smagu ievainojumu, nekavējoties pa telefonu, ar faksu vai rakstiski par to ir jāpaziņo izplatītājam. Ja Jūs iesniedzat sūdzību, lūdzu, uzrādiet komponenta(u) nosaukumu(s)
un numuru(s), sērijas numuru, Jūsu vārdu un adresi un sūdzības būtību, kā arī lūdziet paziņot, vai izplatītājam ir vajadzīga rakstiska atskaite.
ĀRSTA IEVĒRĪBAI: Kaut arī ārsts darbojas kā izglītots starpnieks starp kompāniju un pacientu, šajā dokumentā sniegtā svarīgā medicīniskā informācija ir jānovada arī līdz pacientam.
Tikai ASV auditorijai
BRĪDINĀJUMS: (ASV) FEDERĀLAIS LIKUMS ATĻ
IERĪCES IZŅEMŠANA:
Ja rastos nepieciešamība izņemt INTER FIX™ vītņoto spondilosindēzes ierīci vai INTER FIX™ RP vītņoto spondilosindēzes ierīci, lūdzu, vērsieties pie Medtronic Sofamor Danek.
©
Medtronic Sofamor Danek, 2006.g. Visas tiesības aizsargātas.
AUJ ŠĪS IERĪCES PĀRDOT TIKAI TAD, JA IR ĀRSTA RĪKOJUMS VAI PASŪTĪJUMS.
Page 65

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
LIETUVIŠKAI
Toliau pateikta svarbi medicininė informacija apie INTER FIX™ srieginio sujungimo prietaisą ir INTER FIX™ RP srieginį sujungimo prietaisą.
APRAŠYMAS
INTER FIX™ srieginio sujungimo prietaisą sudaro tuščiaviduris, perforuotas, metalinis cilindras ir galinis dangtelis. INTER FIX™ RP srieginio sujungimo prietaisą sudaro
tuščiaviduris, perforuotas, metalinis cilindras su vienu didelio išoriniu apvaliu grioveliu palei visą išilginę ašį, einančią iki prietaiso vidinio skersmens. Abu INTER FIX™
RP implanto galai yra uždari.
INTER FIX™ ir INTER FIX™ RP implantų skersmuo gali būti nuo 12 mm iki 24 mm, ilgis – nuo 20 mm iki 29 mm. INTER FIX™ implantų galinių dangtelių dydis atitinka
cilindrų skersmenį; jie yra dedami ant atviro cilindrų galo užpildžius juos autogeniniu kaulo transplantu.
INTER FIX™ ir INTER FIX™ RP implantai yra pagaminti iš implantams skirto titano lydinio (Ti-6Al-4V), atitinkančio ASTM F136.
Numanomos perkamumo ir tinkamumo konkrečiai paskirčiai ar naudojimui garantijos yra specialiai neįtraukiamos. Išsamesnės informacijos apie garantijas
ir ribotą atsakomybę rasite Medtronic Sofamor Danek kataloge arba kainoraštyje.
INDIKACIJOS
INTER FIX™ srieginio sujungimo prietaisas ir INTER FIX™ RP srieginio sujungimo prietaisas yra skirti naudoti degeneracine disko liga (DDD) sergančių pacientų, kurių
stuburas yra susiformavęs, stuburo sujungimo procedūroms viename lygyje nuo L2-S1. Degeneracinė disko liga pasireiškia diskogeninės kilmės nugaros skausmu ir
disko degeneracija, patvirtinta ligos istorija ir rentgeno tyrimais. Šiems degeneracine disko liga sergantiems pacientams taip pat pakenktame lygyje gali pasireikšti 1
laipsnio spondilolistezė arba retrolistezė. INTER FIX™ ir INTER FIX™ RP implantai yra skirti naudoti su autogeniniu kaulo transplantu ir yra implantuojamas iš atviros
priekinės pusės.
KONTRAINDIKACIJOS
INTER FIX™ srieginio sujungimo prietaisas ir INTER FIX™ RP srieginio sujungimo prietaisas neturėtų būti implantuojami pacientams, kuriems operuojamoje vietoje yra
aktyvi infekcija arba kuriems pasireiškia alergija titanui arba titano lydiniui.
INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISAS
INTER FIX™ RP SRIEGINIO SUJUNGIMO PRIETAISAS
SVARBI MEDICININĖ INFORMACIJA
Page 66

ĮSPĖJIMAI
• INTER FIX™ srieginio sujungimo prietaisas ir INTER FIX™ RP srieginio sujungimo prietaisas turėtų būti naudojami tik chirurgų, kurie turi stuburo sujungimo procedūrų
patirties ir buvo atitinkamai apmokyti dirbti su šiuo prietaisu.
• Trūkstant tinkamos patirties ir (arba) apmokymo, gali dažniau pasireikšti nepageidaujamos reakcijos, pvz., kraujagyslių pažeidimai, neurologinės ir (arba) urogenitalinės
nepageidaujamos reakcijos (tarp jų retrogradinė ejakuliacija).
ATSARGUMO PRIEMONĖS
• INTER FIX™ srieginio sujungimo prietaiso ir INTER FIX™ RP srieginio sujungimo prietaiso saugumas ir efektyvumas nebuvo nustatytas pacientams, kuriems pasireiškia
bet kuris iš šių:
- 2 laipsnio arba didesnė spondilolistezė arba retrolistezė;
- keli sujungimo reikalaujantys lygiai;
- ankstesnės slankstelių kūnų sujungimo procedūros (procedūrų) revizija;
- pooperaciniai steroidinio ar nesteroidinio priešuždegiminio gydymo reikalavimai;
- didelis nutukimas;
- amžius – iki 18 metų arba virš 65 metų;
- osteoporozė, osteopenija ir (arba) osteomaliacija;
- nėštumas.
• INTER FIX™ srieginio sujungimo prietaiso ir INTER FIX™ RP srieginio sujungimo prietaiso saugumas ir efektyvumas buvo nustatytas tik atliekant priekinio priėjimo
juosmeninės stuburo dalies slankstelių kūnų sujungimo (ALIF) procedūras naudojant autologinio kaulo transplantą.
• Pacientai, kuriems naudojamas INTER FIX™ srieginio sujungimo prietaisas, turėjo būti taikomas ne mažiau nei šešių mėnesių trukmės neoperacinis gydymas.
Pacientai, kuriems naudojamas INTER FIX™ RP srieginio sujungimo prietaisas, turėjo būti taikomas ne mažiau nei šešių mėnesių trukmės neoperacinis gydymas.
• Du INTER FIX™ srieginio sujungimo prietaisai turi būti implantuojami chirurginiame lygyje greta vienas kito. Vienas INTER FIX™ srieginio sujungimo prietaisas ir
vienas INTER FIX™ RP
padeda implantuoti du prietaisus arčiau vieną kito nei įmanoma naudojant du INTER FIX™ prietaisus, taip užtikrinant mažesnį lateralinį profilį.
srieginio sujungimo prietaisas operuojamame lygyje turi būti implantuojami greta vienas kito. Išilginis INTER FIX™ RP prietaiso griovelis
• INTER FIX™ srieginio sujungimo prietaiso ir INTER FIX™ RP srieginio sujungimo prietaiso ilgoji ašis turi būti nukreipta į priekinę-užpakalinę pusę.
• Prieš naudojimą visus implantus ir instrumentus būtina sterilizuoti laikantis šiame informaciniame lapelyje pateiktų sterilizavimo nurodymų, nebent jie būtų pristatyti
sterilūs su aiškiu užrašu ant etiketės, kad jie yra sterilūs.
NEPAGEIDAUJAMAS POVEIKIS
Kaip parodyta 1 lentelėje, nepageidaujamas poveikis buvo nustatytas 185 pacientų, kuriems buvo naudojamas INTER FIX™ prietaisas ir 62 kontroliniams pacientams, užregistruotiems
daugiacentriame klinikiniame INTER FIX™ srieginio sujungimo prietaiso tyrime. Taip pat tyrimo po patvirtinimo metu nepageidaujamas poveikis buvo pastebėtas 116 pacientams po 48
mėnesių ir 109 pacientams po 72 mėnesių. Pateikti duomenys yra paremti bendru tyrime dalyvaujančių pacientų skaičiumi kiekvienu laiko momentu. Bendram dažniui nustatyti apjungiamas
nepageidaujamų reakcijų skaičius randomizuotai ir nerandomizuotai atrinktose gydymo INTER FIX™ prietaisu grupėse.
1 LENTELÐ. NEPAGEIDAUJAMOS REAKCIJOS
3 mÑn.
(t
2-<5 mÑn.)
[SkaiÉius (%)]
INTER FIX
N=185
KONTROL.
N=62
N=62
Po operacijos
(1 diena-<2 mÑn.)
[SkaiÉius (%)]
KONTROL.
INTER FIX
N=185
N=62
Operacijos metu
[SkaiÉius (%)]
KONTROL.
Komplikacija
Kraujagyslič, oper.
metu
Kryžmens
skausmas
NeurologinÑs 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Nugaros skausmas 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
IncizinÑs 11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
Stuburo 1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
UrologinÑs 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Kitos 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Kitas skausmas 4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
Skrandžio ir
žarnyno
RetrogradinÑ
ejakuliacija
KvÑpavimo 3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
Kojč skausmas 2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
Trauma 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
PilvaplÑvÑs 3(1.6) 3(1.6) 0
Kraujagyslič, po
operacijos
Kaulo lćžis 1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
Implanto
pasislinkimas /
atsilaisvinimas
Skausmas
transplanto vietoje
Nesusijungimas 4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
Smukimas 2(1.1) 2(1.1) 0
Nesusijungimas
(LAUKIAMA
PATVIRTINIMO)
Meningitas 1(1.6) 0 1(1.6)
Implanto lćžimas 5(8.1) 0 5(8.1)
VÑžys 1
Mirtis 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
INTER FIX
N=185
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
2(1.1) 3(1.6) 1(0.5) 6(3.2)
2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
1(1.6)2 0 1(1.6) 2(1.7)
(t5-<9 mÑn.)
[SkaiÉius (%)]
INTER FIX
N=185
6 mÑn.
KONTROL.
N=62
9-<19 mÑn.)
(t
[SkaiÉius (%)]
INTER FIX
N=185
(0.5)
12 mÑn.
KONTROL.
N=62
1
1 - Vertinimo praėjus 24 mėn. metu nustatyta, kad šeši (6) iš 81 vyrams, dalyvavusiems INTER FIX™ prietaiso grupėje (7,4%), pasireiškė retrogradinė ejakuliacija.
2 - Vėliau, po operacijos praėjus maždaug po 52 mėnesiams, šiems pacientams (175) buvo nustatytas sujungimas.
Nustatytos dvi svarbios nepageidaujamos reakcijos – kraujagyslių ir neurologiniai pažeidimai operacijos metu. Vertinant po 24 mėn., iš viso INTER FIX™ prietaisu gydytų pacientų grupėje buvo
pastebėta 15 su kraujagyslėmis susijusių nepageidaujamų reakcijų atvejų, kurie operacijos metu pasireiškė 14 pacientų. Pastebėtos šios nepageidaujamos reakcijos: 2 tuščiosios venos pažeidimai,
9 klubinės venos pažeidimai, 1 įplėšta hipogastrinė vena, 1 segmentinis kraujavimas iš venos, 1 sakralinės venos pažeidimas ir 1 paviršinis kraujavimas. Iš viso per tą patį laikotarpį kontrolinėje
grupėje buvo pastebėti du dviejų pacientų kraujagyslių pažeidimo operacijos metu atvejai. Pastebėti šie pažeidimai: 1 klubinės venos pažeidimas ir 1 kraujavimas iš kaulo pagrindo.
24 mėn. vertinimo metu iš viso INTER FIX™ prietaisu gydytų pacientų grupėje buvo pastebėta 30 neurologinių nepageidaujamų reakcijų atvejų, kurie operacijos metu pasireiškė 28
pacientams. Pastebėtos šios nepageidaujamos reakcijos: 1 nukarusios pėdos, 3 nervų šaknelių pažeidimų, 1 angos stenozės, 1 refleksinės simpatinės distrofijos, 7 kojų tirpimo ar šilimo,
2 dizestezijos, 3 parestezijos atvejai, 2 radikulito, 7 nugaros ir (arba) kojų skausmo, 1 nugaros ir (arba) kojų skausmo su kitais simptomais, 1 dieglių apatinėje nugaros dalyje ir 1 karpalinio
tunelio sindromo atvejis.
Iš viso per tą patį laikotarpį kontrolinėje grupėje buvo pastebėta 10 neurologinių nepageidaujamų reakcijų atvejų, pasireiškusių 8 pacientams. Pastebėtos šios nepageidaujamos reakcijos: 1
radikulopatijos su galūnių dilgčiojimu, 1 lėtinio nugaros skausmo su radikulitu, 1 silpnėjančio paskirstymo simptomų atvejis, 2 nugaros ir kojų skausmų su kitais simptomais atvejai, 1 didžiojo
atitraukiamojo raumens denervacijos ir 4 atvejai su kojų tirpimu arba šilimu.
10 pacientų. Pastebėtos šios nepageidaujamos reakcijos: 1 radikulopatijos, 2 kaklo radikulopatijos, 1 rankų tirpimo ir dilgčiojimo, 1 kojų tirpimo ar degimo, 3 nugaros ir (arba) kojų skausmo ir
Tyrimo po patvirtinimo metu buvo pastebėta dar 10 neurologinių nepageidaujamų reakcijų atvejų, pasireiškusių
24 mÑn.
(t19-<30 mÑn.)
[SkaiÉius (%)]
INTER FIX
N=185
(0.5)
KONTROL.
N=62
2 (1.1) 1(0.8) 1(0.9)
BENDRAS
NEPAGEIDAUJAMČ
REAKCIJČ SKAIÈIUS
(PER 24 MÐN.)
KONTROL.
INTER FIX
N=185
(7.4)
N=62
1
48 mÑn.
(>30 - < 60
(t
30<
mÑn.)
INTER FIX
N=116
0
72 mÑn.
60
(t60
mÑn.)
INTER FIX
N=109
Page 67

2 nugaros ir (arba) kojų skausmo su kitais simptomais. Nepageidaujamų reakcijų lentelėje taip pat pateiktos nepageidaujamas pove
iki
iki
stuburui, pvz., disko tarpo mažėjimas, kuris kai kuriais atvejais turėjo neurologinių pasekmių.
Dėl kai kurių neurologinių reakcijų atsiradimo po klinikinio tyrimo operacijos prireikė chirurginės intervencijos. Šią papildomą chirurginę intervenciją galima suskirstyti į revizines, išėmimo,
papildomo fiksavimo ir pakartotines operacijas
ir kontrolinių pacientų grupėse, atliekant IDE tyrimą ir INTER FIX™ prietaisu gydytų pacientų grupėje tyrimo po patvirtinimo metu.
1
Revizija: Procedūra, kurios metu koreguojama arba kaip nors modifikuojama pradinė implanto konfigūracija.
Išėmimas: Procedūra, kurios metu išimamas vienas ar keli pradinės implanto konfigūracijos elementai nekeičiant jų to paties tipo bandomuoju prietaisu.
Papildomas fiksavimas: Procedūra, kurios metu įtaisomi papildomi stuburo prietaisai, nepatvirtinti pagal protokolą.
Pakartotinė operacija: Bet kokia chirurginė procedūra, kurios metu nėra išimami, modifikuojami ar papildomai įtaisomi jokie pradinio implanto elementai.
2 lentelÑ. AntrinÑs chirurginÑs procedćros
IDE tyrimas Tyrimas po patvirtinimo
Per 24 mÑn. laiko
momentÅ
INTER FIX™
prietaisas
N=185
1
. 2 lentelėje apibendrintos antrinės chirurginės intervencijos INTER FIX™ prietaisu gydytų (randomizuotai ir nerandomizuotai atrinktų) pacientų
Kontrol.1
N=62
48 mÑn.
laio momentas
INTER FIX™
prietaisas
N=116
72 mÑn. laiko
momentas
INTER FIX™
prietaisas
N=109
s, susijęs su kojų ir nugarų skausmu ir (arba) pove
Revizijos 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
IšÑmimas 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Papildomas
fiksavimas
PakartotinÑs
operacijos
1 - Kadangi kontrolinių pacientų stebėti po 24 mėnesių nereikėjo, informacijos apie visus šiuos pacientus po 72 mėnesių nėra.
GALIMAS NEPAGEIDAUJAMAS POVEIKIS
Toliau pateiktas galimų nepageidaujamų reakcijų, kurios gali pasireikšti atliekant stuburo sujungimo operacijas naudojant INTER FIX™ srieginio sujungimo prietaisą arba INTER FIX™ RP
srieginio sujungimo prietaisą. Kai kurios iš jau buvo nurodytos nepageidaujamų reakcijų lentelėje.
• Išsiardo, sulinksta, lūžta, atsilaisvina ir (arba) pasislenka elementai.
• (Alerginė) reakcija į svetimkūnius.
• Audinių arba nervų pažeidimas.
• Po operacijos pasikeičia stuburo išlinkimas, prarandama ir (arba) sumažėja korekcija, aukštis.
• Infekcija.
• Kietojo smegenų dangalo plyšimai.
• Neurologinės sistemos pakenkimai.
• Urologinės sistemos pakenkimai.
• Randų formavimasis.
• Kaulo lūžis.
• Nesusijungimas (arba pseudoartrozė), uždelstas susijungimas, netaisyklingas susijungimas.
• Sustojęs bet koks galimas operuotos stuburo dalies augimas. Stuburo judrumo ar funkcionavimo netekimas.
• Komplikacijos transplanto donoro vietoje.
• Kraujagyslių pažeidimai ir širdies bei kraujagyslių sistemos pakenkimai.
• Skrandžio ir žarnyno komplikacijos.
• Reprodukcijos sistemos pakenkimai.
• Vidaus organų ir jungiamųjų audinių pažeidimai.
• Dažnesnės kvėpavimo takų problemos.
• Incizinės komplikacijos.
• Psichinės būklės pokyčiai.
• Mirtis.
Pastaba Kai kuriems iš šių galim
KLINIKINIAI REZULTATAI
JAV buvo atliktas daugiacentris INTER FIX™ srieginio sujungimo prietaiso atitikimo2 klinikinis tyrimas, kurio metu buvo palygintas autogeniniu kaulu užpildyto INTER FIX™ prietaiso naudojimas
iš priekinės stuburo pusės ir autogeniniu kaulu užpildyto šlaunies žiedo alotransplanto (kontrolinio) naudojimas gydant degeneracine disko liga sergančius pacientus. Iš pradžių klinikinis
tyrimas buvo perspektyvinio, randomizuoto, kontroliuojamo pobūdžio. Vėliau tyrimo planas buvo peržiūrėtas leidžiant pacientams, t.y., tik INTER FIX™ prietaisu gydytiems pacientams,
dalyvauti nerandomizuotoje atšakoje.
Vieni iš įtraukimo į klinikinį tyrimą kriterijų buvo simptomatinė degeneracinė disko liga, kuri pasireiškė by nenumalšinamas kojų ir (arba) nugaros skausmas su pozityviais diagnostinio vizualizavimo
rezultatais; didesnis nei 4 mm pasislinkimo stuburo nestabilumas arba didesnis nei 5° kampas galūnių / ekstenzijos rentgenogramose; vieno lygio simptomatinis pakenkimas nuo L2-S1 ir ne
didesnė nei 1 laipsnio spondilolistezė. Į tyrimą nebuvo įtraukti pacientai: kuriems anksčiau buvo atlikta priekinio slankstelių kūnų sujungimo procedūra pakenktame stuburo lygyje, kurie sirgo
ostopenija, osteoporoze arba osteomaliacija arba prireikė kaulų augimo stimuliavimo.
Iš viso INTER FIX™ prietaisu gydytų pacientų tyrimo grupėje buvo užregistruoti 185 pacientai.3 Iš viso randomizuotoje klinikinio trimo atšakoje dalyvavo 62 kontroliniai pacientai.
INTER FIX™ srieginio sujungimo prietaisui buvo taikomas šis reikalavimas po patvirtinimo – įvertinti ilgalaikį prietaiso veikimą, gaunant iš viso 6 metų pooperacinio laikotarpio duomenis
apie 100 pacientų. Ilgalaikis prietaiso veikimas buvo vertinamas tiriant 116 pacientų praėjus 4 metams po operacijos ir 109 pacientus praėjus 6 metams po operacijos. Pacientų populiacija
buvo IDE klinikinio tyrimo dalyviai. Remiantis IDE tyrimo kriterijais, apie šiuos pacientus gauta informacija apėmė visų papildomų chirurginių procedūrų aprašymą, radiografinį sujungimo
vertinimą ir skausmo bei funkcijų vertinimą.
3 lentelėje pateikti klinikiniai tyrimo rezultatai, aprašantys sujungimo, skausmo (Oswestry) ir neurologinius rezultatus praėjus 12, 24, 48 ir 72 mėnesiams po operacijos. Šio klinikinio tyrimo
sėkmė buvo vertinama remiantis INTER FIX™ prietaisu gydytų ir kontrolinių pacientų atitikimo analize.
Šiame klinikiniame tyrime sujungimas buvo apibrėžtas kaip aiškus trabekulinio kaulo sujungimas, pasislinkimo (<3 mm) ir kampinio judėjimo (<5°) stabilumas, ir nepralaidumas spindulinei
energijai aplink daugiau nei 50% kiekvieno implanto dalį. Taip pat skaičiavimuose pacientai, kuriems buvo atliktos antrinės operacijos, buvo priskiriami nepavykusio sujungimo atvejams.
Oswestry skausmo / negalios sėkmė buvo apibrėžta kaip pooperacinės būklės pagerėjimas ne mažiau nei 15 balų palyginti su būkle iki operacijos. Neurologin
kaip ne mažiau nei 3 iš 4 neurologinių kategorijų (judėjimo, jutimo, refleksų ir tiesios kojos kėlimo) išlaikymas ar pagerinimas po operacijos palyginti su būkle iki operacijos. Bendrą sėkmę
rodo paciento demonstruojamas sėkmingas sujungimas, Oswestry skausmo / negalios sėkmė ir neurologinės būklės sėkmė bei nė vienos antrinės chirurginės procedūros, priskirtos prie
„nesėkmių“, nebuvimas.
2
INTER FIX™ prietaisas statistiškai nėra blogesnis už kontrolinį.
3
Neįeina 3 pacientai, kuriems dėl su operacija susijusio nepageidaujamo poveikio gydymas INTER FIX™ prietaisu nebuvo taikomas.
Bendri rezultatai praėjus 12 ir 24 mėnesiams po operacijos taip pat pateikti 3 lentelėje.
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
ų nepageidaujamų poveikių atitaisyti gali prireikti papildomos operacijos.
ės būklės sėkmė buvo apibrėžta
s
Page 68

s
s
ė
o
™
i
a
n
u
a
ų
ą
Sujungimas 96.1%
Oswestry
skausmo /
negalios bćklÑs
gerinimas
Pacientai, kurič
bćklÑ lyginant su
bćkle iki operacijos
pagerÑjo ne
mažiau nei 15 balč
NeurologinÑs
bćklÑs išlaikymas
ar pagerinimas
Bendra sÑkmÑ 42.4%
12 mÑn. dažnis 24 mÑn. dažnis
Tyrimas Kontrol.
(122/127)
53.5%
(76/142)
99.3%
(150/151)
(61/144)
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
(12/56)
Delta
1
(ǻ
)
Tyrimas Kontrol.
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
-0.097 46.1%
(71/154)
(27/52)
(24/46)
(43/45)
(17/57)
Delta
(ǻ1)
51.9%
52.2%
95.6%
29.8%
-0.043 49.5%
48 mÑn.
dažnis
Tyrimas
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
(53/107)
72 mÑn.
dažnis
Tyrimas
97.6% (82/84)
56.3%
(54/96)
1 - Minimali ne blogesnio veikimo riba, rodanti, kad tiriamas produktas nėra blogesnis už kontrolinį.
3 lentelÑ. INTER FIX™ prietaiso tyrimo klinikiniai rezultatai
Visi INTER FIX™ srieginio sujungimo prietaiso klinikiniame tyrime dalyvavę pacientai, nepriklausomai nuo gydymo grupės, buvo įtraukti pagal tuos pačius įtraukimo arba neįtraukimo kriteriju
Randomizuotai atrinktų INTER FIX™ prietaisu gydytų pacientų ir nerandomizuotai atrinktų INTER FIX™ prietaisu gydytų pacientų lyginamumui pagrįsti buvo ištirtos demografinės savybė
priešoperacinė medicininė būklė, degeneracinei disko ligai būdingi diagnostinio vizualizavimo rezultatai, operavimo metodas ir gydytos juosmeninės dalies lygis. Statistiškai gyventojai buv
lygintini, išskyrus šiuos kintamuosius: operavimo metodas, profesinis statusas prieš op eraciją ir priešoperacinio slankstelių galinių plokštelių osteofitų formavimosi diagnostinio vizualizavim
rezultatai; skaidulinio žiedo, geltonojo raiščio ir (arba) faceto sąnario kapsulės randų formavimasis ir (arba) storėjimas bei minkštimin io branduolio išvarža. Tolesnės statistinės analizės parod
kad šie kintamieji nėra susiję su 12 mėn. klinikiniais sujungimo, Oswestry skausmo / negalios būklės gerinimo, neurologinės būklės arba bendros sėkmės rezultatų skirtumais, išskyrus minkštimini
branduolio išvaržą. Minkštiminio branduolio išvarža buvo aptikta daugiausiai nerandomizuotai atrinktos grupės pacientams; buvo nustatyta, kad tai didina bendros sėkmės tikimybę.
Buvo atlikta „ketintų gydyti“ pacientų analizė. Šioje analizėje nesėkmingos antrinės operacijos, mirtys, galimybės stebėti praradimas ir stebėjimo trūkumas dėl kitų priežasčių sąlygojo kintamųj
stebėjimo trūkumą, todėl buvo priskirti prie nustatomo dažnio rodiklių, t.y., laikomi „nesėkmėmis“. Priskiriant šiuos pacientus prie nesėkmingo gydymo atvejų, ketintų gydyti pacientų analizėj
klinikinių pasekmių dažnis buvo daug mažesnis nei užfiksuotas faktiniuose stebėtuose klinikiniuose duomenyse. Toliau pateiktoje lentelėje (4 lentelė) pateikti ketintų gydyti INTER FIX
prietaisu pacientu analizės rezultatai, tačiau neįtraukti kontrolinių pacientų rezultatai.
Mirtys, nesÑkmingos antrinÑs operacijos, galimybÑs stebÑti praradimas ir stebÑjimo trćkumas laikomi
12 mÑn. dažnis 24 mÑn.
Sujungimas 65.9% (122/185) 67.6%
Oswestry skausmo / negalios bćklÑs
gerinimas
Pacientai, kurič bćklÑ lyginant su bćkle iki operacijos
pagerÑjo ne mažiau nei 15 balč
NeurologinÑs bćklÑs išlaikymas ar
pagerinimas
Bendra sÑkmÑ 33.0% (61/185) 38.4% (71/185) 45.7%
NesÑkmingos antrinÑs operacijos
Nesusijungimas
2
Kita
Mirtys 0 1 0 0
1
4 lentelÑ. Ketintč gydyti INTER FIX™ prietaisu pacientč analizÑ
nesÑkmÑmis ir yra priskiriami prie dažnio rodiklič
dažnis
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
5
8
(125/185)
(128/185)
48 mÑn.
dažnis
80.2% (93/116) 77.1%
(66/116)
87.9%
(102/116)
(53 /116)
5
3
4
4
72 mÑn.
dažnis
( 84/109)
56.0%
(61 /109)
75.2%
(82/109)
49.5%
(54/109)
1
2
1 - Šie pacientai yra įtraukti į sujungimo dažnio skaičiavimus, tačiau kitais atžvilgiais klinikinio tyrimo tikslais yra priskiriami prie nesėkmių.
2 - Pacientai, kurie tuo laikotarpiu turi būti stebimi ir kuriems dėl kitų nei nesusijungimas priežasčių buvo atliktos antrinės operacijos, klinikinio tyrimo tikslais yra priskiriami prie nesėkmių.
PAK UOTĖ
Visi elementai turi būti pristatyti nepažeistose pakuotėse. Jei naudojama paskolos ar mokėjimo už prekes, kai jos parduotos, sistema, būtina patikrinti, ar komplekte yra visi elementai,
visus elementus, taip pat ir instrumentus, prieš naudojimą reikia kruopščiai patikrinti, ar jie nepažeisti. Draudžiama naudoti pažeistas pakuotes ar gaminius. Juos reikia grąžinti į Medtron
Sofamor Danek.
VALYMAS IR NUKENKSMINIMAS
Visus implantus ir instrumentus, jei jie nėra pateikti sterilūs, prieš sterilizuojant ir perkeliant į sterilią operacinę patalpą reikia nuvalyti laikantis nustatytos ligoninės tvarkos. Prieš pakartotin
naudojant naudotus instrumentus būtina dezinfekuoti, nuvalyti ir sterilizuoti.
Pastaba: Tirpalų, kurių sudėtyje yra balinančių medžiagų arba formalino, naudoti negalima, nes jie gali pakenkti prietaisui.
Su visais gaminiais reikia elgtis atsargiai. Netinkamai naudojamas ar prižiūrimas įtaisas gali sugesti ir gali pradėti neteisingai funkcionuoti.
STERILIZAVIMAS
Prieš naudojimą visus chirurginius implantus ir instrumentus būtina sterilizuoti ligoninėje, nebent jie būtų pristatyti sterilūs sandarioje sterilioje gamintojo pakuotėje su aiškiu užrašu a
etiketės, kad jie yra sterilūs. Prieš sterilizavimą nuimkite visas įpakavimo medžiagas. Operacinėje patalpoje gali būti tik sterilūs produktai. Pagal 10-6 sterilumo užtikrinimo lygį šiuos produktu
rekomenduojama sterilizuoti garu ligoninėje, taikant vieną iš trijų toliau pateikiamų proceso parametrų:
BŪDAS CIKLAS TEMPERATŪRA POVEIKIO TRUKMĖ
Garas Ikivakuuminis 270°F (132°C) 4 minutės
Garas Gravitacinis 250°F (121°C) 60 minučių
Garas* Gravitacinis* 273°F (134°C)* 20 minučių*
PASTABA Kadangi sterilizacijai taikomi įvairūs parametrai, kiekviena medicininė įstaiga turi sutikrinti ir patvirtinti savo įrangos sterilizacijos procesą (pvz., temperatūrą, laiką).
*Naudojant už JAV ribų kai kurios ne JAV sveikatos apsaugos institucijos rekomenduoja sterilizuoti taikant šiuos parametrus, ypač chirurginiams instrumentams, kurie gali susiliesti su centrin
nervų sistema, kad kiek įmanoma sumažintų galimą Kroicfeldo - Jakobo ligos pernešimo pavojų.
NUSISKUNDIMAI GAMINIU
Bet koks sveikatos priežiūros specialistas (pvz., šių gaminių pirkėjas ar vartotojas), turintis kokių nors nusiskundimų ar nepatenkintas gaminio kokybe, identiškumu, patvarumu, patikimum
saugumu, veiksmingumu ir (arba) eksploatacinėmis savybėmis, privalo apie tai pranešti gamintojui arba Medtronic Sofamor Danek. Be to, jei koks nors implantuotas INTER FIX™ sriegini
sujungimo prietaisas arba INTER FIX™ RP srieginio sujungimo prietaisas „blogai veikia“ (t.y., neatitinka eksploatacinių charakteristikų ar veikia ne taip, kaip turėtų) ar įtariama, kad jis „blog
veikia“, būtina nedelsiant apie tai pranešti tiekėjui. Jei koks nors Medtronic Sofamor Danek gaminys kada nors „blogai dirbo“ ir galėjo sukelti ar prisidėjo prie paciento mirties ar sunkių sužalojim
būtina nedelsiant telefonu, faksu ar laišku pranešti apie tai tiekėjui. Kai rašote skundą, nurodykite elemento(-ų) pavadinimą, detalės numerį, partijos numerį(-ius), savo pavardę ir adres
nusiskundimo pobūdį ir pranešimą, ar reikalaujamas rašytinis aktas tiekėjui.
PASTABA GYDYTOJUI Nors tarp kompanijos ir paciento yra išmanantis gydytojas, tačiau šiame dokumente pateikiamą svarbią medicininę informaciją būtina perteikti pacientui.
TAIKOMA TIK JAV
ĮSPĖJIMAS: FEDERALINIS (JAV) ĮSTATYMAS RIBOJA ŠIŲ ĮTAI SŲ PARDAVIMĄ TIK GYDYTOJAMS AR PAGAL GYDYTOJŲ UŽSAKYMĄ.
PRIETAISO IŠĖMIMAS
Prireikus išimti INTER FIX™ srieginio sujungimo prietaisą arba INTER FIX™ RP srieginio sujungimo prietaisą, kreipkitės į Medtronic Sofamor Danek.
© 2006 Medtronic Sofamor Danek. Visos teisės saugomos.
Page 69

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
POLSKI
Poniżej przedstawiono istotne informacje medyczne dotyczące gwintowanego urządzenia do zespalania INTER FIX™ oraz gwintowanego urządzenia do zespalania INTER FIX™ RP.
OPIS:
Gwintowane urządzenie do zespalania INTER FIX™ składa się z pustego perforowanego metalowego cylindra oraz z nasadki końcowej. Urządzenie do zespalania INTER FIX™ RP składa
się z pustego perforowanego metalowego cylindra z dużym pojedynczym zewnętrznym zaokrąglonym wyżłobieniem wzdłuż całej osi długiej, które ciągnie się do wewnętrznego obwodu
urządzenia. Oba końce implantu INTER FIX™ RP są zamknięte.
Dostępne są implanty INTER FIX™ oraz INTER FIX™ RP o średnicach od 12 do 24mm oraz o długości od 20 do 29mm. Nasadki końcowe implantów INTER FIX™ są dopasowane rozmiarem
do średnicy cylindrów i stosowane są na otwartym końcu cylindrów, po wypełnieniu ich autogennym przeszczepem kostnym.
Implanty INTER FIX™ oraz INTER FIX™ RP wykonane są ze stopu tytanu, używanego do wytwarzania implantów (Ti-6Al-4V), zgodnego ze standardem ASTM F136.
Rękojmia dotycząca przydatności do określonego celu lub sposobu użycia jest wyraźnie wykluczona. Szczegółowe informacje dotyczące warunków gwarancji oraz zobowiązań finansowych
– patrz katalog lub cennik Medtronic Sofamor Danek.
WSKAZANIA:
Gwintowane urządzenie do zespalania INTER FIX™ oraz gwintowane urządzenie do zespalania INTER FIX™ RP przeznaczone są do zabiegów zespalania kręgosłupa u chorych z dojrzałym
układem kostnym ze zwyrodnieniem krążków międzykręgowych (DDD) na poziomie od L2 do S1. Zwyrodnienie krążków międzykręgowych zdefiniowane jest jako ból pleców o podłożu
dyskopatii z degeneracją krążka międzykręgowego, potwierdzone przez wywiad oraz badania radiologiczne. Pacjenci ze zwyrodnieniem krążków międzykręgowych mogą także mieć na
tym samym poziomie kręgozmyk lub tyłozmyk I stopnia. Implanty INTER FIX™ oraz INTER FIX™ RP przeznaczone są do stosowania z autogennym przeszczepem kostnym i do otwartej
implantacji z dostępu przedniego.
PRZECIWWSKAZANIA:
Gwintowane urządzenie do zespalania INTER FIX™ oraz gwintowane urządzenie do zespalania INTER FIX™ RP nie powinny być implantowane u chorych z czynnym procesem infekcyjnym,
toczącym się w miejscu zabiegu operacyjnego oraz u chorych z uczuleniem na tytan lub stop tytanu.
OSTRZEŻENIA:
• Gwintowane urządzenie do zespalania INTER FIX™ oraz gwintowane urządzenie do zespalania INTER FIX™ RP powinny być stosowane wyłącznie przez chirurgów doświadczonych
w zabiegach zespalania kręgosłupa i takich, którzy przeszli odpowiednie szkolenia odnośnie użycia niniejszego urządzenia.
• Brak odpowiedniego doświadczenie i/lub wyszkolenia może prowadzić do zwiększenia częstości występowania zdarzeń niepożądanych, takich jak uszkodzenia struktur naczyniowych,
powikłań neurologicznych i/lub z zakresu układu moczowo-płciowego (obejmujących wsteczną ejakulację).
GWINTOWANE URZĄDZENIE DO ZESPALANIA INTER FIX™
GWINTOWANE URZĄDZENIE DO ZESPALANIA INTER FIX™ RP
ISTOTNE INFORMACJE MEDYCZNE
Page 70

ŚRODKI OSTROŻNOŚCI:
N
• Bezpieczeństwo i skuteczność gwintowanego urządzenia do zespalania INTER FIX™ oraz gwintowanego urządzenia do zespalania INTER FIX™ RP nie zostały ustalone u chorych z
którymkolwiek z podanych poniżej obciążeń:
- kręgozmyk lub tyłozmyk II lub wyższego stopnia;
- planowane wykonanie zespolenia na więcej niż jednym poziomie;
- zabieg rewizyjny uprzedniej(ych) procedur(y) zespolenia międzytrzonowego;
- w okresie pooperacyjnym zapotrzebowanie na steroidy lub niesteroidowe leki przeciwzapalne;
- znaczna otyłość;
- wiek poniżej 18 lub powyżej 65 roku życia;
- osteoporoza, osteopenia i/lub osteomalacja;
- ciąża.
• Bezpieczeństwo i skuteczność gwintowanego urządzenia do zespalania INTER FIX™ oraz gwintowanego urządzenia do zespalania INTER FIX™ RP potwierdzone zostały wyłącznie
w zabiegu międzytrzonowego zespalania kręgosłupa w odcinku lędźwiowym z dostępu przedniego - (ALIF) przy użyciu autogennego przeszczepu kostnego.
• Pacjenci, u których stosowane jest gwintowane urządzenie do zespalania INTER FIX™, powinni być leczeni nieoperacyjnie przez conajmniej sześć miesięcy. Pacjenci, u których
stosowane jest gwintowane urządzenie do zespalania INTER FIX™ RP, powinni także być leczeni nieoperacyjnie przez conajmniej sześć miesięcy.
• Na poziomie na którym wykonywany jest zabieg operacyjny implantowane powinny być dwa gwintowane urządzenia do zespalania INTER FIX™ po obu stronach. Na operowanym
poziomie, po obu stronach, musi być wszczepione jedno gwintowane urządzenie do zespalania INTER FIX™ oraz jedno gwintowane urządzenie do zespalania INTER FIX™ RP
Podłużne wyżłobienie urządzenia INTER FIX™ RP ułatwia implantację dwóch urządzeń w bliższym sąsiedztwie, niż to na które pozwala zastosowanie dwóch urządzeń INTER FIX™,
powodując tym samym redukcję profilu bocznego.
.
• Oś długa gwintowanych urządzeń do zespalania INTER FIX™ oraz gwintowanych urządzeń do zespalania INTER FIX™ RP powinna przebiegać w kierunku przednio - tylnym.
• Implanty i urządzenia muszą przed użyciem zostać wyjałowione, zgodnie z zawartymi w niniejszym opakowaniu instrukcjami, z wyjątkiem urządzeń dostarczonych i wyraźnie
oznaczonych jako sterylne.
EFEKTY UBOCZNE:
Działania niepożądane, przedstawione w tabeli I, odnotowane zostały w wieloośrodkowym badaniu klinicznym dotyczącym zastosowania gwintowanego urządzenia INTER FIX™, w którym
do grupy chorych leczonych przy pomocy urządzenia INTER FIX™ włączono 185 pacjentów a do grupy kontrolnej 62 pacjentów. Efekty uboczne wystąpiły ponadto w ciągu 48 miesięcy u 116
pacjentów, a w ciągu 72 miesięcy u 109 pacjentów biorących udział w badaniu porejestracyjnym. Przedstawione wskaźniki obliczone są na podstawie całkowitej liczby pacjentów włączonych
do badania w poszczególnym przedziale czasowym. W celu przedstawienia całkowitego wskaźnika skumulowano działania niepożądane, które wystąpiły zarówno w grupie pacjentów
zakwalifikowanych w drodze randomizacji, jak również tych, którzy zakwalifikowani zostali bez randomizacji do leczenia przy pomocy urządzenia INTER FIX™.
TABELA I – DZIAèANIA NIEPOđÄDANE
A
3 miesiÅce
(t
miesiÓcy)
[Liczba (%)]
INTER FIX
N=185
2-<5
KONTROL
GRUPA
N=62
Pooperacyjne
(1 dzieë-<2
A
miesiÓcy)
[Liczba (%)]
INTER FIX
N=185
GRUPA
KONTROLN
N=62
üródoperacyjne
[Liczba (%)]
Powikéanie
üródoperacyjne
naczyniowe
Dolegliwoýci
bólowe w okolicy
krzyĒowobiodrowej
Neurologiczne 6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
Ból pleców 1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
DotyczÅce rany
pooperacyjnej
Zdarzenia zwiÅzane
z krÓgoséupem
urologiczne 1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
Inne 2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
Inne dolegliwoýci
bólowe
đoéÅdkowo jelitowe
Wsteczna
ejakulacja
Oddechowe 3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
Ból koëczyn
dolnych
Uraz 2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
Otrzewnowe 3(1.6) 3(1.6) 0
Pooperacyjne
naczyniowe
Zéamania kostne 1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
Przemieszczenie/o
bluzowanie
implantu
Dolegliwoýci
bólowe dotyczÅce
miejsca pobrania
przeszczepu
kostnego
Brak zrostu 4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
Osiadanie 2(1.1) 2(1.1) 0
Brak zrostu
(SKUTEK
NIEROZSTRZYGNI
ÒTY).
Zapalenie opon
mózgowo rdzeniowych
Zéam an ie implant u 5(8.1) 0 5( 8. 1)
Nowotwór 1
INTER FIX
GRUPA
KONTROL
N=185
N=62
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
2(1.1) 3(1.6) 1(0.5) 6(3.2)
2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
1(1.6)
1(1.6) 0 1(1.6)
Zgon 1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
6 miesiÓcy
(t
5-<9
miesiÓcy)
[Liczba (%)]
GRUPA
INTER FIX
KONTROLN
N=185
NA
N=62
12 miesiÓcy
(t
9-<19
miesiÓcy)
[Liczba (%)]
N=185
GRUPA
KONTROLN
N=62
INTER FIX
A
2
0 1(1.6) 2(1.7)
(0.5)
24 miesiÅce
(t
19-< 30
miesiÓcy)
[Liczba (%)]
INTER FIX
KONTROLN
N=185
A
1
(0.5)
GRUPA
A
N=62
CAèKOWITA
LICZBA ZDARZEê
NIEPOđÄDANYCH
(W CZASIE 24
MIESIÒCY)
INTER FIX
KONTROLNA
N=185
1
(7.4)
48 miesiÅc
(t
60
30<
(>30 - < 60
N=62
miesiÓcy)
INTER FIX
N=116 INTER FIX
GRUPA
0
72
miesiÅc
(t
60
miesiÓcy)
N=109
2 (1.1) 1(0.8) 1(0.9)
1 - Wsteczna ejakulacja wystąpiła w ciągu 24 miesięcy obserwacji u sześciu (6) z 81 mężczyzn włączonych do grupy urządzenia INTER FIX™ (7,4%).
2 - U pacjenta tego (175) odnotowano później zrost po upływie około 52 miesięcy od zabiegu operacyjnego, chory ten nie wymagał dodatkowych zabiegów operacyjnych.
Dwa najpoważniejsze zdarzenia niepożądane zaobserwowane w czasie badania klinicznego to uszkodzenia naczyń oraz uszkodzenia neurologiczne. W sumie u 14 pacjentów z grupy chorych
zakwalifikowanych do leczenia przy pomocy urządzenia INTER FIX™ wystąpiło 15 śródoperacyjnych powikłań dotyczących układu naczyniowego, w ciągu 24-miesięcznego okresu obserwacji.
Powikłania te obejmowały: 2 urazy żyły próżnej, 9 urazów żyły biodrowej, 1 rozerwanie żyły podbrzusznej, 1 krwawienie z segmentarnego naczynia żylnego, 1 uraz nerwu krzyżowego oraz
1 powierzchowne krwawienie. U 2 chorych w grupie kontrolnej wystąpiły w tym samym okresie czasu w sumie 2 śródoperacyjne urazy naczyniowe. Obejmowały one: 1 uraz żyły biodrowej
oraz 1 krwawienie z łożyska kostnego.
W sumie u 28 pacjentów z grupy chorych zakwalifikowanych do leczenia przy pomocy urządzenia INTER FIX™ wystąpiło 30 powikłań neurologicznych, w ciągu 24-miesięcznego okresu
obserwacji. Powikłania te obejmowały: 1 przypadek opadania stopy
dystrofii współczulnej, 7 przypadków drętwienia i ocieplenia kończyn dolnych, 2 dysestezje, 3 parestezje, 2 przypadki zapalenia korzeni nerwów, 7 przypadków wystąpienia dolegliwości
bólowych pleców i/lub kończyn dolnych, 1 przypadek dolegliwości bólowych placów i/lub kończyn dolnych wraz z innymi objawami, 1 przypadek przeszywającego bólu w dolnej części pleców
oraz 1 przypadek wystąpienia zespołu cieśni nadgarstka.
W grupie kontrolnej, w tym samy m czasie u 8 pacjentów wystąpiło w sumie 10 powikłań neurologicznych. Powikłania te obejmowały: 1 przypadek radikulopatii z mrowieniami kończyn, 1
przypadek przewlekłych dolegliwości bólowych z zapaleniem korzeni nerwów
dolnych z innymi objawami, 1 odnerwienie mięśnia przywodziciela wielkiego, 4 przypadki drętwienia lub ocieplenia kończyn dolnych. W czasie badania porejestracyjnego odnotowano dodatkowo
, 3 przypadki uszkodzenia korzeni nerwów, 1 przypadek zwężenia otworu międzykręgowego, 1 przypadek odruchowej
, 1 przypadek wystąpienia upośledzających objawów dystrybucyjnych, 2 dolegliwości bólowe pleców oraz kończyn
Page 71

10 neurologicznych zdarzeń niepożądanych u 10 pacjentów. Powikłania te obejmowały: 1 przypadek radikulopatii, 2 przypadki radikulopatii szyjnej, 1 przypadek drętwienia i mrowienia rąk,
1 przypadek drętwienia i pieczenia kończyn dolnych, 3 przypadki dolegliwości bólowych pleców i/lub kończyn dolnych oraz 2 przypadki dolegliwości bólowych pleców i/lub kończyn dolnych
z innymi objawami. Dodatkowo, w tabeli dotyczącej zdarzeń niepożądanych, przedstawiono zdarzenia niepożądane związane z dolegliwościami bólowymi kończyn dolnych i pleców i/lub
zdarzenia związane z kręgosłupem, takie jak zwężenie przestrzeni międzykręgowej, które w niektórych wypadkach posiadały komponentę neurologiczną.
Niektóre z tych zdarzeń niepożądanych prowadziły do interwencji chirurgicznych, stanowiących następstwo operacji związanych z badaniem klinicznym. Dodatkowe interwencje chirurgiczne
sklasyfikowane zostały jako zabiegi rewizji, usunięcia, zainstalowania wspomagającej fiksacji lub reoperacje
przeprowadzonych w grupie pacjentów u których zastosowano urządzenie INTER FIX™ (połączone grupy pacjentów zakwalifikowanych w drodze randomizacji jak również niezrandomizowanych)
oraz w grupie kontrolnej, w badaniu IDE, jak również w grupie pacjentów u których zastosowano urządzenie INTER FIX™ w badaniu porejestracyjnym.
1
Zabieg rewizyjny: Zabieg korekcyjny lub jakikolwiek inny, w wyniku którego modyfikowana jest pierwotna konfiguracja implantu.
Zabiegi usunięcia: Zabieg, w którym usuwany jest jeden lub więcej komponentów wchodzących w skład konfiguracji pierwotnej implantu, bez zastępowania ich innymi komponentami
tego samego, analizowanego w badaniu, urządzenia.
Instalacja wspomagającej fiksacji: Zabieg, w którym umieszczane są dodatkowe, niezgodne z protokołem badania, urządzenia.
Reoperacje: Każdy zabieg operacyjny, w którym nie usuwa się, nie modyfikuje lub nie wszczepia się żadnego dodatkowego implantu.
1
. W tabeli II przedstawiono podsumowanie powtórnych interwencji chirurgicznych,
Tabela II – Powtórne zabiegi operacyjne
UrzÅdzenie
Zabiegi
Badanie IDE Badanie porejestracyjne
Po upéywie 24
INTER FIX™
N=185
miesiÓcy
Grupa
kontrolna
N=62
1
Po upéywie
48 miesiÓcy
UrzÅdzenie
INTER FIX™
N=116
Po upéywie 72
miesiÓcy
UrzÅdzenie
INTER FIX™
N=109
3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
rewizyjne
Zabiegi
2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
usuniÓcia
Instalacja
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
wspomagajÅcej
fiksacji
Reoperacje 42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
1 - Brak informacji dotyczących wszystkich pacjentów, po upływie 72 miesięcy, z uwagi na to, że pacjenci nie byli zobowiązani do uczestniczenia w wizytach kontrolnych po upływie 24 miesięcy.
POTENCJALNE DZIAŁANIA NIEPOŻĄDANE:
Poniżej podano listę potencjalnych działań niepożądanych, które mogą wystąpić w związku z zabiegami zespalania kręgosłupa przy użyciu gwintowanego urządzenia do zespalania INTER
FIX™ lub gwintowanego urządzenia do zespalania INTER FIX™ RP. Niektóre z tych działań niepożądanych mogły być uprzednio wymienione w tabeli dotyczącej zdarzeń niepożądanych.
• Rozłożenie na części, wygięcie, złamanie, obluzowanie i/lub przemieszczenie się komponentów.
• Reakcja na ciało obce (alergiczna).
• Uszkodzenie nerwu lub tkanek.
• Pooperacyjne zmiany krzywizn kręgosłupa, brak korekcji i/lub ograniczenie wzrostu.
• Infekcja.
• Uszkodzenie opony twardej.
• Powikłania ze strony układu nerwowego.
• Powikłania ze strony układu moczowego.
• Tworzenie blizn.
• Złamania kostne.
• Brak zrostu (lub staw rzekomy), zrost opóźniony, nieprawidłowy zrost.
• Zahamowanie potencjalnego wzrostu dotyczące operowanego odcinka kręgosłupa. Utrata ruchomości lub funkcji kręgosłupa.
• Powikłania dotycz
• Uszkodzenie naczyń krwionośnych oraz powikłania ze strony układu sercowo – naczyniowego.
• Powikłania żołądkowo - jelitowe.
• Powikłania ze strony układu rozrodczego.
• Uszkodzenie narządów wewnętrznych i tkanki łącznej.
• Rozwój powikłań ze strony układu oddechowego.
• Powikłania dotyczące rany pooperacyjnej.
• Zmiana stanu psychicznego.
• Zgon.
Uwaga: Korekcja niektórych z wymienionych powyżej zdarzeń niepożądanych może wymagać dodatkowego zabiegu operacyjnego.
WYNIKI KLINICZNE:
W Stanach Zjednoczonych przeprowadzono wieloośrodkowe badanie równoważności2 mające na celu porównanie, wykonanych z dostępu przedniego, u chorych z objawowym zwyrodnieniem
krążków międzykręgowych, zabiegów dotyczących kręgosłupa, z zastosowaniem urządzenia INTER FIX™ wypełnionego autogennym przeszczepem kostnym, ze stanowiącym grupę kontrolną
zabiegami z użyciem allogennego pierścienia wykonanego z kości udowej, wypełnionego również autogennym przeszczepem kostnym. Badanie kliniczne zostało wstępnie zaprojektowane
jako prospektywne randomizowane z grupą kontrolną. Z upływem czasu plan badawczy został zmieniony w taki sposób, aby pacjenci mogli być kwalifikowani do zabiegów bez randomizacji,
t.j. wyłącznie do grupy pacjentów leczonych przy użyciu urządzenia INTER FIX™.
Kryteria włączenia do niniejszego badania klinicznego obejmowały objawowe zwyrodnienie krążków międzykręgowych, określone jako udokumentowany, nie reagujący na leczenie ból
kończyny dolnej i/lub pleców z dodatnim(i) wynikiem(ami) diagnostycznego badania obrazowego; niestabilność kręgosłupa, zdefiniowaną jako przesunięcie o ponad 4mm lub odchylenie o
ponad 5° obserwowane na radiogramach zgięciowo/wyprostnych; jednopoziomowe objawowe zajęcie odcinka od L2-S1; oraz kręgozmyk nie przekraczają
byli, w szczególności, pacjenci: u których w przeszłości wykonano zabieg międzytrzonowego zespolenia kręgosłupa w objętym procesem chorobowym odcinku; z osteopenią, osteoporozą
lub osteomalacją; lub wymagający środków stymulujących wzrost kostny.
W sumie do badanej grupy
Badanie porejestracyjne miało na celu określenie odległych wyników stosowania gwintowanego urządzenia do zespalania INTER FIX™, poprzez zgromadzenie danych z okresu 6 lat po zabiegu
operacyjnym, uzyskanych od minimum 100 pacjentów. Odległe wyniki stosowania urządzenia oceniono w ciągu 4 lat po zabiegu operacyjnym u 116 pacjentów, natomiast w ciągu 6 lat po
zabiegu u 109 pacjentów. Pacjenci ci stanowili część populacji chorych, którzy wzięli udział w badaniu klinicznym IDE. Na podstawie kryteriów ustalonych dla celów badania IDE, informacje
uzyskane od pacjentów stanowiły opis dodatkowych zabiegów operacyjnych, radiologiczną ocenę zespolenia oraz ocenę dolegliwości bólowych oraz czynności.
W tabeli III przedstawiono kliniczne wyniki badania, takie jak parametry uzyskania zespolenia, dolegliwości bólowe (skala Oswestry) oraz korzystne wyniki neurologiczne po 12, 24, 48 i 72
miesiącach od zabiegu operacyjnego. Pozytywny wynik niniejszego badania klinicznego odnotowano na podstawie wyniku analizy równoważności pomiędzy grupą pacjentów leczonych przy
pomocy urządzenia INTER FIX™ oraz grupą kontrolną.
Dla celów niniejszego badania klinicznego zrost zdefiniowano jako potwierdzone wytworzenie mostków kości beleczkowej, przesunięcie (<3mm) oraz stabilność ruchomości kątowej (<5°) a
także brak obszarów przepuszczalnych dla promieni X na przestrzeni ponad 50% wokół każdego z implantu. Ponadto pacjenci, u których wykonano powtórne zabiegi operacyjne, z uwagi na
brak zrostu, zostali zaliczeni do grupy chorych z niepowodzeniem zabiegu zespolenia. Pozytywny wynik w skali bólu/niepełnosprawności Oswestry zdefiniowano jako conajmniej 15 punktową
poprawę wyniku uzyskanego po zabiegu operacyjnym, w porównaniu z wynikiem sprzed operacji. Pozytywny wynik odnośnie stanu neurologicznego okreś
w conajmniej 3 z 4 kategorii neurologicznych (poruszanie się, czucie, odruchy oraz zdolność unoszenia wyprostowanej kończyny dolnej) po zabiegu operacyjnym w porównaniu z wynikiem
sprzed operacji. Wynik oznaczający całkowite powodzenie odnotowywany był u pacjenta, u którego wystąpił zrost, pozytywny wynik w skali bólu/niepełnosprawności Oswestry oraz pozytywny
wynik odnośnie stanu neurologicznego, a także brak konieczności wykonania powtórnego zabiegu operacyjnego, który klasyfikowano jako “niepowodzenie”.
2
Urządzenie INTER FIX™ statystycznie nie gorsze niż kontrola.
3
Nie obejmuje wyników 3 pacjentów, którzy nie byli leczeni przy pomocy urządzenia INTER FIX™ z uwagi na zdarzenia niepożądane związane z zabiegiem operacyjnym
W tabeli III znajdują się także wyniki skuteczności ogólnej w 12 oraz w 24 miesiącu po zabiegu operacyjnym.
ące miejsca pobrania przeszczepu.
cy 1 stopnia. Z badania wykluczeni
3
chorych leczonych przy pomocy urządzenia INTER FIX™ włączono 185 pacjentów. Do grupy kontrolnej włączono w sumie 62 pacjentów.
lono jako utrzymanie lub poprawę
Page 72

Zrost 96.1%
Poprawa w skali
bólu/niepeénospr
awnoýci
OswestryPacjenci
z conajmniej 15
punktowÅ
poprawÅ w
porównaniu z
wynikami
uzyskanymi przed
zabiegiem
Utrzymanie lub
poprawa stanu
neurologicznego
WskaĐnik
caékowitego
powodzenia
WskaĐniki 12 miesiÓczne WskaĐniki 24 miesiÓczne
Badany
(122/127)
53.5%
(76/142)
99.3%
(150/151)
42.4%
(61/144)
Grupa
kontrolna
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
(12/56)
Delta
1
(ǻ
)
Badany
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
-0.097 46.1%
(71/154)
Grupa
kontrolna
51.9%
(27/52)
52.2%
(24/46)
95.6%
(43/45)
29.8%
(17/57)
Delta
WskaĐniki 48
(ǻ1)
miesiÓczne
Badany
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
-0.043 49.5%
(53/107)
WskaĐniki 72
miesiÓczne
Badany
97.6% (82/84)
56.3%
(54/96)
1 - Minimalna wartość przedziału równoważności, konieczna do spełnienia warunku równoważności metody badanej i kontrolnej.
Tabela III – Kliniczne wyniki badania dotyczÅcego urzÅdzenia INTER FIX™
Wszyscy pacjenci zakwalifikowani do leczenia przy użyciu gwintowanego urządzenia do zespalania INTER FIX™, niezależnie od grupy do której zostali zakwalifikowani, włączeni byli na podstawie
takich samych kryteriów włączenia/wykluczenia. Aby potwierdzić porównywalność pacjentów zakwalifikowanych w drodze randomizacji oraz tych, którzy zakwalifikowani zostali do leczenia przy
pomocy urządzenia INTER FIX™ bez randomizacji, zbadano ich charakterystykę demograficzną, obciążenia występujące przed zabiegiem operacyjnym, charakterystykę wyników diagnostycznych
badań obrazowych dotyczących zwyrodnienia krążków międzykręgowych, rodzaju dostępu operacyjnego oraz poddany operacji odcinek kręgosłupa lędźwiowego. Populacje były pod względem
statystycznym porównywalne, z wyjątkiem rodzaju dostępu operacyjnego, przedoperacyjnego statusu zawodowego oraz wyników przedoperacyjnych badań obrazowych dotyczących formowania
osteofitów na blaszkach końcowych kręgów; bliznowacenia i/lub zgrubienia pierścienia włóknistego, wi
Dalsze analizy statystyczne wykazały, że zmienne te nie były powiązane z różnicami dotyczącymi klinicznych wyników związanych z wystąpieniem zrostu w ciągu 12 miesięcy, poprawy w skali bólu/
niesprawności Oswestry, stanu neurologicznego lub wskaźnika powodzenia całkowitego, z wyjątkiem przepukliny jądra miażdżystego. U większej liczby chorych w grupie pacjentów zakwalifikowanych
do leczenia bez randomizacji, stwierdzono przepuklinę jądra miażdżystego, która, jak się okazało, zwiększała prawdopodobieństwo całkowitego powodzenia.
Przeprowadzono analizę zgodną z intencją leczenia. W analizie tej niepowodzenia powtórnych zabiegów operacyjnych, zgony, brak danych odnośnie wizyt kontrolnych oraz liczbę utraconych
z obserwacji pacjentów uznane zostały za brakujące dane dotyczące zmiennych związanych z wynikami i w obliczeniach poszczególnych wskaź
uznane zostały za “niepowodzenie”. Zakwalifikowanie wyżej wymienionej grupy pacjentów do grupy przypadków leczenia zakończonych niepowodzeniem spowodowało, że wskaźniki dotyczące
wyników klinicznych w analizie zgodnej z intencją leczenia były znacznie niższe niż te, które faktycznie uzyskano na podstawie obserwacji aktualnych danych klinicznych.
W poniższej tabeli (tabela IV) przedstawiono wyniki analizy zgodnej z intencją leczenia, dotyczącej pacjentów leczonych przy pomocy urządzenia INTER FIX™. Tabela nie obejmuje wyników
chorych zakwalifikowanych do grupy kontrolnej.
Zgony, niepowodzenia powtórnych zabiegów operacyjnych, brak danych odnoýnie wizyt kontrolnych lub utrata
WskaĐniki 12
Zrost 65.9% (122/185) 67.6%
Poprawa w skali bólu/niepeénosprawnoýci
Oswestry
Pacjenci z conajmniej 15 punktowÅ poprawÅ w
porównaniu z wynikami uzyskanymi przed
zabiegiem
Utrzymanie lub poprawa stanu
neurologicznego
WskaĐnik caékowitego powodzenia 33.0% (61/185) 38.4% (71/185) 45.7%
Niepowodzenia powtórnych zabiegów
Brak zrostu
2
Inne
Zgony 0 1 0 0
1 - Pacjenci ci włączeni są do obliczeń dotyczących wskaźnika uzyskania zrostu, ale z drugiej strony dla celów badania klinicznego uznani zostali za przypadki zakończone
niepowodzeniem.
2 - Pacjenci, którzy poddani byli badaniom kontrolnym i u których w tym okresie wykonano powtórne zabiegi operacyjne z przyczyn innych niż brak zrostu uznani zostali, dla celów badania
klinicznego, za przypadki zakończone niepowodzeniem.
SPOSÓB PAKOWANIA:
Po otrzymaniu przesyłki opakowania każdego z komponentów powinny zostać nienaruszone. Jeśli stosowany jest system pożyczkowy lub przekazowy, wszystkie zestawy powinny być
sprawdzone pod kątem kompletności a wszystkie komponenty, z narzędziami włącznie powinny być starannie sprawdzone, aby upewnić się, że nie są uszkodzone przed użyciem. Zniszczone
opakowania lub urządzenia nie powinny być wykorzystane i powinny zostać zwrócone do firmy Medtronic Sofamor Danek.
CZYSZCZENIE I DEKONTAMINACJA:
Wszystkie implanty oraz narządzia muszą, o ile nie są dostarczane jako jałowe, przed sterylizacją oraz umieszczeniem w jałowym polu operacyjnym, być najpierw oczyszczone przy użyciu
Tabela IV – Analiza zgodna z intencjÅ leczenia dotyczÅca urzÅdzenia INTER FIX™
pacjentów z obserwacji uznane zostaéy za niepowodzenie i wéÅczone sÅ w mianownik wskaĐników
miesiÓczne
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
1
5
8
WskaĐniki 24
miesiÓczne
(125/185)
(128/185)
5
3
WskaĐniki 48
miesiÓczne
80.2% (93/116) 77.1%
(66/116)
87.9%
(102/116)
(53 /116)
ęzadła żółtego i/lub torebki stawu międzykręgowego; oraz przepukliny jądra miażdżystego.
ników włączone były do mianownika, t.j. dane te
WskaĐniki 72
4
4
miesiÓczne
( 84/109)
56.0%
(61 /109)
75.2%
(82/109)
49.5%
(54/109)
1
2
stosowanych w danym ośrodku metod. Wykorzystane narzędzia muszą przed ponownym użyciem zostać zdekontaminowane, oczyszczone oraz wysterylizowane.
Uwaga: Nie należy stosować roztworów czyszczących zawierających środki wybielające lub formalinę, gdyż mogą one spowodować uszkodzenie urządzenia.
Z wszystkimi urządzeniami należy obchodzić się ostrożnie. Nieprawidłowe użycie lub postępowanie może prowadzić do ich uszkodzenia i nieprawidłowego funkcjonowania.
STERYLIZACJA:
Wszystkie implanty i urządzenia stosowane w chirurgii muszą przed użyciem zostać w danym ośrodku wyjałowione, z wyjątkiem urządzeń oznakowanych i wyraźnie oznaczonych jako sterylne,
znajdujących się w nieotwartych opakowaniach dostarczonych przez firmę. Przed sterylizacją należy usunąć materiały stanowiące opakowanie. Tylko produkty jałowe mogą być stosowane w
polu operacyjnym. W celu uzyskania Poziomu Zapewnienia Sterylności (SAL) 10-6, zaleca się, by produkty te wyjaławiane były w danym ośrodku przy użyciu pary wodnej, z wykorzystaniem
jednego z podanych poniżej ustawień procesu sterylizacji.
METODA CYKL / METODA TEMPERATURA CZAS EKSPOZYCJI
Parowa Pre – vacuum – frakcjonowana pulsacyjna próżnia wstępna 270°F (132°C) 4 minuty
Parowa Grawitacyjna 250°F (121°C) 60 minut
Parowa* Grawitacyjna* 273°F (134°C)* 20 minut*
UWAGA: Z uwagi na dużą ilość parametrów dotyczących procesu sterylizacji, każdy ośrodek powinien dokonać kalibracji oraz weryfikacji procesu wyjaławiania (np.: temperatury, czasu),
który używany jest do sterylizacji jego sprzętu.
*Dotyczy stosowania poza Stanami Zjednoczonymi - niektóre nie-amerykańskie Towarzystwa Medyczne rekomendują prowadzenie sterylizacji zgodnie z podanymi parametrami, aby zminimalizować
potencjalne ryzyko przeniesienia choroby Creutzfeldta-Jakoba, dotyczy to zwłaszcza narzędzi chirurgicznych mogących mieć kontakt ze strukturami centralnego systemu nerwowego.
Page 73

SKARGI DOTYCZĄCE PRODUKTU:
Każdy przedstawiciel służby zdrowia (np.: klient lub użytkownik niniejszego systemu produktów), który ma uwagi lub który wyraża niezadowolenie z jakości, jednolitości, trwałości, niezawodności,
bezpieczeństwa, efektywności i/lub skuteczności sprzętu powinien o tym powiadomić dystrybutora lub firmę Medtronic Sofamor Danek. Należy ponadto natychmiast powiadomić dystrybutora,
jeśli zaobserwuje się “nieprawidłowe działanie” jakiegokolwiek wszczepialnego komponentu gwintowanego urządzenia do zespalania INTER FIX™ lub gwintowanego urządzenia do zespalania
INTER FIX™ RP (t.j. nie spełnia ono oczekiwań dotyczących możliwości zastosowania lub mówiąc inaczej nie działa tak jak powinno) albo istnieje podejrzenie jego nieprawidłowego działania.
Dystrybutor powinien być natychmiast poinformowany telefonicznie, faxem lub listownie jeśli jakikolwiek produkt firmy Medtronic Sofamor Danek funkcjonuje nieprawidłowo i mógł spowodować
lub przyczynić się do śmierci lub poważnego obrażenia ciała u pacjenta. Przy wypełnianiu zgł
adres, przyczynę złożenia skargi a także informację czy wymagany jest pisemny raport dla dystrybutora.
UWAGI DLA LEKARZA: Lekarz jest niejako naukowym pośrednikiem pomiędzy firmą a pacjentem i powinien przekazać mu istotne informacje zawarte w niniejszym dokumencie.
Dotyczy tylko użytkowników z USA
ŚRODKI OSTROŻNOŚCI: DO NINIEJSZYCH URZĄDZEŃ MEDYCZNYCH ODNOSZĄ SIĘ PRZEPISY PRAWA FEDERALNEGO (U.S.A) DO SPRZEDAŻY WYŁĄCZNIE PRZEZ LUB NA
ZAMÓWIENIE PERSONELU LEKARSKIEGO.
USUNIĘCIE URZĄDZENIA:
Jeśli zaistnieje konieczność usunięcia gwintowanego urządzenia do zespalania INTER FIX™ lub gwintowanego urządzenia do zespalania INTER FIX™ RP, proszę skontaktować się
telefonicznie z firmą Medtronic Sofamor Danek.
© 2006 Medtronic Sofamor Danek. Wszelkie prawa zastrzeżone.
oszenia skargi proszę podać nazwę i numer podzespołu (ów), numer(y) partii, swoje nazwisko i
Page 74

0381030 Rev. A
INTER FIXTM THREADED FUSION DEVICE
INTER FIX
IMPORTANT MEDICAL INFORMATION INTER FIXTM AND INTER FIXTM RP THREADED FUSION DEVICE
LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ ET LE DISPOSITIF FILETE POUR LA FUSION INTER FIX™ RP
EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX Y EL DISPOSITIVO FILETEADO PARA LA FUSIÓN INTER FIX RP
INTER FIX GEWINDE-INSTRUMENTATION FÜR FUSIONEN INTER FIX RP GEWINDE-INSTRUMENTATION FÜR FUSIONEN
INFORMAZIONI MEDICHE IMPORTANTI RELATIVE AL : DISPOSITIVO FILETTATO DI FUSIONE INTER FIX
INTER FIX
INTER FIX
GÄNGAD FUSIONSANORDNING INTER FIX™ GÄNGAD FUSIONSANORDNING INTER FIX™ RP VIKTIG MEDICINSK INFORMATION
INTER FIX
INTER FIX
DŮLEŽITÉ MEDICÍNSKÉ INFORMACE ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ A ŠROUBOVITÉ FÚZNÍ ZAŘÍZENÍ INTER FIX™ RP
OLULIST MEDITSIINILIST INFORMATSIOONI INTER FIX™ KEERMESTATUD FUSIOONISEADME JA INTER FIX™ RP KEERMESTATUD FUSIOONISEADME
FONTOS ORVOSI INFORMÁCIÓKAT TARTALMAZ AZ INTER FIX™ MENETES FÚZIÓS ESZKÖZRE ÉS AZ INTER FIX™ RP MENETES FÚZIÓS ESZKÖZRE
MEDICĪNISKA INFORMĀCIJA PAR INTER FIX™ VĪTŅOTO SPONDILOSINDĒZES IERĪCI UN PAR INTER FIX™ RP VĪTŅOTO SPONDILOSINDĒZES
SVARBI MEDICININĖ INFORMACIJA APIE INTER FIX™ SRIEGINIO SUJUNGIMO PRIETAISĄS IR INTER FIX™ RP SRIEGINĮ SUJUNGIMO PRIETAISĄS
ISTOTNE INFORMACJE MEDYCZNE DOTYCZĄCE GWINTOWANEGO URZĄDZENIA DO ZESPALANIA INTER FIX™ ORAZ GWINTOWANEGO
LEKÁRSKE INFORMÁCIE O ZARIADENÍ NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ A ZARIADENÍ NA PODPOROVANIE
™
SPONDYLODESESYSTEEM MET SCHROEFDRAAD INTER FIX™ RP SPONDYLODESESYSTEEM MET SCHROEFDRAAD
™
FUSIONSIMPLANTAT MED GEVIND INTER FIX™ RP FUSIONSIMPLANTAT MED GEVIND VIGTIGE MEDICINSKE OPLYSNINGER
™
GJENGET FIKSASJONSUTSTYR INTER FIX™ RP GJENGET FIKSASJONSUTSTYR VIKTIG MEDISINSK INFORMASJON
™
KIERTEITETTY FUUSIOLAITE INTER FIX™ RP KIERTEITETTY FUUSIOLAITE TÄRKEÄ LÄÄKETIETEELLINEN TIEDOTE
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™ RP - Σημαντικές ιατρικές πληροφορίες
INFORMAÇÕES MÉDICAS IMPORTANTES RELATIVAS AO DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™
03/2006
TM
RP THREADED FUSION DEVICE
INFORMATIONS MÉDICALES IMPORTANTES :
INFORMACIONES MÉDICAS IMPORTANTES SOBRE :
WICHTIGE MEDIZINISCHE INFORMATIONEN ÜBER DIE :
DISPOSITIVO FILETTATO DI FUSIONE INTER FIX™ RP
BELANGRIJKE MEDISCHE INFORMATIEM
ΕΛΙΚΟΤΟΜΗΜΕΝΗ ΣΥΣΚΕΥΗ ΣΥΓΧΩΝΕΥΣΗΣ INTER FIX™
DISPOSITIVO DE FUSÃO COM ROSCA INTER FIX™ RP
URZĄDZENIA DO ZESPALANIA INTER FIX™ RP
ZRASTANIA SO ZÁVITOM INTER FIX™ RP
™
Medtronic B.V. 1800 Pyramid Place
Earl Bakkenstraat 10 Memphis, TN 38132
6422 PJ Heerlen Telephone 800 876 3133 (In U.S.A.)
The Netherlands 901 396 3133 (Outside U.S.A.)
Tel: + 31 45 566 80 00 Fax 901 396 0356
0123
SLOVENSKY
Ďalší text obsahuje dôležité lekárske informácie o zariadení na podporovanie zrastania so závitom INTER FIX™ a zariadení na podporovanie zrastania so závitom
INTER FIX™ RP.
POPIS:
Zariadenie na podporovanie zrastania so závitom INTER FIX™ pozostáva z dutého, perforovaného, kovového valca a koncovky. Zariadenie na podporovanie zrastania so
závitom INTER FIX™ RP pozostáva z dutého, perforovaného, kovového valca s jednou veľkou ryhou s vonkajším zaoblením pozdĺž celej pozdĺžnej osi, ktorá zasahuje
do vnútorného priemeru zariadenia. Oba konce implantátu INTER FIX™ RP sú uzavreté.
Implantáty INTER FIX™ a INTER FIX™ RP sú dostupné v priemeroch v rozsahu od 12 mm do 24 mm a v dĺžkach v rozsahu od 20 mm do 29 mm. Koncovky implantátov
INTER FIX™ sú dimenzované podľa priemeru valcov a sú aplikované na otvorený koniec valcov po tom, čo sú vyplnené autogénnym kostným štepom.
Implantáty INTER FIX™ a INTER FIX™ RP sú vyrobené z titánovej ocele s kvalitou vhodnou na implantáty (Ti-6Al-4V), ktorá vyhovuje ASTM F136.
Konkludentné záruky predajnosti a vhodnosti na konkrétny účel alebo použitie sú špecificky vylúčené. Ďalšie informácie o zárukách a obmedzeniach zodpovednosti
nájdete v cenníku alebo katalógu Medtronic Sofamor Danek.
INDIKÁCIE:
Zariadenie na podporovanie zrastania so závitom INTER FIX™ a zariadenie na podporovanie zrastania so závitom INTER FIX™ RP sú indikované pre procedúry
zrastania stavcov u pacientov s vyspelou kostrou s chorobou degeneratívnej platničky (DDD) na jednej úrovni od L2-S1. Choroba degeneratívnej platničky je definovaná
ako diskogénna bolesť chrbtice s degeneráciou platničky potvrdená anamnézou pacienta a rádiografickými štúdiami. Títo pacienti s chorobou degeneratívnej platničky
môžu tiež mať spondylolistézu alebo retrolistézu na predmetnej úrovni do stupňa I. Implantáty INTER FIX™ a INTER FIX™ RP sa majú používať s autogénnym kostným
štepom a implantovať cez otvorený anteriórny prístup.
ZARIADENIE NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™
ZARIADENIE NA PODPOROVANIE ZRASTANIA SO ZÁVITOM INTER FIX™ RP
DÔLEŽITÉ LEKÁRSKE INFORMÁCIE
Page 75

KONTRAINDIKÁCIE:
A
Zariadenie na podporovanie zrastania so závitom INTER FIX™ a zariadenie na podporovanie zrastania so závitom INTER FIX™ RP by sa nemali implantovať u pacientov
s aktívnou infekciou na mieste operácie alebo s alergiou na titán alebo zliatinu titánu.
UPOZORNENIA:
• Zariadenie na podporovanie zrastania so závitom INTER FIX™ a INTER FIX™ RP zariadenie na podporovanie zrastania so závitom by mali používať len chirurgovia,
ktorí majú skúsenosti s procedúrami zrastania stavcov a podstúpili adekvátne vzdelávanie s týmto zariadením.
• Nedostatok adekvátnych skúseností a/alebo vzdelania môže viesť k vyššej miere výskytu nepriaznivých účinkov ako napríklad vaskulárnych poranení, neurologických
poranení, a/alebo urogenitálnych poranení (vrátane retrográdnej ejakulácie).
BEZPEČNOSTNÉ OPATRENIA:
• Bezpečnosť a efektívnosť zariadenia na podporovanie zrastania so závitom INTER FIX™ a zariadenia na podporovanie zrastania so závitom INTER FIX™ RP nebola
stanovená u pacientov s akýmikoľvek z nasledovných podmienok:
- spondylolistéza alebo retrolistéza stupňa II alebo vyššieho;
- viac ako jedna úroveň, ktorá má zrastať;
- revízia predchádzajúcej procedúry (procedúr) zrastania stavcov;
- pooperačné steroidné alebo nesteroidné protizápalové liekové požiadavky;
- silná obezita;
- vek menej ako 18 alebo viac ako 65 rokov;
- osteoporóza, osteopénia, a/alebo osteomalácia;
- tehotenstvo.
• Bezpečnosť and efektívnosť zariadenia na podporovanie zrastania so závitom INTER FIX™ a zariadenia na podporovanie zrastania so závitom INTER FIX™ RP
bola stanovená len pri procedúre anteriórneho lumbárneho zrastania stavcov (ALIF) s využitím vlastného kostného štepu.
• Pacienti, ktorí dostávajú zariadenie na podporovanie zrastania so závitom INTER FIX™ by mali mať aspoň šesťmesačné neoperačné liečenie. Pacienti, ktorí dostávajú
zariadenie na podporovanie zrastania so závitom INTER FIX™ RP by tiež mali mať aspoň šesťmesačné neoperačné liečenie.
• Dve INTER FIX™ zariadenia na podporovanie zrastania so závitom by mali byť implantované vedľa seba na chirurgickej úrovni. Zariadenie na podporovanie zrastania
so závitom INTER FIX™ musí byť implantované na chirurgickej úrovni vedľa zariadenia na podporovanie zrastania so závitom INTER FIX™ RP. Pozdĺžna drážka
na zariadení INTER FIX™ RP umožňuje implantovanie dvoch zariadení v bližšej vzdialenosti, než aká sa dá riadiť pri dvoch zariadeniach INTER FIX™, čo vedie k
zníženému bočnému profilu.
• Dlhá os zariadenia na podporovanie zrastania so závitom INTER FIX™ a zariadenia na podporovanie zrastania so závitom INTER FIX™ RP by mali byť v anteriórnoposteriórnom smere.
• Implantáty a inštrumenty musia byť sterilizované pred použitím podľa pokynov na sterilizáciu uvedených v tomto príbalovom letáku, pokiaľ sa nedodávajú sterilné a
nie sú jasne označené ako také.
NEŽIADÚCE ÚČINKY:
Nežiadúce účinky, ako je uvedené v tabuľke I, boli hlásené od 185 pacientov so zariadením INTER FIX™ a 62 kontrolných pacientov zaradených do klinickej štúdie s
viacerými strediskami pre zariadenie na podporovanie zrastania so závitom INTER FIX™. Ako doplňujúce informácie sú tiež uvádzané nežiadúce účinky hlásené v štúdii
vykonanej 48 mesiacov po schválení, do ktorej bolo zapojených 116 pacientov a v štúdii vykonanej 72 mesiacov, do ktorej bolo zapojených 109 pacientov.
Uvedené početnosti sú založené na celkovom počte pacientov zapojených do štúdie v každom časovom úseku. Nežiadúce účinky, ku ktorým dochádza u randomizovaných
a nerandomizovaných skupín ošetrených zariadením INTER FIX™, sú kombinované, aby sa uviedla celková miera.
Komplikácia
Vaskulárna
intraoperaÉná
Sakroiliakálna
bolesą
Neurologická
Bolesą chrbta
Rezná
Spinálne poranenie
Urologická
Iné
Iná bolesą
Gastrointestinálna
Retrográdna
ejakulácia
RespiraÉná
Bolesą nohy
Trauma
Peritoneálna
Vaskulárna
postoperaÉná
Fraktúra kosti
Posunutie
implantátu /
uvoçnenie
Bolesą v mieste
štepu
Pakãb
Pokles
Pakãb (EŠTE
NEDOSIAHNUTÝ
VÝSLEDOK)
Meningitída
Zlomenie
implantátu
Rakovina
Smrą
PooperaÉné
(1 deï-<2
OperaÉné
[poÉet (%)]
KONTROL
INTER FIX
N=62
N=185
15(8.1) 2(3.2) 15 (8.1) 2(3.2)
5(2.7) 3(1.6) 4(2.2) 1(1.6) 7(3.8) 1(1.6) 4(2.2) 1(1.6) 23(12.4) 3(4.8) 1(0.9)
6(3.2) 2(3.2) 9(4.9) 3(4.8) 5(2.7) 1(1.6) 4(2.2) 3(4.8) 6(3.2) 4(6.5) 30(16.2) 13(21.0) 7(6.0) 3(2.8)
1(0.5) 3(1.6) 2(3.2) 5(2.7) 4(2.2) 3(4.8) 6(3.2) 6(9.7) 19(10.3) 11(17.7) 7(6.0) 6(5.5)
11(5.9) 6(9.7) 1(0.5) 2(1.1) 1(1.6) 2(1.1) 1(0.5) 1(1.6) 17(9.2) 8(12.9) 1(0.9)
1(0.5) 1(0.5) 1(1.6) 1(0.5) 3(1.6) 1(1.6) 10(5.4) 1(1.6) 11(5.9) 3(4.8) 27(14.6) 6(9.7) 13(11.2) 10(9.2)
1(0.5) 8(4.3) 2(3.2) 2(1.1) 1(0.5) 1(1.6) 4(2.2) 16(8.6) 3(4.8) 2(1.7) 1(0.9)
2(1.1) 1(0.5) 2(3.2) 3(1.6) 2(1.1) 3(4.8) 7(3.8) 1(1.6) 7(3.8) 2(3.2) 22(11.9) 8(12.9) 14(12.1) 9(8,3)
4(2.2) 1(1.6) 5(2.7) 3(1.6) 2(3.2) 9(4.9) 6(3.2) 2(3.2) 27(14.6) 5(8.1) 7(6.0) 8(7.3)
7(3.8) 3(4.8) 1(0.5) 1(0.5) 1(1.6) 2(1.1) 2(1.1) 3(4.8) 13(7.0) 7(11.3) 9(7.8) 7(6.4)
2(1.1) 3(1.6) 1(0.5) 6(3.2)
3(1.6) 1(1.6) 1(0.5) 1(0.5) 1(0.5) 3(1.6) 9(4.9) 1(1.6) 5(4.3) 6(5.5)
2(1.1) 1(0.5) 2(1.1) 1(0.5) 6(3.2) 0 2(1.7) 2(1.8)
2(1.1) 1(1.6) 5(2.7) 5(2.7) 6(3.2) 1(1.6) 10(5.4) 1(1.6) 28(15.1) 3(4.8) 11(9.5) 8(7.3)
3(1.6) 3(1.6) 0
2(1.1) 2(3.2) 1(0.5) 3(1.6) 4(2.2) 10(5.4) 2(3.2) 4(3.4) 2(1.8)
1(0.5) 2(1.1) 3(1.6) 0 1(0.9)
1(0.5) 1(1.6) 5(8.1) 1(0.5) 2(1.1) 6(9.7)
1(0.5) 1(1.6) 1(1.6) 1(0.5) 2(3.2)
4(2.2) 2(3.2) 7(3.8) 7(11.3) 4(2.2) 4(6.5) 15(8.1) 13(12.0) 3(2.6)
2(1.1) 2(1.1) 0
1(1.6)2 0 1(1.6) 2(1.7)
1(1.6) 0 1(1.6)
5(8.1) 0 5( 8. 1)
1 (0.5) 1 (0.5) 2 (1.1) 1(0.8) 1(0.9)
1(1.6) 1(0.5) 1(0.5) 1(1.6) 1(0.8)
mesiace)
[poÉet (%)]
INTER FIX
N=185
KONTROLA
N=62
INTER FIX
TABUæKA I - NEŽIADÚCE ÚÈINKY
3 mesiace
(t
2-<5 mesiacov)
[poÉet (%)]
KONTROLA
N=62
N=185
6 mesiacov
mesiacov)
[poÉet (%)]
INTER
FIX
N=185
5-<9
(t
KONTROLA
N=62
12 mesiacov
(t
9-<19 mesiacov)
[poÉet (%)]
INTER FIX
N=185
KONTROLA
N=62
24 mesiacov
(t
mesiacov)
[poÉet (%)]
INTER FIX
N=185
19-< 30
KONTROLA
N=62
CELKOVÉ NEŽIADÚCE
ÚÈINKY (POÈAS 24
MESIACOV))
N=185
(7.4)
KONTROLA
1
INTER FIX
N=62
0
48
mesiacov
(>30 - < 60
30<60
(t
mesiacov)
INTER FIX
N=116
72
mesiacov
(t
60
mesiacov)
INTER FIX
N=109
1 - U šiestich (6) z 81 mužov zaradených do skupiny so zariadením INTER FIX™ (7,4%) došlo počas 24-measčného hodnotenia k retrográdnej ejakulácii.
2 - Tento pacient (175) bol následne hlásený ako pacient so zrastom približne 52 mesiacov po operácii, pričom nebol potrebný žiadna iná chirurgický zásah alebo ošetrenie.
Page 76

Dvomi významnými nežiadúcimi účinkami v klinickej skúške boli vaskulárne a neurologické poranenia počas operácie. Počas 24-mesačného hodnotenia došlo u skupiny 14
pacientov so zariadením INTER FIX™ celkovo k 15 prípadom vaskulárnych príhod počas operácie. Medzi tieto príhody patrili: 2 poranenia dutej žily, 9 poranení iliakálnej
žily, 1 roztrhanú hypogastrickú žilu, 1 segmentálne žilové krvácanie, 1 poranenie krížovej žily a 1 povrchové krvácanie. Celkový počet 2 vaskulárnych intraoperačných
poranení sa vyskytol u 2 pacientov v kontrolnej skupine počas toho istého obdobia. Medzi ne patrilo: 1 poranenie iliakálnej žily a 1 krvácanie z kostného lôžka.
Celkový počet 30 neurologických príhod sa vyskytol u 28 pacientov v skupine zariadení INTER FIX™ počas 24-mesačného hodnotenia. Medzi tieto príhody patrilo: 1
plantárne ohnutie nohy spôsobené paralýzou svalov, 3 poranenia nervových korienkov, 1 perforovaná stenóza, 1 reflexná sympatická dystrofia, 7 pocitov necitlivosti
alebo tepla v nohách, 2 dysestézia, 3 parestézie, 2 radikulitídy, 7 bolestí chrbta a/alebo nohy, 1 bolesť chrbta a/alebo nohy s inými symptómami, 1 bolesť vystreľujúca
do spodnej časti chrbta a 1 syndróm zápästného kanálika.
Celkový počet 10 neurologických príhod sa vyskytol u 8 pacientov v kontrolnej skupine počas toho istého obdobia. Medzi tieto príhody patrili: 1 radikulopatia s mravenčením
v končatinách, 1 chronická bolesť chrbta s ratikulitídou, 1 systémy slabnúcej distribúcie, 2 bolesti chrbta a nohy s inými symptómami, 1 denervovaný sval abductor magnus,
a 4 s pocitom necitlivosti alebo teplom v nohách. Počas štúdie vykonanej po schválení bolo u 10 pacientov zaznamenaných ďalších 10 neurologických príhod. Medzi tieto
príhody patrilo: 1 radikulopatia, 2 cervikálne radikulopatie, 1 pocit necitlivosti a mravenčenie v rukách, 1 pocit necitlivosti alebo pálenia v nohách, 3 bolesti chrbta a/alebo
nohy a 2 bolesti chrbta a/alebo nohy s inými symptómami. Okrem toho, tabuľka s nežiadúcimi účinkami uvádza nežiadúce účinky v podobe bolesti nôh a chrbta a/alebo
spinálne príhody, ako napríklad kolaps platničkového priestoru, ktoré v niektorých prípadoch mali neurologický komponent.
Niektoré nežiadúce účinky viedli k chirurgickým intervenciám po chirurgickom zákroku v rámci klinickej štúdie. Tieto dodatočné chirurgické intervencie môžu byť klasifikované
ako revízie, odstránenia, doplnkové fixácie alebo opakované operácie
a nerandomizované kombinované) a skupiny s kontrolným vyšetrením počas štúdie IDE a skupiny so zariadením INTER FIX™ počas štúdie vykonanej po schválení.
1
Revízia: Procedúra, ktorá upravuje, alebo inak modifikuje pôvodnú konfiguráciu implantátu.
Odstránenie: Procedúra, ktorá odstraňuje jeden alebo viac komponentov pôvodnej konfigurácie implantátu bez jeho nahradenia tým istým typom skúšobného zariadenia.
Doplnková fixácia: Procedúra, pri ktorej sa umiestňujú dodatočné spinálne zariadenia neschválené ako súčasť protokolu.
Opakovaná operácia: Akákoľvek chirurgická procedúra, ktorá neodstraňuje, nemodifikuje alebo nepridáva žiadne originálne komponenty implantátu.
Tabuçka II - Sekundárne chirurgické procedúry
1
. Tabuľka II sumarizuje sekundárne chirurgické intervencie v zariadení INTER FIX™ (randomizované
Zariadenie
Štúdia IDE Štúdia vykonaná po schválení
Èasový úsek do 24
mesiacov
INTER FIX™
N=185
Kontrola1
N=62
Èasový úsek 48
mesiacov
Zariadenie
INTER FIX™
N=116
Èasový úsek 72
mesiacov
Zariadenie
INTER FIX™
N=109
Revízie 3 (1.6%) 3 (4.8%) 1 (0.9%) 0 (0.0%)
Odstránenia 2 (1.1%) 0 (0.0%) 0 (0.0%) 0 (0.0%)
Doplnkové
21 (11.4%) 17 (27.4%) 7 (6.0%) 3 (2.8%)
fixácie
Opakované
42 (22.7%) 17 (27.4%) 33 (28.4%) 17 (15.6%)
operácie
1 - Pretože sa nepožadovalo ďalšie sledovanie kontrolných pacientov po 24 mesiacoch, tieto informácie nie sú dostupné pre všetkých týchto pacientov počas 72 mesiacov.
POTENCIÁLNE NEŽIADÚCE ÚČINKY:
Ďalej je uvedený zoznam potenciálnych nežiadúcich účinkov, ktoré sa môžu vyskytnúť pri chirurgickom zákroku so zrastaním stavcov s využitím zariadenia na podporovanie
zrastania so závitom INTER FIX™ alebo zariadenia na podporovanie zrastania so závitom INTER FIX™ RP. Niektoré z týchto nežiadúcich účinkov boli predtým uvedené
v tabuľke s nežiadúcimi účinkami.
• Demontáž, ohnutie, zlomenie, uvoľnenie a/alebo migrácia komponentov.
• (Alergická) reakcia na cudzie teleso.
• Tkanivové alebo nervové poškodenie.
• Pooperačná zmena v zakrivení chrbtice, strata korekcie, výšky a/alebo redukcie.
• Infekcia.
• Durálne slzy.
• Oslabenie neurologického systému.
• Oslabenie urologického systému.
• Tvorba jaziev.
• Fraktúra kosti.
• Pakĺb (alebo pseudoartróza), oneskorené zhojenie zlomeniny, zlé zahojenie zlomeniny.
• Zastavenie akéhokoľvek potenciálneho rastu operovanej časti chrbtice. Strata spinálnej mobility alebo funkcie.
• Komplikácie na mieste darcovstva štepu.
• Poškodenie krvných ciev a oslabenie kardiovaskulárneho systému.
• Gastrointestinálne komplikácie.
• Oslabenie reprodukčného systému.
• Poškodenie interných orgánov a spojivových tkanív.
• Rozvinutie respiračných problémov.
• Rezné komplikácie.
• Zmena duševného stavu.
• Smrť.
Poznámka: Na korekciu niektorých týchto potenciálnych nežiadúcich udalostí môže byť nutný ďalší chirurgický zásah.
KLINICKÉ VÝSLEDKY:
Klinická skúška ekvivalencie s viacerými strediskami2 zariadenia na podporovanie zrastania so závitom INTER FIX™ bola vykonaná v Spojených štátoch amerických porovnávaním anteriórneho
používania chrbtice zariadenia INTER FIX™ vyplneného autogénnou kosťou s femorálnym krúžkovým alotransplantátom vyplneným autogénnou kosťou, kontrolou, a to pri ošetrovaní pacientov
so symptomatickou chorobou degeneratívnej platničky. Najskôr klinická skúška mala perspektívny, randomizovaný, kontrolovaný dizajn. Následne bol plán skúmania revidovaný, aby umožnil
pacientom ich zaradenie do nerandomizovaného ramena, t.j. len pacientov ošetrených pomocou zariadenia INTER FIX™.
Kritériá zaradenia pre klinickú skúšku zahŕňali symptomatickú chorobu degeneratívnej platničky, ako je zaznamenané nepoddajnou nohou a/alebo bolesťou chrbta so zistením (zisteniami)
pozitívneho diagnostického zobrazovania; spinálnou nestabilitou, ako je definované transláciou väčšou ako 4 mm alebo zakrivením viac ako 5° na rádiografoch flexie/extenzie; jedinej úrovne
symptomatického postihnutia od L2-S1; a spondolistézy stupňa nie väčšieho ako stupeň 1. Špecificky vylúčení zo štúdie boli pacienti, ktorí: mali predchádzajúcu procedúru anteriórneho
zrastania stavcov na príslušnej spinálnej úrovni; mali osteopéniu, osteoporózu, alebo osteomaláciu; alebo požadovanú stimuláciu rastu kosti.
Celkový počet 185 pacientov bol zapísaný do investigačnej skupiny ošetrenia zariadením INTER FIX™3. Celkový počet 62 kontrolných pacientov bolo zadaných do randomizovaného
ramena klinickej skúšky.
Požiadavkou po schválení zariadenia na podporovanie zrastania so závitom INTER FIX™ bolo zhodnotiť dlhodobú funkčnú charakteristiku zariadenia získaním údajov za obdobie 6 rokov
po operácii za minimálne 100 pacientov. Dlhodobá funkčná charakteristika zariadenia bola hodnotená u 116 pacientov 4 roky po operácii a u 109 pacientov 6 rokov po operácii. Populácia
pacientov bola získaná spomedzi účastníkov klinickej skúšky IDE. Na základe kritérií zo štúdie IDE, medzi informácie získané o týchto pacientoch patril popis všetkých doplňujúcich chirurgických
procedúr, rádiografické hodnotenie zrastu a hodnotenie bolesti a funkcie.
Ta bu ľka III uvádza klinické výsledky štúdie, pričom popisuje parametre úspechu pri zraste, úspechu pri zvládaní bolesti (Oswestry) a neurologickom úspechu 12, 24, 48 a 72 mesiacov po
chirurgickom ošetrení. Úspech tejto klinickej skúšky bol zaznamenaný na základe analýzy ekvivalencie medzi zariadením INTER FIX™ a kontrolnou skupinou.
Page 77

Pri tejto klinickej skúške bol zrast definovaný ako dôkaz premostenia trabekulárnej kosti, stability translácie (<3mm) a uhlového pohybu (<5°), a bez priesvitnosti pri snímkovaní obklopujúcej
viac ako 50% ktoréhokoľvek implantátu. Aj pacienti so sekundárnym chirurgickým zákrokom v dôsledku pakĺbov boli vo výpočtoch považovaní za neúspešné zrasty. Oswestryho
bolesť/úspech pri postihnutí bola definovaný ako aspoň 15-bodové zlepšenie v pooperačnom skóre v porovnaní s predoperačným skóre. Úspech neurologického statusu bol definovaný
ako udržiavanie alebo zlepšenie v aspoň 3 zo 4 neurologických kategórií (pohybová, zmyslová, reflexy, a zdvíhanie natiahnutej nohy) po operácii, v porovnaní s predoperačným stavom.
Celkový úspech bol založený na pacientoch, u ktorých došlo k zrastaniu, Oswestryho bolesti/úspechu s postihnutím, a úspechu s neurologickým statusom, a žiadna sekundárna chirurgická
procedúra nebola klasifikovaná ako „neúspech“.
2
Zariadenie INTER FIX™ nie je štatisticky horšie ako kontrola.
3
Nezahŕňa 3 pacientov, ktorým sa nedostali ošetrenia zariadením INTER FIX™ v dôsledku chirurgických nežiadúcich účinkov.
V tabuľke III je tiež možné nájsť úspešné výsledky 12 a 24 mesiacov po operácii.
Zrast 96.1%
Oswestrova
bolesą/zlepšenie
postihnutia
Pacienti aspoï
s 15-bodovým
zlepšením od
predoperaÉného
stavu
Neurologický
stav udržiavania
alebo zlepšenia
Celkový úspech 42.4%
12-mesaÉné miery 24-mesaÉné miery
Hodnotené
zariadenie Kontrola
(122/127)
53.5%
(76/142)
99.3%
(150/151)
(61/144)
Tabuçka III - Klinické výsledky štúdie zariadenia INTER FIX™
Delta
Hodnotené
1
(ǻ
)
40.4%
(21/52)
51.9%
(27/52)
100%
(52/52)
21.4%
(12/56)
zariadenie Kontrola
92.6%
(125/135)
57.7%
(79/137)
95.5%
(128/134)
-0.097 46.1%
(71/154)
(27/52)
(24/46)
(43/45)
(17/57)
Delta
(ǻ1)
51.9%
52.2%
95.6%
29.8%
-0.043 49.5%
48-mesaÉné
miery
Hodnotené
zariadenie
94.9% (93/98) 94.4% (84/89)
59.5% (66/111) 68.5% (61/89)
99.0%
(102/103)
(53/107)
72-mesaÉné
miery
Hodnotené
zariadenie
97.6% (82/84)
56.3%
(54/96)
1 - Minimálny okraj bez zhoršenej akosti požadovaný pre tvrdenie, že hodnotené údaje nemajú horšiu akosť než kontrolná skupina.
Všetci pacienti zapojení do klinickej skúšky zariadenia na podporovanie zrastania so závitom INTER FIX™, bez ohľadu na skupinu ošetrenia, boli zapísaní za tých istých kritérií zradenia/vylúčenia.
Aby sa dokázala porovnateľnosť randomizovaných pacientov so zariadením INTER FIX™ a nerandomizovaných pacientov so zariadením INTER FIX™, boli preskúmané demografické
charakteristiky, preoperačné zdravotnícke podmienky, zistenia diagnostického zobrazovania charakteristiky choroby degeneratívnej platničky, operačný prístup a umiestnenie lumbárnej
ošetrovanej oblasti. Obyvateľstvo bolo štatisticky porovnateľné s výnimkou operačného prístupu, profesionálneho stavu pred operačným zákrokom a zistení diagnostického zobrazovania
tvorby osteofytov stavcových koncových platničiek; zjazvenie a/alebo zhrubnutie annulus fibrosus, ligamentum flavum, a/alebo puzdra facetového kĺbu; a pretrhnutý nucleus pulposus. Ďalšie
štatistické analýzy dokázali, že tieto premenné neboli spojené s rozdielmi v 12-mesačných klinických výsledkoch zrastu, Oswestryho bolesťou/zlepšením postihnutia, neurologického stavu,
alebo celkovým úspechom, s výnimkou pretrhnutého nucleus pulposus. Viac pacientov v nerandomizovanej skupine malo zistenie pretrhnutého nucleus pulposus, o ktorom sa zistilo, že
zvýši pravdepodobnosť celkového úspechu.
Bola vykonaná analýza „plánovaných ošetrení“. Pri tejto analýze viedli neúspechy pri sekundárnom chirurgickom zákroku, úmrtia, strata ďalšieho sledovania pacientov a chýbajúce pozorovania
v dôsledku iných príčin k chýbajúcim pozorovaniam pre výsledné premenné a preto boli zahrnuté do menovateľov vypočítaných mier, t.j. považovali sa za „neúspechy“. Zaobchádzaním s týmito
pacientmi ako s neúspešnými ošetreniami boli miery klinického výsledku v analýze plánovaných ošetrení výrazne nižšie než miery v skutočne pozorovaných klinických údajoch.
Nasledovná tabuľka (Tabuľka IV) uvádza výsledky analýzy plánovaných ošetrení pacientov so zariadením INTER FIX™, ale nezahŕňa výsledky kontrolných pacientov.
Úmrtia, neúspechy sekundárneho chirurgického zákroku, strata Ëalšieho sledovania a chýbajúce pozorovania
12-mesaÉné miery 24-mesaÉné
Zrast 65.9% (122/185) 67.6%
Oswestrova bolesą/zlepšenie postihnutia
Pacienti aspoï s 15-bodovým zlepšením od
predoperaÉného stavu
Neurologický stav udržiavania alebo
zlepšenia
Celkový úspech 33.0% (61/185) 38.4% (71/185) 45.7%
Neúspechy sekundárneho chirurgického
zákroku
1
Pakãby
2
Iné
Úmrtia 0 1 0 0
Tabuçka IV - Analýza plánovaných ošetrení pre zariadenie INTER FIX™
sa považujú za neúspechy a sú zahrnuté do menovateça mier
miery
41.1% (76/185) 42.7% (79/185) 56.9%
81.1% (150/185) 69.2%
5
8
(125/185)
(128/185)
5
3
48-mesaÉné
miery
80.2% (93/116) 77.1%
(66/116)
87.9%
(102/116)
(53 /116)
4
4
72-mesaÉné miery
( 84/109)
56.0%
(61 /109)
75.2%
(82/109)
49.5%
(54/109)
1
2
1 - Títo pacienti sú zahrnutí do výpočtov miery zrastania, ale inak sa považujú za neúspechy na účely klinických skúšok.
2 - Pacienti pred ďalším sledovaním v danom období, ktorí mali sekundárne chirurgické zákroky z dôvodov iných než nespojenia, sa považujú za neúspechy na účely klinických skúšok.
BALENIE:
Obal všetkých komponentov musia byť pri prijatí neporušení. Ak sa používa systém zapožičaných predmetov alebo dodávok, je nutné dôkladne skontrolovať úplnosť obsahu sád a všetky
komponenty vrátane inštrumentov sa musia pred použitím dôkladne skontrolovať, aby sa zabezpečilo, že nie sú poškodené. Poškodené obaly alebo výrobky sa nesmú používať a musia sa
vrátiť spoločnosti Medtronic Sofamor Danek.
ČISTENIE A DEKONTAMINÁCIA:
Ak sa nedodávajú sterilné, všetky implantáty a inštrumenty musia byť pred sterilizáciou a zavedením do sterilného poľa chirurgického zákroku najskôr vyčistené pomocou metód uznávaných
nemocnicou. Použité inštrumenty musia byť pred opakovaným použitím dekontaminované, vyčistené a sterilizované.
Poznámka: Nepoužívajte čistiace roztoky, ktoré obsahujú bieliace prostriedky alebo formalín, pretože môžu poškodiť zariadenie.
So všetkými výrobkami by sa malo zaobchádzať so starostlivosťou. Nesprávne použitie alebo manipulácia môže viest k poškodeniu a možnému nesprávnemu fungovaniu zariadenia.
STERILIZÁCIA:
Pokiaľ nie sú inštrumenty označené ako sterilné a pokiaľ nie je jasne na ich neotvorenom sterilnom obale dodanom spoločnosťou uvedené, že sú sterilné, všetky implantáty a inštrumenty
používané v chirurgii musia byť v nemocnici pred použitím sterilizované. Pred sterilizáciou odstráňte všetok obalový materiál. V priestore vykonávania chirurgického zákroku musia byť uložené
len sterilné produkty. Pri úrovni zabezpečenia sterility 10-6 sa odporúča tieto výrobky sterilizovať parou v nemocnici s použitím jednej z troch sád parametrov procesov uvedených nižšie:
METÓDA CYKLUS TEPLOTA DOBA EXPOZÍCIE
Parná predsterilizačné vákuum 270°F (132°C) 4 minúty
Parná gravitačný 250°F (121°C) 60 minút
Parná * gravitačný* 273°F (134°C)* 20 minút*
POZNÁMKA: Kvôli množstvu premenných pri sterilizácii by sa malo kalibrovať každé lekárske zariadenie a mal by sa preverovať proces sterilizácie (napr. teploty, doby) použité pre ich
zariadenia.
*Pre použitie mimo územia Spojených štátov amerických niektoré neamerické zdravotnícke orgány odporúčajú sterilizáciu podľa týchto parametrov, aby sa minimalizovalo potenciálne riziko
prenosu Creutzfeldt-Jakobovej choroby, najmä pri chirurgických inštrumentoch, ktoré sa dostávajú do kontaktu s centrálnym nervovým systémom.
Page 78

REKLAMÁCIA PRODUKTU:
Akýkoľvek kvalifikovaný pracovník v zdravotníctve (napr. zákazník alebo používateľ tohto systému výrobkov), ktorý má akúkoľvek reklamáciu, alebo ktorý nie je spokojný s kvalitou výrobku,
jeho identitou, trvanlivosťou, spoľahlivosťou, bezpečnosťou, účinnosťou a/alebo výkonom, by mal informovať distributéra alebo spoločnosť Medtronic Sofamor Danek. Ďalej, ak nejaké
komponenty implantovaného zariadenia na podporovanie zrastania so závitom INTER FIX™ alebo zariadenia na podporovanie zrastania so závitom INTER FIX™ RP „nepracuje správne” (t.j.
nevyhovuje akýmkoľvek špecifikáciám výkonu alebo inak nefunguje, ako by mal), alebo ak existuje podozrenie na jeho nesprávne fungovanie, distributér o tom musí byť okamžite informovaný.
Ak akýkoľvek výrobok spoločnosti Medtronic Sofamor Danek niekedy „nepracoval správne“ a mohol zapríčiniť smrť alebo vážne poranenie pacienta, alebo ak k nim prispel, distributér musí
byť o tom informovaný hneď, ako je to možné, a to telefonicky, faxom alebo písomnou korešpondenciou. Pri podávaní reklamácie uveďte názov a číslo komponentu alebo komponentov, číslo
alebo čísla šarže, vaše meno a adresu, charakter reklamácie a oznámenie, či je požadovaná písomná správa pre distributéra.
POZNÁMKA LEKÁRA: Hoci je lekár erudovaný prostredník medzi spoločnosťou a pacientom, je potrebné pacientovi oznámiť dôležité zdravotnícke informácie uvedené v tomto
dokumente.
Len pre použivatel’ov v USA
VÝSTRAHA: FEDERÁLNE ZÁKONY (USA) OBMEDZUJÚ PREDAJ TÝCHTO ZARIADENÍ LEN PRE LEKÁROV A NA ICH OBJEDNÁVKU.
VYBRATIE ZARIADENIA:
Ak je nevyhnutné vybrať zariadenie na podporovanie zrastania so závitom INTER FIX™ alebo zariadenie na podporovanie zrastania so závitom INTER FIX™ RP, zavolajte spoločnosť
Medtronic Sofamor Danek.
© 2006 Medtronic Sofamor Danek. Všetky práva sú vyhradené.
 Loading...
Loading...