

Page 1

BRYAN® CERVICAL DISC 0381326E Rev. C
2020-05-11
IMPORTANT INFORMATION ON THE BRYAN® CERVICAL DISC
DESCRIPTION
The BRYAN® Cervical Disc is a cervical disc replacement device comprised of two titanium shells, two titanium retaining wires,
a polycarbonate polyurethane nucleus, a polyether polyurethane sheath, and two titanium seal plugs. The articulating surfaces
of the device are polyurethane and titanium.
The nucleus is designed to fit between the two shells. The bone-contacting side of each shell includes a sintered titanium
porous coating to provide bony ingrowth. The nucleus-contacting side of each shell has a center pin which interacts with a
central hole in the nucleus to control the range of motion and help prevent nucleus expulsion. A stop or wing on the anterior
aspect of the device, which extends superiorly on the cephalad shell and inferiorly on the caudal shell, is intended to prevent
migration of the device into the spinal canal. A polyurethane sheath surrounds the nucleus and is attached to each shell with
titanium retaining wires, forming a closed compartment. The device is supplied pre-assembled with the exception of saline and
two seal plugs. Prior to implantation, the surgeon fills the BRYAN
function as an initial lubricant for the prosthesis. The surgeon screws titanium alloy seal plugs into one hole in each shell to
initially retain the saline.
The prosthesis is held in the intervertebral disc space by the fit of each shell’s outside diameter and convex outer surfaces into a
custom-milled cavity created in each vertebral endplate by the surgeon prior to device implantation. The prosthesis was
designed to allow for the following motions from the neutral position ex vivo: approximately ±11° flexion/extension, ±11° lateral
bending, ±7° rotation, and ±1 mm translation for all cervical disc sizes. The available components are shown in Table 1.
Table 1: BRYAN
Catalog Number Diameter (mm)
6470314 14
6470315 15
6470316 16
6470317 17
6470318 18
®
Cervical Disc Device Sizes
®
Cervical Disc with sterile saline. The saline is intended to
After implantation of the device, the resultant interbody height is approximately 6mm.
Implied warranties of merchantability and fitness for a particular purpose or use are specifically excluded.
INDICATIONS
The BRYAN® Cervical Disc is indicated in skeletally mature patients for reconstruction of the disc from C3-C7 following singlelevel discectomy for intractable radiculopathy and/or myelopathy. The BRYAN
approach. Intractable radiculopathy and/or myelopathy is defined as any combination of the following: disc herniation with
radiculopathy, spondylotic radiculopathy, disc herniation with myelopathy, or spondylotic myelopathy resulting in impaired
function and at least one clinical neurological sign associated with the cervical level to be treated, and necessitating surgery as
demonstrated using computed tomography (CT), myelography and CT, and/or magnetic resonance imaging (MRI). Patients
receiving the BRYAN
BRYAN
®
Cervical Disc.
®
Cervical Disc should have failed at least six weeks of non-operative treatment prior to implantation of the
®
device is implanted via an open anterior
CONTRAINDICATIONS
The BRYAN® Cervical Disc should not be implanted in patients with the following conditions:
▪ Active systemic infection or infection at the operating site.
▪ Allergy to titanium, polyurethane, or ethylene oxide residues.
▪ Osteoporosis defined as a DEXA bone mineral density T-score equal to or worse than -2.5.
▪ Moderate to advanced spondylosis characterized by bridging osteophytes, marked reduction or absence of motion, or
collapse of the intervertebral disc space of greater than 50% of its normal height.
▪ Marked cervical instability on radiographs (e.g., radiographic signs of subluxation greater than 3.5mm or angulation of the
disc space more than 11° greater than adjacent segments).
Page 2

▪ Significant cervical anatomical deformity or compromised vertebral bodies at the index level (e.g., ankylosing spondylitis,
rheumatoid arthritis, or compromise due to current or past trauma).
▪ Significant kyphotic deformity or significant reversal of lordosis.
▪ Symptoms necessitating surgical treatment at more than one cervical level.
WARNINGS
Correct sizing and placement of the device is essential to optimal performance. The BRYAN® Cervical Disc should only be used
by surgeons experienced in the surgical procedure and have undergone adequate hands-on training with this specific device.
Medtronic will offer hand-on training to physicians prior to the first surgical treatment. A lack of adequate experience and/or
training may lead to a higher incidence of adverse events, such as neurological complications.
Due to the proximity of vascular and neurological structures to the implantation site, there are risks of serious or fatal
hemorrhage and risks of neurological damage with the use of this device. Serious or fatal hemorrhage may also occur if the
major cervical blood vessels are eroded or punctured during implantation and are subsequently damaged due to breakage of
implants, migration of implants, or if pulsatile erosion of the vessels occurs because of close apposition of the implants.
PRECAUTIONS
The safety and effectiveness of this device has not been established in patients with the following conditions:
▪ Axial neck pain as solitary symptom.
▪ Not skeletally mature.
▪ Prior cervical spine surgery, including prior surgery at the index level.
▪ Facet joint pathology of involved vertebral bodies.
▪ Active malignancy.
▪ Paget’s disease, osteomalacia, or other metabolic bone disease.
▪ Chronic or acute renal failure or history of renal disease.
▪ Taking medications known to potentially interfere with bone/soft tissue healing (e.g., steroids).
▪ Pregnant.
▪ Unstable cardiac disease.
▪ Diabetes mellitus requiring daily insulin management.
▪ Extreme obesity as defined by the NIH Clinical Guidelines Body Mass Index (i.e., BMI ≥40).
There were no patients in the pivotal study who were less than 21 years of age. The safety and effectiveness of this device has
not been studied in the pediatric or adolescent age group (<21 years old).
The safety and effectiveness of this device has not been established in patients who were not refractory to at least six weeks of
unsuccessful conservative treatment or had signs of progression or spinal cord/nerve root compression with continued nonoperative care.
Implanted metal alloys release metallic ions into the body. The long term effect of these ions on the body is not known.
In order to minimize the risk of periprosthetic vertebral fractures, surgeons must consider all comorbidities, past and present,
medications, previous treatments, etc. Surgeons should screen patients to determine if a DEXA bone mineral density
measurement is necessary. If DEXA is performed, the patient should not receive the BRYAN® Cervical Disc (per the
Contraindications) if the DEXA bone mineral density T-score is ≤ -2.5, as the patient may be osteoporotic. It may also be
advisable to exclude patients with a T score ≤ -1.0, as those patients may be osteopenic.
Patients in the clinical study were instructed to use non-steroidal anti-inflammatory drugs (NSAIDs) for two weeks
postoperatively. Dosing and frequency were left to the discretion of the physician. It has been reported in the literature that
short-term postoperative use of NSAIDs may reduce the instance of heterotopic ossification.
Correct selection of the appropriate implant size is extremely important to ensure the placement and function of the disc.
Patient selection is extremely important. In selecting patients for a total disc replacement, the following factors can be of
extreme importance to the success of the procedure: the patient’s occupation or activity level; a condition of senility, mental
illness, alcoholism or drug abuse; certain degenerative diseases (e.g., degenerative scoliosis or ankylosing spondylitis) that may
be so advanced at the time of implantation that the expected useful life of the device is substantially decreased, and medical
conditions that may affect postoperative management, such as Alzheimer’s disease and emphysema.
Surgical implants must never be re-used or re-implanted. Even though the device appears undamaged, it may have small
defects and internal stress patterns that may lead to early breakage.
This system was designed for single patient use only. Do not reuse, reprocess, or resterilize this product. Reuse, reprocessing,
or resterilization may compromise the structural integrity of the device and/or create a risk of contamination of the device, which
could result in patient injury, illness, or death.
Use aseptic technique when removing the BRYAN
Use care when handling a BRYAN
implant. Damaged implants are no longer functionally reliable.
BRYAN
manufacturers.
Patients should be instructed in postoperative care procedures and should be advised of the importance of adhering to these
procedures for successful treatment with the device. Patients should be advised to avoid any activities that require repeated
®
Cervical Disc components should not be used with components or instruments of spinal systems from other
®
disc component to ensure it does not come in contact with objects that could damage the
®
Cervical Disc device from the innermost packaging.
Page 3

bending, lifting, and twisting, such as athletic activities. Gradual increase in physical activity will depend on individual patient
progress.
PHYSICIAN NOTE: Although the physician is the learned intermediary between the company and the patient, the important
medical information given in this document should be conveyed to the patient.
FOR US AUDIENCES ONLY
CAUTION: FEDERAL LAW (USA) RESTRICTS THESE DEVICES TO SALE BY OR ON THE ORDER OF A PHYSICIAN.
MRI Information for the BRYAN® Cervical Disc Implant
MR Conditional
Non-clinical testing demonstrated that the BRYAN
following conditions:
®
Cervical Disc is MR Conditional. It can be scanned safely under the
▪ Static magnetic field of 1.5 Tesla and 3.0 Tesla
▪ Highest spatial gradient magnetic field of 720 Gauss/cm
▪ Maximum whole body average specific absorption rate (SAR) of 2.0 W/kg or less under Normal Operating Mode only, for 15
minutes of scanning (per pulse sequence).
In non-clinical testing, the BRYAN
averaged SAR of 2.1 W/kg, as assessed by calorimetry for 15 minutes of MR scanning (per pulse sequence) in a 1.5 Tesla MR
system (1.5 Tesla/64 MHz, Magnetom, Siemens Medical Solutions, Malvern, PA. Software Numaris/4,Version Syngo MR 2002B
DHHS). Additionally, the BRYAN
body averaged SAR of 2.7 W/kg, as assessed by calorimetry for 15 minutes of MR scanning (per pulse sequence) in a 3 Tesla
MR system (3 Tesla/128 MHz, Excite, G3.0-052B, General Electric Healthcare, Milwaukee, WI).
®
Cervical Disc produced a maximum temperature rise of 1.7º C at a maximum whole body
®
Cervical Disc System produced a maximum temperature rise of 2.0ºC at a maximum whole
Artifact Information
MR image quality may be compromised if the area of interest is in the same area or relatively close to the position of the
BRYAN
®
Cervical Disc. The artifact size information is as follows:
Pulse Sequence T1-SE T1-SE GRE GRE
Signal Void Size
Imaging Plane Parallel Perpendicular Parallel Perpendicular
The image artifact extends approximately 25 mm from the device when scanned in non-clinical testing using a gradient echo
sequence in a 3 Tesla MR system (Excite, HDx, Software 14X.M5, General Electric Healthcare, Milwaukee, WI) using a
transmit/receive RF body coil.
582 mm
2
557 mm
2
1,350 mm
2
1,287 mm
2
MRI Patient Counseling Information
Physicians should communicate with the patient the following information about MR with respect to the BRYAN® Cervical Disc:
▪ BRYAN
®
Cervical Disc performance has been established for MRI systems at field strengths of 1.5 and 3.0 Tesla.
▪ During an MRI, the patient may notice a warming sensation around the implant or feel a tingling sensation. If the warming or
tingling sensation is uncomfortable, the patient should communicate this to the MR technologist, the MRI should be
stopped, and the settings adjusted to reduce or eliminate the sensation. The highest temperature change observed in nonclinical testing was +2.0ºC (associated with specific conditions listed).
▪ Additionally, the metal in the implant may cause the MRI image to be distorted in the area around the implant. The MRI can
be adjusted to minimize the image distortion.
Physicians should instruct patients to:
▪ Inform any healthcare personnel (e.g., doctor or MR technologist) that they have an implanted cervical disc prior to
receiving an MRI.
▪ The patient’s doctor will recommend whether or not an MRI is appropriate.
POTENTIAL ADVERSE EVENTS
Risks associated with the use of the BRYAN® Cervical Disc include those commonly associated with any surgery, those
specifically associated with cervical spinal surgery using an anterior approach, and those associated with a spinal implant, as
well as those pertaining to the BRYAN
categories. There is also the risk that this surgical procedure will not be effective, and may not relieve or may cause worsening
of preoperative symptoms. Some of these effects were observed in the clinical study and therefore have been previously
reported in the adverse events tables.
1. Risks associated with any surgical procedure are those such as abscess; cellulitis; wound dehiscence; wound necrosis;
edema; hematoma; heart and vascular complications; hypertension; thrombosis; ischemia; embolism; thromboembolism;
hemorrhage; thrombophlebitis; adverse reactions to anesthesia; pulmonary complications; gastrointestinal complications;
organ, nerve or muscular damage; seizure, convulsion, or changes to mental status; and complications of pregnancy
including miscarriage and fetal birth defects.
2. Risks associated with anterior interbody surgery of the cervical spine include dysphagia; dysphasia; dysphonia; hoarseness;
vocal cord paralysis; laryngeal palsy; sore throat; recurring aspirations; nerve deficits or damage; tracheal, esophageal, and
®
Cervical Disc. However, the causality of these adverse events is not exclusive to these
Page 4

pharyngeal perforation; airway obstruction; external chylorrhea; warmth or tingling in the extremities; deficit or damage to
the spinal cord, nerve roots, or nerves possibly resulting in paralysis or pain; dural tears or leaking; cerebrospinal fistula;
discitis, arachnoiditis, and/or other types of inflammation; loss of disc height; loss of proper curvature, correction, height or
reduction of the spine; vertebral slipping; scarring, herniation or degeneration of adjacent discs; surrounding soft tissue
damage, spinal stenosis; spondylolysis; otitis media; fistula; vascular damage and/or rupture; and headache.
3.
Risks associated with implants in the spine, including the BRYAN
®
device, are early or late loosening of the components;
disassembly; bending or breakage of any or all of the components; implant migration; malpositioning of implant; loss of
purchase; sizing issues with components; anatomical or technical difficulties; implant fracture; bone fracture; skin
penetration, irritation, pain, bursitis resulting from pressure on the skin from component parts in patients with inadequate
tissue coverage over the implant; foreign body reaction to the implants including possible tumor formation, autoimmune
disease, metallosis, and/or scarring; possible tissue reaction; bone resorption; bone formation that may reduce spinal
motion or result in a fusion, either at the treated level or at adjacent levels; development of new radiculopathy; myelopathy
or pain; tissue or nerve damage caused by improper positioning and placement of implants or instruments; loss of
neurological function; decreased strength of extremities; decreased reflexes; appearance of cord or nerve root injury; loss of
bowel and/or bladder control; and interference with radiographic imaging because of the presence of the implant.
4. Wound, local, and/or systemic infections.
5. Surgical instrument bending or breakage, as well as the possibility of a fragment of a broken instrument remaining in the
patient.
6. Inability to resume activities of normal daily living, including loss of consortium.
7. Death.
NOTE: Additional surgery may be necessary to correct some of the adverse effects.
CLINICAL STUDIES
A multi-center, prospective, randomized, non-inferiority clinical trial of the BRYAN® Cervical Disc was conducted in the US
comparing the anterior spinal use of the BRYAN
plating stabilization, the control, for reconstruction of the disc from C3-C7 following single-level discectomy for intractable
radiculopathy and/or myelopathy.
The IDE study was approved for a total of 470 patients. Medtronic performed a pre-specified interim statistical analysis when
approximately 300 implanted subjects had completed their 24-month follow-up visit. As predetermined at the time of the IDE
study initiation, if the results of this interim analysis demonstrated non-inferiority of the subjects receiving the BRYAN
compared to controls, the sponsor would submit a marketing application.
A total of 463 patients were treated at 30 investigational centers in the clinical trial: 242 patients in the investigational BRYAN
device treatment group and 221 patients in the control group. A post-approval study required as a condition of PMA approval
was conducted to follow the original IDE subject cohort through 120 months postoperatively. Additionally, an enhanced
surveillance study was also conducted collect adverse event and complaint information for five (5) years after device approval.
®
device to anterior cervical discectomy and fusion (ACDF) using allograft and
®
device
®
SUMMARY OF THE IDE AND POST-APPROVAL STUDY (PAS) METHODS
Study Objectives
IDE Study
The primary objective of the IDE study was to demonstrate that the overall success rate for the investigational BRYAN® Cervical
Disc treatment is statistically non-inferior to the overall success rate of the control treatment at 24 months following surgery. If
statistical non-inferiority was established, the investigational treatment is considered to be safe and effective, and the study was
considered a success.
Other secondary objectives were to compare the success rates of individual effectiveness endpoints and neurological status at
24 months following surgery.
Post-approval Study
The primary objective of the post-approval study was to demonstrate that the overall success rate for the investigational
BRYAN
following surgery. If statistical non-inferiority was established, the investigational treatment is considered to be safe and
effective, and the study was considered a success.
Other secondary objectives were to compare the success rates of individual effectiveness endpoints and neurological status at
120 months following surgery.
®
Cervical Disc treatment is statistically non-inferior to the overall success rate of the control treatment at 120 months
Enhanced Surveillance Study
The study objective was to collect the information for any adverse event and complaint, including those not associated with any
adverse clinical effect, received by the company for the BRYAN
®
Cervical Disc device following 5 years after device approval.
Page 5

Study Designs
IDE Study
The IDE study was a multi-center, prospective, randomized, controlled clinical trial comparing the BRYAN® Cervical Disc
treatment to the control treatment. Data were collected at pre-operative, discharge, 6 weeks, 3 months, 6 months, 12 months (1
year), and at 24 months (2 years). Additionally, 36 month data (3 years) was also collected prior to the start of the post-approval
study.
Post-approval Study
The post-approval study was a prospective study to continue follow-up on the subjects who participated in the IDE study. Data
were collected at 48 months (4 years), 60 months (5 years), 84 months (7 years), and at 120 months (10 years).
Enhanced Surveillance Study
Enhanced Surveillance data was collected annually on the post-market device for five (5) years after PMA approval.
Inclusion and Exclusion Criteria
To qualify for enrollment in the IDE study, subjects met all of the following inclusion criteria and none of the following exclusion
criteria.
Inclusion Criteria
▪ Requires surgical treatment at any one level (C3-4, C4-5, C5-6, or C6-7) that failed conservative treatment (by the
investigator or referring physician) lasting at least six weeks; for any combination of the following: disc herniation with
radiculopathy, spondylotic radiculopathy, disc herniation with myelopathy, or spondylotic myelopathy. The six-week
conservative treatment period may be waived in cases of myelopathy requiring immediate treatment (e.g., acute onset of
clinically significant signs).
▪ The requirement for surgical treatment must be demonstrated using computed tomography (CT), or myelography and CT,
and/or magnetic resonance imaging (MRI).
▪ Patient must score 30 or more points on the NDI questionnaire and exhibit at least one clinical sign associated with the
cervical level to be treated (i.e., abnormal reflex, decreased motor strength, or abnormal dermatome sensitivity).
▪ Skeletally mature (at least 21 years old).
▪ Willing and likely to follow the requirements of the protocol.
▪ Voluntarily signs the Patient Informed Consent.
Exclusion Criteria
A patient meeting any of the following criteria was to be excluded from the study:
▪ Active systemic infection or infection at the operating site.
▪ Metabolic bone disease, such as osteoporosis, which is defined as a bone mineral density T-score equal to or worse than –
2.5.
Note: If the investigator detects the presence of significant radiolucence, bone mineral density (BMD) scan in the spine,
wrist, and femoral neck must be obtained to verify the absence of osteoporosis.
▪ Known allergy to titanium, polyurethane, or ethylene oxide residuals.
▪ Concomitant conditions requiring steroid treatment.
▪ Diabetes mellitus requiring daily insulin management.
▪ Extreme obesity, as defined by NIH Clinical Guidelines Body Mass Index.
▪ Pregnancy.
▪ Axial neck pain as the solitary symptom.
▪ Previous cervical spine surgery.
▪ A medical condition that may interfere with the postoperative management program, such as advanced emphysema, or
Alzheimer’s disease.
▪ A medical condition that may result in patient death prior to study completion.
▪ Unstable cardiac disease.
▪ Active malignancy.
▪ Current or recent history of substance abuse (alcoholism and/or narcotic addiction) requiring intervention.
▪ Signs of being geographically unstable, such as recent or pending divorce, or high level of job dissatisfaction.
Patients with the following radiographic features at the symptomatic level were excluded from the study. These features at
adjacent levels did not disqualify the patient from the study.
▪ Significant cervical anatomical deformity (e.g., ankylosing spondylitis, rheumatoid arthritis, etc.).
▪ Moderate to advanced spondylosis. Patients who demonstrate advanced degenerative changes. Such advanced changes
are characterized by any one or combination of the following:
▪ Bridging osteophytes.
▪ Marked reduction or absence of motion.
▪ Collapse of the intervertebral disc space of greater than 50% of its normal height.
▪ Radiographic signs of subluxation greater than 3.5mm.
▪ Angulation of the disc space more than 11° greater than adjacent segments.
Page 6

▪ Significant kyphotic deformity or significant reversal of lordosis.
Study Population
IDE and Post-Approval Studies
The studies included 463 enrolled subjects who were at least 21 years old at the time of the surgery and met the study inclusion
and exclusion criteria.
Enhanced Surveillance Study
The study population was as per device indication. This device is indicated in skeletally mature patients for reconstruction of the
disc from C3-C7 following single-level discectomy for intractable radiculopathy and/or myelopathy.
Post-Operative Care
The recommended post-operative care included avoidance of heavy physical activity and limitations on extended automobile
rides, working, lifting, bending and twisting. The recommended post-operative regimen also included avoidance of physically
demanding sports or recreational activities for 3 months post-operatively. The decision whether to use a post-operative orthosis
was left to the discretion of the investigator. Investigational patients were instructed to use NSAIDs for the first two weeks
postoperatively.
IDE and PAS Follow-up Schedules
For the IDE study, patients were evaluated preoperatively (within 2 months of surgery), intraoperatively, and post-operatively at
6 weeks, 3 months, 6 months, 12 months (1 year), and at 24 months (2 years). Additionally, patients were also evaluated at 36
months (3 years) prior to the beginning of the post-approval study. For the post-approval study, patients were evaluated at 48
months, 60 months, 84 months, and 120 months. Complications and adverse events, device-related or not, were evaluated over
the course of the clinical trial. At each evaluation timepoint, the primary and secondary clinical and radiographic outcome
parameters were evaluated. For the IDE, success was determined from data collected during the initial 24 months of follow-up.
For the post-approval study, success was determined from the data collected up to 120 months.
Data Source
New data collection
Study Endpoints
IDE and PAS Study Endpoints
The primary endpoint was determined at 24 months (for the IDE) and 120 months (for the PAS) as a composite variable termed
overall success which included key safety and efficacy considerations. The efficacy variable was Neck Disability Index (NDI)
success, which was defined as at least 15-points improvement in the postoperative NDI score compared to the pre-operative
score. Safety variables included:
▪ Maintenance or improvement in neurological status from preoperative
▪ No serious adverse events classified as implant associated or implant/surgical procedure associated, and
▪ No additional surgical procedure classified as a “failure.”
Secondary endpoints for both the IDE and post-approval studies included neck pain, arm pain, general heatlh status, patient
satisfaction, patient global perceived effect, and gait assessments.
Enhanced Surveillance Study Endpoint
Complaints and adverse events for up to five (5) years after BRYAN® Cervical Disc device approval.
Strengths and Limitations of the Clinical Study Design
The following are important study design strengths of the IDE, post-approval, and enhanced surveillance studies:
▪ The PAS provided extended term profile of safety and effectiveness as well as functional data established on a multicenter,
randomized, controlled clincal trial.
▪ The consistency of data gathering approaches and endpoints was maintained through both the IDE and post-approval
studies.
▪ The strength and reliability of the final IDE and PAS conclusions were confirmed by several sensitivity analyses.
▪ The study design minimized potential biases by integrating a wide variety of assessments including subjective data,
physician-judged and core laboratory-assessed evaluations.
▪ The duration of the enhanced surveillance study was five (5) years after PMA approval which allowed time to monitor longer
term adverse events and complaints received by the sponsor for the device in the post-marketing setting.
The following potential study design limitations should also be considered when interpreting the results of the IDE, postapproval, and enhanced surveillance studies:
▪ Though there were no significant differences in Adverse Event (AE) rates between the groups, categorization of AE is
suboptimal in that all systemic AEs were considered non-device or non-procedure related, which may or may not be the
case. AE rates should be interpreted accordingly.
▪ Data on neurologic outcomes is incomplete in that relatedness to the device and procedure could not be determined and
should be interpreted accordingly.
Page 7

▪ There were a total of 37 reported cases of dysphagia or dysphonia in the investigational group. Of those reported cases, 10
were deemed unrelated to the device or surgical procedure. Of those 10 cases, eight were deemed unrelated to the
BRYAN
cases were undetermined but could potentially be related to the surgical procedure.
®
device or procedure, although definitive lack of device or procedure-relatedness cannot be determined. Two other
▪ Avoidance of adjacent level degeneration (device vs ACDF control) could not be demonstrated. There was also a lack of
significance shown for surrogate measures.
▪ Fusion outcomes on those subjects requiring a secondary surgical intervention (SSI) for BRYAN
captured during the study. As such, fusion outcomes after SSI are not known (e.g., the possibility of an incomplete fusion or
delayed fusion is not known.
®
device removal was not
▪ There may have been under-reporting of adverse events and complaints to the company during the enhanced surveillance
study due to the spontaneous nature of the post-market reporting. Therefore, the true number of the post-market adverse
events and complaints duing the 5-year study period may not be known.
SUMMARY OF THE IDE AND POST-APPROVAL STUDY RESULTS
Subject Demographics and Baseline Paremeters
Table 2 summarizes the study patient demographics and baseline characteristics.
Table 2: Study Patient Demographics and Baseline Characteristics
Variables Investigational
(N=242)
Age (years) 44.4 ± 7.9 44.7 ± 8.6 0.723
Height (inches) 67.6 ± 3.8 67.6 ± 3.8 0.991
Weight (lbs.) 173.3 ± 37.7 180.0 ± 38.9 0.061
BMI 26.6 ± 4.8 27.6 ± 5.0 0.027
Sex (% male) 45.5% 51.1% 0.228
Race
Caucasian
Black
Asian
Hispanic
Other
Marital Status
Single
Married
Divorced
Separated
Widowed
Education Level
< High School
High School
> High School
Worker’s Compensation 6.2% 5.0% 0.687
Unresolved Spinal Litigation 2.5% 2.7% 1.000
Current Tobacco Use 25.5% 24.0% 0.746
Current Alcohol Use 8.4% 4.1% 0.083
Preoperative Work Status 64.5% 65.0% 0.923
Preoperative NDI Score 51.4 ± 15.3 50.2 ± 15.9 0.392
Duration of Symptoms
< 6 wks.
6 wks. – 3 mos.
3 – 6 mos
6 mos – 1 yr.
1 – 2 yrs.
> 2 yrs.
231
3
1
3
4
29
184
19
6
4
16
63
161
10
36
47
52
38
59
Control
(N=221)
204
5
2
7
3
29
169
20
1
2
15
65
141
13
52
39
37
28
52
p-value
0.527
0.437
0.743
0.180
In addition to the study patients described in Table 2, 117 patients were randomized but declined participation in the study prior
to receiving the assigned treatment. Of these patients, 37 would have received the BRYAN
potential control patients. The demographic and preoperative characteristics of the patients who declined to participate were
comparable to the study patients.
There were 12 patients in this study who were randomized to the investigational treatment but received the control treatment,
and one patient who was randomized to the control treatment but received the investigational treatment. Most of these were
®
disc treatment, while 80 were
Page 8

intraoperative conversions due to sizing issues or difficulty visualizing the target disc space. Table 3 summarizes the surgical
and hospitalization information.
Table 3: Surgical Results
Investigational Control Probability that the surgical measurements of the
investigational group are less than that of the control
group
(%)
Mean operative time (hrs) 2.2 (n=241) 1.4 (n=221) 0.0
Mean EBL (ml) 91.5 (n=240) 59.6 (n=221) 0.0
Hospitalization (days) 1.1 (n=242) 1.0 (n=221) 4.7
Spinal level treated
(%) 3 (1.2) 0 (0.0) N/A
C
3-4
C
(%) 12 (5.0) 17 (7.7) N/A
4-5
C
(%) 140 (57.9) 110 (49.8) N/A
5-6
C
(%) 87 (36.0) 94 (42.5) N/A
6-7
®
BRYAN
Cervical Disc Size Used
14mm (%) 55 (22.7) N/A N/A
15mm (%) 66 (27.3) N/A N/A
16mm (%) 57 (23.6) N/A N/A
17mm (%) 36 (14.8) N/A N/A
18mm (%) 28 (11.6) N/A N/A
Safety Results
The safety of the BRYAN® Cervical Disc was assessed by monitoring intraoperative and postoperative complications.
Radiographs were examined for device subsidence, functional spinal unit height maintenance, device migration, and breakage.
All radiographic endpoints were evaluated independently by a core laboratory and reviewed by independent radiographic
reviewers. In addition, some radiographic observations, such as implant malposition, reported by investigators were handled as
adverse events.
Adverse Events
The adverse effects, as shown in Table 4, were reported from the 242 BRYAN® disc patients and 221 control patients enrolled
in the multi-center clinical study. Adverse event rates presented are based on the number of patients having at least one
occurrence for a particular adverse event divided by the total number of patients in that treatment group. Patients experiencing
adverse events in more than one category are represented in each category in which they experienced an adverse event. As
shown in Table 5, a minority of the adverse events were deemed related to the study treatment. Relationship determinations
were approved by a physician reviewer.
Table 4: Adverse Events for US IDE
Complication Surgery Postoperative 6 Weeks 3 Months 6 Months 12 Months 24 Months # of Patients Reporting &
3
Inves.
Anatomical/Technical
Difficulty
Cancer 00 0 0 1000101011 4 (1.7)41 (0.5)
Cardiovascular 10 1 0 0110103037 8 (3.3)107 (3.2)
Carpal Tunnel
Syndrome
Death 00 0 0 0000000100 0 (0.0)01 (0.5)
Dysphagia/ Dysphonia 15 5 13 12 2 1 0 3 0 0 0 0 2 2 29 (12)
Gastrointestinal 02 2 1 0210312295 12 (5.0)1711 (5.0)
Implant Displacement/
Loosening
Infection 0 0 8 3 4010512246 22 (9.1)2412 (5.4)
Malpositioned Implant 1 0 0 0 0 0 0 0 1 0 0 0 0 0 2 (0.8)
01 0 0 0000000000 0 (0.0)01 (0.5)
00 0 1 2053114111 12 (5.0)137 (3.2)
00 0 0 0000000000 0 (0.0)00 (0.0)
Control
Inves.
1
and Post-Approval Studies
Control
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Total adverse events
Investig. #
Patients (% of
242) Total #
Events
32
2
Control # Patients
(% of 221) Total #
Events
1
1
8
7
1
20 (9.0)
23
13
0
12
0 (0.0)
0
Page 9
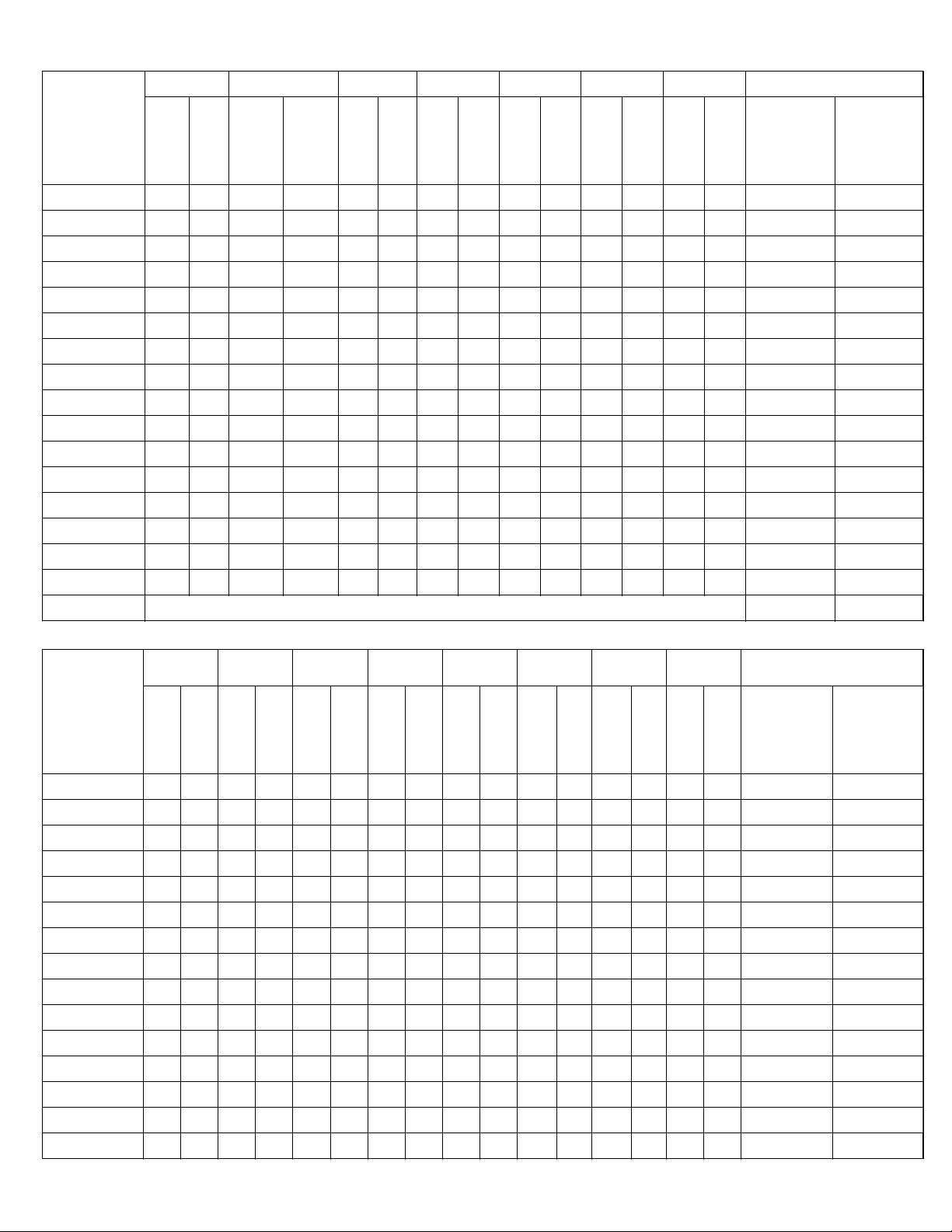
Table 4: Adverse Events for US IDE1 and Post-Approval Studies (continued)
Complication Surgery Postoperative 6 Weeks 3 Months 6 Months 12 Months 24 Months # of Patients Reporting &
3
Inves.
Control
Inves.
Control
Inves.
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Control
Total adverse events
Investig. #
Patients (% of
242) Total #
Events
Control # Patients
(% of 221) Total #
Events
Neck and/or Arm Pain 1 3 25 15 25 18 24 26 22 19 34 22 11 19 110 (45.5)
Neurological 1 1 6 5 3 7 17 11 10 10 21 11 11 10 55 (22.7)
142
69
Non-Union 00 0 0 0102010102 0 (0.0)07 (3.2)
Non-Union – Outcome
Pending
4
Other
5
Other Pain
00 0 0 0102000100 0 (0.0)04 (1.8)
11 7 19 5 9 5 10 8 13 9 26 12 31 21 79 (32.6)
0 1 7 5 6 4 10 12 9 7 16 9 17 13 54 (22.3)
119
65
Respiratory 2 0 1 4 1002003330 10 (4.1)109 (4.1)
Spinal Event
Cervical Adjacent
Non Contiguous
Cervical Adjacent
Contiguous
6
Cervical Target Level 1 1 0 0 0 0 4 0 0 1 5 1 4 1 13 (5.4)
Non-Cervical Level 0 0 1 0 3 2 4 1 1 2 5 8 5 7 18 (7.4)
1 1 1 3 4 4 12 4 3 7 15 13 15 22 44 (18.2)
51
00 0 1 0020000112 3 (1.2)34 (1.8)
00 0 2 12232453512 15 (6.2)1526 (11.8)
14
19
Subsidence 00 0 0 0001000000 0 (0.0)01 (0.5)
Trauma 1 0 2 3 2 1 5 5 12 4 15 8 14 9 40 (16.5)
Urogenital 0 0 0 0 0 0 0 1 2 0 5 4 6 2 10 (4.1)
Vascular Injury IntraOp
21 0 2 0000000000 2 (0.8)23 (1.4)
Any Adverse Event 208 (86.0)
51
13
624
90 (40.7)
122
48 (21.7)
55
7
4
51 (23.1)
67
45 (20.4)
51
9
44 (19.9)
54
4
26
3 (1.4)
4
19 (8.6)
20
1
23 (10.4)
30
7 (3.2)
7
3
185 (83.7)
476
Complication 36 Months 48 Months 60 Months 72 Months 84 Months 96 Months 108 Months 120 Months # of Patients Reporting &
3
Anatomical/Technical
Difficulty
Inves.
0000000000 000000 0 (0.0)
Control
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Cancer 1201112011 110010 9 (3.7)
Cardiovascular 65106446066 274312 40 (16.5)4931 (14.0)
Carpal Tunnel
Syndrome
1023000002 300000 17 (7.0)1910 (4.5)
Death 0001002000 000100 2 (0.8)
Dysphagia/Dysphonia 0 0 1 0 1 1 0 0 2 1 0 1 0 0 1 0 32 (13.2)
Gastrointestinal 9 11 13 6 7 7 13 5 6 5 4 4 3 1 0 2 49 (20.2)
Implant Displacement/
Loosening
0010100000 000000 2 (0.8)
Infection 38811778463345700 43 (17.8)6540 (18.1)
Malpositioned Implant 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 2 (0.8)
Neck and/or Arm Pain 17 8 17 19 6 6 7 5 9 4 0 4 8 3 0 4 136 (56.2)
Neurological 6 5 14 6 14 12 3 10 11 6 4 3 7 3 0 0 88 (36.4)
Non-Union 0000001000 000000 1 (0.4)
Non-Union – Outcome
Pending
4
Other
0000000000 000000 0 (0.0)
41 24 54 26 30 21 18 11 21 19 11 8 12 4 5 7 127 (52.5)
Total adverse events
Investig. # Patients
(% of 242) Total #
(≤120 Months)
Events
0
11
2
37
72
2
2
206
128
1
0
311
Control # Patients
(% of 221) Total #
Events
1 (0.5)
1
7 (3.2)
7
41
12
3 (1.4)
3
23 (10.4)
26
34 (15.4)
54
0 (0.0)
0
56
0 (0.0)
0
112 (50.7)
175
74 (33.5)
100
7 (3.2)
7
4 (1.8)
4
90 (40.7)
187
Page 10

Complication 36 Months 48 Months 60 Months 72 Months 84 Months 96 Months 108 Months 120 Months # of Patients Reporting &
3
Inves.
Inves.
Control
Control
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Total adverse events
Investig. # Patients
(% of 242) Total #
(≤120 Months)
Events
Control # Patients
(% of 221) Total #
Events
5
Other Pain
Respiratory 4153432111 212201 24 (9.9)3021 (9.5)
Spinal Event
Cervical Adjacent
Non Contiguous
Cervical Adjacent
Contiguous
Cervical Target Level 2 2 7 2 4 4 1 1 6 0 1 0 0 0 3 0 33 (13.6)
Non-Cervical Level 2 1 8 9 5 2 5 3 4 3 4 3 0 4 0 0 40 (16.5)
Subsidence 0000000000 000000 0 (0.0)
Trauma 14 10 21 9 14 8 7 5 13 4 5 2 5 4 0 0 78 (32.2)
Urogenital 42912446452451300 36 (14.9)4629 (13.1)
Vascular Injury Intra-Op0000000000 000000 2 (0.8)
Any Adverse Event 231 (95.5)
6
21 14 17 27 18 23 10 8 8 13 1 7 2 9 8 4 97 (40.1)
9 8 24 18 16 14 9 8 13 9 7 6 4 8 4 2 95 (39.3)
1202001001 000100 6 (2.5)
4395782435 234402 41 (16.9)4651 (21.3)
150
137
6
38
47
0
130
2
1400
99 (44.8)
156
22
82 (37.1)
127
9 (4.1)
9
60
12 (5.4)
13
37 (16.7)
45
1 (0.5)
1
46 (20.8)
72
39
3 (1.4)
3
199 (90.0)
1093
_________________________________________________________
1
Some adverse events may lead to additional surgeries or interventions. Refer to Table 7 for more information.
3
Control=Single-level anterior interbody fusion procedure with allograft and plate stabilization.
4
Other consists of various events that do not fit into another category, such as rash, depression, or hypertension. This category
also consists of three events related to an investigator’s report of lack of motion of the prosthesis. A formal evaluation of
heterotopic ossification was not included in the IDE protocol.
5
Other Pain consists of non-neck and/or arm pain events such as headache, lower back pain, or leg pain.
6
Spinal event consists of events reported as a spinal diagnosis/disorder (e.g., degenerative disc disease, disc herniation,
stenosis, scoliosis).
1
Table 5: Adverse Events Classified as Device-Related or Device/Surgical Procedure-Related in US IDE
and Post-
Approval Studies
Complication Surgery Postoperative 6 Weeks 3 Months 6 Months 12 Months 24 Months # of Patients Reporting &
3
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Inves.
Control
Total adverse events
Invest. #
Patients (% of
242) Total #
Events
Control #
Patients (% of
221) Total #
Events
Implant
Displacement/
Loosening
Malpositioned
Implant
Neck and/or Arm
Pain
Non-Union 0 0 0 0 0102000102 0 (0.0)06 (2.7)
Non-Union –
Outcome Pending
4
Other
Spinal Event 00 0 0 0020013021 6 (2.5)72 (0.9)
Cervical Adjacent
Contiguous
Cervical Target
Level
Subsidence 00 0 0 0001000000 0 (0.0)01 (0.5)
Trauma 00 0 0 0000100000 1 (0.4)10 (0.0)
Any Adverse Event 13 (5.4)
00 0 0 0000000000 0 (0.0)00 (0.0)
10 0 0 0000100000 1 (0.4)10 (0.0)
00 2 0 0100000000 2 (0.8)21 (0.5)
00 0 0 0102000100 0 (0.0)04 (1.8)
00 0 0 0000101010 3 (1.2)30 (0.0)
00 0 0 0000010000 0 (0.0)01 (0.5)
00 0 0 0020003021 6 (2.5)71 (0.5)
14
0
0
1
6
4
0
2
1
1
1
0
14 (6.3)
14
Page 11

Complication 36 Months 48 Months 60 Months 72 Months 84 Months 96 Months 108 Months 120 Months # of Patients Reporting &
3
Total adverse events
Invest. # Patients
(% of 242) Total #
Events
Control # Patients
(% of 221) Total #
Events
Invest.
Implant
Displacement/
Loosening
Malpositioned
Implant
Neck and/or Arm
Pain
Non-Union 0000000000000000 0 (0.0)06 (2.7)
Non-Union –
Outcome Pending
Other 0000000000000000 3 (1.2)30 (0.0)
Spinal Event 0011102030000020 15 (6.2)163 (1.4)
Cervical Adjacent
Contiguous
Cervical Target
Level
Subsidence 0000000000000000 0 (0.0)01 (0.5)
Trauma 0000000000000000 1 (0.4)10 (0.0)
Any Adverse Event 24 (9.9)
0010100000000000 2 (0.8)20 (0.0)
0000000000000000 1 (0.4)10 (0.0)
0000000010000000 3 (1.2)31 (0.5)
0000000000000000 0 (0.0)04 (1.8)
0001001000000000 1 (1.4)12 (0.9)
0010101030000020 14 (5.8)151 (0.5)
Control
Invest.
Invest.
Control
Invest.
Control
Invest.
Control
Invest.
Control
Invest.
Control
Invest.
Control
Control
0
0
1
6
4
0
3
2
1
1
0
26
15 (6.8)
25
* denotes WHO Grade 3 or 4 serious adverse events.
Two deaths were reported in the investigational group. One reported investigational death was due to end stage COPD
approximately 75 months postoperatively, and the other was reported as secondary to prescription drug medication
approximately 78 months postoperatively. Neither of the deaths were considered to be associated with the investigational
implant or implantation procedure.
Three deaths were reported in the control group. One reported control death was due to injuries sustained in a motor vehicle
crash approximately 17 months postoperatively. The second death was due to a drug overdose approximately 54 months
postoperatively. The third death was reported as exacerbation of cardiovascular and ischemic heart disease approximately 108
months postoperatively. None of these deaths were considered to be associated with the control group implant or implantation
procedure.
The total number of spinal events (overall and those classified as device-related or device/surgical procedure-related) were
numerically greater in the investigational group than the control group. Although not statistically significant, the findings are
clinically significant.
An time to event anlaysis was conducted on adverse events up to 120 months. The results are presented in Table 6.
Table 6: Time to Event Analysis of Adverse Events in Main Categories Up to 120 Month Interval
INV (N=242) CTR (N=221) P-Value
(Log-Rank Test)
Total Subjects Experiencing
Events Subjects (n) (cumulative
rate %)
Events Subjects (n) (cumulative
rate %)
1400 231 (98.4%) 1093 199 (98.8%) 0.166
Adverse Events
Anatomical/Technical Difficulty 0 0 (0.0%) 1 1 (0.5%) 0.295
Cancer 11 9 (8.1%) 7 7 (4.8%) 0.860
Cardiovascular 49 40 (27.6%) 41 31 (26.1%) 0.653
Carpal Tunnel Syndrome 19 17 (8.0%) 12 10 (5.4%) 0.309
Death 2 2 (1.4%) 3 3 (2.4%) 0.507
Dysphagia/Dysphonia 37 32 (17.4%) 26 23 (11.7%) 0.341
Gastrointestinal 72 49 (27.8%) 54 34 (27.2%) 0.389
Implant Displacement/
22 (1.0%)00 (0.0%) 0.200
Loosening
Infection 65 43 (23.1%) 56 40 (28.1%) 0.669
Malpositioned Implant 2 2 (0.8%) 0 0 (0.0%) 0.179
Neck and/or Arm Pain 206 136 (61.0%) 175 112 (72.6%) 0.411
Neurological 128 88 (43.1%) 100 74 (43.8%) 0.888
Page 12

Table 6: Time to Event Analysis of Adverse Events in Main Categories Up to 120 Month Interval (continued)
INV (N=242) CTR (N=221) P-Value
Events Subjects (n) (cumulative
rate %)
Events Subjects (n) (cumulative
rate %)
(Log-Rank Test)
Non-Union 1 1 (0.7%) 7 7 (3.4%) 0.019
Non-Union – Outcome Pending 0 0 (0.0%) 4 4 (1.9%) 0.034
Other 311 127 (60.8%) 187 90 (62.1%) 0.015
Other Pain 150 97 (51.2%) 156 99 (60.9%) 0.147
Respiratory 30 24 (12.8%) 22 21 (16.9%) 0.984
Spinal Event 137 95 (50.2%) 127 82 (54.6%) 0.792
Cervical Adjacent Non
66 (6.4%)99 (5.2%) 0.266
Contiguous
Cervical Adjacent Contiguous 46 41 (22.5%) 60 51 (38.5%) 0.033
Cervical Target Level 38 33 (24.1%) 13 12 (7.3%) 0.006
Non-Cervical Level 47 40 (20.8%) 45 37 (24.4%) 0.713
Subsidence 0 0 (0.0%) 1 1 (0.5%) 0.293
Trauma 130 78 (40.3%) 72 46 (26.1%) 0.012
Urogenital 46 36 (20.9%) 39 29 (19.4%) 0.873
Vascular Injury Intra-Op 2 2 (0.8%) 3 3 (1.4%) 0.583
Table 7 summarizes the secondary interventions in the BRYAN® device and control treatment groups. Revisions, removals, and
supplemental fixations occurring at or before the 24-month and 120-month follow-up visits were respectively considered second
surgery failures for the IDE primary endpoint and PAS primary endpoint in the clinical study. Reoperations were not considered
second surgery failures in the study. Table 8 also presents the Time to Event Analysis of Secondary Surgeries comparison
between the BRYAN
®
device and control treatment groups. As shown in this table, the rate of secondary surgeries at the index
level was 6.7% in the investigational group and 11.5% in the control group. For these safety comparisons, p-values less than
0.05 are considered statistically significant.
Table 7: Secondary Interventions and Surgical Procedures
Total ≤ 24 Months
Control
60 months
Control
6 Weeks
Invest.
Control
72 months
Invest.
Invest.
Control
3 Months
Control
Invest.
Invest.
84 months
Control
6 Months
Control
Invest.
Invest.
96 months
Control
12 Months
Control
Invest.
Invest.
108 months
Control
24 Months
Control
Invest.
Invest.
≤ 120 months
36 Months
Control
Control
Invest.
# Patients
(% of 242)
Total # Events
Invest.
# Patients
(% of 242)
Total #
Events
Total ≤ 120 Months
Control
# Patients
(% of 221)
Total #
Events
Control
# Patients
(% of 221)
Total # Events
Surgery
Invest.
Control
Revisions 10000000000000001 (0.4)10 (0.0)
Removals 00000010111002113 (1.2)33 (1.4)
Reoperations00000000002110103 (1.2)31 (0.5)
Supplemental
Fixations
Other –
Cervical
Surgery
Other - Noncervical Spinal
Surgery
00000001010004010 (0.0)06 (2.7)
00100010004225317 (2.9)87 (3.2)
000000121221650210 (4.1)109 (4.1)
48 months
Invest.
Control
Postoperative
Invest.
Invest.
0
3
1
6
7
10
Revisions 10000000000000 1 (0.4)10 (0.0)
Removals 00131102010000 6 (2.5)611 (5.0)
0
11
Page 13

Total ≤ 120 Months
48 months
Invest.
Reoperations101000000000006 (2.5)61 (0.5)
Supplemental
Fixations
Other – Cervical
Surgery
Other - Noncervical Spinal
Surgery
Control
011000000000001 (0.4)17 (3.2)
1423322116100018 (7.4)2120 (9.0)
2465124401260120 (8.3)2529 (13.1)
60 months
Invest.
Control
72 months
Invest.
Control
84 months
Invest.
Control
96 months
Invest.
Control
108 months
Invest.
Control
≤ 120 months
Invest.
Control
Invest.
# Patients
(% of 242)
Total # Events
Control
# Patients
(% of 221)
Total # Events
Table 8: Time to Event Analysis of Secondary Surgeries Up to 120 Month Interval
INV (N=242) CTR (N=221) P-Value
(Log-Rank Test)
Any Second Surgery at Index
Events Subjects (n) (cumulative
rate %)
Events Subjects (n) (cumulative
rate %)
14 14 (6.7%) 20 18 (11.5%) 0.214
Level
Revision 1 1 (0.4%) 0 0 (0.0%) 0.339
Removal 6 6 (2.9%) 11 11 (7.9%) 0.103
Supplemental Fixation 1 1 (0.5%) 8 7 (3.6%) 0.017
Reoperations 6 6 (2.8%) 1 1 (0.5%) 0.091
Supplemental Fixation - External
22 (1.0%)
Bone Growth Stimulator
Other 245 109 (54.4%) 236 92 (60.4%) 0.933
Cervical Surgery 21 18 (9.8%) 24 20 (13.1%) 0.352
Non-cervical Spinal Surgery 25 20 (10.2%) 35 29 (21.1%) 0.050
Non-Spinal Surgery 199 92 (47.0%) 177 76 (51.8%) 0.771
Second surgeries involved with
25 21 (11.3%) 37 29 (18.8%) 0.054
adjacent levels and/or levels
second to adjacent Levels
Second surgeries involved with
22 19 (9.7%) 32 24 (15.8%) 0.146
adjacent levels
1
8
24
35
Enhanced Surveillance Safety Results
Information on the cumulative total MDRs (n=25) and device sales has been collected and evaluated for the five-year time
period after PMA approval. During the 60-month study period, no unanticipated adverse events were identified, and the device
has not been withdrawn from any of the markets due to safety concerns.
Effectiveness Results
Primary Effectiveness Analysis
The primary endpoint for both the IDE and post-approval studies was a composite of the following parameters: pain and
functional disability, neurological status, adverse events, and secondary surgical interventions. This was termed overall
success.
In the approved protocol, individual subject success (i.e., overall success) was defined as attainment of all of the following:
▪ An improvement of at least 15 points from the baseline Neck Disability Index (NDI) score.
▪ Maintenance or improvement in each evaluated neurological status parameter as compared to the pre-operative baseline
score for motor function, sensory function and reflexes.
▪ No serious adverse event classified as implant-associated or implant/surgical procedure-associated.
▪ No additional surgical procedure classified as “Failure”.
Overall success was determined at the 24-month time point for the IDE and the 120-month time point for the post-approval
study.
Overall Success Summary
Study success was expressed as the number of individual subjects categorized as a success divided by the total number of
subjects evaluated. Table 9 describes the success rates and p-values for the comparison of individual outcome parameters and
Page 14

overall success. Observed success rates are the 24-month and 120-month outcomes of the clinical trial. The p-values were
calculated using the z-test of the normal approximation to the binomial distributions with the standard error derived from using
the Farrington-Manning Method to assess both the non-inferiority and superiority hypotheses. The conclusions were based on
the analysis which was pre-defined in the protocol.
Table 9: Observed Success Rates at 24 and 120 Months and P-Values for the Comparison of Success Rates Between
the Two Groups
Primary Outcome
Variable
NDI 197/229
Neurological 215/229
Serious, Related Adverse
Event Failure*
2nd Intervention Failure* 4 5 n/a n/a 8 8 n/a n/a
Overall Success 190/230
24-Month Observed Success Rate P-Values for the Comparison of 24-Month Results 120-Month Observed Success Rate 120-Month Posterior Probabilities
Inv Ctrl Non-inferiority Superiority Inv Ctrl Non-inferiority Superiority
(86.0%)
(93.9%)
45 n/a n/a 910n/an/a
(82.6%)
153/194
(78.9%)
175/194
(90.2%)
142/194
(73.2%)
< 0.001 0.026 114/126
< 0.001 0.08 116/126
< 0.001 0.01 104/128
(90.5%)
(92.1%)
(81.3%)
78/103
(75.7%)
98/103
(95.1%)
69/104
(66.3%)
< 0.001 0.001
0.023 0.826
< 0.001 0.005
* The demonimators for these rates are given as the total number of patients due to the cumulative nature of these events. In
Table 9, the clinical evaluation visit dates were used as the back end of the windows, whereas Tables 4 and 5 utilized wider,
continuous time windows. Therefore, the events included in Table 9 may appear to differ from those in Tables 4 and 5.
Non-inferiority of the BRYAN
success at both 24 and 120 months as listed in Table 9. Statistical superiority of the BRYAN
®
disc group to the control group was demonstrated for NDI, neurological status, and overall
®
disc group to the control group
was demonstrated for overall success and the NDI variable for the specifically defined population studied in the clinical trial at
both 24 months and 120 months postoperatively. The neurological component was not found to be statistically superior in the
BRYAN
®
group at these time points even though it did achieve non-inferiority at both time points.
Sensitivity Analyses
To examine the effect of unknown outcomes on study conclusions, sensitivity analyses were conducted by imputing possible
overall success outcomes for the missing patients. All analyses, even the worst-case assumptions, demonstrated at least non-
inferiority of the BRYAN
®
Cervical Disc to the fusion control in Overall Success.
Secondary Effectiveness Analysis
The secondary endpoints assessed were Neck Pain, Arm Pain, SF-36 Health Survey, Gait Assessment, Foraminal
Compression, Work Status, Patient Satisfaction, Radiographic endpoints, Global Perceived Effect (patient’s overall assessment
of study treatment effectiveness as a function of pain), and Doctor’s Perception (investigator’s perception of patient’s condition).
Radiographic endpoints include FSU height/implant subsidence; anteroposterior implant migration; treated level angular motion
in flexion, extension, and side bending; and translational motion. Fusion measurements replaced motion measurements in
control patients.
Radiographic Assessments
For patients receiving the BRYAN® device, the mean angular range of motion values at 24 and 120 months postoperative,
respectively, were 8.08° (n=222) and 8.69° (n=126) as compared to a preoperative value of 6.45° (n=215). The range of motion
values measured from flexion/extension radiographs at 24 and 120 months for the BRYAN
histograms below. This histograms used values obtained by rounding recorded range of motion for each subject to the nearest
integer.
Figure 1: Histogram of BRYAN
®
Cervical Disc Angular Range of Motion at 24 Months
®
device patients are presented in the
Page 15

Figure 2: Histogram of BRYAN® Cervical Disc Angular Range of Motion at 120 Months
An analysis of the correlation between the degree of segmental motion and pain was also performed, and no statistically
significant correlations were noted.
At 120 months, 7.4% (9/122) investigational subjects had Grade III heterotopic ossification (HO), and 6.6% (8/122) had Grade
IV HO. The incidence of HO increased and worsened over time with resultant loss motion. BRYAN
observed in two investigational subjects (one at 60 months and another at 120 months).
®
implant migration was
Conclusions Drawn from the Study Data
In summary, the overall success rate for the investigational group at 24 and 120 months was found to be not only statistically
non-inferior to the control group rate, but also superior. Therefore, the primary clinical study objective was met, thus indicating
that the BRYAN
intractable radiculopathy or myelopathy or the cervical spine.
®
Cervical Disc is as safe and effective in the long term as the current standard of care for treating patients with
PACKAGING
Packages for each of the components should be intact upon receipt. If a loaner or consignment system is used, all sets should
be carefully checked for completeness and all components including instruments should be carefully checked to ensure there is
no damage prior to use. Damaged packages or products should not be used, and should be returned to Medtronic.
CLEANING AND DECONTAMINATION
Implants are supplied sterile from Medtronic and should be used directly from the sterile package. Implants should not be
cleaned or decontaminated by the user. Unless just removed from an unopened Medtronic package, all instruments must be
disassembled (if applicable), cleaned using neutral or enzymatic cleaners prepared per manufacturer’s instructions before
sterilization, introduction into a sterile surgical field, or (if applicable) before returning the product to Medtronic. Cleaning and
disinfecting of instruments can be performed with aldehyde-free solvents at higher temperatures. Cleaning and decontamination
must include the use of neutral cleaners followed by a deionized water rinse. This should be performed as soon as reasonably
practical following use to minimize the potential of drying prior to cleaning.
Instrument Cleaning
When cleaning instruments by hand, the following should be observed:
1. Clear any corners or recesses of all debris (Note: extra care should be taken to clean out any cannulated areas by using an
appropriate cleaning stylet and rinsing immediately).
2. Remove all traces of blood and other such residues as soon as reasonably practical following use to minimize the potential
of drying.
3. The instruments should be submerged and cleaned with a commercially available manual cleaner (i.e., Terg-A-Zyme from
ALCONOX, Inc.) prepared according to the manufacturer’s recommendation.
4. Scrub the device while immersed in the cleaning solution with a soft nylon brush for a minimum of one minute, giving special
attention to threads, crevices, and hard to reach areas until visibly clean. During cleaning, actuate device.
5. The instrument should be thoroughly rinsed after cleaning for a minimum of one minute with fresh deionized or tap water.
Repeat process if visual inspection warrants repeat cleaning.
Note: certain cleaning solutions such as those containing formalin, glutaraldehyde, bleach and/or other alkaline cleaners may
damage some devices, particularly instruments. These solutions should not be used. Also, many instruments require
disassembly before cleaning.
All products should be treated with care. Improper use or handling may lead to damage and/or possible improper functioning of
the device.
Page 16

STERILIZATION
Implants are supplied sterile from Medtronic and should be used directly from the sterile package. Unless marked sterile and
clearly labeled as such in an unopened sterile package provided by the company, instruments used in surgery
must be sterilized
by the hospital prior to use. Remove all packaging materials prior to sterilization. Only sterile products should be placed in the
operative field. Unless specified elsewhere, these products are recommended to be steam sterilized by the hospital using the
process parameters listed in Table 10.
Table 10: Sterilization Cycle Parameters for the United States and Its Territories
METHOD CYCLE TEMPERATURE EXPOSURE TIME DRY TIME
Steam Pre-Vacuum 270°F (132°C) 4 Minutes 30 Minutes
NOTE: Because of the many variables involved in sterilization, each medical facility should calibrate and verify the sterilization
process (e.g., temperatures, times) used for their equipment. It is the responsibility of the end user to ensure the available
equipment (sterilizer and accessories including appropriate sterilization indicators and wraps, as applicable) is compatible with
the set of parameters listed in Table 10.
Remove all packaging material prior to sterilization. Only sterile implants and instruments should be used in surgery. No implant
should be re-used once it comes into contact with human tissue or body fluid. Always immediately clean and re-sterilize
instruments used in surgery. This process must be performed before handling or (if applicable) returning the product to
Medtronic.
PRODUCT COMPLAINTS
Any health care professional (e.g., customer or user of this system of products) who has any complaints or who has
experienced any dissatisfaction in the product quality, identity, durability, reliability, safety, effectiveness, and/or performance,
should notify the distributor or Medtronic. Further, if any of the implanted spinal system component(s) ever “malfunctions” (i.e.,
does not meet any of its performance specifications or otherwise does not perform as intended), or is suspected of doing so, the
distributor should be notified immediately. If any Medtronic product ever malfunctions and may have caused or contributed to
the death or serious injury of a patient, the distributor should be notified immediately by telephone, fax, or written
correspondence. When filing a complaint, provide the component(s) name and number, lot number(s), your name and address,
the nature of the complaint, and notification of whether or not a written report from the distributor is requested.
FURTHER INFORMATION
Recommended directions for use of this system (surgical operative techniques) are available at no charge upon request. If
further information is needed or required, contact Medtronic.
DEVICE RETRIEVALS
Should it be necessary to remove a BRYAN® Cervical Disc device, call Medtronic prior to the scheduled surgery for product/
tissue retrieval information. Refer to the BRYAN
technique for device retrieval and instructions for returning the explanted device to Medtronic. All explanted devices must be
returned to Medtronic for analysis.
©2020 Medtronic Sofamor Danek USA, Inc. All rights reserved.
Medtronic Sofamor Danek USA, Inc.
1800 Pyramid Place
Memphis, TN 38132
Telephone: 800 933 2635 (USA)
901 396 3133 (Outside USA)
Fax: 901 396 0356
EXPLANATION OF SYMBOLS
For US audiences only
CAUTION: Federal law (USA) restricts these devices to
sale by or on the order of a physician.
Consult instructions for use
®
Cervical Disc Surgical Technique for step-by-step instructions on the required
Do not re-use
Batch code
Manufacturer
Page 17
Catalogue number
Sterilized using ethylene oxide
Sterilized using irradiation
Use-by date
MR Conditional
 Loading...
Loading...