

Page 1

DiamondTemp™
Ablation Catheter
Instructions for Use
Caution: Federal law (USA) restricts this device to sale by or on the order of a physician.
Page 2

Medtronic, Medtronic with rising man logo, and Medtronic logo are trademarks of Medtronic. Third-party trademarks (“TM*”) belong to their respective owners. The
following list includes trademarks or registered trademarks of a Medtronic entity in the United States and/or in other countries.
DiamondTemp™
Page 3

Contents
1 Glossary of symbols . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2 Device description . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
3 Indications for use . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
4 Contraindications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
5 Warnings and precautions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
6 Potential adverse events . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
7 Clinical data . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
7.1 Study Design, Study Population, Study Visits, and Length of Follow-up Methods . . . . . . . . . . . . . . . . . . . . . . . 9
7.2 Study Endpoints . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
7.3 Total Number of Enrolled Study Sites and Subjects, Subject Accountability, and Follow-up Rate . . . . . . . . . 11
7.4 Baseline Characteristics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
7.5 Procedural Characteristics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
7.6 Rhythm Monitoring Compliance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
7.7 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
7.8 Study Strengths and Weaknesses . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
7.9 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
8 Directions for use . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
9 Connection to other equipment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
10 How supplied . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
11 Packaging and shelf life . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
12 Limited warranty . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
3
Page 4

1 Glossary of symbols
The following table defines symbols that are used on packaging and product labeling. Refer to the labels to determine
which symbols apply to this product and for the product-specific information, such as the date of manufacture.
Symbol
Standard/Standard Title
or Reference
21 CFR 801.109
a
Symbol title/Reference
number Explanatory Text
Prescription only USA Federal law restricts
this device to sale by or on
the order of a licensed
healthcare practitioner.
ISO 15223-1
b
Consult instructions for use
(clause 5.4.3)
Consult instructions for use
at this website:
www.medtronic.com/manuals
ISO 15223-1
b
Catalog number
(clause 5.1.6)
Indicates the manufacturer’s catalog number so the
device can be identified
ISO 15223-1
b
Batch code
(clause 5.1.5)
Indicates the manufacturer’s batch code so that the
batch or lot can be identified
ISO 15223-1
b
Use by date
(clause 5.1.4)
Indicates the date after
which the device is not to be
used
ISO 15223-1
N/A Manufactured in/ manufac-
ISO 15223-1
b
Manufacturer
(clause 5.1.1)
Indicates the medical
device manufacturer
Indicates where the device
turing site
b
Date of manufacture
(clause 5.1.3)
was manufactured
Indicates the date when the
medical device was manufactured
ISO 15223-1
b
Keep Dry
(clause 5.3.4)
Indicates a medical device
that needs to be protected
from moisture
ISO 15223-1
b
Do not use if package is
damaged
(clause 5.2.8)
Indicates a medical device
that should not be used if
the package has been damaged or opened
ISO 15223-1
b
Caution
(clause 5.4.4)
Indicates there is important
cautionary information for
the medical device.
ISO 15223-1
b
Do not re-use
(clause 5.4.2)
Indicates a medical device
that is intended for one use,
or for use on a single patient
during a single procedure
ISO 15223-1
ISO 15223-1
ISO 15223-1
ISO 15223-1
b
b
b
b
Do not resterilize
(clause 5.2.6)
Sterilized using ethylene
oxide
(clause 5.2.3)
Non-pyrogenic
(clause 5.6.3)
Keep away from sunlight
(clause 5.3.2)
Indicates a medical device
that is not to be resterilized
Indicates a medical device
that has been sterilized
using ethylene oxide
Indicates a medical device
that is non-pyrogenic.
Indicates a device that
needs protection from light
sources.
N/A Storage temperature limit Indicates the required tem-
perature range for storing
the device
N/A Transit temperature limit Indicates the required tem-
perature range for transporting the device
N/A Package contents Indicates the components
included in the device package
4
Page 5

Symbol
Standard/Standard Title
or Reference
Symbol title/Reference
number Explanatory Text
N/A Unidirectional catheter Indicates that a unidirec-
tional catheter is included in
the device package
N/A Bidirectional catheter Indicates that a bidirec-
tional catheter is included in
the device package
ISO 7000
c
Product documentation
(symbol 0419)
Indicates that product documentation is included in
the device package
N/A Unidirectional Indicates a catheter that
operates only in one direction
N/A Bidirectional Indicates a catheter that
operates in two directions
ISO 15223-1
b
Sterile barrier
(clause 5.2.13)
Single sterile barrier system
with protective packaging
inside
a
21 CFR 801.109: United States Code of Federal Regulations, Title 21, Food and Drugs
b
ISO 15223-1: Medical devices – Symbols to be used with medical device labels, labeling and information to be supplied
c
ISO 7000: Graphical symbols for use on equipment
2 Device description
The Medtronic DiamondTemp ablation catheter is a sterile, single use, externally irrigated ablation catheter designed
to deliver radiofrequency (RF) energy for cardiac ablation. It is designed to be used in conjunction with the
DiamondTemp generator, DiamondTemp catheter-to-RFG cable, DiamondTemp GenConnect cable, DiamondTemp
EGM cable, DiamondTemp irrigation pump, and DiamondTemp tubing set.
The DiamondTemp ablation catheter is available as unidirectional (Figure 1) or bidirectional (Figure 2) with a distal
electrode segment and a proximal handle that are connected by a torquable shaft. The unidirectional catheter has an
actuation piston to actuate the curve in one direction. The bidirectional catheter has a steering knob to actuate the
curve in either direction and a tension knob to lock the curve. (Refer to Table 1 for model information.) The distal tip and
ring electrodes of the catheter are designed to record intracardiac electrocardiogram (ECG) signals for mapping and
stimulus delivery. RF energy delivery by catheter tip-to-tissue contact is similar to other commercially available,
externally irrigated catheters. Refer to the catheter package labeling for electrode spacing details.
Table 1. Catheter models and specifications
Description Specification
Catheter shaft size (outer diameter) 2.67 mm (8 Fr)
Catheter ablation tip size (tip electrode) 2.50 mm (7.5 Fr)
Length 110 cm (43.3 in)
Compatible sheath 2.83 mm (8.5 Fr)
Model CEDT100S Unidirectional, small curve (45 mm)
Model CEDT200L Unidirectional, large curve (63 mm)
Model CEDTB300S Bidirectional, small curve (45 mm)
Model CEDTB400L Bidirectional, large curve (63 mm)
The catheter is constructed of thermoplastic elastomer materials and noble metal electrodes. Thermocouples are
incorporated at the distal and proximal end of the tip electrode for temperature sensing during RF ablation. The highest
temperature from all thermocouples is displayed on the generator. The generator modulates the delivered RF energy
to maintain tissue temperature at a user-defined temperature set-point.
The catheter, when connected to the tubing set and irrigation pump, delivers normal saline (0.9%) with Heparin at 1
IU mL via a lumen in the catheter. Saline is delivered to the tissue and through 6 ports located on the catheter tip. The
saline cools the catheter tip and tip-tissue interface. One luer connection at the proximal end of the handle connects
to the tubing set, allowing the irrigation pump to generate the flow of normal saline to the catheter.
5
Page 6

Figure 1. DiamondTemp Ablation Catheter (unidirectional)
1 Catheter shaft
2 Shaft strain relief
3 Actuation knob
Figure 2. DiamondTemp Ablation Catheter (bidirectional)
1 Catheter shaft
2 Strain relief
3 Steering knob
4 Handle
5 Cooling lumen
4 Tension knob
5 Cooling lumen
3 Indications for use
The DiamondTemp catheter is indicated for use in cardiac electrophysiological mapping (stimulation and recording)
and for treatment of drug refractory, recurrent, symptomatic paroxysmal atrial fibrillation when used in conjunction with
the DiamondTemp RF generator and accessories (DiamondTemp catheter-to-RF generator cable, DiamondTemp
GenConnect cable, DiamondTemp EGM cable, DiamondTemp irrigation pump, DiamondTemp irrigation tubing set)
and compatible mapping system.
4 Contraindications
• Patients with active systemic infection
• Patients with prosthetic valves
• Patients with intracardiac thrombus or myxoma, or interatrial baffle or patch via transseptal approach
• Patients unable to receive heparin or an acceptable alternative to achieve adequate anticoagulation
• Pregnant women and children <18 years of age
• Patients who are hemodynamically unstable
5 Warnings and precautions
• Before use, inspect the catheter for any foreign particles or material, defects, or physical damage, including
electrical insulation of the cable. If the catheter is defective or damaged, do not use and contact a Medtronic
representative. Replace damaged devices or equipment as necessary.
• Carefully read all DiamondTemp ablation system (catheter, generator, catheter-to-RFG cable, GenConnect cable,
EGM cable, irrigation pump, tubing set) instructions before use. Observe all contraindications, warnings, and
precautions noted in the directions. Failure to do so may result in patient complications.
• If the DiamondTemp system is used in conjunction with a compatible mapping system (such as the Abbott
EnSite™ Velocity™ or Precision™ Mapping System), consult the respective instructions to ensure correct
connectivity and usage. Construct an anatomic map of the region of interest only after all mapping catheters and
electrodes, the DiamondTemp catheter, respective cables, and neutral electrodes (including the ablation return
pad) are completely and properly connected. The subsequent addition of catheters or electrodes may render the
anatomic map inaccurate and require remapping.
• Carefully read the instructions for all ancillary devices or products used in the procedure before use.
• The catheter is for single use only. Do not reprocess or resterilize. Reusing, reprocessing, or resterilizing may
compromise the structural integrity of the device or lead to product failure, which may result in patient injury, illness,
or death. Reuse, reprocessing, or resterilizing may also create a risk of contamination of the device. Contamination
may lead to injury, illness, or death of the patient.
• Use the catheter before the “Use by” date on the device package.
• Cardiac ablation procedures should be performed only by physicians trained in the techniques of RF catheter
ablation in a fully equipped electrophysiology (EP) laboratory.
• Pacemakers and implantable cardioverter defibrillators (ICDs) can be adversely affected by RF signals and
appropriate precautions should be taken before, during, and after ablation to minimize risk.
6
Page 7

• Implantable devices such as ICDs should be deactivated or programmed to the OFF mode before ablation.
Perform complete ICD system analysis on all patients after ablation.
• Use careful catheter manipulation and awareness when in close proximity to atrial or ventricular leads.
• Anticoagulation therapy is required before device introduction to prevent thrombus formation.
• Catheter ablation procedures present a risk of significant x-ray exposure, which can result in acute radiation injury
as well as increased risk for somatic and genetic effects to both patients and laboratory staff. Catheter ablation
should only be performed after adequate attention has been given to the potential exposure associated with the
procedure and steps taken to minimize this exposure.
• Long-term risks of RF ablation lesions have not been established.
• To avoid risk of injury, do not ablate near the phrenic nerve.
• Ablation too close to the esophageal area can result in esophageal fistula.
• Use caution when placing lesions in the proximity of the cardiac conduction system.
• Ablation near the AV node can cause permanent or partial conduction block.
• Ablation within and in close proximity to the coronary arterial vasculature has been associated with myocardial
infarction and death.
• Catheter materials are not compatible with magnetic resonance imaging (MRI).
• In accordance with hospital protocol, monitor the patient’s fluid balance throughout the procedure to avoid fluid
overload.
• Catheter entrapment within the heart or blood vessels is a possible complication of EP procedures.
• Do not use excessive force when inserting, advancing, or removing the catheter.
• Before insertion into the patient, flush the catheter by pressing the Purge Flow button on the irrigation pump, at a
flow rate of 60 mL/min, and ensure there are no air bubbles.
• To ensure proper operation of the tissue contact impedance measurement function, all 4 electrodes and 6
thermocouples on the catheter tip must protrude from the distal tip of the sheath.
• Ablation over other catheter electrodes (on a diagnostic catheter, for example) has been associated with
unintended EGM noise.
• Carefully monitor the tissue contact impedance before delivery of RF energy. Do not place the RF electrode in
proximity to any other mapping or ablation electrodes, as this may cause inadvertent, ineffective, or unsafe tissue
ablation and may increase chances of char, coagulum, or steam pops.
• If the contact impedance reads less than 35 Ω, the generator will not allow delivery of RF energy. In such
circumstances, replace the catheter. If after replacing the catheter the condition persists, replace the
catheter-to-RFG cable and the GenConnect cable (if used). If the condition still persists, power down the
generator and contact a Medtronic representative.
• Although a high contact impedance value typically indicates acceptable tissue contact, and low contact
impedance values typically indicate lack of tissue contact, caution should be exercised. Areas of previously
ablated tissue may display a low contact impedance value. Other parameters, such as EGM, fluoroscopic images,
and intracardiac ultrasound should be monitored before deciding to apply RF.
• Do not deliver RF energy if the catheter is outside the target site. The RF generator can deliver significant electrical
energy and may cause patient injury.
• Use caution during multiple sheath and catheter exchanges through the transseptal puncture. Caution is
necessary to avoid causing a residual atrial septal defect that would require repair.
• The transseptal procedure presents the potential risk for an air embolus, which may involve the coronary arteries.
Aspiration and flushing of the sheath, dilator, and needle should be performed during insertion or exchange, to
minimize this risk. Refer to the individual sheath, dilator, and needle instructions.
• Fibrin may accumulate in or on the sheath and catheter assembly during the procedure. Aspirate when removing
the dilator or catheter. Follow the sheath instructions for maintaining sheath patency during use.
• Stimulation of cardiac tissues caused by pacing stimulus or RF energy may lead to inadvertent induction of
arrhythmias. These arrhythmias may require defibrillation that could also result in skin burns.
• Do not use the catheter for epicardial ablation.
• Using ablation parameters (such as temperature set-point, ablation duration, or irrigation flow rate) other than
those recommended by Medtronic may be hazardous to patients. Exercise caution and sound medical reasoning
when deciding to deviate from recommended parameters.
• Delivery of RF energy may result in neuromuscular stimulation. Monitor patient reactions.
• The power, temperature, and impedance display of the generator should be continuously monitored during RF
energy delivery.
• Irrigated ablation systems have been shown to create larger lesions than standard RF ablation catheters. Use
caution when ablating near electrically vulnerable, thin-walled, or other arterial structures.
• During use of the RF generator and irrigation pump, pay attention to all messages, error codes, warnings, and
tones, and exercise caution as needed.
• Perform catheter advancement under fluoroscopic guidance in conjunction with careful manipulation,
electrograms, and impedance monitoring to minimize the risk of cardiac damage, perforation, or tamponade.
7
Page 8

• Tip-to-tissue contact impedance is actively monitored only before and after ablation. During ablation, use caution
when the temperature drops suddenly. A drop in temperature may be associated with loss of tissue contact.
• In case of steam pop or automatic shut off, discontinue RF energy. Remove the catheter for visual inspection and
check for coagulum, charring, or other catheter defects.
• Always straighten the catheter before insertion or withdrawal from the patient.
• Always maintain irrigation flow to prevent coagulation within and around electrodes.
• If the irrigation flow is interrupted, withdraw the catheter from the patient, visually inspect the tip, and then flush the
catheter before reinsertion and before resuming the procedure.
• Do not use the catheter if it appears damaged or kinked, or if there is any difficulty in deflecting the distal section
to achieve the desired curve.
• Do not use if the catheter irrigation ports are blocked.
• Use fluoroscopy or information from a mapping system to confirm the position of the catheter during the procedure.
• Manual prebending of the distal curve may damage the steering mechanism and may cause patient injury.
• Do not immerse the proximal handle or cable connectors in fluid. Ensure that the cable and catheter connections
remain dry throughout the procedure.
• Position connecting cables to avoid contact with the patient and other electrical leads.
• Do not attempt ablation with the catheter without the use of the DiamondTemp irrigation pump, the DiamondTemp
generator, and approved accessories.
• After use, dispose of the catheter and packaging in accordance with hospital, administrative, and local
government policy.
• Do not expose the catheter to organic solvents such as alcohol.
6 Potential adverse events
The following potential adverse events are associated with cardiac ablation procedures:
• Abnormal vision
• Air embolism
• Anaphylaxis
• Anemia
• Aneurysm
• Angina
• Arrhythmia (including new or worsening of existing condition, or requiring cardioversion)
• Arterial or venous thrombus
• Atrial septal defect
• AV fistula
• Cardiac arrest
• Cardiac tamponade
• Catheter entrapment leading to valve or heart wall damage
• Catheter insertion site hematoma
• Chest pain (non-specific)
• Congestive heart failure exacerbation
• Component damage to ICD or pacemaker
• Coronary artery dissection
• Death
• Dislodgement of implantable device or permanent pacing lead
• Dizziness
• Embolic events, including infarction of other tissues, coronary, pulmonary, and bowel structures
• Endocarditis
• Esophageal damage or necrosis
• Exacerbation of COPD
• Exacerbation of pre-existing atrial fibrillation
• Fluid overload
• Gastroparesis or GI event
• Hemorrhage
• Hemothorax
• Hypotension
• Hypoxia
• Inadvertent AV block
8
Page 9

• Infection
• Myocardial infarction
• Neck, back, or groin pain
• Palpitations
• Perforation (cardiac)
• Pericardial effusion
• Pericarditis
• Peripheral venous thrombosis
• Phrenic nerve damage
• Pleural effusion
• Pneumonia
• Pneumothorax
• Pseudoaneurysm
• Pulmonary edema
• Pulmonary vein stenosis
• Radiation injury resulting in dermatitis, erythema, etc.
• Renal insufficiency or failure
• Respiratory failure
• Seizure
• Sepsis
• Skin burns
• Stroke or cerebrovascular incident
• Syncope
• Thromboembolic event
• Transient ischemic attack
• Vasovagal reaction
• Ventricular arrhythmia
• Vessel wall or valvular damage or insufficiency
7 Clinical data
Study title – DIAMOND-AF: A Randomized Controlled Clinical Evaluation of the DiamondTemp Ablation System for
the Treatment of Paroxysmal Atrial Fibrillation
Number of centers – 23 centers in the United States, Canada, and Europe
Number of subjects – 482 enrolled/randomized subjects globally. There were 243 enrolled/randomized subjects in
the control (TactiCath) group and 239 enrolled/randomized subjects in the investigational (DiamondTemp) group.
Study purpose – The purpose of DIAMOND-AF was to provide data demonstrating the safety and effectiveness of
the DiamondTemp Ablation System for the treatment of drug refractory, recurrent, symptomatic paroxysmal atrial
fibrillation (PAF). The study was considered successful if the investigational device was considered non-inferior to the
control device for the primary safety and effectiveness endpoints.
7.1 Study Design, Study Population, Study Visits, and Length of Follow-up Methods
The Diamond-AF Study was a prospective, single-blind, 1:1 randomized, controlled trial performed at multiple centers
in the United States, Canada and Europe. The primary focus of the left atrial ablation procedure was to create a series
of point-by-point RF lesions encircling the left and right PVs to achieve electrical PVI from the rest of the left atrium (LA).
All subjects were followed per protocol in relation to the date of the index ablation procedure. Follow-up was required
prior to hospital discharge and at 7 days, 1 month, 3 months, 6 months, and 12 months post-ablation. Subjects were
provided a cardiac event monitor at the hospital pre-discharge visit to be used throughout the duration of the study. This
data was transmitted to and read at an ECG core lab.
9
Page 10

7.2 Study Endpoints
7.2.1 Primary Endpoints
7.2.1.1 Primary Effectiveness Endpoint
The primary effectiveness endpoint was defined as freedom from documented AF, atrial flutter1 (AFL) and atrial
tachycardia (AT) episodes following the blanking period (3-month follow-up post-ablation procedure) through the end
of the effectiveness evaluation period (12-month follow-up post-ablation procedure).
An effectiveness failure was defined by any of the following events:
• Inability to electrically isolate all accessible targeted pulmonary veins during the ablation procedure2.
• Documented episodes of AF, AFL or AT lasting ≥ 30 seconds in duration as evidenced by electrocardiographic
data during the effectiveness evaluation period
• DC cardioversion for AF, AFL or AT during the effectiveness evaluation period
• A repeat ablation procedure to treat AF, AFL or AT during the effectiveness evaluation period
• Use of a new or modification to existing Class I-IV anti-arrhythmic drug (AAD) regimen to treat AF, AFL or AT
recurrence during the effectiveness evaluation period
• Use of a non-study device for ablation of any AF targets during the index or repeat ablation procedure during the
blanking period
• More than one (1) repeat ablation procedure during the blanking period.
7.2.1.2 Primary Safety Endpoint
The primary safety endpoint was defined as freedom from a composite of serious adverse events (SAE) occurring
within 30 days and clinically symptomatic pulmonary vein stenosis through 6 months post-index ablation procedure,
as adjudicated by an independent Clinical Events Committee (CEC) for relatedness to the procedure or device.
The primary safety device- or procedure-related SAE composite was a combined rate of the following events:
• Atrioesophageal fistula
• Bleeding complication
• Cardiac tamponade / perforation
• Death
• Extended hospitalization
• Myocardial infarction
• Pericarditis
• Phrenic nerve paralysis
• Pulmonary edema
• Pulmonary vein stenosis
• Stroke post-ablation
• Thromboembolism
• Transient ischemic attack (TIA) post-ablation
• Vagal nerve injury
• Vascular access complications
7.2.2 Secondary Endpoints
Secondary Endpoints and Results
There were seventeen (17) pre-defined secondary endpoints in the Diamond-AF Study, four (4) of which included
hypothesis testing.
Secondary endpoints to characterize the performance of the DiamondTemp Ablation System, relative to the control
device, include:
• Mean duration of individual RF ablations (seconds).
• Mean cumulative RF time per procedure (minutes).
• Freedom from a composite of SAE occurring within 7 days post-index ablation procedure as adjudicated by an
independent CEC for relatedness to the procedure or device.
• Freedom from documented AF, AT and AFL3 episodes following the blanking period through 12 month follow-up
post-ablation procedure in the absence of class I and III anti-arrhythmic drug therapy.
1
Occurrence and/or ablation of cavotricuspid isthmus (CTI)-dependent AFL, as confirmed by entrainment maneuvers during EP
testing at any time during this study was not a primary effectiveness failure because it was not considered an iatrogenic arrhythmia
following a left atrial ablation procedure for AF.
2
Electrical isolation as confirmed by demonstration of exit and/or entrance conduction block.
10
Page 11

• Rate of acute procedural success, defined as confirmation of electrical isolation of PVs via assessment of
entrance block at least 20 minutes following the last ablation around the respective PV.
• Rate of single procedure success defined as the rate of subjects treated with one single ablation procedure during
study participation and with freedom from documented AF, AT and AFL3 at 12 months.
• Rate of single procedure success defined as the rate of subjects treated with one single ablation procedure during
study participation and with freedom from ALL primary effectiveness endpoint failure criteria.
• Rate of occurrence of electrically reconnected PVs following a 20-minute waiting period assessed by entrance
block at index procedure.
• Accumulated changes in Quality of Life (QOL) using the AF QOL Survey (AFEQT Questionnaire) from baseline
through 6 and 12 months following ablation procedure.
• Neurological changes measured using the NIH stroke scale between baseline and post-ablation (pre-discharge
visit) and at 12 months post-ablation procedure.
• Total procedure time (minutes), defined as time of first assigned ablation catheter insertion into the vasculature to
time of last procedural ablation catheter removed.
• Time to achieve initial PVI at index procedure (minutes), defined as time of delivery of first RF ablation with the
assigned ablation catheter until confirmation of PVI.
• Total treatment device time (minutes), defined as time of delivery of first RF ablation with the assigned ablation
treatment catheter to removal of the treatment catheter.
• Total number of RF ablations per procedure.
• Total fluid infused through the assigned ablation catheter (mL).
• Total fluoroscopy time (minutes).
• Number of re-hospitalizations due to atrial fibrillation recurrence after blanking period.
7.3 Total Number of Enrolled Study Sites and Subjects, Subject Accountability, and Follow-up Rate
Investigators at 23 participating study sites in the United States, Canada, and Europe enrolled/randomized a total of
482 subjects.
Four hundred eighty five (485) subjects signed an informed consent to participate in the DIAMOND-AF clinical study.
Four hundred eighty two (482) subjects were enrolled/ randomized in the Diamond-AF clinical study (Intention-to-Treat
Cohort). Two hundred thirty nine (239) subjects were randomized to treatment with the investigational DiamondTemp
catheter and two hundred forty three (243) subjects were randomized to treatment with the control TactiCath catheter
system.
Enrolled/randomized subject accountability is summarized in Table 2, Table 3, and Figure 3.
• Consented subjects: All subjects who signed a consent form.
• Intention-to-Treat (ITT) analysis cohort: The ITT cohort comprised of all randomized subjects regardless of
whether they received study treatment, with analyses conducted according to the randomized treatment
assignment.
• Safety analysis cohort: The 476 subjects who had a study ablation catheter inserted comprise the Safety Analysis
cohort.
Of the 239 subjects randomized to the DiamondTemp group, 225 completed the study with 14 (5.9%) exited
prematurely. Likewise, of the 243 subjects randomized to the control group, 230 completed the study and 13 (5.3%)
exited prematurely. Of the 482 subjects in ITT, 476 subjects had exposure to a study device (ablation catheter).
Table 2. Subject Disposition
Subject Disposition Number (N)
Number of Subjects with Signed Consent 485
Number of Subjects Not Randomized 3
Documented Stroke, CVA, TIA or Suspected Neurological Event
Enrollment Cap Met
Regularly Prescribed Amiodarone
a
b
c
1
1
1
Number of Subjects Enrolled/Randomized 482
3
Occurrence and/or ablation of cavotricuspid isthmus (CTI)-dependent AFL, as confirmed by entrainment maneuvers during EP
testing at any time during this study was not a primary effectiveness failure because it was not considered an iatrogenic arrhythmia
following a left atrial ablation procedure for AF.
11
Page 12

Table 2. Subject Disposition (continued)
Investigational/DiamondTemp
N=239
Treatment Not Attempted/
Study Exit
N=4 [a]
Treatment Not Delivered
N=0
Treatment Not Attempted/
Study Exit
N=2 [b]
Treatment Not Delivered
N=2 [c]
Control/TactiCath
N=243
Enrolled/Randomized
N=482
Treatment Attempted
N=235
Treatment Attempted
N=241
Pre-Discharge Visit
N=235
7-Day Visit
N=234
1-Month Visit
N=227
Study Exit
N=1
Study Exit
N=7
Study Exit
N=2
Study Exit
N=4
Study Exit
N=1
Study Exit
N=3
Study Exit
N=3
3-Month Visit
N=226
6-Month Visit
N=222
12-Month Visit
N=225
Study Completed/Study Exit
N=225
Pre-Discharge Visit
N=241
7-Day Visit
N=238
1-Month Visit
N=236
3-Month Visit
N=230
6-Month Visit
N=223
12-Month Visit
N=230
Study Completed/Study Exit
N=230
Subject Disposition Number (N)
(Intention-to-Treat Analysis Cohort)
a
Subject 15-021 met exclusion criterion #24
b
Subject 15-010 met all criteria but was never randomized prior to study exit (30-Oct-2018), with a reason of “Subject’s ablation was not
scheduled and patient was not randomized prior to enrollment number being met”
c
Subject 17-001 met exclusion criterion #19
Table 3. Scheduled Visit Compliance
Visit Control (N=243) DiamondTemp (N=239) All Subjects (N=482)
Enrolled/Randomized 243 (100%) 239 (100%) 482 (100%)
Ablation Procedure 241 (99.2%) 235 (98.3%) 476 (98.8%)
Pre-Discharge Visit 241 (99.2%) 235 (98.3%) 476 (98.8%)
7 Day Visit 238 (97.9%) 234 (97.9%) 472 (97.9%)
1 Month Visit 236 (97.1%) 227 (95.0%) 463 (96.1%)
3 Month Visit 230 (94.7%) 226 (94.6%) 456 (94.6%)
6 Month Visit 223 (91.8%) 222 (92.9%) 445 (92.3%)
12 Month Visit 230 (94.7%) 225 (94.1%) 455 (94.4%)
Study Completion
Completion of Study as
230 (94.7%) 225 (94.1%) 455 (94.4%)
Planned
Discontinued Prematurely 13 (5.3%) 14 (5.9%) 27 (5.6%)
Figure 3 study flowchart shows subject accountability from enrollment to 12 months follow-up/study completion.
Figure 3. Study Flowchart of Subject Population, from Enrollment to 12 Month Visit
12
Page 13

[a] Subject withdrew consent (06-013); Subject progressed to persistent AF (11-001); Enrollment closure (09-003,
13-015).
[b] Enrollment closure (10-004); Physician no longer believed the subject was a good candidate for the study (11-004).
[c] Treatment attempted but not delivered with TactiCath due to technical difficulties (09-001); Treatment attempted but
not delivered with TactiCath due to procedure failure (22-005).
Follow-up visit compliance was 91.8% or higher for all follow-up visits, with 94.7% of control subjects and 94.1% of
DiamondTemp subjects completing the study as planned through the 12 month follow-up visit.
7.4 Baseline Characteristics
Table 4 shows demographic information for subjects by treatment group in the Intention-to-Treat cohort. Demographic
data was balanced with no significant differences between the treatment groups.
Table 4. Demographic Characteristics, Intention-to-Treat Cohort, Control vs DiamondTemp
Control
Demographics
Age, years
Mean (SEM / SD) 63.0 (0.67 / 10.42) 62.3 (0.72 / 11.13)
Median 64.0 65.0
Min, Max 27.0, 84.0 22.0, 82.0
N (N Missing) 243 (0) 239 (0)
Sex, n (%)
Male 143 (58.8%) 136 (56.9%)
Female 100 (41.2%) 103 (43.1%)
Race, n (%)
American Indian or Alaska Native 2 (0.8%) 3 (1.3%)
Asian 2 (0.8%) 0 (0%)
Black or African American 4 (1.6%) 4 (1.7%)
Other, specify: Caribbean 0 (0%) 1 (0.4%)
Other, specify: Ecuadorian 0 (0%) 1 (0.4%)
Prefer Not to Say 68 (28.0%) 66 (27.6%)
Unknown 6 (2.5%) 5 (2.1%)
White 161 (66.3%) 159 (66.5%)
Ethnicity, n (%)
Hispanic or Latino 5 (2.1%) 6 (2.5%)
Not Hispanic or Latino 169 (69.5%) 167 (69.9%)
Prefer Not to Say 69 (28.4%) 66 (27.6%)
(N=243)
DiamondTemp
(N=239)
Min = Minimum, Max = Maximum; SD= Standard Deviation; SEM=Standard Error of the Mean.
Notes: N = Number of subjects in the population. n = Number of subjects in the specific category. Percentages are
calculated as 100 x (n/N).
Age is derived from the date of informed consent.
Table 5 shows baseline information for subjects by treatment group in the Intention-to-Treat cohort. Baseline
characteristics were well balanced with no significant differences between the treatment groups.
Table 5. Baseline Characteristics, Intention-to-Treat Cohort, Control vs DiamondTemp
Control
Baseline Characteristics
Height, cm
Mean (SEM / SD) 172.7 (0.63 / 9.89) 172.9 (0.63 / 9.69)
Median 172.7 172.7
Min, Max 152.0, 196.0 147.3, 205.7
N (N Missing) 243 (0) 239 (0)
Weight, kg
Mean (SEM / SD) 85.4 (1.03 / 16.01) 84.1 (1.17 / 18.06)
Median 85.0 83.0
Min, Max 51.0, 146.0 45.8, 131.7
N (N Missing) 243 (0) 239 (0)
(N=243)
13
DiamondTemp
(N=239)
Page 14

Table 5. Baseline Characteristics, Intention-to-Treat Cohort, Control vs DiamondTemp (continued)
Baseline Characteristics
BMI, kg/m
2
Control
(N=243)
DiamondTemp
(N=239)
Mean (SEM / SD) 28.6 (0.29 / 4.48) 28.0 (0.32 / 5.00)
Median 28.1 27.5
Min, Max 19.8, 42.9 14.2, 44.1
N (N Missing) 243 (0) 239 (0)
Serum Creatinine, mg/dL
Mean (SEM / SD) 0.9 (0.01 / 0.22) 0.9 (0.02 / 0.23)
Median 0.9 0.9
Min, Max 0.1, 1.6 0.5, 2.2
N (N Missing) 227 (16) 223 (16)
LVEF, %
Mean (SEM / SD) 60.1 (0.45 / 7.08) 59.8 (0.47 / 7.19)
Median 60.0 60.0
Min, Max 38.0, 80.0 44.0, 82.0
N (N Missing) 243 (0) 235 (4)
LA Diameter, cm
Mean (SEM / SD) 4.1 (0.04 / 0.67) 4.0 (0.04 / 0.59)
Median 4.0 4.0
Min, Max 2.2, 5.5 2.5, 5.5
N (N Missing) 243 (0) 233 (6)
NYHA Functional Class, n (%)
Class I 36 (14.8%) 32 (13.4%)
Class II 20 (8.2%) 22 (9.2%)
Class III 0 (0%) 0 (0%)
Class IV 0 (0%) 0 (0%)
NA (1) 117 (48.1%) 116 (48.5%)
Unknown (2) 70 (28.8%) 69 (28.9%)
Heart Rate, bpm
Mean (SEM / SD) 69.3
(1.23 / 19.23)
69.0
(1.12 / 17.37)
Median 64.0 65.0
Min, Max 36.0, 169.0 35.0, 140.0
N (N Missing) 243 (0) 239 (0)
Systolic BP, mmHg
Mean (SEM / SD) 136.3
(1.25 / 19.41)
138.1
(1.28 / 19.82)
Median 133.0 135.0
Min, Max 92.0, 206.0 77.0, 199.0
N (N Missing) 243 (0) 239 (0)
Diastolic BP, mmHg
Mean (SEM / SD) 77.4
(0.70 / 10.94)
77.5
(0.69 / 10.73)
Median 78.0 78.0
Min, Max 46.0, 110.0 43.0, 104.0
N (N Missing) 243 (0) 239 (0)
CHA2DS2-VASc Score
Mean (SEM / SD) 2.11 (0.10 / 1.50) 1.92 (0.09 / 1.38)
Median 2.0 2.0
Min, Max 0.0, 7.0 0.0, 6.0
N (N Missing) 243 (0) 239 (0)
BMI=Body Mass Index; BP=Blood Pressure; LA=Left Atrium; LVEF=Left Ventricular Ejection Fraction; NYHA=New
York Heart Association.
14
Page 15

Min = Minimum, Max = Maximum; SD = Standard Deviation; SEM=Standard Error of the Mean.
(1) Subjects without heart failure, will have an NYHA result that is not applicable (NA).
(2) NYHA score is missing/not available in source documents.
Table 6 shows years since first diagnosis of atrial fibrillation and history of AAD therapy for subjects by treatment group
in the Intention-to-Treat cohort.
Table 6. History of Atrial Fibrillation and AAD Therapy, Intention-to-Treat Cohort, Control vs DiamondTemp
Medical History
Control
(N=243)
DiamondTemp
(N=239)
Years Since First Diagnosis (years)
Mean (SEM / SD) 4.0 (0.34 / 4.85) 3.5 (0.33 / 4.68)
Median 2 2
Min, Max 0, 26 0, 28
N (N Missing) 205 (38) 207 (32)
AAD Use History
Subjects with History of AAD Use and
243 (100.0%) 239 (100.0%)
Failed/Not Tolerate, n(%)
Subjects with History of Class I/III
191 (78.6%) 187 (78.2%)
AAD Use and Failed/Not Tolerate, (*),
n(%)
Subjects with History of Class II/IV
121 (49.8%) 117 (49.0%)
AAD Use and Failed/Not Tolerate, (*),
n(%)
AAD=Anti-arrhythmic drugs; AF=Atrial Fibrillation; PAF=Paroxysmal Atrial Fibrillation
Min = Minimum, Max = Maximum; SD=Standard Deviation; SEM=Standard Error of the Mean.
Notes: N = Number of subjects in the population. n = Number of subjects in the specific category. Percentages are
calculated as 100 x (n/N).
All other percentages are calculated as 100 x (n/N1). N1 = Number of subjects in category.
(*) Categories are not mutually exclusive and subjects may count in more than one category.
Table 7 shows medical history information for subjects by treatment group in the Intention-to-Treat cohort. Medical
history was well-balanced between treatment groups with the exception of ‘Non-PAF/AFL Arrhythmias or conduction
disturbances’. Medical history details related to a previous left atrial ablation, receipt of a septal closure device or mitral
valve surgical procedure was not collected in this study, as these were study exclusion criterion if they occurred at any
time prior to enrollment.
Table 7. Medical and Smoking History, Intention-to-Treat Cohort, Control vs DiamondTemp
Medical History
Control
(N=243)
DiamondTemp
(N=239)
Atrial Flutter 51 (21.0%) 46 (19.2%)
Hypertension Requiring Medica-
137 (56.4%) 124 (51.9%)
tion
Hypertension Regardless of Medi-
138 (56.8%) 125 (52.3%)
cations Required
Diabetes 28 (11.5%) 20 (8.4%)
Structural Heart Disease 7 (2.9%) 7 (2.9%)
Cerebrovascular Accident/Transi-
20 (8.2%) 12 (5.0%)
ent Ischemic Attack
Thromboembolic Events 6 (2.5%) 4 (1.7%)
Coronary Artery Disease 29 (11.9%) 27 (11.3%)
Myocardial Infarction 2 (0.8%) 4 (1.7%)
Non-PAF/AFL Arrhythmias or con-
42 (17.3%) 22 (9.2%)
duction disturbances
Vascular Disease 20 (8.2%) 13 (5.4%)
Congestive Heart Failure 3 (1.2%) 6 (2.5%)
Previous CABG Procedure 1 (0.4%) 0 (0%)
Previous ICD/CRT/Pacemaker
0 (0%) 1 (0.4%)
Implant
Gastrointestinal (GI) Disease 25 (10.3%) 28 (11.7%)
15
Page 16

Table 7. Medical and Smoking History, Intention-to-Treat Cohort, Control vs DiamondTemp (continued)
Medical History
Pulmonary Disease with Different
Control
(N=243)
4 (1.6%) 6 (2.5%)
DiamondTemp
(N=239)
Etiologies
Other 159 (65.4%) 149 (62.3%)
Smoking History – Yes 75 (30.9%) 71 (29.7%)
Smoking History - Current Smoker 16 (6.6%) 17 (7.1%)
Smoking History - Previously
59 (24.3%) 54 (22.6%)
Smoked
CABG=Coronary Artery Bypass Graft; CRT= Cardiac resynchronization therapy; ICD= implantable cardioverter
defibrillator;
PAF= Paroxysmal Atrial Fibrillation
Notes: N = Number of subjects in the population. n = Number of subjects in the specific category. Percentages are
calculated as 100 x (n/N).
Categories are not mutually exclusive and subjects may count in more than one category.
7.5 Procedural Characteristics
Table 8 shows a summary of index ablation procedure details.
There were 5 (2.1%) subjects with steam pops reported in the control group and 7 (3.0%) subjects with steam pops
reported in the DiamondTemp group. No adverse events were reported as a result of a steam pop that occurred in
either treatment group.
Table 8. Index Ablation Procedure Characteristics, Intention-to-Treat Cohort, Control vs DiamondTemp
Control
Procedural Characteristics
Total Number of AF Index Ablation
Procedures
N1
TEE for LA Thrombus Screening Per-
(N=243)
241 235
236 (97.9%) 234 (99.6%)
formed
Esophageal Monitoring/Protection:
Esophageal Deviation, n (%) 22 (9.1%) 19 (8.1%)
Use of Esophageal Temperature
191 (79.3%) 186 (79.1%)
Probe, n (%)
Ablation Parameter Settings – Max Power Set Point
Mean (SEM/SD) 33.3 (0.39/6.01) 49.7 (0.24/3.74)
Median 30.0 50.0
Min, Max 5.0, 70.0 0.0, 56.0
N (N Missing) 239 (2) 233 (2)
Ablation Parameter Settings – Max Temperature Set Point
Mean (SEM/SD) 44.1 (0.32/4.94) 59.9 (0.10/1.60)
Median 43.0 60.0
Min, Max 30.0, 70.0 43.0, 65.0
N (N Missing) 233 (8) 234 (1)
Non-PVI Ablation Targets
(*)
:
Type I CTI Flutter 69 (28.6%) 74 (31.5%)
Other 5 (2.1%) 3 (1.3%)
Posterior Wall 3 (1.2%) 4 (1.7%)
Focal Triggers 2 (0.8%) 4 (1.7%)
Roof 2 (0.8%) 2 (0.9%)
SVC 3 (1.2%) 1 (0.4%)
CFAE 2 (0.8%) 1 (0.4%)
Flutter Line 2 (0.8%) 1 (0.4%)
Mitral Isthmus Line 2 (0.8%) 1 (0.4%)
AVRT/AVNRT 1 (0.4%) 1 (0.4%)
Incidence of Steam Pops, n (%) 5 (2.1%) 7 (3.0%)
Incidence of Char or Coagulum, n (%) 0 (0%) 0 (0%)
DiamondTemp
(N=239)
16
Page 17

AF=Atrial Fibrillation; CTI=Cavotricuspid Isthmus; LA= Left Atrium; PV=Pulmonary Vein; TEE =Transesophageal
echocardiography.
Min = Minimum, Max = Maximum; SD=Standard Deviation; SEM=Standard Error of the Mean.
Notes: N = Number of subjects in the Intention-to-Treat Population.
n = Number of subjects in the specific category. Percentages are calculated as 100 x (n/N1). N1 = Number of subjects
in category.
(*) Categories are not mutually exclusive and subjects may count in more than one category.
Table 9 presents the additional intervention performed to maintain sinus rhythm during the blanking
period. Comparable number of subjects between the two arms received repeat ablation or cardioversion before the
evaluation period.
Table 9. Additional Treatment During the Blanking Period
Subjects with Additional
Treatment during Blank-
ing Period Control (N=243) DiamondTemp (N=239) P-value
a
Repeat ablation 11 (4.53%) 10 (4.18%) 1.00
Cardioversion 11 (4.53%) 7 (2.93%) 0.47
a
P-value is calculated using two-sided Fisher’s exact test
7.6 Rhythm Monitoring Compliance
Post-ablation rhythm monitoring included symptomatic and twice monthly symptomatic/asymptomatic event monitor
transmissions during the evaluation period, ECG at 3, 6, and 12 months, and 24-hour Holter monitor at 6 and 12
months.
The rhythm monitoring compliance was similar between the two treatment groups with regard to 12-lead ECGs,
24-hour Holter monitors, and trans-telephone monitors (Table 10 and Table 11).
Table 10. Rhythm Monitoring Compliance, 12-Lead ECGs and Holter from CRF Data, Intention-to-Treat Cohort, Control vs DiamondTemp
Rhythm Monitoring
Method
Control
(N=243)
n (%)
DiamondTemp
(N=239)
n (%)
All Subjects
(N=482)
n (%)
12-Lead ECGs
3 Month Visit 12-Lead
219/237 (92.4%) 209/235 (88.9%) 428/472 (90.7%)
ECG
6 Month Visit 12-Lead
206/234 (88.0%) 207/228 (90.8%) 413/462 (89.4%)
ECG
12 Month Visit 12-Lead
224/231 (97.0%) 223/227 (98.2%) 447/458 (97.6%)
ECG
24-hour Holter Monitor
6 Month Visit 24-hour
202/234 (86.3%) 199/228 (87.3%) 401/462 (86.8%)
Holter Monitor
12 Month Visit 24-hour
213/231 (92.2%) 204/227 (89.9%) 417/458 (91.0%)
Holter Monitor
ECG= Electrocardiogram;
Notes: N = Number of subjects in the Intention-to-Treat Population. n = Number of subjects in the specific category. For
ECG and Holter, percentages for populations are calculated as 100 x (n/expected number of measurements at that
visit).
Six subjects were randomized/enrolled but did not undergo index ablation procedure (06-013, 09-003, 10-004,
11-001, 11-004, 13-015).
Table 11. Rhythm Monitoring Compliance, Trans-telephonic Monitor (TTM), Intention-to- Treat Cohort, Control vs. DiamondTemp
Rhythm Monitoring - TTM
Control
(N=243)
DiamondTemp
(N=239)
All Subjects
(N=482)
TTMs
(Total) Transmitted TTMs 5419 4557 9976
Expected TTMs
Overall TTM Compliance (Subject) (%)
[1]
4373 4288 8661
[2]
Mean (SEM/SD) 60.5 (2.02/31.01) 61.3 (2.09/32.03) 60.9 (1.45/31.49)
17
Page 18

Table 11. Rhythm Monitoring Compliance, Trans-telephonic Monitor (TTM), Intention-to- Treat Cohort, Control vs.
DiamondTemp (continued)
Rhythm Monitoring - TTM
Control
(N=243)
DiamondTemp
(N=239)
All Subjects
(N=482)
Median 70.0 72.2 72.2
Min, Max 0.0, 100.0 0.0, 100.0 0.0, 100.0
N (N Missing) 236 (7) 235 (4) 471 (11)
TTM= Trans-telephonic Monitor.
Six subjects were randomized/enrolled but did not undergo index ablation procedure (06-013, 09-003, 10-004,
11-001, 11-004, 13-015).
[1] Subject Expected TTM is 2 if the subject’s study participation in a given month is longer than 15 days, otherwise it
is 1 for that month.
[2] Overall compliance is defined on a per subject basis and is based on a subject average monthly compliance rates
over months 1 through 10 after the blanking period. A subject’s monthly compliance rate is defined as minimum (TTM
transmitted for that month, TTM expected for that month)/TTM expected for that month, over months 1 through 10 after
the blanking period.
7.7 Results
7.7.1 Safety Results
7.7.1.1 Primary Safety Endpoint
The primary safety analysis includes all randomized ITT subjects (243 Control and 239 DiamondTemp). There were
16 (6.6%) Control subjects and 8 (3.3%) DiamondTemp subjects that experienced at least one CEC-adjudicated
primary safety endpoint event that contributed to the primary safety endpoint. The primary safety event freedom rate
was 96.7% for the DiamondTemp group and 93.4% for the Control group. The difference (DiamondTemp – Control) in
the primary safety endpoint freedom was 3.24% (95% CI: -1.32%, 7.79%), and the lower 97.5% confidence bound
of -1.32% exceeded the pre-specified non-inferiority margin (NIM) of -6.5%. The primary safety endpoint was met (p
< 0.0001, Table 12).
Table 12. Summary of CEC Adjudicated Adverse Events Contributing to the Primary Safety Endpoint, Intention-toTreat Cohort, Control vs DiamondTemp
Control DiamondTemp
CEC Adjudicated Adverse Events
Contributing to the Primary Safety
Endpoint
By Subject
(Ns=243)
n (%)
By Subject
(Ns=239)
n (%)
Atrioesophageal Fistula 0 (0%) 0 (0%)
Bleeding Complication 0 (0%) 0 (0%)
Cardiac Tamponade/Perforation 2 (0.8%) 2 (0.8%)
Cardiovascular-Related Death
0 (0%) 0 (0%)
Post-Ablation
Clinically Symptomatic Pulmonary
0 (0%) 0 (0%)
Vein Stenosis at 6 Months PostIndex Ablation Procedure
Extended Hospitalization 6 (2.5%)
a
0 (0%)
Myocardial Infarction 0 (0%) 0 (0%)
Pericarditis 1 (0.4%) 0 (0%)
Phrenic Nerve Paralysis 0 (0%) 1 (0.4%)
Pulmonary Edema 1 (0.4%) 0 (0%)
Stroke Post Ablation 1 (0.4%) 0 (0%)
Thromboembolism 0 (0%) 0 (0%)
Transient Ischemic Attack 1 (0.4%) 2 (0.8%)
Vagal Nerve Injury 0 (0%) 1 (0.4%)
Vascular Access Complication 4 (1.6%) 2 (0.8%)
Total 16 (6.6%) 8 (3.3%)
a
Reasons for extended hospitalization include hematoma, pericardial effusion (< 1 cm), fever and chill, bladder outlet obstruction with
UTI, hypotension, chest pain.
Notes: Ns = Number of subjects in the population. n = Number of subjects in the specific category.
Subject based percentages are calculated as 100 x (n/Ns).
18
Page 19
For the ‘by Subject’ columns, subjects reporting a particular adverse event more than once are only counted once by
X: Between Treatment Difference and 95% Confidence Interval: 3.24% (-1.32%, 7.79%)
[DiamonTemp is better]0-6.5%[Control better]
Confidence Intervals are based on Farrington and Manning’s Likelihood Score Test
the event category.
Table 13 and Figure 4 display the primary safety objective results for the ITT cohort. The primary safety event freedom
rate was 96.7% for the DiamondTemp group and 93.4% for the control group. The DiamondTemp minus control group
primary safety endpoint freedom rate was 3.24% with a two-sided 95% confidence interval of -1.32% to 7.79%. Since
the lower two-sided 95% confidence limit of -1.32% exceeded the non-inferiority margin of -6.5%, the primary safety
objective was met (p<0.0001).
Table 13. Primary Safety Result, Intention-to-Treat Cohort
Primary Safety
Endpoint: Freedom
from Primary
Safety Event as
Adjudicated by the
CEC
Total, By Subject 227 (93.4%)
Control
(N=243)
Number (%) (95%
CI)
(89.5%, 96.2%)
DiamondTemp
(N=239)
Number (%)
(95% CI)
231 (96.7%)
(93.5%, 98.5%)
Difference
(95% CI)
3.24%
(-1.32%, 7.79%)
Farrington- Manning p-value
(Non-inferiority
Test)
<0.0001
Figure 4. Primary Safety Freedom Rate: Between Treatment Difference
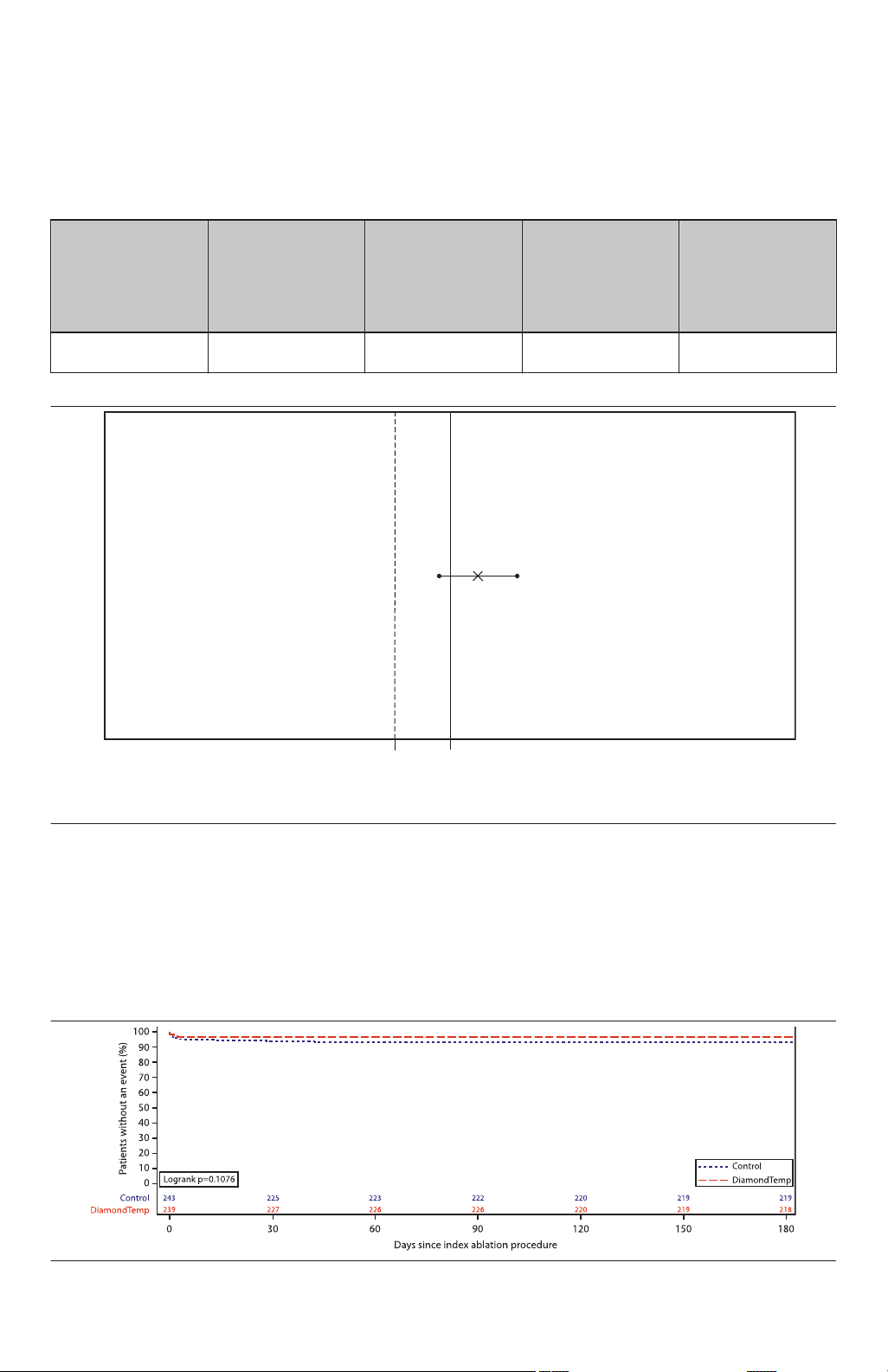
A Kaplan-Meier analysis was also performed to evaluate the primary safety endpoint as a sensitivity analysis. The
Kaplan-Meier method allows subjects to be included in the analysis up until the time they fail the primary safety
endpoint or are censored due to premature study exit.
Figure 5 displays the Kaplan-Meier estimates for the freedom from primary safety event through 6 months (180 days)
post-index ablation procedure; the entire time period for which subjects were at risk for a primary safety event. Based
on the Kaplan-Meier methodology the freedom from primary safety event at 6 months was 97% for the DiamondTemp
group and 93% for the control group. The log-rank test indicated that there was no difference in the freedom from
primary safety event between groups (p=0.11).
Figure 5. Kaplan-Meier Survival: Time to Failure of the Primary Safety Endpoint, Intention-to-Treat Cohort
19
Page 20
Kaplan-Meier Survival: Time to Failure of the Primary Safety Endpoint, Intention-to-Treat Cohort
Month 1 Month 3 Month 6
Number at Risk 225 222 219
Control
Kaplan-Meier Estimate
0.94 0.93 0.93
Standard Error 0.0155 0.0160 0.0160
Number at Risk 227 226 218
DiamondTemp
Kaplan-Meier Estimate
0.97 0.97 0.97
Standard Error 0.0117 0.0117 0.0117
Month 1 (day 30), Month 3 (day 90), Month 6 (day 180)
Table 14 displays the primary safety endpoint status by treatment group and geography and indicates that the primary
safety endpoint results were consistent by geography (Breslow-Day p-value = 0.54).
Table 14. Primary Safety Event Outcome: Relative Risk of Success; Overall and Stratified by Geographic Region and Treatment, Intention-to-Treat Cohort
Geographic
Region
Treatment
PSE Success PSE Failure Total
Relative Risk
of Success
Breslow Day
Test
p-value
Europe DiamondTemp 124 5 129 1.02 0.5425
Control 124 8 132
DiamondTemp 107 3 110 1.05
North America Control 103 8 111
Overall DiamondTemp 231 8 239 1.03
Control 227 16 243
Pooled / Adjusted (CMH)
a
CMH= Cochran-Mantel-Haenszel
a
1.03
The DIAMOND-AF Clinical Study met its primary safety objective (Intention-to-Treat Cohort). Primary safety endpoint
success was observed in 227 (93.4%) control (TactiCath) subjects and 231 (96.7%) DiamondTemp subjects (95% CI
for difference: -1.3% to 7.8%; p<0.0001 for non-inferiority). There was no evidence of heterogeneity in primary
effectiveness outcome between treatment groups by geography (p=0.54). The DiamondTemp Ablation System
demonstrated a reasonable assurance of safety for the treatment of drug refractory, recurrent, symptomatic PAF.
7.7.1.2 Summary of Adverse Events
Adverse events occurring during the study were continuously monitored and collected. There were no Unanticipated
Adverse Device Effects or deaths reported in the DIAMOND-AF Clinical Study.
Table 15 summarizes all adverse events by seriousness and relatedness. In the ITT cohort, there were 171 adverse
events reported in 98 (41.0%) of the 239 subjects randomized to the DiamondTemp group. Of these events, 21 events
in 18 (7.5%) subjects were considered device or procedure related regardless of severity. There were 199 total
adverse events reported in 103 (42.4%) of the 243 subjects randomized to the control group. Of these events, 35
events in 31 (12.8%) subjects were considered device or procedure related regardless of severity.
Table 15. AE Overall Summary Table, Intention-to-Treat Cohort, Control vs DiamondTemp
Number of Events (Number of subjects, % of Subjects)
Adverse Event Classification
Control
(N=243)
DiamondTemp
(N=239)
Total Adverse Events 199 (103, 42.4%) 171 (98, 41.0%)
Primary Safety Events
Serious
[2]
[1]
16 (16, 6.6%) 8 (8, 3.3%)
Yes 61 (43, 17.7%) 53 (34, 14.2%)
No 138 (80, 32.9%) 118 (79, 33.1%)
Relatedness
Device and/or Procedure Related
[2],[4]
[3]
Related 35 (31, 12.8%) 21 (18, 7.5%)
Possibly Related 23 (18, 7.4%) 16 (15, 6.3%)
Unknown 2 (2, 0.8%) 0 (0, 0%)
Not Related 139 (79, 32.5%) 134 (81, 33.9%)
Device Relatedness
20
Page 21
Table 15. AE Overall Summary Table, Intention-to-Treat Cohort, Control vs DiamondTemp (continued)
Number of Events (Number of subjects, % of Subjects)
Adverse Event Classification
Control
(N=243)
DiamondTemp
(N=239)
Related 3 (3, 1.2%) 2 (2, 0.8%)
Possibly Related 4 (4, 1.6%) 3 (3, 1.3%)
Unknown 2 (2, 0.8%) 2 (2, 0.8%)
Not Related 190 (99, 40.7%) 164 (95, 39.7%)
Procedure Relatedness
Related 35 (31, 12.8%) 21 (18, 7.5%)
Possibly Related 23 (18, 7.4%) 16 (15, 6.3%)
Unknown 2 (2, 0.8%) 0 (0, 0%)
Not Related 139 (79, 32.5%) 134 (81, 33.9%)
[1] Primary safety events are based on CEC adjudication.
[2] Seriousness and Relatedness are based on investigator assessment.
[3] Device and/or Procedure Relatedness- the strongest relationship with device or procedure is used in this category.
[4] A subject may count in more than one relatedness category.
7.7.2 Effectiveness Results
7.7.2.1 Primary Effectiveness Endpoint
A summary of Diamond-AF Study primary effectiveness endpoint results are noted below. The primary effectiveness
endpoint analysis was performed on the Intention-to-Treat (ITT) Cohort (i.e. all enrolled/randomized subjects). The
primary effectiveness endpoint was met. Table 16, Table 17 and Figure 6 below show the primary effectiveness results
for subjects by treatment group in the Intention-to-Treat cohort. Of the 243 enrolled/randomized control subjects (ITT
cohort), 184 (75.7%) were free of all primary effectiveness endpoint failure criteria. Of the 239 enrolled/ randomized
DiamondTemp subjects (ITT cohort), 189 (79.1%) were free of all primary effectiveness endpoint failure criteria. The
DiamondTemp minus control freedom rate was 3.4% with a two-sided 95% confidence interval ranging from -4.2% to
10.9%. The lower two-sided 95% confidence limit exceeded the non-inferiority margin of -12.5%, and the
Farrington-Manning Score test for non-inferiority yielded a p-value of <0.0001. Therefore, the primary effectiveness
objective was met.
Table 16. Primary Effectiveness Endpoint, Intention-to-Treat Cohort, Control vs DiamondTemp
Criteria
Inability to electrically isolate all accessible targeted pulmonary veins during the ablation procedure.
a
Documented episodes of AF, AFL or AT lasting
Control
(N=243)
Success
(No/Absent)
n (%)
Failure
(Yes/Present)
n (%)
241 (99.2%) 2 (0.8%) 239 (100.0%) 0 (0%)
197 (81.1%) 46 (18.9%) 198 (82.8%) 41 (17.2%)
DiamondTemp
(N=239)
Success
(No/Absent)
n (%)
≥30 seconds in duration as evidenced by electrocardiographic data during the effectiveness
evaluation period.
DC cardioversion for AF, AFL, or AT during the
238 (97.9%) 5 (2.1%) 231 (96.7%) 8 (3.3%)
effectiveness evaluation period.
A repeat ablation procedure to treat AF, AFL or
229 (94.2%) 14 (5.8%) 226 (94.6%) 13 (5.4%)
AT during the effectiveness evaluation period.
Use of a new or modification to existing Class I-IV
217 (89.3%) 26 (10.7%) 218 (91.2%) 21 (8.8%)
anti-arrhythmic drug (AAD) regimen to treat AF,
AFL or AT during the effectiveness evaluation
period.
Use of a non- study device for ablation of any AF
242 (99.6%) 1 (0.4%) 239 (100.0%) 0 (0%)
targets during the index or the repeat ablation
procedure during the blanking period.
More than one (1) repeat ablation procedure dur-
243 (100.0%) 0 (0%) 239 (100.0%) 0 (0%)
ing the blanking period.
a
Electrical isolation as confirmed by demonstration of exit and/or entrance conduction block.
Failure
(Yes/Present)
n (%)
AAD=Anti-arrhythmic drugs; AF=Atrial Fibrillation; AFL=Atrial Flutter; AT=Atrial Tachycardia; DC=Direct Current.
21
Page 22

Notes: N = Number of subjects in the Intent-to-Treat Population. n = Number of subjects in the specific category.
X: Between Treatment Difference and 95% Confidence Interval: 3.36% (-4.20%, 10.92%)
[DiamonTemp is better]0-12.5%[Control better]
Confidence Intervals are based on Farrington and Manning’s Likelihood Score Test
Percentages are calculated as 100 x (n/N).
Subjects failing a particular criterion more than once are counted only once for that predefined category.
Table 17. Primary Effectiveness Result, Intention-to-Treat Cohort
Primary Effectiveness Endpoint:
Freedom from AF,
AFL, AT During the
Effectiveness
Period
Total, By Sub-
[1],[2]
ject
Control
(N=243)
Number (%) (95%
CI)
184 (75.7%)
(69.8%, 81.0%)
DiamondTemp
(N=239)
Number (%)
(95% CI)
189 (79.1%)
(73.4%, 84.1%)
Difference
(95% CI)
3.4%
(-4.2%, 10.9%)
Farrington- Manning p-value
(Non-inferiority
Test)
<0.0001
[1] In the total row, success is the absence of any of the criteria, while failure is the presence of one or more of the
criteria.
[2] The Farrington-Manning Score test for non-inferiority (DiamondTemp minus control) is used with a non-inferiority
margin of -12.5%.
Figure 6. Primary Effectiveness: Between Treatment Difference
A Kaplan-Meier analysis was also performed to evaluate the primary effectiveness endpoint as a sensitivity analysis.
The Kaplan-Meier method allows subjects to be included in the analysis up until the time they fail the primary
effectiveness endpoint or are censored due to premature study exit.
Figure 7 displays the Kaplan-Meier estimates for the freedom from primary effectiveness endpoint failure through 12
months post-ablation by treatment arm. Based on the Kaplan-Meier methodology the freedom from primary
effectiveness endpoint failure at the end of the primary effectiveness period (day 410) post-ablation was 76% for the
DiamondTemp group and 70% for the control group. The log-rank test suggested there was no difference in the
freedom rate between groups (p=0.47).
Figure 7. Kaplan-Meier Survival: Time to Failure of the Primary Effectiveness Endpoint, Intention-to-Treat Cohort
22
Page 23
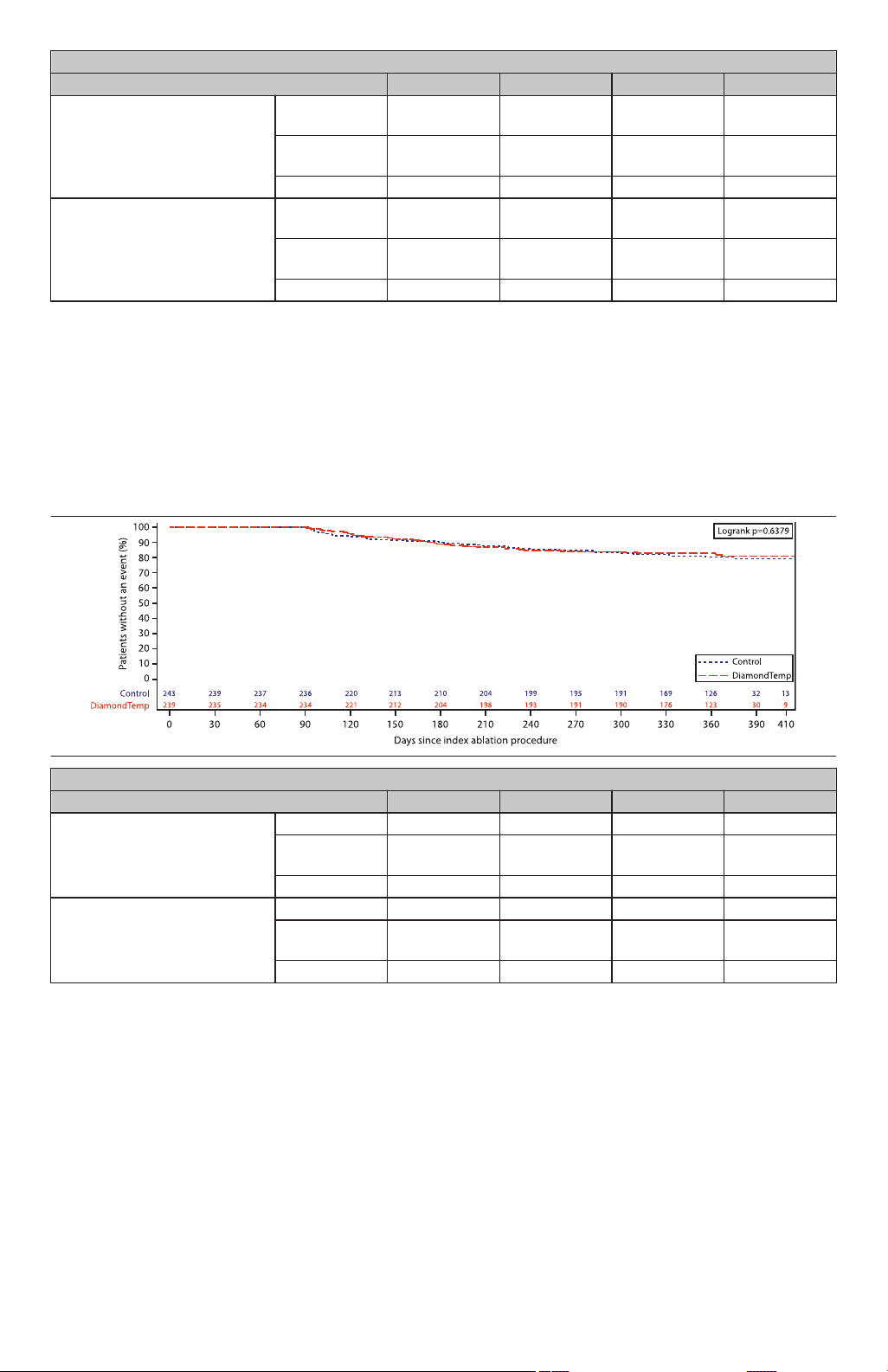
Kaplan-Meier Survival: Time to Failure of the Primary Effectiveness Endpoint, Intention-to-Treat Cohort
Month 3 Month 6 Month 9 Month 12
Control Number at
236 210 189 13
Risk
Kaplan-Meier
0.99 0.89 0.81 0.70
Estimate
Standard Error 0.0058 0.0199 0.0256 0.0419
DiamondTemp Number at
234 200 185 9
Risk
Kaplan-Meier
1.00 0.87 0.82 0.76
Estimate
Standard Error 0.0000 0.0219 0.0256 0.0309
3 Months (day 90), 6 Months (day 180), 9 Months (day 270), 12 Months (day 410)
An important component of the primary efficacy endpoint was the freedom from documented AF/AFL/AT episodes
lasting 30 or more seconds following the 3-month blanking period through 12 months as identified on 24-hour Holter
recordings, 12-lead ECG, or event monitor recordings (twice a month plus symptom driven). Figure 8 displays
Kaplan-Meier estimates for the freedom from documented AF/AFL/AT episodes lasting 30 or more seconds by
treatment group. At 12 months post-ablation, Kaplan-Meier estimates for the freedom from documented AF/AFL/AT
was 81% for the DiamondTemp group and 79% for the control group. The log-rank test indicated that there was no
difference between treatment groups in the risk for AF/AFL/AT recurrence during the effectiveness evaluation period
(p=0.64).
Figure 8. Kaplan-Meier Survival: Time to Documented AF/AFL/AT ≥ 30 Seconds, Intention-to-Treat Cohort
Kaplan-Meier Survival: Time to Documented AF/AFL/AT ≥ 30 Seconds, Intention-to-Treat Cohort
Month 3 Month 6 Month 9 Month 12
Control # at Risk 236 210 195 13
Kaplan-Meier
1.00 0.90 0.85 0.79
Estimate
Standard Error 0.0000 0.0194 0.0236 0.0290
DiamondTemp # at Risk 234 204 191 9
Kaplan-Meier
1.00 0.89 0.84 0.81
Estimate
Standard Error 0.0000 0.0205 0.0240 0.0270
3 Months (day 90), 6 Months (day 180), 9 Months (day 270), 12 Months (day 410)
Table 18 displays the primary effectiveness endpoint outcome by treatment group and geography. The Breslow-Day
test indicated that there was no evidence for heterogeneity in primary effectiveness endpoint success rate between
treatment groups by geography (p=0.36).
23
Page 24
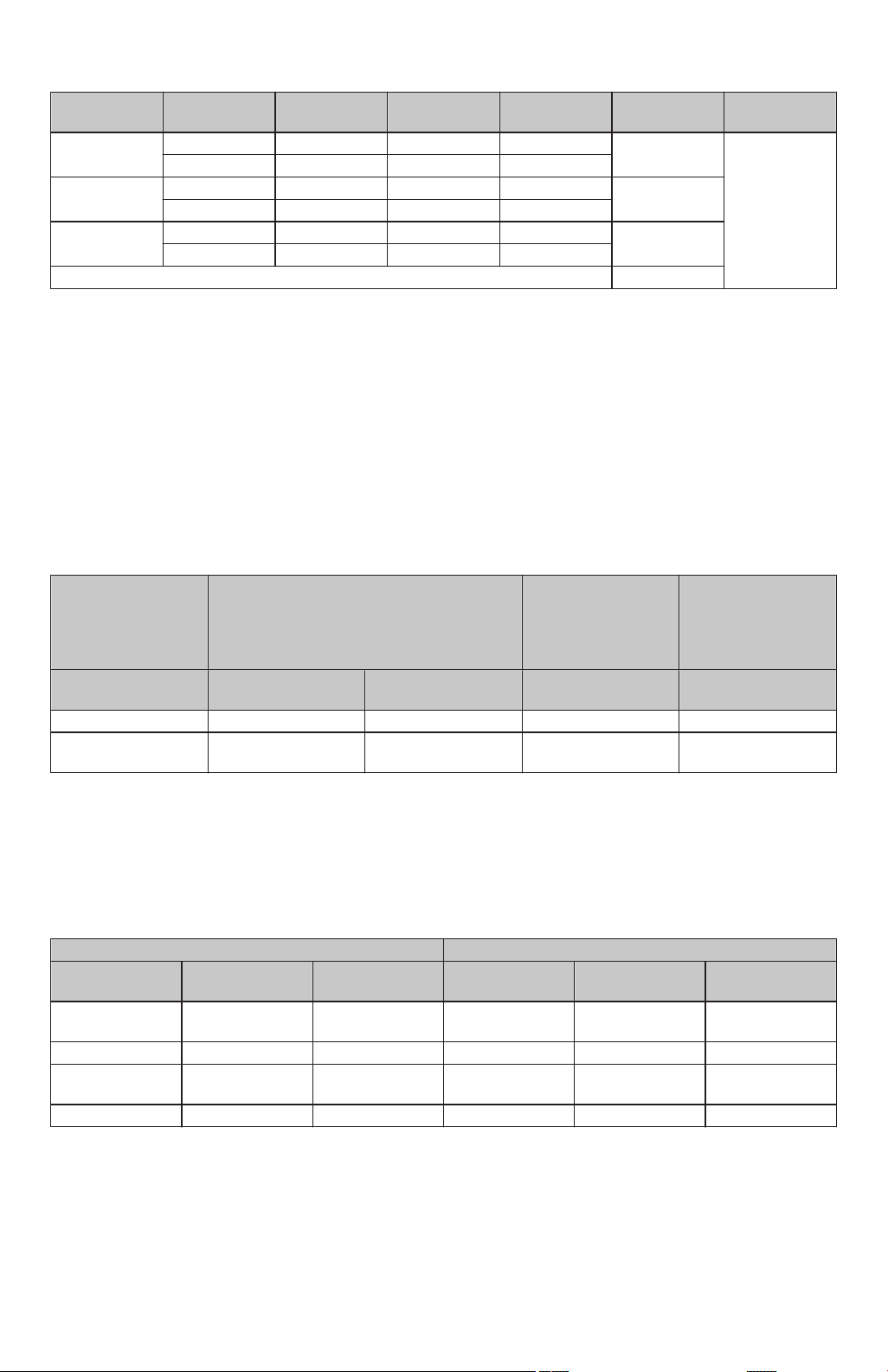
Table 18. Primary Effectiveness Endpoint: Relative Risk of Success; Overall and Stratified by Treatment and Geographic Region, Intention-to-Treat Cohort
Geographic
Region Treatment PEE Success PEE Failure Total
Relative Risk
of Success
Breslow Day
Test p-value
Europe DiamondTemp 93 36 129 1.01 0.3580
Control 94 38 132
North America DiamondTemp 96 14 110 1.08
Control 90 21 111
Overall DiamondTemp 189 50 239 1.04
Control 184 59 243
Pooled / Adjusted (CMH) 1.04
CMH= Cochran–Mantel–Haenszel
The DIAMOND-AF Clinical Study met its primary effectiveness objective (Intention-to-Treat Cohort). Primary
effectiveness endpoint success was observed in 184 (75.7%) control (TactiCath) subjects and 189 (79.1%)
DiamondTemp subjects (95% CI for difference: -4.2% to 10.9%; p<0.0001 for non-inferiority). There was no evidence
of heterogeneity in primary effectiveness outcome between treatment groups by geography (p=0.36). The
DiamondTemp Ablation System demonstrated a reasonable assurance of effectiveness for the treatment of drug
refractory, recurrent, symptomatic PAF.
Worst Case Scenario
Sensitivity analysis was performed using a worst case scenario in the safety cohort, in which all missing DiamondTemp
data (n=8) were considered as primary effectiveness failures and all missing Control data (n=8) were considered as
primary effectiveness successes. The difference of primary effectiveness success rate was -0.2% (95% CI: -8.0%,
7.6%), and the 97.5% lower confidence bound of -8.0% still met the predetermined NIM of -12.5% (Table 19).
Table 19. Primary Effectiveness Endpoint Sensitivity Analysis
Farrington-Manning
p-value
Analysis
Primary Effectiveness
Endpoint Successes
Control
(N=241)
DiamondTemp
(N=235)
95% CI for
Difference
(non-inferiority
test)
Safety Cohort 182 (75.5%) 185 (78.7%) (-4.4%, 10.8%) <0.0001
Worst Case Sce-
a
nario
a
The worst case scenario considered all Control subjects who had not failed the primary effectiveness endpoint at the time of their
pre-mature exit as successes and all DiamondTemp subjects who had not failed the primary effectiveness endpoint prior to pre-mature
exit as failures.
182 (75.5%) 177 (75.3%) (-8.0%, 7.6%) 0.001
TTM Compliance Analysis
Table 20 presents the distribution of TTM compliance and the primary effectiveness success at each quartile between
the two groups.
Table 20. TTM Monitoring Compliance and Primary Effectiveness Endpoint (PEE) Success per Quartile
PEE Status TTM Compliance
Frequency (Per-
(0-25%) (25-50%) (50-75%) (75-100%) Total
centage)
DiamondTemp
Success
38
(84.44%)
23
(76.67%)
36
(65.45%)
88
(83.81%)
185
DiamondTemp N 45 30 55 105 235
Control
Success
41
(82.00%)
28
(73.68%)
37
(68.52%)
76
(76.77%)
182
Control N 50 38 54 99 241
7.7.3 Secondary Endpoint Results
A summary of Diamond-AF Study secondary endpoint results is noted below in Table 21, and the first four (4)
secondary endpoints in the summary included hierarchical hypothesis testing and therefore have p-values reported
if applicable.
24
Page 25
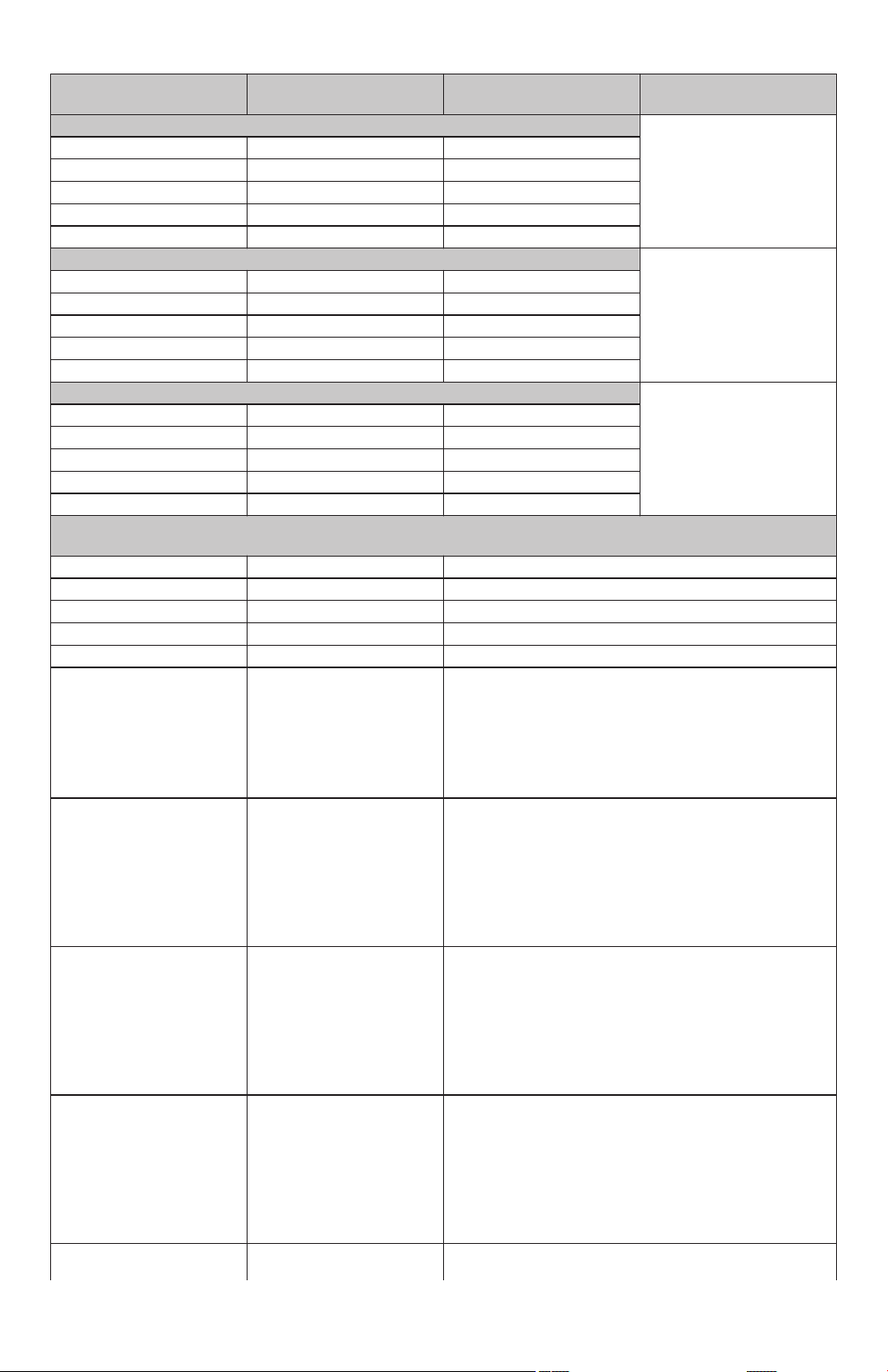
Table 21. Secondary Endpoints, Intention-to-Treat Cohort, Control vs DiamondTemp
Secondary Endpoints
Results
Control
(N=243)
DiamondTemp
(N=239) p-value
[1]
Mean Duration of Individual RF Ablations (Seconds) <0.0001
Mean (SEM / SD) 32.59 (1.642/25.34) 14.67 (0.343/5.260)
95% CI (29.4, 35.8) (14.0, 15.4)
Median 26.2 14.0
Min, Max 8.9, 193.0 7.0, 47.4
N (N missing) 238 (5) 235 (4)
Mean Cumulative RF Time Per Procedure (minutes) <0.0001
Mean (SEM / SD) 29.80 (0.908/14.00) 17.93 (0.527/8.085)
95% CI (28.0, 31.6) (16.9, 19.0)
Median 25.9 16.0
Min, Max 8.4, 83.2 2.5, 54.9
N (N missing) 238 (5) 235 (4)
Total Fluoroscopy Time (minutes) 0.8528
Mean (SEM / SD) 12.83 (0.611/9.439) 12.66 (0.669/10.19)
95% CI (11.6, 14.0) (11.3, 14.0)
Median 10.58 9.95
Min, Max 0.00, 60.05 0.00, 54.90
N (N missing) 239 (4) 232 (7)
Total Procedure Time (minutes), Defined as Time of First Assigned Ablation Catheter Insertion Into the Vasculature
to Time of Last Procedural Ablation Catheter Removed
Mean (SEM / SD) 115.4 (3.28/50.84) 109.7 (3.01/46.18)
95% CI (108.9, 121.8) (103.8, 115.6)
Median 100.0 97.0
Min, Max 37, 314 48, 389
N (N missing) 240 (3) 235 (4)
Freedom from a composite
230/243 (94.7%) 231/239 (96.7%)
of SAE occurring within
7-days post-index ablation
procedure as adjudicated
by an independent CEC for
relatedness to the procedure or device
Freedom from documented
120/243 (49.4%) 142/239 (59.4%)
AF, AT and AFL episodes
following the blanking
period through 12 month
follow-up post-ablation procedure in the absence of
class I and III anti-arrhythmic drug therapy
Rate of acute procedural
228/243 (93.8%) 228/239 (95.4%)
success, defined as confirmation of electrical isolation of PVs via assessment
of entrance block at least 20
minutes following the last
ablation around the respective PV
Rate of single procedure
185/243 (76.1%) 183/239 (76.6%)
success defined as the rate
of subjects treated with one
single ablation procedure
during study participation
and with freedom from
documented AF, AT and
AFL at 12 months
Rate of single procedure
173/243 (71.2%) 175/239 (73.2%)
success defined as the rate
25
Page 26

Table 21. Secondary Endpoints, Intention-to-Treat Cohort, Control vs DiamondTemp (continued)
Secondary Endpoints
Results
Control
(N=243)
DiamondTemp
(N=239) p-value
[1]
of subjects treated with one
single ablation procedure
during study participation
and with freedom from ALL
primary effectiveness endpoint failure criteria
Rate of occurrence of elec-
45/243 (18.5%) 45/239 (18.8%)
trically reconnected PVs
following a 20-minute waiting period assessed by
entrance block at index procedure
Accumulated Changes in QOL Using the AF QOL Survey (AFEQT Questionnaire) from Baseline Through 6 and 12
Months Following Ablation Procedure
At 6 Months:
Mean (SEM / SD) 25.54 (1.569/22.68) 27.79 (1.606/23.10)
Median 22.2 25.0
Min, Max -28.7, 86.1 -26.9, 98.1
N (N missing) 209 (34) 207 (32)
At 12 Months:
Mean (SEM / SD) 30.15 (1.570/23.23) 31.07 (1.599/23.44)
Median 26.9 26.9
Min, Max -35.2, 86.1 -50.9, 98.1
N (N missing) 219 (24) 215 (24)
Neurological Changes Measured Using the NIH Stroke Scale Between Baseline and Post-Ablation (Pre-Discharge
Visit) and at 12 Months Post-Ablation Procedure
At Discharge:
Mean (SEM / SD) 0.0 (0.02/0.32) 0.0 (0.02/0.30)
Median 0.0 0.0
Min, Max -1, 3 -2, 2
N (N missing) 222 (21) 216 (23)
At 12 Months:
Mean (SEM / SD) -0.1 (0.03/0.38) -0.1 (0.02/0.36)
Median 0.0 0.0
Min, Max -4, 1 -3, 0
N (N missing) 212 (31) 214 (25)
Time to Achieve Initial PVI at Index Procedure (minutes), Defined as Time of Delivery of First RF Ablation with the
Assigned Ablation Catheter Until Confirmation of PVI
Mean (SEM / SD) 69.4 (2.28/35.15) 65.7 (1.95/29.89)
Median 56.5 56.0
Min, Max 21, 218 24, 192
N (N missing) 238 (5) 235 (4)
Total Treatment Device Time (minutes), Defined as Time of Delivery of First RF Ablation with the Assigned Ablation
Treatment Catheter to Removal of the Treatment Catheter
Mean (SEM / SD) 91.4 (3.94/60.91) 83.1 (2.22/33.99)
Median 75.0 71.0
Min, Max 22, 802 30, 196
N (N missing) 239 (4) 235 (4)
Total Number of RF Ablations Per Procedure
Mean (SEM / SD) 71.1 (2.58/39.78) 74.2 (2.16/32.95)
Median 63.0 67.5
Min, Max 11, 264 17, 279
N (N missing) 238 (5) 232 (7)
Total Fluid Infused Through the Assigned Ablation Catheter (mL)
Mean (SEM / SD) 785.2 (22.83/351.5) 332.2 (7.88/120.8)
26
Page 27

Table 21. Secondary Endpoints, Intention-to-Treat Cohort, Control vs DiamondTemp (continued)
Secondary Endpoints
Results
Control
(N=243)
DiamondTemp
(N=239) p-value
[1]
Median 721.7 307.0
Min, Max 3.8, 2095 57.0, 800.0
N (N missing) 237 (6) 235 (4)
Number of Re-Hospitalizations Due to Atrial Fibrillation Recurrence After Blanking Period
Mean (SEM / SD) 0.1 (0.02/0.26) 0.1 (0.02/0.29)
Median 0.0 0.0
Min, Max 0.0, 2.0 0.0, 2.0
N (N missing) 243 (0) 239 (0)
0 Re-Hospitalization 229/243 (94.2%) 221/239 (92.5%)
1 Re-Hospitalization 13/243 (5.3%) 17/239 (7.1%)
2 Re-Hospitalizations 1/243 (0.4%) 1/239 (0.4%)
AF=Atrial Fibrillation, AFL=Atrial Flutter, AT=Atrial Tachycardia, CEC=Clinical Events Committee, NIH=National
Institute of Health, PV=Pulmonary Vein, PVI=Pulmonary Vein Isolation, QOL=Quality of Life, RF=Radiofrequency
Min=Minimum, Max=Maximum, SEM=Standard Error of the Mean, SD=Standard Deviation
Notes: N=Number of subjects in the Intent-to-Treat Population. n=Number of subjects in the specific category.
Percentages are calculated as 100 x (n/N).
[1] The top four specific secondary endpoints were evaluated for superiority over Control using a priori hierarchical
hypotheses with a two-tailed alpha of 0.05, and testing stopped when the first non-significant result was reached.
7.7.4 Blinding Assessment
The DIAMOND AF trial is a randomized, controlled, single-blind study. The success of subject blinding to treatment
assignment was evaluated by asking subjects whether they know they were in which treatment group at the 12-month
follow-up visit. Table 22 summarizes the blinding assessment results. In both treatment groups, majority of subjects did
not know the treatment assignment.
Table 22. Blinding Assessment
Which Treatment Group Does the Subject Believe He/She was
Number of Subjects (Percentage)
Actual Treatment
Does Not Know Control Device Investigational
Group Assignment
Control 121
(53.1%)
Investigational 120
(53.3%)
Total 241 60 152 453
a
Of all 455 subjects who completed the 12-month visit, 453 provided a response to this blinding question.
Assigned To? (At 12-Month Follow-up Visit)
Device
49
(21.5%)
11
(4.9%)
58
(25.4%)
94
(41.8%)
Total
228
225
a
7.8 Study Strengths and Weaknesses
The following points cover the major strengths and weaknesses of the study.
Strengths:
• Randomized control trial design
• Independent adjudication of primary safety and effectiveness endpoints
• This large, prospective, global, multicenter randomized study had sufficient statistical power to test the primary
safety and effectiveness hypotheses
Weaknesses:
• This study provided effectiveness and safety data limited through 12 months
7.9 Conclusion
The DIAMOND-AF Clinical Study met its primary objectives for effectiveness and safety. The DiamondTemp Ablation
System demonstrated a reasonable assurance of effectiveness and safety for the treatment of drug refractory,
recurrent, symptomatic PAF, with 79.1% of subjects randomized to the DiamondTemp Ablation System free from all
primary effectiveness endpoint failure criteria at 12 months compared to 75.7% of subjects randomized to the control
group (non-inferiority p-value <0.0001). Furthermore, 96.7% of subjects randomized to the DiamondTemp Ablation
System were free from any primary safety endpoint event relative to 93.4% of subjects randomized to the control group,
as adjudicated by the CEC (non-inferiority p-value <0.0001). Of the four (4) secondary endpoints with hypothesis
27
Page 28

testing, a statistically significant difference in favor of the DiamondTemp Ablation System was demonstrated with
regard to lower mean duration of individual RF ablations (p-value <0.0001) and lower mean cumulative RF time per
procedure (p-value <0.0001) relative to the control group ablation system.
8 Directions for use
1. Verify that the generator and irrigation pump are set up per instructions in the respective user manuals.
2. Inspect the catheter packaging before use. If the packaging is open, damaged, or expired, do not use and
contact a Medtronic representative.
3. Remove the catheter from its package. Carefully inspect the catheter, including the ablation tip and electrodes,
for integrity and overall condition. Activate the steering mechanism to confirm that the desired curve is achieved.
4. Do not use if the catheter is damaged.
5. Power ON the generator and irrigation pump. Refer to the generator and irrigation pump user manuals for a
complete description of generator and pump set-up and communication between the devices. The
recommended generator and irrigation pump settings are summarized in Table 23.
Table 23. Recommended Generator and Irrigation Pump Settings
GENERATOR SETTINGS
Operational Mode Temperature Control
Maximum Temperature Set-Point 60°C
Maximum Power Setting 50 W
Maximum Ablation Duration 45 s
IRRIGATION PUMP SETTINGS
Irrigation flow rate during ablation 8 mL/min
Minimum continuous flow rate 2 mL/min
6. Connect an IV bag with sterile, normal (0.9%) saline heparinized at 1 unit/mL to the tubing set.
7. Connect the catheter to the luer fitting of the tubing set.
8. Flush the catheter by pressing and holding the Purge Flow button on the irrigation pump (refer to the irrigation
pump user manual for complete instructions).
Caution: Purge the tubing set and catheter of air bubbles before inserting the device into the patient.
9. Connect the catheter-to-RFG cable to the catheter. To do this, align the blue strain-relief end of the
catheter-to-RFG cable connector key to the catheter receptacle key, and then push the connector into the
catheter receptacle firmly until it stops. Do not force connectors or pin damage can occur. To disconnect, pull the
connector body until it separates from the receptacle.
10. Connect the catheter-to-RFG cable to the generator or GenConnect cable (refer to Chapter 9). To do this, align
the catheter-to-RFG cable green connector to the green receptacle key of the generator or GenConnect cable,
and then push the connector into the receptacle firmly until it stops. Do not force the connectors or pin damage
can occur. To disconnect, pull the connector body until it separates from the receptacle.
11. Connect the return pad cable to the return pad icon on the generator front panel.
12. Start minimum continuous irrigation of the catheter at a flow rate of 2 mL/min.
Caution: To avoid occlusion of the irrigation conduits, the catheter must be continuously irrigated when within
the vasculature. Irrigation should only be stopped after removing the catheter from the body.
13. Make sure the catheter tip orientation is in the neutral (straight) position before insertion into the sheath. To aid
in insertion, the catheter may be used with a compatible 2.83 mm (8.5 Fr) inner diameter sheath.
14. The catheter should be passed from a peripheral vessel to the desired endocardial position with the aid of
fluoroscopy.
15. To adjust the curve of the distal tip on the unidirectional catheter, push or pull the actuation knob located on the
handle. Do not pull on the saline luer or connector. A notch on the actuation knob provides orientation to the plane
of the tip curvature. To adjust the curve of the distal tip of the bidirectional catheter, actuate the steering knob in
either direction and turn the tension knob to lock the curve.
16. Set the generator to the desired temperature control set-point. The generator will automatically adjust the power
output. Refer to the generator user manual for more detailed information.
17. Press the START button on the generator to begin the RF ablation. The irrigation pump will automatically
increase from minimum flow rate to ablation flow rate.
Caution: While creating a lesion, do not leave the power on for more than 45 seconds continuously.
18. Monitor the temperature, power, and impedance display on the generator during RF energy delivery.
Caution: If a sudden rise in impedance is noted during RF delivery that does not exceed the impedance cut-off
preset limit, manually discontinue the power delivery by pressing the STOP button. To assist in this regard, the
generator screen displays the real-time relative impedance change in the green impedance field. When a large,
positive-sign, percent increase is displayed, consider stopping RF delivery. Clinically assess the situation. If
necessary, remove the catheter and inspect the tip electrode for any char or coagulum. Clean the distal tip to
eliminate any coagulum, if present. Flush the catheter before reinsertion.
Note: Temperature represents the temperature of the tissue surface in contact with the catheter.
28
Page 29

19. The temperature set-point should not exceed 60°C. Assess intracardiac electrograms and impedance before
changing the temperature set-point.
Caution: In case of a steam pop or automatic shut off, remove the catheter for visual inspection and check for
coagulum, charring, or defects. Flush the ports before reinsertion into the patient. If the catheter has defects,
exchange it for a new one. Relocate the new catheter within the cardiac anatomy and attempt another RF
application.
Caution: If the pump alarms and stops the irrigation, immediately remove the catheter from the patient and
inspect. Flush the catheter. At the end of each ablation period, the irrigation pump will automatically return to the
minimum continuous flow rate.
20. To stop ablation, press the STOP button on the generator.
Note: Alternatively, the foot switch may be used to initiate and stop RF energy delivery.
21. The catheter may be repositioned for additional ablation. When the procedure is finished, bring the distal tip of
the catheter to a neutral position (straight) before removing the catheter from the patient.
9 Connection to other equipment
The DiamondTemp ablation catheter may be connected to a commercially available EP recording system and cardiac
stimulator using a connection cable with connectors in the pin configuration corresponding to the DiamondTemp
catheter. The use of cables with shrouded pins is recommended and is required in some countries, such as the United
States. Such equipment must have an isolated patient cable. Only EP recording systems that show proof of certified
compliance with all applicable requirements of IEC 60601-1, IEC 60601-1-1, and IEC 60601-1-2 should be used,
including, but not limited to, compliance with requirements for patient isolation, patient auxiliary currents, leakage
currents, and EMC/EMI.
The DiamondTemp ablation system can be used with a compatible mapping system (for example, the Abbott EnSite™
Velocity™ or Precision™ Mapping System). When connecting the DiamondTemp system to the mapping system, use
a GenConnect box (or similar connection box) (Figure 9) with the DiamondTemp GenConnect cable. Refer to the
GenConnect cable instructions for additional information.
1. Connect the distal end (26-pin female receptacle) of the GenConnect cable to the catheter-to-RFG cable. To do
this, align the green connector end of the catheter-to-RFG cable to the green receptacle key of the GenConnect
cable, and then push the connector into the receptacle firmly until it stops. Do not force the connectors or pin
damage can occur. To disconnect, pull the connector body until it separates from the receptacle.
2. Connect the proximal end (26-pin male connector) of the GenConnect cable to the generator. To do this, align
the green strain-relief end of the GenConnect cable connector key to the generator receptacle key, and then push
the connector into the receptacle firmly until it stops. Do not force connectors or pin damage can occur. To
disconnect, pull the connector body until it separates from the receptacle.
3. Connect the GenConnect cable grey 9-pin connector to the catheter input of the GenConnect box.
4. Connect the GenConnect cable black 14-pin connector to the generator output of the GenConnect box.
5. Confirm correct connectivity with the mapping system.
6. Connect the return pad directly to the generator.
Figure 9. DiamondTemp Generator Connection to Mapping and Navigation System
1 DiamondTemp ablation catheter
2 Catheter-to-RFG cable
3 9-pin quick connector
4 GenConnect cable
5 DiamondTemp RF generator
6 Ablation return pad
7 14-pin twist connector
8 GenConnect box (oriented upside down, for
purposes of illustration)
9 Amplifier
29
Page 30

10 How supplied
The DiamondTemp ablation catheter is supplied along with the required product documentation. The contents were
sterilized with ethylene oxide (EtO) and are sterile if the package is unopened and undamaged. Do not resterilize the
catheter.
11 Packaging and shelf life
The DiamondTemp ablation catheter packaging is designed to protect the product from damage, minimize product
exposure to the atmosphere, and provide aseptic product transfer. It is recommended that the product remain in the
unopened package until time of use. Contents are sterile if the package is unopened and undamaged. Do not
resterilize the catheter. Do not use the catheter if the packaging sterile barrier is open or damaged.
The product expiration date (“Use by”) is stated on the product labeling. The product must be stored in a cool and dry
location, in a 15°C to 30°C (59°F to 86°F) noncondensing environment. Dispose of the product and packaging
according to standard procedures for solid biohazard waste products.
12 Limited warranty
The following Limited Warranty applies to customers within the United States only:
A. This Limited Warranty provides the following assurance to the purchaser of a Medtronic catheter, hereafter referred
to as Product:
(1) Should the Product fail to function within normal tolerances due to a defect in materials or workmanship on or before
its “Use By” or "Use Before" date, Medtronic will at its option: (a) issue a credit to the purchaser equal to the Purchase
Price, as defined in Subsection A(2), against the purchase of the replacement product or (b) provide a functionally
comparable replacement product at no charge.
(2) As used herein, Purchase Price shall mean the lesser of the net invoiced price of the original, or current functionally
comparable, or replacement product.
B. To qualify for this Limited Warranty, these conditions must be met:
(1) The Product must be used on or before its “Use By” or "Use Before" date.
(2) The Product must be returned to Medtronic within 60 days and shall be the property of Medtronic.
(3) The Product must not have been used for any other patient.
(4) The Product must be used in accordance with the labeling and not altered or subjected to misuse, abuse, accident,
or improper handling.
C. This Limited Warranty is limited to its express terms. In particular:
(1) Except as expressly provided by this Limited Warranty, MEDTRONIC IS NOT RESPONSIBLE FOR ANY DIRECT,
INCIDENTAL, OR CONSEQUENTIAL DAMAGES BASED ON ANY DEFECT,FAILURE, OR MALFUNCTION OF THE
Product, WHETHER THE CLAIM IS BASED ON WARRANTY, CONTRACT, TORT, OR OTHERWISE.
(2) This Limited Warranty is made only to the purchaser of the Product. AS TO ALL OTHERS, MEDTRONIC MAKES
NO WARRANTY, EXPRESS OR IMPLIED, INCLUDING, BUT NOT LIMITED TO, ANY IMPLIED WARRANTY OF
MERCHANTABILITY, OR FITNESS FORA PARTICULAR PURPOSE WHETHER ARISING FROM STATUTE,
COMMON LAW, CUSTOM, OR OTHERWISE. NO EXPRESS OR IMPLIED WARRANTY TO THE PATIENT SHALL
EXTEND BEYONDTHE PERIOD SPECIFIED IN A(1) ABOVE. THIS LIMITED WARRANTY SHALL BE THE
EXCLUSIVE REMEDY AVAILABLE TO ANY PERSON.
(3) The exclusions and limitations set out above are not intended to, and should not be construed so as to, contravene
mandatory provisions of applicable law. If any part or term of this Limited Warranty is held to be illegal, unenforceable,
or in conflict with applicable law by a court of competent jurisdiction, the validity of the remaining portions of the Limited
Warranty shall not be affected, and all rights and obligations shall be construed and enforced as if this Limited Warranty
did not contain the particular part or term held to be invalid. This Limited Warranty gives the purchaser specific legal
rights. The purchaser may also have other rights which vary from state to state.
(4) No person has any authority to bind Medtronic to any representation,condition, or warranty except this Limited
Warranty. This Limited Warranty is provided by Medtronic, Inc., 710 Medtronic Parkway, Minneapolis, MN
55432-5604. It applies only in the United States. Areas outside the United States should contact their local Medtronic
representative for exact terms of the Limited Warranty.
General warning
Medtronic catheters are used in the extremely hostile environment of the human body. Catheters may be easily
damaged by improper handling or use due to their unavoidably fragile character, which is dictated by the unusual
requirements of their application. Consequently, no representation or warranty is made that failure or cessation of
function of the catheter will not occur, or that the body will not react adversely to the catheter, or that medical
complications will not follow.
30
Page 31

Page 32

Medtronic, Inc.
*M005667C001*
710 Medtronic Parkway
Minneapolis, MN 55432
USA
www.medtronic.com
+1 763 514 4000
© 2021 Medtronic
M005667C001 B
2021-03-30
 Loading...
Loading...