


INSTRUCTIONS FOR USE FOR:
en
English
hu
Magyar

1
INSTRUCTIONS FOR USE FOR:
GORE® CARDIOFORM Septal Occluder
Carefully read all instructions prior to use. Observe all warnings and
precautions noted throughout these instructions.
Failure to do so may result in complications.
DESCRIPTION
The GORE® CARDIOFORM Septal Occluder consists of an implantable Occluder
and a Delivery System. The Occluder is comprised of a platinum-filled nickeltitanium (Nitinol) wire frame covered with expanded polytetrafluoroethylene
(ePTFE). The ePTFE includes a hydrophilic surface treatment to facilitate
echocardiographic imaging of the Occluder and surrounding tissue during
implantation. When fully deployed, the Occluder assumes a double-disc
configuration to prevent shunting of blood between the right and left atria. The
Delivery System consists of a 75 cm working length 10 Fr outer diameter Delivery
Catheter that is coupled to a Handle. The Handle facilitates loading, deployment,
and locking of the Occluder. The Handle also allows repositioning and retrieval of
the Occluder via the Retrieval Cord, if necessary.
The Occluder is available in diameters of 15, 20, 25, and 30 mm. The Occluder is
delivered using conventional catheter delivery techniques and may be delivered
with the aid of a 0.035" guidewire (or smaller), if desired.
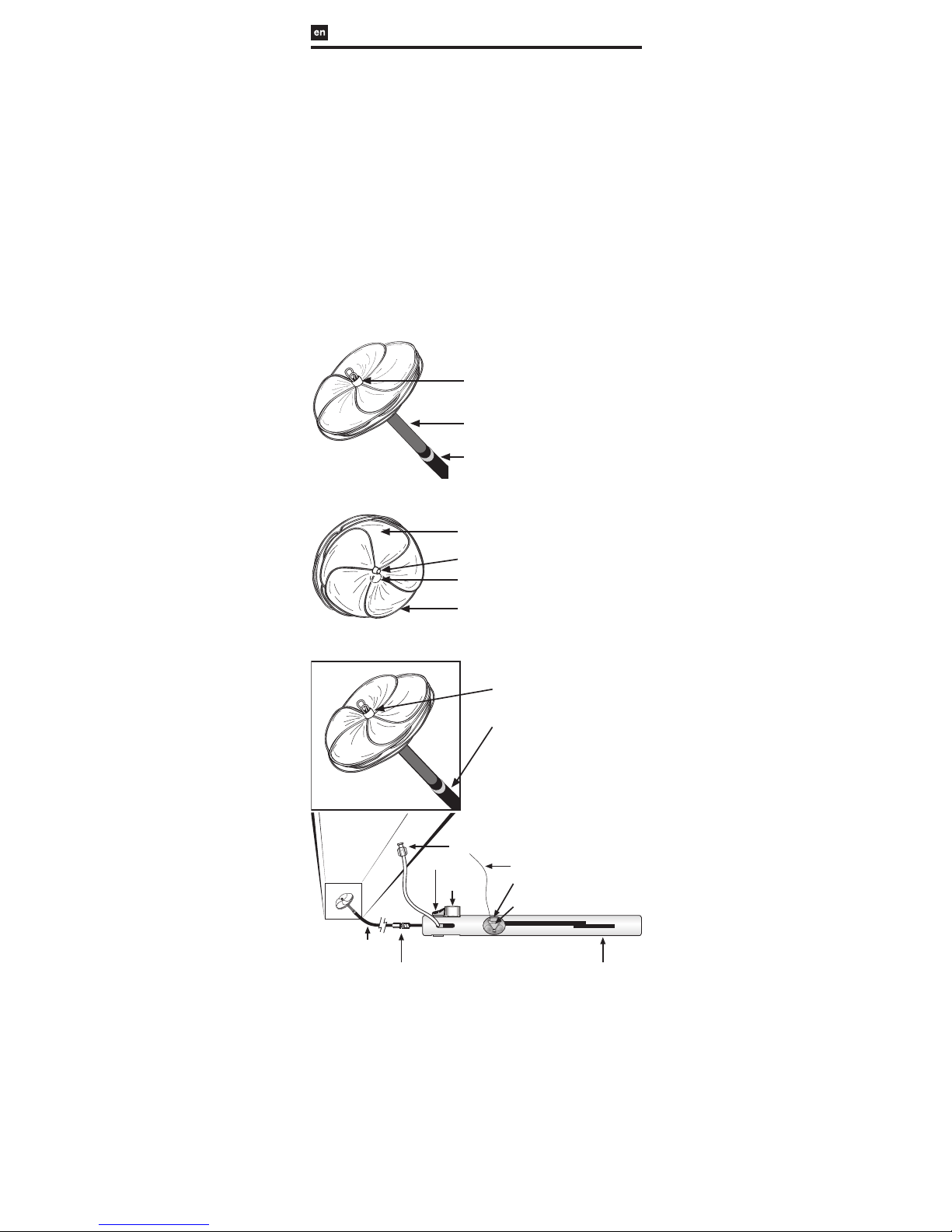
FIGURE 1: GORE® CARDIOFORM Septal Occluder
FIGURE 1a: Left Atrial View
Left Atrial Eyelet
Control Catheter (Gray)
Delivery Catheter (Blue)
FIGURE 1b: Right Atrial View
Occluder Leaflet
Platinum-Filled
Nitinol Wire Frame
Lock Loop
Right Atrial Eyelet
FIGURE 2: GORE® CARDIOFORM Septal Occluder Delivery System
Left Atrial Eyelet
Flush Port
Packaging
Insert (clear)
Occluder Lock
(red)
Delivery Catheter (Blue)
Retrieval Cord
Slider (gray)
Retrieval Cord Lock (red)
Handle
Retrieval Luer
Delivery Catheter (blue)
- 75 cm Working Length -
INDICATIONS / INTENDED USE
The GORE® CARDIOFORM Septal Occluder is a permanently implanted device
indicated for the percutaneous, transcatheter closure of ostium secundum atrial
septal defects (ASDs).
CONTRAINDICATIONS
The GORE® CARDIOFORM Septal Occluder is contraindicated for use in patients:
• Unable to take anti-platelet or anticoagulant medications such as aspirin,
heparin, or warfarin.
• With anatomy where the GORE® CARDIOFORM Septal Occluder size or
position would interfere with other intracardiac or intravascular structures,
such as cardiac valves or pulmonary veins.
• With active endocarditis, or other infections producing bacteremia, or
patients with known sepsis within one month of planned implantation,
or any other infection that cannot be treated successfully prior to device
placement.
• With known intracardiac thrombi.

2
WARNINGS
• The GORE® CARDIOFORM Septal Occluder is not recommended for, and has
not been studied in, patients with other anatomical types of ASDs that are
eccentrically located on the septum (e.g. sinus venosus ASD and ostium
primum ASD), or fenestrated Fontan.
• The GORE® CARDIOFORM Septal Occluder has not been studied in patients
with multiple defects requiring placement of more than one device.
• The GORE® CARDIOFORM Septal Occluder is not recommended for defects
larger than 17 mm.
• Regarding device sizing:
- The defect and atrial chamber size should be evaluated by
Transesophageal (TEE) or Intracardiac Echo (ICE) with color flow
Doppler measurement to confirm that there is adequate space to
accommodate the selected occluder size without impinging on
adjacent cardiac structures (e.g., A-V valves, ostia of the pulmonary
veins, coronary sinus, or other critical features).
- There must be adequate room in the atrial chambers to allow the
right and left atrial discs to lie flat against the septum with disc
spacing equal to the septal thickness, and without interference with
critical cardiac structures or the free wall of the atria.
- An occluder that pulls through the defect after disc conformation
may be too small and should be removed and replaced with a
larger size.
• Embolized devices must be removed. An embolized device should not be
withdrawn through intracardiac structures unless the occluder has been
adequately collapsed within a sheath.
• The GORE® CARDIOFORM Septal Occluder should be used only by
physicians trained in its use, and in transcatheter defect closure techniques.
• Patients allergic to nickel may suffer an allergic reaction to this device.
Certain allergic reactions can be serious; patients should be instructed to
notify their physicians immediately if they suspect they are experiencing an
allergic reaction such as difficulty breathing or inflammation of the face or
throat. Some patients may also develop an allergy to nickel if this device is
implanted.
PRECAUTIONS
Handling
• The GORE® CARDIOFORM Septal Occluder is intended for single use only.
An unlocked and removed occluder cannot be reused. Gore does not have
data regarding reuse of this device. Reuse may cause device failure or
procedural complications including device damage, compromised device
biocompatibility, and device contamination. Reuse may result in infection,
serious injury, or patient death.
• Inspect the package before opening. If seal is broken, contents may not be
sterile.
• Inspect the product prior to use in the patient. Do not use if the product
has been damaged.
• Do not use after the labeled “use by” (expiration) date.
• Do not resterilize.
Procedure
• The GORE® CARDIOFORM Septal Occluder should only be used in patients
whose vasculature is adequate to accommodate a 10 Fr delivery sheath (or
12 Fr delivery sheath when a guidewire is used).
• Retrieval equipment such as large diameter sheaths, loop snares, and
retrieval forceps should be available for emergency or elective removal of
the occluder.
• An Activated Clotting Time (ACT) greater than 200 seconds should be
maintained throughout the procedure.
• The GORE® CARDIOFORM Septal Occluder should be used only in
conjunction with appropriate imaging techniques to assess the septal
anatomy and to visualize the wire frame.
• If successful deployment cannot be achieved after three attempts,
an alternative device or treatment for ASD closure is recommended.
Consideration should be given to the patient’s total exposure to radiation
and anesthesia if prolonged or multiple attempts are required for the
placement of the GORE® CARDIOFORM Septal Occluder.
• Expansion of an occluder disc may occur in the periprocedural time period.
If there is uncertainty that an expanded device remains locked, fluoroscopic
examination is recommended in order to identify if the Lock Loop captures
all three eyelets.
• Removal of the Occluder should be considered if:
- The Lock Loop does not capture all three eyelets
- The Occluder will not come to rest in a planar position apposing the
septal tissue
- The selected Occluder allows excessive shunting
- There is impingement on adjacent cardiac structures
Post Procedure
• Patients should take appropriate prophylactic antibiotic therapy consistent
with the physician’s routine procedures following device implantation.
• Patients should be treated with antiplatelet therapy for six months postimplant. The decision to continue antiplatelet therapy beyond six months is
at the discretion of the physician.
• In patients sensitive to antiplatelet therapy, alternative therapies, such as
anticoagulants, should be considered.
• Patients should be advised to avoid strenuous physical activity for a period
of at least two weeks after occluder placement.
• Patients should have Transthoracic Echocardiographic (TTE) exams prior to
discharge, and at 1, 6, and 12 months after occluder placement to assess
defect closure. Attention should be given to the stability of the device on
the atrial septum during these assessments, as a lack of device stability may
be indicative of wire frame fractures. In instances where device stability is
questionable, fluoroscopic examination without contrast is recommended
in order to identify and assess wire frame fractures.

3
Adverse Events
Clinical Summary
The GORE® CARDIOFORM Septal Occluder was evaluated in a multi-center,
non-randomized, Pivotal Study that included 50 subjects. An Independent
Data Reviewer provided external oversight and review of subject safety data,
including evaluation of all reported adverse events for accuracy of event coding,
seriousness, and relationship to the device. An event was considered a Serious
Adverse Event if it led to death or serious deterioration in health that resulted in
a life threatening illness or injury or in permanent impairment. Device Events, a
type of Serious Adverse Event, were defined as any post-procedure embolization,
post-procedural device removal, or any other reintervention to the septal defect.
Deaths
No deaths have been reported in study subjects.
Serious Adverse Events
No Serious Adverse Events, including Device Events, were observed in any study
subjects through the 6-month follow-up.
Non-Serious Adverse Events
Non-Serious Adverse Events reported through the 6-month follow-up for Pivotal
Study subjects and determined to be potentially or definitely related to the
implant procedure or the device are presented in Table 1.
Table 1. Subjects with Non-Serious Adverse Events Through 6 Months
– Pivotal Study
Subjects Evaluable for Safety 50
Subjects With One or More Non-Serious Adverse Events 12 (24.0%)
Anesthesia or Procedural 8 (16.0%)
Incision site complication 4 (8.0%)
Anesthesia complication 3 (6.0%)
Procedural pain 2 (4.0%)
Nervous System 2 (4.0%)
Burning sensation 1 (2.0%)
Migraine 1 (2.0%)
Other 3 (6.0%)
Respiratory, thoracic and mediastinal 2 (4.0%)
Gastrointestinal 1 (2.0%)
POTENTIAL DEVICE OR PROCEDURERELATED ADVERSE EVENTS
Adverse Events associated with the use of the Occluder may include, but are not
limited to:
• Repeat procedure to the septal defect
• Device embolization
• New arrhythmia requiring treatment
• Intervention for device failure or ineffectiveness
• Access site complications requiring surgery, interventional procedure,
transfusion, or prescription medication
• Thrombosis or thromboembolic event resulting in clinical sequelae
• Perforation of a cardiovascular structure by the device
• Device fracture resulting in clinical sequelae or surgical intervention
• Occluder disc expansion resulting in clinical sequelae or intervention
• Air embolism
• Myocardial infarction
• Pericardial tamponade
• Cardiac arrest
• Renal failure
• Sepsis
• Significant pleural or pericardial effusion requiring drainage
• Significant bleeding
• Endocarditis
• Headache or migraine
• TIA or stroke
• Death
CLINICAL STUDIES
The GORE® CARDIOFORM Septal Occluder was evaluated for safety and
effectiveness in a multicenter, non-randomized Pivotal Study with 50 subjects
enrolled for closure of ostium secundum atrial septal defects.
Design
Patient Selection
Subjects enrolled in the Pivotal Study were required to have an ostium secundum
atrial septal defect with evidence of right heart volume overload. Subjects
were eligible for enrollment if their defect measured ≤ 17 mm in diameter by
stop-flow balloon sizing and had adequate septal rims to successfully retain the
occluder. Exclusion criteria included:
• Significant known pre-existing electrophysiologic, structural cardiovascular
defect, or other comorbidities that could elevate morbidity / mortality
beyond what is common for ASD or would be expected to require surgical
treatment within three years of device placement.
• Systemic or inherited conditions that would significantly increase subject
risk of major morbidity and mortality during the term of the study.
• Anatomy where the size or position of the occluder would interfere with
other intracardiac or intravascular structures, such as cardiac valves or
pulmonary veins.
• Active endocarditis, other infections producing bacteremia, or known
sepsis within one month of planned implantation, or any other infection
that could not be treated successfully prior to device placement.
• One or more known intracardiac thrombi.
• Uncontrolled arrhythmia.
• History of stroke resulting in a significant morbidity or disability.
• Pregnant or lactating at time of enrollment.
• Contraindication to antiplatelet therapy.

4
• Pulmonary artery systolic pressure greater than half the systemic systolic
arterial pressure unless the indexed pulmonary arteriolar resistance was
<5 Woods units.
• Multiple defects based on screening imaging and stop-flow balloon sizing
that would require placement of more than one device.
Subjects provided informed consent prior to enrollment. No training cases were
required by study investigators prior to enrollment in the Pivotal Study.
Procedure and Follow-up
Dimensional verification and characterization of the ASD, interatrial septum, and
surrounding cardiac structures were performed during the implant procedure. The
measurement of ASD size was taken utilizing the stop-flow technique
(a balloon was placed across the defect and slowly expanded until it filled
the defect space and blood flow through the defect was prevented). The
measurement of the balloon’s waist (i.e. the narrowest portion) was recorded
as the defect diameter and used to determine the appropriate size GORE®
CARDIOFORM Septal Occluder. Fluoroscopic and echocardiographic guidance
were used throughout the procedure for placement and assessment of the GORE®
CARDIOFORM Septal Occluder. All subjects were placed on the investigator’s
choice of antiplatelet therapy for six months following device implantation,
and on prophylactic, post-procedure antibiotic therapy consistent with the
investigator’s routine procedure.
Follow-up evaluations, which included a physical exam, ECG, and an assessment
of residual shunt status by echocardiography, were performed at hospital
discharge, and at 1 and 6 months post-procedure. At the 6-month follow-up visit,
fluoroscopic examination was performed to assess device integrity.
Endpoints
The primary endpoint for the study was Composite Clinical Success, evaluated at
6 months post-procedure. Composite Clinical Success was defined as:
1) Successful deployment and retention of a GORE® CARDIOFORM Septal
Occluder, 2) No Serious Adverse Events during 30-day follow-up, 3) No Device
Events through 6-month follow-up, and 4) Clinical closure success, where
the defect was classified as either completely occluded or having a clinically
insignificant shunt at the 6-month follow-up as determined by echocardiography
core lab evaluation. An event was considered a Serious Adverse Event if it led
to death or serious deterioration in health that resulted in a life threatening
illness or injury or in permanent impairment. Device Events were defined as
any post-procedure embolization, post-procedural device removal, or any other
reintervention to the septal defect.
Secondary endpoints evaluated specific safety and efficacy results in study
subjects. Safety endpoints were defined as the proportion of subjects who
experienced a Serious Adverse Event in the first 30 days or a Device Event
through the 6-month follow-up. Technical Success was defined as successful
deployment and retention of a GORE® CARDIOFORM Septal Occluder. Closure
success endpoints were evaluated for those subjects with Technical Success.
Procedure Success was defined as a residual shunt of ≤ 2 mm at the end of the
implant procedure as measured by the investigational site. Closure Success was
defined as a residual shunt ≤ 2 mm at 6-month follow-up as measured by the
echocardiography core lab.
Results
Demographics and Defect Characteristics
Subject demographics at enrollment and defect characteristics assessed at the
implant procedure by the investigational site are listed in Table 2. Subject medical
history is shown in Table 3.

5
Table 2. Subject Demographics and Defect Characteristics – Pivotal
Study
Number of Subjects 50
Patient Demographics
Gender
Male 23 (46.0%)
Female 27 (54.0%)
Race
Black or African American 8 (16.0%)
White or Caucasian 39 (78.0%)
Other Race 4 (8.0%)
Age (years) N = 50
Mean (Std Dev) 19.7 (21.0)
Median 7.4
(Min, Max) (3.4, 68.3)
Height (cm) N = 50
Mean (Std Dev) 133.0 (33.6)
Median 121.5
(Min, Max) (40.5, 188.0)
Weight (kg) N = 50
Mean (Std Dev) 45.1 (32.3)
Median 27.6
(Min, Max) (11.9, 133.6)
Defect Characteristics
Stop Flow Balloon Defect Size (mm) N=49
Mean (Std Dev) 11.9 (3.4)
Median 12.0
(Min, Max) (5.7, 17)
Atrial Septal Aneurysm
1
14.0% (7/50)
Deficient Retroaortic Rim
2
26.0% (13/50)
Multiple Fenestrations 20.0% (10/50)
1
Protrusion of the septum ≥10 mm from baseline in either direction or ≥15 mm total septal
excursion
2
Measured as <5 mm
Table 3. Subject Medical History – Pivotal Study
Number of Subjects 50
Cardiac Arrhythmia 8 (16.0%)
Hypertension 5 (10.0%)
Migraines 8 (16.0%)
Diabetes Mellitus 4 (8.0%)
Previous Cardiac Surgeries 2 (4.0%)
Non-ASD Cardiac Disorders 27 (54.0%)
Vascular Disorders 3 (6.0%)
History of Stroke and/or TIA 4 (8.0%)
Birth/Genetic Defects 9 (18.0%)
Neurological Disorders 7 (14.0%)
Pulmonary/Respiratory Disorders 14 (28.0%)
A wire frame fracture was observed in 9.3% (4/43) of subjects with fluoroscopic
evaluation completed at 6 months. No fractures were associated with device
instability or clinical sequelae.
Procedure and Endpoint Outcomes
Primary, safety, and efficacy endpoint results are shown in Table 4. All subjects
with an atrial septal aneurysm, multiple fenestrations or deficient retroaortic
rim who received a GORE® CARDIOFORM Septal Occluder had complete clinical
closure and no Serious Adverse Events at 6 months.

6
Table 4. Primary, Safety, and Efficacy Endpoints – Pivotal Study
Primary Endpoint
Composite Clinical Success
1
40/43 (93.0%)
Safety Endpoints
30-Day Serious Adverse Events 0.0% (0/50)
6-Month Serious Adverse Events 0.0% (0/50)
6-Month Device Events
2
0.0% (0/50)
Efficacy Endpoints
Technical Success
3
47/50 (94.0%)
Procedure Success
4
46/47 (97.9%)
6-Month Closure Success
5
43/45 (95.6%)
6-Month Clinical Closure Success
6
40/40 (100%)
1
Technical Success and 6-Month Clinical Closure Success without Serious Adverse Events through
30 days or Device Events through 6 months
2
Post-procedural device embolization, post-procedural device removal, or any reintervention to the
septal defect
3
Successful delivery and retention of the device in subjects with a delivery attempted
4
Technical Success with completely occluded defect or residual shunt ≤ 2 mm at the completion of the
implant procedure
5
Technical Success with completely occluded defect or residual shunt ≤ 2 mm at 6 months
6
Technical Success with completely occluded defect or clinically insignificant residual shunt at 6 months
HOW SUPPLIED
The GORE® CARDIOFORM Septal Occluder is supplied sterile in a protective tray
and pouch. Provided that the integrity of the pouch is not compromised in any
way, it will serve as an effective barrier until the “use by” (expiration) date printed
on the box.
REQUIRED ACCESSORIES
• 10 Fr Introducer Sheath
• Heparinized saline
• Flushing syringe
• Stopcock
• Sizing balloon
• Sterile bowl for flushing catheter
OPTIONAL ACCESSORIES
0.035" / 0.89 mm guidewire, or smaller (if necessary for defect access)
12 Fr Introducer Sheath when a guidewire is utilized.
RECOMMENDED PROCEDURES
A. Sizing the Defect and Selecting the Proper Occluder Size
1. Use echocardiography to measure the septal length.
2. Measure the septal defect using fluoroscopy or echocardiography; the stop
flow balloon technique is recommended, as described below:
a. Place a contrast filled, compliant balloon across the defect and gently
inflate until shunting through the defect has stopped.
b. Measure the diameter of the defect using either echocardiography or
calibrated fluoroscopy.
3. Select the appropriate occluder size for the defect, taking the following
recommendations into consideration:
• A minimum occluder to defect size ratio of 1.75:1 is recommended
(reference Table 5). The defect size should be no greater than 17 mm.
An occluder that pulls through the defect after disc conformation
may be too small and should be removed and replaced with a larger
size.
• There must be adequate space to accommodate the discs within
the atrial chambers. To assure that there is adequate space to
accommodate the discs within the atrial chambers, the selected
occluder diameter should be less than 90% of the measured septal
length.
• The septal tissue margins surrounding the defect must be of
sufficient size and integrity to prevent disc prolapse through the
defect and Occluder embolization.
Table 5: GORE® CARDIOFORM Septal Occluder Device Sizing
Labeled Occluder Diameter (mm) Maximum Recommended Defect Size
Measured with Stop Flow Balloon
Sizing (mm)
15 8.5
20 11
25 14
30 17
B. Access Site Preparation
1. Prepare the venous access site according to standard practice.
2. Place appropriately sized Introducer Sheath.
C. Occluder Preparation and Loading
1. Check the “use by” (expiration date) and the condition of the package.
2. Using aseptic technique, remove the sterile tray from the pouch, and
remove the packaging tray lid.
3. Remove the device from the package and visually inspect the device for
shipping damage. Ensure that the Retrieval Luer is tight.
4. Remove the Packaging Insert from the handle (Figure 3).
5. Loading and Flushing the Occluder:
a. Submerge the Occluder and catheter tip in a heparinized saline bath
during loading to reduce the chance of air entrapment in the delivery
system.
b. Fill a syringe with heparinized saline.

7
c. Attach the syringe to a stopcock and the Flush Port.
d. Flush the device until air no longer exits the tip of the Delivery
Catheter.
e. When the initial flushing is completed, begin loading the Occluder
by pushing the Slider up and then to the right until the Slider stops
(Figure 4a).
f. Complete Occluder loading by pushing the Slider down and then to
the right until it stops (Figure 4b).
g. Flush the device again until air no longer exits the tip of the Delivery
Catheter.
h. If additional air removal is desired, it is recommended to deploy the
Occluder (refer to Section E "Occluder Deployment") and repeat steps
d - g above.
The Occluder Lock should not be moved before or during Occluder loading
or deployment. Partial or complete Occluder locking may prevent Occluder
loading and deployment.
FIGURE 3: Packaging Insert Removal
FIGURE 4: Occluder Loading
FIGURE 4a: Initial Occluder Loading
FIGURE 4b: Completion of Occluder Loading
D. Occluder Delivery
1. If applicable, load the Delivery Catheter onto the guidewire by threading
the guidewire into the lumen of the Delivery Catheter from the tip and out
the Guidewire Slot (Figure 5).
2. While flushing the device, load the Delivery Catheter into the appropriately
sized introducer sheath. Close the stopcock and remove the flushing
syringe from the stopcock.
FIGURE 5
E. Occluder Deployment
1. Advance the Delivery Catheter across the atrial septum until the tip is
positioned within the left atrium.
2. If a guidewire was utilized, remove the guidewire before attempting to
deploy the Occluder.
3. Begin deploying the Occluder left disc by pushing the Slider to the left until
it stops (Figure 6a).
4. Complete Occluder left disc deployment by pushing the Slider up and
then to the left until a flat left disc has formed (Figure 6b). This step may
be performed while simultaneously retracting the Delivery System to
minimize advancement of the Occluder within the left atrial chamber.

8
5. Gently pull on the Handle to bring the left atrial disc onto the surface of the
left atrial septum.
6. Deploy the right atrial disc by pushing the Slider to the left until it stops
and then down. Confirm that the Slider has moved completely to the left
and down position (Figure 6c). Failure to move the Slider completely to the
left and down position may prevent Occluder locking.
7. Confirm that both left and right discs appear planar and apposed to the
septum with septal tissue between the discs.
If the position is not correct, refer to Section G, “Reloading the
Occluder”. Note that the Occluder can only be Reloaded prior to
Occluder Locking.
FIGURE 6: Occluder Deployment
FIGURE 6a: Initial Occluder Deployment
FIGURE 6b: Left Atrial Disc Deployment
FIGURE 6c: Right Atrial Disc Deployment
F. Occluder Locking and Delivery System Removal
1. Prior to Occluder locking, assess that the Occluder position and defect
closure are acceptable and that the Delivery System is not exerting tension
on the septum and Occluder.
2. Lock the Occluder by holding the Handle in a fixed position to prevent
applying tension on the Occluder. Note that excessive compression of
the handle may prevent Occluder locking. Next, squeeze and then slide
the Occluder Lock decisively and with a consistent amount of force to the
right (Figure 7). At the completion of Occluder locking, the Occluder is still
attached to the Delivery System by the Retrieval Cord.
During the Occluder locking step, the Delivery Catheter moves
proximally and may exert minimal tension on the introducer sheath.
It is recommended to confirm adequate introducer sheath insertion
prior to Occluder locking.
3. If the Occluder position is not acceptable, refer to Section H, “Removing the
Occluder with the Retrieval Cord After Occluder Locking”.
4. If the Occluder position is acceptable, hold the Handle in a fixed position,
pull up on the red Retrieval Cord Lock (Figure 8a), disengage it from the
Slider, and gently pull the Retrieval Cord Lock until the Retrieval Cord has
been completely removed from the Handle (Figure 8b).
5. The Occluder is now released from the Delivery System and the Delivery
System can be removed.
6. Once the Retrieval Cord is removed, the Occluder cannot be removed using
the Delivery System, refer to Section I, "Recapture".

9
FIGURE 7: Occluder Locking
FIGURE 8: Occluder Release
FIGURE 8a: Retrieval Cord Lock Release
FIGURE 8b: Retrieval Cord Removal
G. Reloading the Occluder Before Occluder Locking
1. Reload the Occluder by pushing the Slider up and then to the right until the
desired portion of the Occluder discs is reloaded or until the Slider stops, if
complete disc reloading is desired (Figure 4a).
2. If desired, complete Occluder reloading by pushing the Slider down and
then to the right until it stops (Figure 4b). Ensure that the Delivery Catheter
tip remains across the defect to maintain defect access.
3. Refer to Section E, “Occluder Deployment” to re-deploy the Occluder.
• If desired device placement cannot be achieved after multiple
deployment attempts, consideration should be given to minimize the
patient’s exposure to radiation and prolonged anesthesia time. If the
patient’s septal anatomy is determined to be unsuitable for the GORE®
CARDIOFORM Septal Occluder, alternative treatment options such as
other devices or surgical closure of the defect should be considered.
H. Removing the Occluder with the Retrieval Cord After Occluder
Locking
1. Unscrew the Retrieval Luer, hold the Delivery Catheter in place and
withdraw the Handle until the Occluder has unlocked (Figure 9). This step
requires that the Delivery Catheter is sufficiently spaced away from the
Occluder to permit full extension of the Lock Loop.
2. Continue to withdraw the Handle to pull the entire Occluder into the
Delivery Catheter. Do not use excessive force in an attempt to withdraw
all of the Occluder into the Delivery Catheter. Doing so could cause the
Retrieval Cord to break or result in Occluder damage.
• The operator must ensure that the Occluder does not catch on the
Delivery Catheter tip or introducer sheath. If the Lock Loop or eyelet
catch and the Delivery System is forcibly retracted, the Retrieval Cord
or Occluder frame is at risk of damage.
3. If necessary, remove the introducer sheath and Occluder together.
• If the Occluder is removed, it should be disposed of and a new
Occluder should be used.
Note that without a hemostatic valve at the Delivery Catheter
proximal end, care should be taken to avoid air entry or blood loss if
the Occluder is completely removed from the Delivery Catheter.

10
FIGURE 9: Occluder Retrieval
I. Recapture
1. In the event that the Occluder is malpositioned, embolized, or otherwise
requires removal, it may be recaptured with the aid of a loop snare or other
suitable means. A long sheath (11 Fr or greater) positioned close to the
device is recommended for recapture.
2. Attempt to recapture the device by first snaring the left or right atrial eyelet
to facilitate Occluder retraction into the sheath. If necessary, the loop snare
may be placed around any portion of the Occluder frame.
3. Pull the Occluder into the long sheath using the snare. If a portion of
the Occluder frame cannot be retracted into the long sheath, it may be
necessary to remove the Occluder, loop snare, and long sheath as one unit.
Do not use excessive force in an attempt to withdraw all of the Occluder
into the long sheath. Doing so could result in Occluder damage.
4. Bring the recaptured Occluder into the sheath to avoid pulling the unlocked
device across valve tissue.
MR CONDITIONAL
MR Conditional
J. MRI Information
The GORE® CARDIOFORM Septal Occluder has been determined to be
MR-conditional.
Non-clinical testing demonstrated that the GORE® CARDIOFORM Septal Occluder
is MR Conditional. A patient with this device can be scanned safely immediately
after placement under the following conditions:
Static Magnetic Field
-Static magnetic field of 3-Tesla or 1.5 Tesla
-Maximum spatial gradient magnetic field of 720-Gauss/cm or less
-Maximum scanner displayed whole body averaged specific absorption rate
(WB-SAR) of 3.0W/kg for 15 minutes of scanning.
MRI-Related Heating
In non-clinical testing, the GORE® CARDIOFORM Septal Occluder produced the
following temperature rise during MRI performed for 15 minutes of scanning
(i.e., per pulse sequence) in 1.5-Tesla/64-MHz (Magnetom, Siemens Medical
Solutions, Malvern, PA. Software Numaris/4, Version Syngo MR 2002B DHHS
Active-shielded, horizontal field scanner) and 3-Tesla/128-MHz (Excite, HDx,
Software 14X.M5, General Electric Healthcare, Milwaukee, WI) MR systems:
Highest temperature change +1.8°C
The MRI-related heating experiments for the GORE® CARDIOFORM Septal
Occluder at 1.5-Tesla using a transmit / receive RF body coil at an MR system
reported whole body averaged SAR of 2.9-W/kg (i.e., associated with a calorimetry
measured whole body averaged value of 2.1-W/kg) indicated that the greatest
amount of heating that occurred in association with these specific conditions was
equal to or less than +1.8°C.
The MRI-related heating experiments for the GORE® CARDIOFORM Septal
Occluder at 3- Tesla using a transmit / receive RF body coil at an MR system
reported whole body averaged SAR of 3.0 -W/kg (i.e., associated with a
calorimetry measured whole body averaged value of 2.8-W/kg) indicated that
the greatest amount of heating that occurred in association with these specific
conditions was equal to or less than +1.6ºC.
Multiple overlapping implants
MRI- related heating effects with multiple overlapping GORE® CARDIOFORM
Septal Occluder implants is unknown.
Artifact Information
MR image quality may be compromised if the area of interest is in the exact
same area or relatively close to the position of the GORE® CARDIOFORM Septal
Occluder. Therefore, optimization of MR imaging parameters to compensate for
the presence of this device may be necessary.
Pulse Sequence T1-SE T1-SE GRE GRE
Signal Void Size 981-mm2483-mm
2
1,308-mm2724-mm
2
Plane Orientation Parallel Perpendicular Parallel Perpendicular

11
DEFINITIONS
Use By
Caution
Consult Instructions for Use
STERILIZE
2
Do Not Resterilize
Do Not Reuse
Catalogue Number
Batch Code
Serial Number
MR Conditional
CAUTION: USA Federal Law restricts the sale, distribution, or use of this device to, by,
or on the order of a physician.
Sterile
Sterilized using Ethylene Oxide
Do Not Use if Package is Damaged
Keep Dry
Store in a Cool Place
Catheter Working Length
Delivery Profile
Guidewire Compatibility
Diameter
Manufacturer
Date of Manufacture



GORE®, CARDIOFORM and designs are trademarks of W. L. Gore & Associates.
© 2012, 2014-2015 W. L. Gore & Associates, Inc. APRIL 2015
Printed on recyclable paper 20024247
Manufacturer
W. L. Gore & AssociAtes, inc.
1505 North Fourth Street
Flagstaff, Arizona 86004
United States
Order Information: Tel.: 928.526.3030 • Tel.: 800.528.8763
Technical Information: Tel.: 928.779.2771 • Tel.: 800.437.8181
For international contact and additional product information,
visit www.goremedical.com
20024246
 Loading...
Loading...