


EN
23
CS
36
DA
50
DE
64
EL
78
ES
92
FR
106
HU
120
IT
134
NL
148
NO
162
PL
175
PT
189
SV
203
MEDICAL
Zenith Flex® AAA Endovascular Graft with the H&L-B One-Shot™ Introduction System
Instructions for Use
Endovaskulární graft Zenith Flex® AAA se zaváděcím systémem H&L-B One-Shot™
Návod k použití
Zenith Flex® AAA endovaskulær protese med H&L-B One-Shot™ indføringssystem
Brugsanvisning
Endovaskuläre AAA-Prothese Zenith Flex® mit H&L-B One-Shot™ Einführsystem
Gebrauchsanweisung
Ενδαγγειακό μόσχευμα AAA Zenith Flex® με το σύστημα εισαγωγής H&L-B One-Shot™
Οδηγίες χρήσης
Endoprótesis vascular para AAA Zenith Flex® con el sistema de introducción H&L-B One-Shot™
Instrucciones de uso
Endoprothèse vasculaire Zenith Flex® AAA à système d’introduction H&L-B One-Shot™
Mode d’emploi
Zenith Flex® AAA endovaszkuláris graft H&L-B One-Shot™ felvezetőrendszerrel
Használati utasítás
Endoprotesi addominale Zenith Flex® con sistema di introduzione H&L-B One-Shot™
Istruzioni per l’uso
Zenith Flex® AAA endovasculaire prothese met H&L-B One-Shot™ introductiesysteem
Gebruiksaanwijzing
Zenith Flex® AAA endovaskulært implantat med H&L-B One-Shot™ innføringssystem
Bruksanvisning
Stent-graft wewnątrznaczyniowy Zenith Flex® AAA z systemem wprowadzającym H&L-B One-Shot™
Instrukcja użycia
Prótese endovascular AAA Zenith Flex® com o sistema de introdução H&L-B One-Shot™
Instruções de utilização
Zenith Flex® AAA endovaskulära transplantat med H&L-B One-Shot™ införingssystem
Bruksanvisning
*T_ZAAAF36_REV7*


ENGLISH
TABLE OF CONTENTS
1 DEVICE DESCRIPTION ................................................23
1.1 Aortic Main Body and Iliac Leg Components ........................23
1.2 Main Body Delivery System .........................................23
1.3 Iliac Leg Delivery System ...........................................23
1.4 Zenith AAA Endovascular Graft Ancillary Components ..............23
2 INDICATIONS FOR USE ...............................................23
3 CONTRAINDICATIONS ................................................23
4 WARNINGS AND PRECAUTIONS ......................................23
4.1 General ...........................................................23
4.2 Patient Selection, Treatment and Follow-Up .......................23
4.3 Pre-Procedure Measurement Techniques and Imaging ............23
4.4 Device Selection .................................................24
4.5 Implant Procedure ............................................... 24
4.6 Molding Balloon Use ............................................. 24
4.7 MRI Safety and Compatibility ......................................24
5 ADVERSE EVENTS ....................................................24
5.1 Observed Adverse Events ..........................................24
Table 5.1.1 Death and Rupture from Clinical Study .................24
Table 5.1.2 Adverse Events in Clinical Study ........................25
5.2 Potential Adverse Events ...........................................25
Device Related Adverse Event Reporting ..........................25
6 SUMMARY OF CLINICAL STUDIES .....................................25
6.1 Objectives .........................................................25
6.2 Study Design .......................................................25
Table 6.2.1 Patient Follow-Up and Accountability ..................26
6.3 Patient Demographics ..............................................26
Table 6.3.1 Comparison of Subject Characteristics ..................26
Table 6.3.2 Aneurysm Diameter Distribution .......................26
6.4 Results .............................................................26
Table 6.4.1 Devices Implanted .....................................26
Table 6.4.2 Primary Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .27
Table 6.4.3 Success Measures .....................................28
Table 6.4.4 Abdominal Radiographic Findings – Device Integrity ...28
Table 6.4.5 CT Findings – Graft Patency ............................28
Table 6.4.6 CT Findings – Graft (Main Body) Migration ..............28
Table 6.4.7 Abdominal Radiograph Findings – Limb Separation ....29
6.5 Endoleak Management .............................................29
Table 6.5.1 Endoleaks (All Types, New and Persistent) ..............29
Table 6.5.2 First Occurrence of Endoleak for Standard Risk Patients .29
Table 6.5.3 First Occurrence of Endoleak for High Risk Patients .....29
Table 6.5.4 First Occurrence of Endoleak for Roll-in Patients ........29
6.6 Aneurysm Change .................................................30
Table 6.6.1 Change in Maximum Aneurysm Diameter by Interval ...30
Table 6.6.2 Change in Aneurysm Size and Endoleak at 12 Months ..30
Table 6.6.3 Change in Aneurysm Size and Endoleak at 24 Months ..30
6.7 AAA-Related Secondary Interventions ..............................30
Table 6.7.1 Secondary Interventions (to 12 Months) ................30
Table 6.7.2 Secondary Interventions (>12 to 24 Months) ...........30
6.8 Secondary Outcome Measures .....................................31
Table 6.8.1 Secondary Outcomes by Treatment Group .............31
7 PATIENT SELECTION AND TREATMENT ................................31
Individualization of Treatment .........................................31
8 PATIENT COUNSELING INFORMATION .................................31
9 HOW SUPPLIED ......................................................31
10 CLINICAL USE INFORMATION ........................................31
10.1 Physician Training .................................................31
10.2 Inspection Prior to Use ............................................31
10.3 Materials Required ...............................................31
10.4 Materials Recommended .........................................31
10.5 Device Diameter Sizing Guidelines ................................32
Table 10.5.1 Main Body Graft Diameter Sizing Guide ...............32
Table 10.5.2 Iliac Leg Graft Diameter Sizing Guide
11 DIRECTIONS FOR USE ...............................................32
Anatomical Requirements .............................................32
General Use Information ...............................................32
Pre-Implant Determinants .............................................32
Patient Preparation ....................................................32
11.1 Bifurcated System ................................................32
11.1.1 Bifurcated Main Body Preparation/Flush ....................32
11.1.2 Contralateral Iliac Leg Preparation/Flush ....................32
11.1.3 Ipsilateral Iliac Leg Preparation/Flush .......................32
11.1.4 Vascular Access and Angiography ..........................32
11.1.5 Main Body Placement ......................................32
11.1.6 Contralateral Iliac Wire Guide Placement ....................32
11.1.7 Main Body Proximal (Top) Deployment .....................33
11.1.8 Contralateral Iliac Leg Placement and Deployment ..........33
11.1.9 Main Body Distal (Bottom) Deployment .....................33
11.1.10 Docking of Top Cap .......................................33
11.1.11 Ipsilateral Iliac Leg Placement and Deployment ...........33
11.1.12 Molding Balloon Insertion ................................33
12 IMAGING GUIDELINES AND POSTOPERATIVE FOLLOW-UP ............33
12.1 General ..........................................................33
12.2 Contrast and Non-Contrast CT Recommendations .................34
12.3 Abdominal Radiographs ..........................................34
12.4 Ultrasound. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .34
12.5 MRI Safety and Compatibility ......................................34
12.6 Additional Surveillance and Treatment ............................34
13 PATIENT TRACKING INFORMATION ................................ 34
14 TROUBLESHOOTING ..............................................34
14.1 Trigger-Wire Release Troubleshooting ........................... 34
14.2 Suprarenal Stent Deployment Troubleshooting ....................35
Final Angiogram ..........................................33
Table 12.1 Recommended Imaging Schedule
for Endograft Patients .............................................34
Table 12.2 Acceptable Imaging Protocols ..........................34
14.1.1 Main Body Proximal (Top) Deployment
14.1.2 Docking of Top Cap
14.1.3 Ipsilateral Iliac Leg Placement and Deployment
14.1.4 Contralateral Iliac Leg Placement and Deployment
14.2.1 Main Body Proximal (Top) Deployment .....................35
14.2.2 Docking of Top Cap ........................................35
14.2.3 Ipsilateral Iliac Leg Placement and Deployment .............35
14.2.4 Contralateral Iliac Leg Placement and Deployment ..........35
........................................35
..................32
.....................34
............35
.........35
ČESKY
OBSAH
1 POPIS ZAŘÍZENÍ ...................................................36
1.1 Komponenty aortálního hlavního těla a iliakálního ramena ........36
1.2 Aplikační systém hlavního těla ....................................36
1.3 Aplikační systém pro iliakální rameno .............................36
1.4 Pomocné komponenty endovaskulárního graftu Zenith AAA ......36
2 URČENÉ POUŽITÍ ..................................................36
3 KONTRAINDIKACE .................................................36
4 VAROVÁNÍ A UPOZORNĚNÍ .........................................36
4.1 Obecně ...........................................................36
4.2 Výběr, léčba a následné kontroly pacienta .........................36
4.3 Měřicí techniky a zobrazování před výkonem .....................36
4.4 Volba zařízení ....................................................37
4.5 Postup implantace ...............................................37
4.6 Použití tvarovacího balónku ......................................37
4.7 Bezpečnost a kompatibilita vyšetření MRI ...........................37
5 NEŽÁDOUCÍ PŘÍHODY ............................................. 37
5.1 Zaznamenané nežádoucí příhody ................................37
Tabulka 5.1.1 Úmrtí a ruptura v klinické studii ....................37
Tabulka 5.1.2 Nežádoucí příhody v klinické studii ................ 38
5.2 Potenciální nežádoucí příhody ....................................38
Hlášení nežádoucích příhod souvisejících se zařízením ............. 38
6 SOUHRN KLINICKÝCH STUDIÍ .......................................38
6.1 Cíle ..............................................................38
6.2 Uspořádání studie ................................................38
Tabulka 6.2.1 Kontrolní vyšetření a dostupnost pacienta ......... 39
6.3 Data o pacientech ................................................39
Tabulka 6.3.1 Porovnání charakteristik subjektů ..................39
Tabulka 6.3.2 Distribuce průměru aneuryzmatu ..................39
6.4 Výsledky ......................................................... 39
Tabulka 6.4.1 Implantované zařízení .............................39
Tabulka 6.4.2 Primární výsledky. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
Tabulka 6.4.3 Měřítka úspěšnosti ................................ 41
Tabulka 6.4.4 Radiografické nálezy v břišní oblasti –
integrita zařízení ................................................41
Tabulka 6.4.5 Nálezy na CT – průchodnost graftu ................41
Tabulka 6.4.6 Nálezy na CT – migrace graftu (hlavního těla) ...... 41
Tabulka 6.4.7 Radiografické nálezy v břišní oblasti –
separace větve ..................................................42
6.5 Léčba endoleaku .................................................42
Tabulka 6.5.1 Endoleaky (všechny typy, nové a perzistující) .......42
Tabulka 6.5.2 První výskyt endoleaku u pacientů se
standardním rizikem ............................................42
Tabulka 6.5.3 První výskyt endoleaku u pacientů s vysokým
rizikem .........................................................42
Tabulka 6.5.4 První výskyt endoleaku u roll-in pacientů ..........42
6.6 Změna aneuryzmatu .............................................43
Tabulka 6.6.1 Změna maximálního průměru aneuryzmatu
podle časového intervalu .......................................43
Tabulka 6.6.2 Změna velikosti aneuryzmatu a endoleaku
po 12 měsících ..................................................43
Tabulka 6.6.3 Změna velikosti aneuryzmatu a endoleaku
po 24 měsících ..................................................43
6.7 Sekundární intervence související s AAA .......................... 43
Tabulka 6.7.1 Sekundární intervence (do 12 měsíců) .............43
Tabulka 6.7.2 Sekundární intervence (>12 až 24 měsíců) .........43
6.8 Měření sekundárních výsledků. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
Tabulka 6.8.1 Sekundární výsledky podle léčených skupin ........44
7 VÝBĚR A LÉČBA PACIENTA ..........................................44
Individualizace léčby .................................................44
8 PORADENSTVÍ PRO PACIENTY ......................................44
9 STAV PŘI DODÁNÍ ..................................................44
10 INFORMACE O KLINICKÉM POUŽITÍ ................................44
10.1 Školení lékařů ...................................................44
10.2 Kontrola před použitím .........................................44
10.3 Požadovaný materiál ............................................44
10.4 Doporučený materiál ............................................44
10.5 Pokyny k určení průměru zařízení. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
Tabulka 10.5.1 Návod k měření průměru hlavního těla graftu .....45
Tabulka 10.5.2 Návod k měření průměru iliakálního ramena
graftu. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
11 POKYNY K POUŽITÍ ...............................................45
Anatomické požadavky ................................................45
Obecné informace o použití ............................................45
Rozhodující činitele před implantací ....................................45
Příprava pacienta ......................................................45
11.1 Systém s bifurkací ...............................................45
11.1.1 Příprava a propláchnutí hlavního těla s bifurkací ...........45
11.1.2 Příprava a propláchnutí kontralaterálního iliakálního
ramena .........................................................45
11.1.3 Příprava a propláchnutí ipsilaterálního iliakálního
ramena .........................................................45
11.1.4 Cévní přístup a angiografie ............................... 45
11.1.5 Umístění hlavního těla ....................................45
11.1.6 Umístění vodicího drátu kontralaterální iliakální větve .....46
11.1.7 Rozvinutí proximální (horní) části hlavního těla ............46
11.1.8 Umístění a rozvinutí kontralaterálního iliakálního ramena ..46
11.1.9 Rozvinutí distální (dolní) části hlavního těla ...............46
11.1.10 Aretace horní čepičky ...................................46
11.1.11
Umístění a rozvinutí ipsilaterálního iliakálního ramena
11.1.12 Zavedení tvarovacího balónku ...........................46
Finální angiogram .......................................46
12 POKYNY K ZOBRAZOVÁNÍ A POOPERAČNÍ KONTROLE ..............46
12.1 Obecně .........................................................46
Tabulka 12.1 Doporučený plán zobrazovacích vyšetření pro
pacienty s endograftem .........................................47
12.2 Doporučení pro kontrastní a nekontrastní CT ....................47
Tabulka 12.2 Akceptovatelné zobrazovací protokoly .............47
12.3 Abdominální radiogramy ........................................47
12.4 Ultrazvuk ....................................................... 47
12.5 Bezpečnost a kompatibilita vyšetření MRI ........................47
12.6 Další sledování a léčba ..........................................47
13 INFORMACE PRO SLEDOVÁNÍ PACIENTŮ ...........................47
14 ŘEŠENÍ PROBLÉMŮ ..................................................47
14.1 Řešení problémů s uvolňovacím drátem ............................ 47
14.1.1 Rozvinutí proximální (horní) části hlavního těla ...................47
14.1.2 Aretace horní čepičky .............................................48
14.1.3 Umístění a rozvinutí ipsilaterálního iliakálního ramena ...........48
14.1.4 Umístění a rozvinutí kontralaterálního iliakálního ramena ........48
14.2 Řešení problémů s rozvinováním suprarenálního stentu .............48
14.2.1 Rozvinutí proximální (horní) části hlavního těla ...................48
14.2.2 Aretace horní čepičky .............................................48
14.2.3 Umístění a rozvinutí ipsilaterálního iliakálního ramena ...........48
14.2.4 Umístění a rozvinutí kontralaterálního iliakálního ramena ........48
......46

DANSK
INDHOLDSFORTEGNELSE
1 BESKRIVELSE AF PRODUKTET ........................................50
1.1 Komponenter til aortisk hovedprotese og til iliaca-ben ..............50
1.2 Hovedprotesens fremføringssystem ................................50
1.3 Fremføringssystem for iliaca-ben ...................................50
1.4 Tilbehørskomponenter til Zenith AAA endovaskulær protese ........50
2 TILSIGTET ANVENDELSE. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .50
3 KONTRAINDIKATIONER ..............................................50
4 ADVARSLER OG FORHOLDSREGLER ...................................50
4.1 Generelle ..........................................................50
4.2 Patientudvælgelse, behandling og opfølgning ....................
4.3 Måleteknik og billeddiagnostik før proceduren ....................
4.4 Udvælgelse af produkt ........................................... 51
4.5 Implantationsprocedure ..........................................51
4.6 Brug af formningsballon ..........................................51
4.7 Sikkerhed og kompatibilitet ved MR-scanning ......................
5 UØNSKEDE HÆNDELSER .............................................51
5.1 Observerede uønskede hændelser ..................................
Tabel 5.1.1 Død og ruptur fra klinisk undersøgelse .................
Tabel 5.1.2 Uønskede hændelser i klinisk undersøgelse ............52
5.2 Mulige uønskede hændelser. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .52
Rapportering af uønskede hændelser, relateret til anordning .........52
6 RESUME OVER KLINISKE UNDERSØGELSER ............................52
6.1 Målsætning ........................................................52
6.2 Undersøgelsens design .............................................52
Tabel 6.2.1 Patientopfølgning og ansvarlighed .....................53
6.3 Demografiske oplysninger om patienter ............................53
Tabel 6.3.1 Sammenligning af forsøgspersoners karakteristika ......53
Tabel 6.3.2 Fordeling af aneurismets diameter .....................53
6.4 Resultater ..........................................................53
Tabel 6.4.1 Implanterede anordninger .............................53
Tabel 6.4.2 Primære resultater .....................................54
Tabel 6.4.3 Måltal for succes .......................................55
Tabel 6.4.4 Abdominale fund under røntgen –
Anordningens integritet ..........................................55
Tabel 6.4.5 CT fund – Åbenhed af protese .........................55
Tabel 6.4.6 CT fund – Migration af protese (hovedprotese) .........55
Tabel 6.4.7 Abdominale fund under røntgen – separation af lem ...56
6.5 Styring af endolækage .............................................56
Tabel 6.5.1 Endolækager (alle typer, nye og vedvarende) ...........56
Tabel 6.5.2 Første tilfælde af endolækage for
standardrisikopatienter ...........................................56
Tabel 6.5.3 Første tilfælde af endolækage for højrisikopatienter ....56
Tabel 6.5.4 Første tilfælde af endolækage for indlæringspatienter ..56
6.6 Aneurismeændring ................................................57
Tabel 6.6.1 Ændring i aneurismets maksimale diameter
efter interval ......................................................57
Tabel 6.6.2 Ændring af aneurismets størrelse og endolækage
ved 12 måneder ..................................................57
Tabel 6.6.3 Ændring af aneurismets størrelse og endolækage
ved 24 måneder ..................................................57
6.7 AAA-relaterede sekundære interventioner ..........................57
Tabel 6.7.1 Sekundære interventioner (til 12 måneder) .............57
Tabel 6.7.2 Sekundære interventioner (> 12 til 24 måneder) ........57
6.8 Måltal for sekundære resultater .....................................58
Tabel 6.8.1 Sekundære resultater efter behandlingsgruppe ........58
7 PATIENTUDVÆLGELSE OG BEHANDLING ..............................58
Individualisering af behandling ........................................58
8 PATIENTRÅDGIVNINGSINFORMATION .................................58
9 LEVERING ...........................................................58
10 INFORMATION OM KLINISK ANVENDELSE ............................58
10.1 Lægeuddannelse .................................................58
10.2 Inspektion inden brug ............................................58
10.3 Nødvendige materialer ............................................58
10.4 Anbefalede materialer ............................................58
10.5 Retningslinjer for størrelsesbestemmelse af produktdiameter ......59
Tabel 10.5.1 Størrelsesguide for hovedprotesens diameter .........59
Tabel 10.5.2 Størrelsesguide for iliaca-benprotesens diameter ......59
11 VEJLEDNING .......................................................59
Anatomiske krav .......................................................59
Generel information om anvendelse ...................................59
Afgørende faktorer før implantation ....................................59
Klargøring af patienten ................................................59
11.1 Bifurkationssystem ................................................59
11.1.1 Forberedelse/skylning af bifurkationshovedprotese .........59
11.1.2 Forberedelse/skylning af kontralateral iliaca-ben ............59
11.1.3 Forberedelse/skylning af ipsilateral iliaca-ben ...............59
11.1.4 Vaskulær adgang og angiografi .............................59
11.1.5 Placering af hovedprotese ..................................59
11.1.6 Placering af den kontralaterale iliaca-kateterleder ...........59
11.1.7 Proksimal (top) anlæggelse af hovedprotese ................60
11.1.8 Placering og anlæggelse af kontralateral iliaca-ben ..........60
11.1.9 Distal (bund) anlæggelse af hovedprotese ..................60
11.1.10 Sammenkobling af tophætten .............................60
11.1.11 Placering og anlæggelse af ipsilateral iliaca-ben ............60
11.1.12 Indføring af formningsballon ..............................60
Slutangiogram .............................................60
12 RETNINGSLINJER FOR BILLEDDIAGNOSTIK OG POSTOPERATIV
OPFØLGNING ......................................................60
12.1 Generelle .........................................................60
Tabel 12.1 Anbefalet billeddiagnostisk plan for
endoprotesepatienter .............................................61
12.2 Anbefalinger for CT-scanning med og uden kontrast ...............61
Tabel 12.2 Acceptable billeddiagnostiske protokoller ..............61
12.3 Abdominale røntgenbilleder ......................................61
12.4 Ultralydsscanning .................................................61
12.5 Sikkerhed og kompatibilitet ved MR-scanning .....................61
12.6 Yderligere kontrol og behandling ..................................61
13 OPLYSNINGER OM SPORING AF PATIENTER ..........................61
14 FEJLSØGNING .......................................................61
14.1 Fejlfinding ved udløser-wirens frigørelse ............................61
14.1.1 Proksimal (top) anlæggelse af hovedprotese. . . . . . . . . . . . . . . . . . . . . .61
14.1.2 Sammenkobling af tophætten ....................................62
14.1.3 Placering og anlæggelse af ipsilateral iliaca-ben ..................62
14.1.4 Placering og anlæggelse af kontralateral iliaca-ben ...............62
14.2 Fejlfinding ved anlæggelse af den suprarenale stent ................ 62
14.2.1 Proksimal (top) anlæggelse af hovedprotese. . . . . . . . . . . . . . . . . . . . . .62
14.2.2 Sammenkobling af tophætten ....................................62
14.2.3 Placering og anlæggelse af ipsilateral iliaca-ben ..................62
14.2.4 Placering og anlæggelse af kontralateral iliaca-ben ...............62
DEUTSCH
INHALTSVERZEICHNIS
1 BESCHREIBUNG DES INSTRUMENTS ..................................64
1.1 Hauptteil (Aortenteil) und iliakale Schenkel .........................64
1.2 Einführsystem für Hauptteil ........................................64
1.3 Einführsystem für iliakale Schenkel .................................64
1.4 Hilfskomponenten für die endovaskuläre AAA-Prothese Zenith ......64
2 VERWENDUNGSZWECK ..............................................64
3 KONTRAINDIKATIONEN ..............................................64
4 WARNHINWEISE UND VORSICHTSMASSNAHMEN ......................64
4.1 Allgemeines .......................................................64
50
4.2 Auswahl, Behandlung und Nachsorge der Patienten ..............
50
4.3 Messtechniken und Bildgebung vor dem Eingriff ..................
4.4 Auswahl der Prothese ............................................65
4.5 Implantationsverfahren ..........................................65
4.6 Verwendung des Modellierungsballons ...........................65
51
4.7 Sicherheit und Kompatibilität mit MRT ..............................65
5 UNERWÜNSCHTE EREIGNISSE ........................................65
51
5.1 Beobachtete unerwünschte Ereignisse ..............................65
51
Tabelle 5.1.1 Todesfälle und Rupturen in der klinischen Studie .....65
Tabelle 5.1.2 Unerwünschte Ereignisse in der klinischen Studie .....66
5.2 Mögliche unerwünschte Ereignisse .................................66
Bericht zu prothesenbezogenen unerwünschten Ereignissen .........66
6 ZUSAMMENFASSUNG DER KLINISCHEN STUDIEN ......................66
6.1 Ziele ...............................................................66
6.2 Studiendesign .....................................................66
Tabelle 6.2.1 Patientennachsorge und Verantwortlichkeiten ........67
6.3 Patientendemographische Daten ...................................67
Tabelle 6.3.1 Vergleich der Patientenmerkmale ....................67
Tabelle 6.3.2 Ver teilung der Aneurysmadurchmesser ...............67
6.4 Ergebnisse .........................................................67
Tabelle 6.4.1 Implantierte Geräte ..................................68
Tabelle 6.4.2 Primärergebnisse ....................................68
Tabelle 6.4.3 Erfolgsmaßstäbe .....................................69
Tabelle 6.4.4 Abdominale Röntgenbefunde – Unversehrtheit
der Prothese ......................................................69
Tabelle 6.4.5 CT-Befunde – Durchgängigkeit der Prothese ..........69
Tabelle 6.4.6 CT-Befunde – Migration der Prothese (Hauptteil) ......69
Tabelle 6.4.7 Abdominale Röntgenbefunde – Ansatzablösung. . . . . .70
6.5 Endoleak-Management. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .70
Tabelle 6.5.1 Endoleaks (alle Typen, neu und persistierend) .........70
Tabelle 6.5.2 Erstes Auftreten eines Endoleaks bei
Standardrisikopatienten ..........................................70
Tabelle 6.5.3 Erstes Auftreten eines Endoleaks bei
Hochrisikopatienten ..............................................70
Tabelle 6.5.4 Erstes Auftreten eines Endoleaks bei Studienpatienten ...70
6.6 Aneurysmaänderungen ............................................71
Tabelle 6.6.1 Änderungen des maximalen
Aneurysmadurchmessers nach Intervall ...........................71
Tabelle 6.6.2 Änderung der Aneurysmagröße und
Endoleak nach 12 Monaten .......................................71
Tabelle 6.6.3 Änderung der Aneurysmagröße und
Endoleak nach 24 Monaten .......................................71
6.7 AAA-bezogene Sekundärinterventionen ............................71
Tabelle 6.7.1 Sekundäre Interventionen (bis 12 Monate) ............71
Tabelle 6.7.2 Sekundäre Interventionen (> 12 bis 24 Monate) .......71
6.8 Maßstäbe für sekundäre Ergebnisse ................................72
Tabelle 6.8.1 Sekundäre Ergebnisse nach Behandlungsgruppe .....72
7 AUSWAHL UND BEHANDLUNG DER PATIENTEN. . . . . . . . . . . . . . . . . . . . . . . .72
Individuelle Gestaltung der Behandlung ...............................72
8 INFORMATIONEN ZUR AUFKLÄRUNG DES PATIENTEN ..................72
9 LIEFERFORM ........................................................72
10 INFORMATIONEN ZUM KLINISCHEN EINSATZ .........................72
10.1 Ärzteschulung ....................................................72
10.2 Überprüfung vor dem Gebrauch ..................................72
10.3 Erforderliche Materialien ..........................................72
10.4 Empfohlene Materialien ...........................................72
10.5 Richtlinien zur Bestimmung des Prothesendurchmessers ...........73
Tabelle 10.5.1 Anleitung zur Bestimmung des Durchmessers
des Prothesenhauptteils ...........................................73
Tabelle 10.5.2 Anleitung zur Bestimmung des Durchmessers
des iliakalen Prothesenschenkels ..................................73
11 GEBRAUCHSANWEISUNG ...........................................73
Anatomische Voraussetzungen. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .73
Allgemeine Informationen zum Gebrauch ..............................73
Die Präimplantationsphase bestimmende Faktoren .....................73
Vorbereitung des Patienten ............................................73
11.1 Gegabeltes System ................................................73
11.1.1 Vorbereitung/Spülen des gegabelten Hauptteils ............73
11.1.2 Vorbereitung/Spülen des kontralateralen iliakalen
Schenkels ........................................................73
11.1.3 Vorbereitung/Spülen des ipsilateralen iliakalen
Schenkels ........................................................73
11.1.4 Gefäßzugang und Angiographie ............................73
11.1.5 Positionieren des Hauptteils ................................73
11.1.6 Positionieren des kontralateralen iliakalen Führungsdrahts ..74
11.1.7
Entfalten des proximalen (oberen) Hauptkörpers
11.1.8 Positionieren und Entfalten des kontralateralen iliakalen
Schenkels ........................................................74
11.1.9 Entfalten des distalen (untersten) Stents des Hauptteils .....74
11.1.10 Andocken der oberen Kappe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .74
11.1.11 Positionieren und Entfalten des ipsilateralen iliakalen
Schenkels ........................................................74
11.1.12 Einführen des Modellierungsballons .......................74
Abschließendes Angiogramm. . . . . . . . . . . . . . . . . . . . . . . . . . . . . .75
12 RICHTLINIEN FÜR DIE BILDGEBUNG UND POSTOPERATIVE
NACHSORGE .......................................................75
12.1 Allgemeines ......................................................75
Tabelle 12.1 Empfohlener Bildgebungsplan für Patienten mit
Endoprothese ....................................................75
12.2 Empfehlungen für CT mit und ohne Kontrastmittel ................75
Tabelle 12.2 Akzeptable Bildgebungsprotokolle ...................75
12.3 Röntgenaufnahmen des Abdomens ...............................75
12.4 Ultraschall ........................................................75
12.5 Sicherheit und Kompatibilität mit MRT ............................75
12.6 Zusätzliche Überwachung und Behandlung .......................76
13 INFORMATIONEN ZUR PATIENTENVERFOLGUNG .....................76
14 FEHLERBEHEBUNG ...................................................76
14.1 Fehlerbehebung am Auslösedrahtmechanismus ....................76
14.1.1 Entfalten des proximalen (oberen) Hauptkörpers .................76
14.1.2 Andocken der oberen Kappe .....................................76
14.1.3 Positionieren und Entfalten des ipsilateralen iliakalen Schenkels .76
14.1.4 Positionieren und Entfalten des kontralateralen iliakalen
Schenkels ................................................................76
14.2 Fehlerbehebung bei der suprarenalen Stententfaltung ..............76
14.2.1 Entfalten des proximalen (oberen) Hauptkörpers .................76
14.2.2 Andocken der oberen Kappe .....................................77
14.2.3 Positionieren und Entfalten des ipsilateralen iliakalen Schenkels .77
14.2.4 Positionieren und Entfalten des kontralateralen iliakalen
Schenkels ................................................................77
.............74
64
64

ΕΛΛΗΝΙΚΑ
ΠΙΝΑΚΑΣ ΠΕΡΙΕΧΟΜΕΝΩΝ
1 ΠΕΡΙΓΡΑΦΗ ΤΗΣ ΣΥΣΚΕΥΗΣ ..........................................78
1.1 Εξαρτήματα αορτικού κύριου σώματος και λαγόνιου σκέλους .......78
1.2 Σύστημα τοποθέτησης κύριου σώματος ............................78
1.3 Σύστημα τοποθέτησης λαγόνιου σκέλους ...........................78
1.4 Βοηθητικά εξαρτήματα ενδαγγειακού μοσχεύματος AAA Zenith .....78
2 ΧΡΗΣΗ ΓΙΑ ΤΗΝ ΟΠΟΙΑ ΠΡΟΟΡΙΖΕΤΑΙ .................................78
3 ΑΝΤΕΝΔΕΙΞΕΙΣ ......................................................78
4 ΠΡΟΕΙΔΟΠΟΙΗΣΕΙΣ ΚΑΙ ΠΡΟΦΥΛΑΞΕΙΣ ................................78
4.1 Γενικά .............................................................78
4.2 Επιλογή, θεραπεία και παρακολούθηση ασθενούς .................
4.3 Τεχνικές μέτρησης και απεικόνιση πριν από τη διαδικασία .........
4.4 Επιλογή συσκευής ................................................79
4.5 Διαδικασία εμφύτευσης .......................................... 79
4.6 Χρήση μπαλονιού διαμόρφωσης .................................79
4.7 Ασφάλεια και συμβατότητα με μαγνητική τομογραφία (MRI) .........
5 ΑΝΕΠΙΘΥΜΗΤΕΣ ΕΝΕΡΓΕΙΕΣ ..........................................79
5.1 Παρατηρούμενες ανεπιθύμητες ενέργειες ...........................
Πίνακας 5.1.1 Θάνατος και ρήξη από την κλινική μελέτη ...........80
Πίνακας 5.1.2 Ανεπιθύμητες ενέργειες στην κλινική μελέτη .........80
5.2 Δυνητικές ανεπιθύμητες ενέργειες ..................................80
Αναφορά ανεπιθύμητων ενεργειών που σχετίζονται με
τη συσκευή .........................................................80
6 ΠΕΡΙΛΗΨΗ ΤΩΝ ΚΛΙΝΙΚΩΝ ΜΕΛΕΤΩΝ. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .80
6.1 Στόχοι .............................................................80
6.2 Σχεδιασμός της μελέτης ............................................80
Πίνακας 6.2.1 Παρακολούθηση και υπευθυνότητα ασθενών ........81
6.3 Δημογραφικά στοιχεία των ασθενών ................................81
Πίνακας 6.3.1 Σύγκριση των χαρακτηριστικών των ασθενών .......81
Πίνακας 6.3.2 Κατανομή διαμέτρων ανευρύσματος ................81
6.4 Αποτελέσματα .....................................................81
Πίνακας 6.4.1 Συσκευές που εμφυτεύτηκαν ........................82
Πίνακας 6.4.2 Πρωτεύοντα αποτελέσματα .........................82
Πίνακας 6.4.3 Μετρήσεις επιτυχίας. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .83
Πίνακας 6.4.4 Κοιλιακά ακτινογραφικά ευρήματα –
Ακεραιότητα της συσκευής. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .83
Πίνακας 6.4.5 Ευρήματα αξονικής τομογραφίας –
Βατότητα μοσχεύματος ...........................................83
Πίνακας 6.4.6 Ευρήματα αξονικής τομογραφίας –
Μετανάστευση μοσχεύματος (κύριο σώμα) ........................83
Πίνακας 6.4.7 Κοιλιακά ακτινογραφικά ευρήματα –
Διαχωρισμός μέλους ..............................................84
6.5 Διαχείριση ενδοδιαφυγής ..........................................84
Πίνακας 6.5.1 Ενδοδιαφυγές (Όλοι οι τύποι, νέες και επίμονες). . . . . .84
Πίνακας 6.5.2 Πρώτη εμφάνιση ενδοδιαφυγής για ασθενείς
τυπικού κινδύνου .................................................84
Πίνακας 6.5.3 Πρώτη εμφάνιση ενδοδιαφυγής για ασθενείς
υψηλού κινδύνου .................................................84
Πίνακας 6.5.4 Πρώτη εμφάνιση ενδοδιαφυγής για ασθενείς
εισαγωγικής περιόδου ............................................84
6.6 Μεταβολή ανευρύσματος ..........................................85
Πίνακας 6.6.1 Μεταβολή στη μέγιστη διάμετρο ανευρύσματος
κατά διάστημα ....................................................85
Πίνακας 6.6.2 Μεταβολή στο μέγεθος του ανευρύσματος και
ενδοδιαφυγή στους 12 μήνες .....................................85
Πίνακας 6.6.3 Μεταβολή στο μέγεθος του ανευρύσματος και
ενδοδιαφυγή στους 24 μήνες .....................................85
6.7 Δευτερεύουσες επεμβάσεις που σχετίζονται με AAA ................85
Πίνακας 6.7.1 Δευτερεύουσες επεμβάσεις (έως 12 μήνες) ..........85
Πίνακας 6.7.2 Δευτερεύουσες επεμβάσεις (>12 έως 24 μήνες) ......85
6.8 Μετρήσεις δευτερεύουσας έκβασης ................................86
Πίνακας 6.8.1 Δευτερεύουσες εκβάσεις κατά ομάδα θεραπείας .....86
7 ΕΠΙΛΟΓΗ ΚΑΙ ΘΕΡΑΠΕΙΑ ΑΣΘΕΝΩΝ ...................................86
Εξατομίκευση της θεραπείας ...........................................86
8 ΠΛΗΡΟΦΟΡΙΕΣ ΓΙΑ ΤΙΣ ΣΥΣΤΑΣΕΙΣ ΠΡΟΣ ΤΟΝ ΑΣΘΕΝΗ ................86
9 ΤΡΟΠΟΣ ΔΙΑΘΕΣΗΣ ..................................................86
10 ΠΛΗΡΟΦΟΡΙΕΣ ΓΙΑ ΤΗΝ ΚΛΙΝΙΚΗ ΧΡΗΣΗ ............................86
10.1 Εκπαίδευση ιατρού ...............................................86
10.2 Επιθεώρηση πριν από τη χρήση ...................................86
10.3 Απαιτούμενα υλικά ................................................86
10.4 Συνιστώμενα υλικά ................................................86
10.5 Κατευθυντήριες οδηγίες προσδιορισμού μεγέθους διαμέτρου
συσκευής ..............................................................87
Πίνακας 10.5.1 Οδηγός προσδιορισμού μεγέθους διαμέτρου
μοσχεύματος κύριου σώματος ....................................87
Πίνακας 10.5.2 Οδηγός προσδιορισμού μεγέθους διαμέτρου
μοσχεύματος λαγόνιου σκέλους ...................................87
11 ΟΔΗΓΙΕΣ ΧΡΗΣΗΣ ..................................................87
Ανατομικές απαιτήσεις .................................................87
Γενικές πληροφορίες χρήσης ...........................................87
Προσδιοριστικοί παράγοντες προ της εμφύτευσης .....................87
Προετοιμασία ασθενούς ...............................................87
11.1 Διχαλωτό σύστημα ................................................87
11.1.1 Προετοιμασία/Έκπλυση διχαλωτού κύριου σώματος ........87
11.1.2 Προετοιμασία/Έκπλυση ετερόπλευρου λαγόνιου σκέλους ...87
11.1.3 Προετοιμασία/Έκπλυση σύστοιχου λαγόνιου σκέλους .......87
11.1.4 Αγγειακή προσπέλαση και αγγειογραφία ....................87
11.1.5 Τοποθέτηση κύριου σώματος ...............................87
11.1.6 Τοποθέτηση του ετερόπλευρου λαγόνιου συρμάτινου
οδηγού ...........................................................88
11.1.7 Εγγύς (άνω) απελευθέρωση του κύριου σώματος ............88
11.1.8 Τοποθέτηση και απελευθέρωση του ετερόπλευρου λαγόνιου
σκέλους ..........................................................88
11.1.9 Περιφερική (κάτω) απελευθέρωση του κύριου σώματος .....88
11.1.10 Σύνδεση του άνω πώματος ................................88
11.1.11 Τοποθέτηση και απελευθέρωση του σύστοιχου λαγόνιου
σκέλους ..........................................................88
11.1.12 Εισαγωγή μπαλονιού διαμόρφωσης .......................88
Τελικό αγγειόγραμμα ......................................89
12 ΚΑΤΕΥΘΥΝΤΗΡΙΕΣ ΟΔΗΓΙΕΣ ΑΠΕΙΚΟΝΙΣΗΣ ΚΑΙ ΜΕΤΕΓΧΕΙΡΗΤΙΚΗ
ΠΑΡΑΚΟΛΟΥΘΗΣΗ .................................................89
12.1 Γενικά ............................................................89
Πίνακας 12.1 Συνιστώμενο πρόγραμμα απεικόνισης για
ασθενείς με ενδαγγειακό μόσχευμα ...............................89
12.2 Συστάσεις αξονικής τομογραφίας με και χωρίς
σκιαγραφικό μέσο ................................................89
Πίνακας 12.2 Αποδεκτά πρωτόκολλα απεικόνισης ..................89
12.3 Κοιλιακές ακτινογραφίες ..........................................89
12.4 Υπέρηχος .........................................................89
12.5 Ασφάλεια και συμβατότητα με μαγνητική τομογραφία (MRI) .......89
12.6 Επιπλέον παρακολούθηση και θεραπεία ...........................90
13 ΠΛΗΡΟΦΟΡΙΕΣ ΠΑΡΑΚΟΛΟΥΘΗΣΗΣ ΑΣΘΕΝΩΝ ......................90
14 ΑΝΤΙΜΕΤΩΠΙΣΗ ΠΡΟΒΛΗΜΑΤΩΝ .................................... 90
14.1 Αντιμετώπιση προβλημάτων με την απελευθέρωση του σύρματος
ενεργοποίησης .......................................................... 90
14.1.1 Εγγύς (άνω) απελευθέρωση του κύριου σώματος ............. 90
14.1.2 Σύνδεση του άνω πώματος ..................................90
14.1.3 Τοποθέτηση και απελευθέρωση σύστοιχου λαγόνιου σκέλους .. 90
14.1.4 Τοποθέτηση και απελευθέρωση ετερόπλευρου λαγόνιου
σκέλους ............................................................ 90
14.2 Αντιμετώπιση προβλημάτων απελευθέρωσης της επινεφρικής
ενδοπρόσθεσης ......................................................... 91
14.2.1 Εγγύς (άνω) απελευθέρωση του κύριου σώματος ............. 91
14.2.2 Σύνδεση του άνω πώματος ..................................91
14.2.3 Τοποθέτηση και απελευθέρωση σύστοιχου λαγόνιου σκέλους 91
14.2.4 Τοποθέτηση και απελευθέρωση ετερόπλευρου λαγόνιου
σκέλους ............................................................ 91
ESPAÑOL
ÍNDICE
1 DESCRIPCIÓN DEL DISPOSITIVO ......................................92
1.1 Cuerpo principal aórtico y ramas ilíacas .............................92
1.2 Sistema de implantación del cuerpo principal .......................92
1.3 Sistema de implantación de la rama ilíaca ...........................92
1.4 Componentes auxiliares de la endoprótesis vascular para
AAA Zenith ............................................................92
2 INDICACIONES ......................................................92
3 CONTRAINDICACIONES ..............................................92
4 ADVERTENCIAS Y PRECAUCIONES ....................................92
78
4.1 Generales .........................................................92
78
4.2 Selección, tratamiento y seguimiento de los pacientes ............
4.3 Técnicas de medición y estudios de imagen previos
al procedimiento ....................................................
4.4 Selección de los dispositivos ......................................93
79
4.5 Procedimiento de implantación ..................................93
4.6 Uso del balón moldeador .........................................93
79
4.7 Seguridad y compatibilidad con MRI ................................
5 REACCIONES ADVERSAS .............................................93
5.1 Reacciones adversas observadas ....................................
Tabla 5.1.1 Muertes y roturas observadas en el estudio clínico ......
Tabla 5.1.2 Reacciones adversas observadas en el estudio clínico ...94
5.2 Reacciones adversas posibles .......................................94
Informes de reacciones adversas relacionadas con el dispositivo ......94
6 RESUMEN DE ESTUDIOS CLÍNICOS ....................................94
6.1 Objetivos ..........................................................94
6.2 Diseño del estudio .................................................94
Tabla 6.2.1 Seguimiento y disponibilidad de los pacientes. . . . . . . . . .95
6.3 Datos demográficos de los pacientes ...............................95
Tabla 6.3.1 Comparación de las características de los sujetos .......95
Tabla 6.3.2 Distribución de los diámetros de los aneurismas ........95
6.4 Resultados .........................................................95
Tabla 6.4.1 Dispositivos implantados ..............................96
Tabla 6.4.2 Resultados primarios. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .96
Tabla 6.4.3 Medidas de éxito ......................................97
Tabla 6.4.4 Resultados radiográficos abdominales: integridad
del dispositivo ....................................................97
Tabla 6.4.5 Resultados de las TAC: permeabilidad de la
endoprótesis vascular .............................................97
Tabla 6.4.6 Resultados de las TAC: migración de la endoprótesis
vascular (cuerpo principal) ........................................97
Tabla 6.4.7 Resultados radiográficos abdominales: separación
de las ramificaciones ..............................................98
6.5 Tratamiento de las endofugas ......................................98
Tabla 6.5.1 Endofugas (todos los tipos, nuevas y persistentes) ......98
Tabla 6.5.2 Primera aparición de una endofuga en pacientes
de riesgo normal. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .98
Tabla 6.5.3 Primera aparición de una endofuga en pacientes
de alto riesgo .....................................................98
Tabla 6.5.4 Primera aparición de una endofuga en pacientes
de prueba ........................................................98
6.6 Cambio del aneurisma .............................................99
Tabla 6.6.1 Cambio del diámetro máximo del aneurisma
por intervalos .....................................................99
Tabla 6.6.2 Cambio del tamaño del aneurisma y endofugas
a los 12 meses ....................................................99
Tabla 6.6.3 Cambio del tamaño del aneurisma y endofugas
a los 24 meses ....................................................99
6.7 Intervenciones secundarias relacionadas con el AAA ................99
Tabla 6.7.1 Intervenciones secundarias (hasta los 12 meses) ........99
Tabla 6.7.2 Intervenciones secundarias (desde los 12 hasta
los 24 meses) .....................................................99
6.8 Medidas de los resultados secundarios ........................... 100
Tabla 6.8.1 Resultados de las intervenciones secundarias por
grupo de tratamiento ........................................... 100
7 SELECCIÓN Y TRATAMIENTO DE LOS PACIENTES ..................... 100
Individualización del tratamiento ....................................100
8 INFORMACIÓN PARA EL ASESORAMIENTO DE LOS PACIENTES. . . . . . . . 100
9 PRESENTACIÓN ....................................................100
10 INFORMACIÓN SOBRE EL USO CLÍNICO ............................100
10.1 Formación de médicos .......................................... 100
10.2 Inspección previa al uso ......................................... 100
10.3 Material necesario ..............................................100
10.4 Material recomendado .......................................... 100
10.5 Pautas para la selección del diámetro de los dispositivos ......... 101
Tabla 10.5.1 Guía para la selección del diámetro del cuerpo
principal ........................................................ 101
Tabla 10.5.2 Guía para la selección del diámetro de las ramas
ilíacas. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
11 MODO DE EMPLEO ............................................... 101
Requisitos anatómicos ............................................... 101
Información general sobre el uso ..................................... 101
Factores determinantes previos al implante .......................... 101
Preparación del paciente ............................................. 101
11.1 Sistema bifurcado ............................................... 101
11.1.1 Preparación y lavado del cuerpo principal bifurcado ....... 101
11.1.2 Preparación y lavado de la rama ilíaca contralateral. . . . . . . . 101
11.1.3 Preparación y lavado de la rama ilíaca ipsilateral. . . . . . . . . . . 101
11.1.4 Acceso vascular y angiografía ............................. 101
11.1.5 Colocación del cuerpo principal ..........................101
11.1.6 Colocación de la guía ilíaca contralateral .................. 102
11.1.7 Despliegue proximal (parte superior) del cuerpo principal . 102
11.1.8 Colocación y despliegue de la rama ilíaca contralateral .... 102
11.1.9 Despliegue distal (parte inferior) del cuerpo principal .....102
11.1.10 Acoplamiento de la cápsula superior ....................102
11.1.11 Colocación y despliegue de la rama ilíaca ipsilateral ...... 102
11.1.12 Introducción del balón moldeador. . . . . . . . . . . . . . . . . . . . . . . 102
Angiografía final ......................................... 102
12 PAUTAS PARA LOS ESTUDIOS DE IMAGEN Y SEGUIMIENTO
POSOPERATORIO .................................................103
12.1 Generales ....................................................... 103
Tabla 12.1 Programa de estudios de imagen recomendado para
pacientes con endoprótesis vasculares .......................... 103
12.2 Recomendaciones para la TAC con contraste y sin él ............. 103
Tabla 12.2 Protocolos válidos de estudios de imagen ............ 103
12.3 Radiografías abdominales ....................................... 103
12.4 Ecografía .......................................................103
12.5 Seguridad y compatibilidad con MRI ............................ 103
12.6 Vigilancia y tratamiento adicionales ............................. 104
13 INFORMACIÓN PARA LA LOCALIZACIÓN DEL PACIENTE .............104
14 SOLUCIÓN DE PROBLEMAS .........................................104
14.1 Solución de problemas de liberación mediante alambre disparador . 104
14.1.1 Despliegue proximal (parte superior) del cuerpo principal ...104
14.1.2 Acoplamiento de la cápsula superior ........................104
14.1.3 Colocación y despliegue de la rama ilíaca ipsilateral .........104
14.1.4 Colocación y despliegue de la rama ilíaca contralateral ......104
14.2 Solución de problemas de despliegue del stent suprarrenal ........104
14.2.1 Despliegue proximal (parte superior) del cuerpo principal ...104
14.2.2 Acoplamiento de la cápsula superior ........................105
14.2.3 Colocación y despliegue de la rama ilíaca ipsilateral .........105
14.2.4 Colocación y despliegue de la rama ilíaca contralateral ......105
92
92
93
93
93

FRANÇAIS
TABLE DES MATIÈRES
1 DESCRIPTION DU DISPOSITIF ....................................... 106
1.1 Corps principal aortique et composants de branches iliaques ..... 106
1.2 Système de largage du corps principal ............................ 106
1.3 Système de largage de la branche iliaque ......................... 106
1.4 Composants auxiliaires de l’endoprothèse Zenith AAA ............106
2 UTILISATION. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 106
3 CONTRE-INDICATIONS .............................................106
4 AVERTISSEMENTS ET MISES EN GARDE .............................106
4.1 Généralités ....................................................... 106
4.2 Sélection, traitement et suivi des patients ........................
4.3 Techniques de mesure et imagerie avant l’intervention ...........
4.4 Sélection des dispositifs .........................................107
4.5 Méthode d’implantation .........................................107
4.6 Utilisation du ballonnet de modelage ............................107
4.7 Sécurité d’emploi et compatibilité IRM ............................
5 ÉVÉNEMENTS INDÉSIRABLES ....................................... 107
5.1 Événements indésirables observés ................................
Tableau 5.1.1 Décès et ruptures survenus dans l’étude clinique ... 108
Tableau 5.1.2 Événements indésirables dans l’étude clinique ..... 108
5.2 Événements indésirables possibles ............................... 108
Rapport d’événement indésirable associé au dispositif. . . . . . . . . . . . . . .108
6 SOMMAIRE DES ÉTUDES CLINIQUES ................................ 108
6.1 Objectifs ......................................................... 108
6.2 Modèle de l’étude ................................................ 108
Tableau 6.2.1 Suivi et bilan post-opératoire des patients ......... 109
6.3 Données démographiques des patients. . . . . . . . . . . . . . . . . . . . . . . . . . . 109
Tableau 6.3.1 Comparaison des caractéristiques des patients .....109
Tableau 6.3.2 Distribution des diamètres d’anévrisme ............109
6.4 Résultats ......................................................... 109
Tableau 6.4.1 Dispositifs implantés .............................. 110
Tableau 6.4.2 Résultats principaux ............................... 110
Tableau 6.4.3 Critères de réussite ................................111
Tableau 6.4.4 Résultats des radiographies abdominales -
Intégrité du dispositif ........................................... 111
Tableau 6.4.5 Résultats TDM - Perméabilité de l’endoprothèse ....111
Tableau 6.4.6 Résultats TDM - Migration de l’endoprothèse
(corps principal) ................................................ 111
Tableau 6.4.7 Résultats des radiographies abdominales -
Séparation d’un membre. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112
6.5 Prise en charge des endofuites ................................... 112
Tableau 6.5.1 Endofuites (tous types confondus, nouvelles
et persistantes) ................................................. 112
Tableau 6.5.2 Première survenue d’endofuite chez les patients
à risque standard ............................................... 112
Tableau 6.5.3 Première survenue d’endofuite chez les patients
à haut risque ................................................... 112
Tableau 6.5.4 Première survenue d’endofuite chez les patients
non randomisés ................................................112
6.6 Changement de taille de l’anévrisme ............................. 113
Tableau 6.6.1 Changement de diamètre maximum de
l’anévrisme par intervalle .......................................113
Tableau 6.6.2 Changement de taille de l’anévrisme et
endofuite à 12 mois ............................................. 113
Tableau 6.6.3 Changement de taille de l’anévrisme et
endofuite à 24 mois ............................................. 113
6.7 Interventions de seconde intention en rapport avec l’AAA .........113
Tableau 6.7.1 Interventions de seconde intention
(jusqu’à 12 mois). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113
Tableau 6.7.2 Interventions de seconde intention
(entre 12 et 24 mois) ............................................ 113
6.8 Mesures des résultats secondaires ................................ 114
Tableau 6.8.1 Résultats secondaires par groupe de traitement ....114
7 SÉLECTION ET TRAITEMENT DES PATIENTS .......................... 114
Individualisation du traitement ...................................... 114
8 CONSEILS AUX PATIENTS ...........................................114
9 PRÉSENTATION ....................................................114
10 UTILISATION CLINIQUE ........................................... 114
10.1 Formation clinique .............................................. 114
10.2 Inspection avant l’utilisation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 114
10.3 Matériel requis .................................................. 114
10.4 Matériel recommandé ........................................... 114
10.5 Directives de mesures relatives au diamètre des dispositifs ....... 115
Tableau 10.5.1 Guide de mesures du diamètre du corps principal 115
Tableau 10.5.2 Guide de mesure du diamètre de la branche
iliaque .......................................................... 115
11 DIRECTIVES D’UTILISATION ....................................... 115
Exigences anatomiques .............................................. 115
Informations générales sur l’utilisation ............................... 115
Facteurs déterminants avant l’implantation ...........................115
Préparation du patient ............................................... 115
11.1 Système bifurqué ............................................... 115
11.1.1 Préparation et rinçage du corps principal bifurqué ........ 115
11.1.2 Préparation et rinçage de la branche iliaque controlatérale ..
11.1.3 Préparation et rinçage de la branche iliaque homolatérale ..
11.1.4 Accès vasculaire et angiographie ......................... 115
11.1.5 Mise en place du corps principal .......................... 115
11.1.6 Mise en place du guide iliaque controlatéral .............. 116
11.1.7 Déploiement du stent proximal (supérieur) du corps
principal ........................................................ 116
11.1.8
Mise en place et déploiement du jambage iliaque
controlatéral
11.1.9 Déploiement du stent distal (inférieur) du corps principal . 116
11.1.10 Raccordement du capuchon supérieur ................... 116
11.1.11
homolatéral
11.1.12 Insertion du ballonnet de modelage ..................... 116
Angiographie finale .....................................117
12 DIRECTIVES D’IMAGERIE ET SUIVI POST-OPÉRATOIRE ............... 117
12.1 Généralités ..................................................... 117
Tableau 12.1 Planification d’imagerie recommandée pour
les patients porteurs d’une endoprothèse ....................... 117
12.2 Recommandations de TDM avec et sans injection de produit
de contraste .................................................... 117
Tableau 12.2 Protocoles d’imagerie agréés ....................... 117
12.3 Radiographies abdominales ..................................... 117
12.4 Échographie .................................................... 117
12.5 Sécurité d’emploi et compatibilité IRM ...........................117
12.6 Surveillance et traitement complémentaires ..................... 118
13 INFORMATIONS RELATIVES AU SUIVI DU PATIENT .................. 118
14 DÉPANNAGE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .118
14.1 Dépannage du mécanisme de largage des fils de sécurité ..........118
14.1.1 Déploiement du stent proximal (supérieur) du corps principal . 118
14.1.2 Raccordement du capuchon supérieur ......................... 118
14.1.3 Mise en place et déploiement du jambage iliaque homolatéral 118
14.1.4 Mise en place et déploiement du jambage iliaque
controlatéral .......................................................... 118
14.2 Dépannage lors du déploiement du stent suprarénal ..............118
14.2.1 Déploiement du stent proximal (supérieur) du corps principal . 118
14.2.2 Raccordement du capuchon supérieur ......................... 119
14.2.3 Mise en place et déploiement du jambage iliaque homolatéral 119
14.2.4 Mise en place et déploiement du jambage iliaque controlatéral 119
.................................................... 116
Mise en place et déploiement du jambage iliaque
..................................................... 116
106
106
107
107
115
115
MAGYAR
TARTALOMJEGYZÉK
1 AZ ESZKÖZ LEÍRÁSA ..............................................120
1.1 Aortikus fő graft-törzs és iliaca-szár komponenesek ..............120
1.2 A fő graft-törzs bejuttatórendszere ..............................120
1.3 Az iliaca-szár bejuttatórendszere .................................120
1.4 A Zenith AAA endovaszkuláris graft tartozékai ...................120
2 HASZNÁLATI JAVALLATOK ........................................120
3 ELLENJAVALLATOK ...............................................120
4 FIGYELMEZTETÉSEK ÉS ÓVINTÉZKEDÉSEK .........................120
4.1 Általános. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.2 A betegek kiválasztása, kezelése és utánkövetése ................120
4.3 A beavatkozást megelőzően alkalmazott mérési technikák
és leképezés ........................................................120
4.4 Az eszköz kiválasztása ...........................................121
4.5 Beültetési eljárás ................................................121
4.6 A formázóballon használata .....................................121
4.7 MRI-biztonság és -kompatibilitás .................................
5 NEMKÍVÁNATOS ESEMÉNYEK ......................................121
5.1 Megfigyelt nemkívánatos események ............................121
5.1.1. táblázat. A klinikai vizsgálatban előfordult halálesetek
és ruptúrák ....................................................121
5.1.2. táblázat. A klinikai vizsgálat során jelentkező
nemkívánatos események ......................................122
5.2 Lehetséges nemkívánatos események ...........................122
Az eszközzel kapcsolatos nemkívánatos események bejelentése ...122
6 KLINIKAI VIZSGÁLATOK ÖSSZEFOGLALÁSA ........................122
6.1 Célpontok .......................................................122
6.2 Vizsgálati elrendezés ............................................122
6.2.1. táblázat. A betegek utánkövetése és
beszámoltathatósága ..........................................123
6.3 A betegek demográfiai adatai ...................................123
6.3.1. táblázat. Egyéni jellemzők összehasonlítása ...............123
6.3.2. táblázat. Az aneurysma-átmérők megoszlása ..............123
6.4 Eredmények ....................................................123
6.4.1. táblázat. Beültetett eszközök .............................124
6.4.2. táblázat. Elsődleges eredmények .........................124
6.4.3. táblázat. Sikermutatók ....................................125
6.4.4. táblázat. Abdominális röntgenfelvételek eredményei –
az eszköz épsége ..............................................125
6.4.5. táblázat. CT-felvételek eredményei – a graft
átjárhatósága ..................................................125
6.4.6. táblázat. CT-felvételek eredményei – graft
(fő graft-törzs) migrációja ......................................125
6.4.7. táblázat. Abdominális röntgenfelvételek eredményei –
ág leválása .....................................................126
6.5 Endoleak kezelése ...............................................126
6.5.1. táblázat. Endoleak-ek (valamennyi típus, új és perzisztens) 126
6.5.2. táblázat. Endoleak első előfordulása standard kockázatú
betegeknél ....................................................126
6.5.3. táblázat. Endoleak első előfordulása magas kockázatú
betegeknél ....................................................126
6.5.4. táblázat. Endoleak első előfordulása roll-in
betegeknél ....................................................126
6.6 Aneurysma változás .............................................127
6.6.1. táblázat. Az aneurysma maximális átmérőjében
bekövetkezett változások időintervallum szerint ................127
6.6.2. táblázat. Az aneurysma méretében és az endoleak-
státuszban bekövetkezett változások 12 hónapnál ..............127
6.6.3. táblázat. Az aneurysma méretében és az endoleak-
státuszban bekövetkezett változások 24 hónapnál ..............127
6.7 Az AAA-val összefüggő másodlagos beavatkozások ..............127
6.7.1. táblázat. Másodlagos beavatkozások (1-12. hónap) ........127
6.7.2. táblázat. Másodlagos beavatkozások
(>12-24. hónap) ................................................127
6.8 Másodlagos végpontok mérőszámai .............................128
6.8.1. táblázat. Másodlagos végpontok kezelési
csoportok szerint ..............................................128
7 A BETEGEK KIVÁLASZTÁSA ÉS KEZELÉSE ...........................128
A kezelés egyénivé tétele ...........................................128
8 BETEGTÁJÉKOZTATÁS .............................................128
9 KISZERELÉS ......................................................128
10 KLINIKAI FELHASZNÁLÁSSAL KAPCSOLATOS INFORMÁCIÓ ........128
10.1 Orvosképzés ...................................................128
10.2 Használat előtti szemle .........................................128
10.3 Szükséges anyagok ............................................128
10.4 Ajánlott anyagok ...............................................128
10.5 Az eszköz átmérője méretezésének irányelvei ...................129
10.5.1. táblázat. Útmutató a fő graft-törzs átmérőjének
méretezéséhez .................................................129
10.5.2. táblázat. Útmutató az iliaca-szár átmérőjének
méretezéséhez .................................................129
11 HASZNÁLATI UTASÍTÁS ..........................................129
Anatómiai követelmények ..........................................129
A felhasználással kapcsolatos általános információk .................129
Implantáció előtti meghatározó elemek .............................129
A beteg előkészítése ................................................129
11.1 Bifurcatiós rendszer ............................................129
11.1.1 A bifurcatiós fő grafttörzs előkészítése és öblítése ........129
11.1.2 A kontralaterális iliacaszár előkészítése és öblítése ........129
11.1.3 Az ipsilaterális iliacaszár előkészítése és öblítése ..........129
11.1.4 Vaszkuláris hozzáférés és angiográfia .....................129
11.1.5 A fő grafttörzs elhelyezése ...............................129
11.1.6 Kontralaterális iliaca vezetődrót elhelyezése ..............130
11.1.7 A fő grafttörzs proximális (csúcsi) telepítése ..............130
11.1.8 A kontralaterális iliacaszár elhelyezése és telepítése ......130
11.1.9 Fő grafttörzs disztális (alsó) telepítése ....................130
11.1.10 A csúcsi sapka összekapcsolása .........................130
11.1.11 Az ipsilaterális iliacaszár elhelyezése és telepítése .......130
11.1.12 A formázó ballon felvezetése ...........................130
Végső angiogram ......................................130
12 LEKÉPEZÉSI IRÁNYELVEK ÉS POSZTOPERATÍV UTÁNKÖVETÉS ......130
12.1 Általános ......................................................130
12.1. táblázat. Endografttal rendelkező betegek számára
ajánlott képalkotó vizsgálati program ..........................131
12.2 Kontrasztanyaggal és anélkül végzett CT-vizsgálattal
kapcsolatos ajánlások ..........................................131
12.2. táblázat. Elfogadható leképezési protokollok ..............131
12.3 Hasi röntgenfelvételek .........................................131
12.4 Ultrahang ......................................................131
12.5 MRI-kompatibilitás és -biztonságosság .........................131
12.6 További felügyelet és kezelés ...................................131
13 A BETEGEK NYOMONKÖVETÉSÉRE VONATKOZÓ INFORMÁCIÓ .....131
14 HIBAKERESÉS .......................................................131
14.1 Elsütődrótos kioldószerkezet hibakeresése .......................131
14.1.1 A fő grafttörzs proximális (csúcsi) telepítése .................. 131
14.1.2 A csúcsi sapka összekapcsolása .............................. 132
14.1.3 Az ipsilateralis iliacaszár elhelyezése és telepítése ............. 132
14.1.4 A kontralaterális iliacaszár elhelyezése és telepítése ........... 132
14.2 A suprarenalis sztent telepítésének hibakeresése .................132
14.2.1 A fő grafttörzs proximális (csúcsi) telepítése .................. 132
14.2.2 A csúcsi sapka összekapcsolása .............................. 132
14.2.3 Az ipsilateralis iliacaszár elhelyezése és telepítése .............132
14.2.4 A kontralaterális iliacaszár elhelyezése és telepítése ...........133
120
121

ITALIANO
INDICE
1 DESCRIZIONE DEL DISPOSITIVO .................................... 134
1.1 Corpo aortico principale e branche iliache della protesi ........... 134
1.2 Sistema di inserimento del corpo principale ...................... 134
1.3 Sistema di inserimento della branca iliaca ......................... 134
1.4 Componenti ausiliari dell’endoprotesi addominale Zenith .........134
2 INDICAZIONI PER L’USO ............................................134
3 CONTROINDICAZIONI .............................................. 134
4 AVVERTENZE E PRECAUZIONI ...................................... 134
4.1 Informazioni generali ............................................. 134
4.2 Selezione, trattamento e follow-up del paziente ..................
4.3 Tecniche di misurazione e imaging pre-procedura ................
4.4 Selezione del dispositivo ........................................135
4.5 Procedura di impianto ...........................................135
4.6 Uso del palloncino dilatatore ....................................135
4.7 Sicurezza e compatibilità in ambito MRI ...........................
5 EVENTI NEGATIVI .................................................. 135
5.1 Eventi negativi osservati ..........................................
Tabella 5.1.1 - Decesso e rottura nel corso dello studio clinico ....
Tabella 5.1.2 - Eventi negativi nel corso dello studio clinico .......136
5.2 Possibili eventi negativi ..........................................136
Notifica degli eventi negativi correlati al dispositivo .................136
6 RIEPILOGO DEGLI STUDI CLINICI ....................................136
6.1 Obiettivo ........................................................ 136
6.2 Struttura dello studio ............................................. 136
Tabella 6.2.1 - Follow-up dei pazienti e statistiche ................ 137
6.3 Caratteristiche demografiche dei pazienti ......................... 137
Tabella 6.3.1 - Confronto delle caratteristiche dei soggetti ........137
Tabella 6.3.2 - Distribuzione degli aneurismi in base al diametro. . 137
6.4 Risultati .......................................................... 137
Tabella 6.4.1 - Dispositivi impiantati ............................. 137
Tabella 6.4.2 - Risultati principali. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 138
Tabella 6.4.3 - Parametri di valutazione del successo ............. 139
Tabella 6.4.4 - Eventi evidenziati mediante lastre radiografiche
addominali - Integrità del dispositivo ............................ 139
Tabella 6.4.5 - Eventi evidenziati mediante TC - Pervietà
dell’endoprotesi ................................................ 139
Tabella 6.4.6 - Eventi evidenziati mediante TC - Migrazione
dell’endoprotesi (corpo principale) .............................. 139
Tabella 6.4.7 - Eventi evidenziati mediante lastre radiografiche
addominali - Separazione delle estremità ........................ 140
6.5 Gestione degli endoleak .......................................... 140
Tabella 6.5.1 - Endoleak (tutti i tipi, nuovi e recidivi) ..............140
Tabella 6.5.2 - Prima occorrenza di endoleak per i pazienti a
rischio normale ................................................. 140
Tabella 6.5.3 - Prima occorrenza di endoleak per i pazienti ad
alto rischio. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 140
Tabella 6.5.4 - Prima occorrenza di endoleak per i pazienti
di pratica ....................................................... 140
6.6 Mutazione dell’aneurisma ........................................ 141
Tabella 6.6.1 - Variazione del diametro massimo dell’aneurisma
per intervallo di tempo ......................................... 141
Tabella 6.6.2 - Variazione della dimensione dell’aneurisma e
endoleak a 12 mesi ............................................. 141
Tabella 6.6.3 - Variazione della dimensione dell’aneurisma e
endoleak a 24 mesi ............................................. 141
6.7 Interventi secondari correlati all’AAA ..............................141
Tabella 6.7.1 - Interventi secondari (da 0 a 12 mesi) .............. 141
Tabella 6.7.2 - Interventi secondari (da >12 a 24 mesi) ............ 141
6.8 Parametri per la valutazione degli esiti secondari .................. 142
Tabella 6.8.1 - Esiti secondari per gruppo di trattamento ......... 142
7 SELEZIONE E TRATTAMENTO DEI PAZIENTI ..........................142
Requisiti per il trattamento ........................................... 142
8 INFORMAZIONI DA FORNIRE AI PAZIENTI ........................... 142
9 CONFEZIONAMENTO ..............................................142
10 INFORMAZIONI PER USO CLINICO ................................. 142
10.1 Programma di formazione per il medico ......................... 142
10.2 Esame prima dell’uso ........................................... 142
10.3 Materiali necessari .............................................. 142
10.4 Materiali consigliati ............................................. 142
10.5 Linee guida per la determinazione del diametro idoneo
del dispositivo ....................................................... 143
Tabella 10.5.1 - Guida alla determinazione del diametro idoneo
del corpo principale dell’endoprotesi ............................143
Tabella 10.5.2 - Guida alla determinazione del diametro idoneo
della branca iliaca dell’endoprotesi ..............................143
11 ISTRUZIONI PER L’USO ............................................143
Requisiti anatomici .................................................. 143
Informazioni generali sull’impiego ................................... 143
Fattori da considerare in sede preliminare ............................143
Preparazione del paziente ...........................................143
11.1 Sistema biforcato ............................................... 143
11.1.1 Preparazione/lavaggio del corpo principale biforcato ......143
11.1.2 Preparazione/lavaggio della branca iliaca controlaterale ...143
11.1.3 Preparazione/lavaggio della branca iliaca ipsilaterale ...... 143
11.1.4 Accesso al sistema vascolare e angiografia ................ 143
11.1.5 Posizionamento del corpo principale .....................143
11.1.6 Posizionamento della guida iliaca controlaterale .......... 144
11.1.7 Rilascio dell’estremità prossimale (superiore) del corpo
principale ...................................................... 144
11.1.8 Posizionamento e rilascio della branca iliaca controlaterale 144
11.1.9 Rilascio dell’estremità distale (inferiore) del corpo
principale ...................................................... 144
11.1.10 Innesto della calotta superiore ........................... 144
11.1.11 Posizionamento e rilascio della branca iliaca ipsilaterale .. 144
11.1.12 Inserimento del palloncino dilatatore .................... 144
Angiogramma conclusivo ................................ 145
12 LINEE GUIDA PER LE TECNICHE DI IMAGING E IL FOLLOW-UP
POSTOPERATORIO ................................................145
12.1 Generali ........................................................ 145
Tabella 12.1 - Programma consigliato per le procedure di
imaging per i pazienti portatori di endoprotesi .................. 145
12.2 Consigli per la TC con e senza mezzo di contrasto ................ 145
Tabella 12.2 Protocolli di imaging accettabili ..................... 145
12.3 Lastre radiografiche addominali ................................. 145
12.4 Ecografia .......................................................145
12.5 Sicurezza e compatibilità in ambito MRI ......................... 145
12.6 Ulteriori esami di controllo e trattamento ........................ 146
13 INFORMAZIONI DOCUMENTANTI IL DISPOSITIVO .................. 146
14 GUIDA ALLA RISOLUZIONE DEI PROBLEMI ...........................146
14.1 Risoluzione dei problemi relativi al meccanismo di rilascio a filo di
sicurezza ...............................................................146
14.1.1 Rilascio dell’estremità prossimale (superiore) del corpo
principale ..............................................................146
14.1.2 Innesto della calotta superiore ..................................146
14.1.3 Posizionamento e rilascio della branca iliaca ipsilaterale ........146
14.1.4 Posizionamento e rilascio della branca iliaca controlaterale .....146
14.2 Risoluzione dei problemi relativi al rilascio dello stent soprarenale. . 146
14.2.1 Rilascio dell’estremità prossimale (superiore) del corpo
principale ..............................................................146
14.2.2 Innesto della calotta superiore ..................................147
14.2.3 Posizionamento e rilascio della branca iliaca ipsilaterale ........147
14.2.4 Posizionamento e rilascio della branca iliaca controlaterale .....147
134
134
135
135
135
NEDERLANDS
INHOUD
1 BESCHRIJVING VAN HET HULPMIDDEL ..............................148
1.1 Componenten voor main body in de aorta en voor iliacale poten .. 148
1.2 Introductiesysteem van de main body ............................ 148
1.3 Introductiesysteem van de iliacale poot ........................... 148
1.4 Zenith hulpcomponenten voor de AAA endovasculaire prothese .. 148
2 INDICATIES VOOR GEBRUIK .......................................148
3 CONTRA-INDICATIES ...............................................148
4 WAARSCHUWINGEN EN VOORZORGSMAATREGELEN ................ 148
4.1 Algemeen. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 148
4.2 Selectie, behandeling en controle van de patiënt ................
4.3 Preprocedurele meettechnieken en beeldvorming ...............
4.4 Selectie van het hulpmiddel .....................................149
4.5 De implantatieprocedure ........................................149
4.6 Gebruik van de modelleerballon .................................149
4.7 MRI-veiligheid en -compatibiliteit .................................
5 ONGEWENSTE VOORVALLEN ....................................... 149
5.1 Waargenomen ongewenste voorvallen ...........................
Tabel 5.1.1 Overlijden en ruptuur bij klinische studie .............
Tabel 5.1.2 Ongewenste voorvallen in klinische studie ........... 150
5.2 Mogelijke ongewenste voorvallen ................................ 150
Melding van prothesegerelateerde ongewenste voorvallen ..........150
6 SAMENVAT TING VAN KLINISCHE STUDIES ........................... 150
6.1 Doelstellingen ...................................................150
6.2 Opzet van de studie .............................................. 150
Tabel 6.2.1 Patiëntcontrole en controleerbaarheid ............... 151
6.3 Demografische patiëntgegevens ................................. 151
Tabel 6.3.1 Vergelijking van kenmerken van proefpersonen ...... 151
Tabel 6.3.2 Verdeling diameter aneurysma ....................... 151
6.4 Resultaten ....................................................... 151
Tabel 6.4.1 Gebruikte implantaten ...............................152
Tabel 6.4.2 Primaire resultaten ...................................152
Tabel 6.4.3 Resultaatmetingen .................................. 153
Tabel 6.4.4 Abdominale radiografische bevindingen –
Integriteit van de prothese ...................................... 153
Tabel 6.4.5 CT-bevindingen – Afwezigheid van obstructies
in prothese ..................................................... 153
Tabel 6.4.6 CT-bevindingen – Migratie van main body ........... 153
Tabel 6.4.7 Abdominale radiografische bevindingen –
losraken van de stompen ....................................... 154
6.5 Behandeling van endolekkages ................................... 154
Tabel 6.5.1 Endolekkages (alle typen, nieuw en persistent) ....... 154
Tabel 6.5.2 Eerste voorkomen van endolekkage bij patiënten
met standaardrisico ............................................. 154
Tabel 6.5.3 Eerste voorkomen van endolekkage bij patiënten
met hoog risico ................................................. 154
Tabel 6.5.4 Eerste voorkomen van endolekkage bij
oefenpatiënten ................................................. 154
6.6 Veranderingen van het aneurysma. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 155
Tabel 6.6.1 Verandering van de maximale diameter van het
aneurysma per interval ......................................... 155
Tabel 6.6.2 Veranderingen in afmeting van het aneur ysma en de
endolekkage na 12 maanden ................................... 155
Tabel 6.6.3 Veranderingen in afmeting van het aneur ysma en de
endolekkage na 24 maanden ................................... 155
6.7 AAA-gerelateerde secundaire interventies ........................ 155
Tabel 6.7.1 Secundaire interventies (tot 12 maanden) ............ 155
Tabel 6.7.2 Secundaire interventies (> 12 tot 24 maanden) ....... 155
6.8 Metingen secundaire resultaten .................................. 156
Tabel 6.8.1 Secundaire resultaten per behandelgroep ............156
7 SELECTIE EN BEHANDELING VAN DE PATIËNT .......................156
Individualisering van de behandeling ................................ 156
8 DE PATIËNT INFORMEREN .......................................... 156
9 WIJZE VAN LEVERING .............................................. 156
10 INFORMATIE OVER HET KLINISCHE GEBRUIK .......................156
10.1 Opleiding van de arts ........................................... 156
10.2 Inspectie voorafgaand aan gebruik .............................. 156
10.3 Benodigde materialen ..........................................156
10.4 Aanbevolen materialen ......................................... 156
10.5 Richtlijnen voor het bepalen van de diameter van
het hulpmiddel ...................................................... 157
Tabel 10.5.1 Handleiding voor het bepalen van de diameter
van de main body ............................................... 157
Tabel 10.5.2 Handleiding voor het bepalen van de diameter
van de iliacale poot ............................................. 157
11 GEBRUIKSAANWIJZING ........................................... 157
Anatomische vereisten ............................................... 157
Algemene gebruiksinformatie ........................................ 157
Bepalende factoren vóór de implantatie .............................. 157
De patiënt voorbereiden ............................................. 157
11.1 Bifurcatiesysteem ............................................... 157
11.1.1 Voorbereiding/spoelen van de main body met bifurcatie ..157
11.1.2 Voorbereiding/spoelen van de contralaterale iliacale poot ..157
11.1.3 Voorbereiding/spoelen van de ipsilaterale iliacale poot ....157
11.1.4 Vasculaire introductie en angiografie. . . . . . . . . . . . . . . . . . . . . . 157
11.1.5 Plaatsing van de main body ..............................157
11.1.6 De contralaterale iliacale voerdraad plaatsen .............. 158
11.1.7 Ontplooiing van het proximale deel (top) van de main
body ........................................................... 158
11.1.8 Plaatsing en ontplooiing van de contralaterale iliacale
poot ............................................................ 158
11.1.9 Ontplooiing van het distale deel (onderkant) main body ..158
11.1.10 Koppelen van de topkap ................................ 158
11.1.11 Plaatsing en ontplooiing van de ipsilaterale iliacale poot . 158
11.1.12 Introductie van de modelleerballon ..................... 158
Afrondend angiogram ................................... 158
12 RICHTLIJNEN VOOR BEELDVORMEND ONDERZOEK EN
POSTOPERATIEVE CONTROLE ..................................... 159
12.1 Algemeen ...................................................... 159
Tabel 12.1 Aanbevolen schema voor beeldvorming bij patiënten
met een endovasculaire prothese ............................... 159
12.2 Aanbevelingen voor CT-onderzoek met en zonder
contrastmiddel .................................................159
Tabel 12.2 Geaccepteerde beeldvormingsprotocollen ............ 159
12.3 Röntgenopnamen van de buik .................................. 159
12.4 Echografie ...................................................... 159
12.5 Veiligheid bij en compatibiliteit met MRI .........................159
12.6 Extra controle en behandeling ..................................159
13 INFORMATIE PATIËNTVOLGSYSTEEM ..............................159
14 OPLOSSEN VAN PROBLEMEN ........................................160
14.1 Problemen met de ontkoppeling van de trigger wire oplossen .....160
14.1.1 Ontplooiing van het proximale deel (top) van de main body . 160
14.1.2 Koppelen van de topkap .....................................160
14.1.3 Plaatsing en ontplooiing van de ipsilaterale iliacale poot .....160
14.1.4 Plaatsing en ontplooiing van de contralaterale iliacale poot ..160
14.2 Problemen met de ontplooiing van de suprarenale stent oplossen . 160
14.2.1 Ontplooiing van het proximale deel (top) van de main body . 160
14.2.2 Koppelen van de topkap .....................................160
14.2.3 Plaatsing en ontplooiing van de ipsilaterale iliacale poot .....160
14.2.4 Plaatsing en ontplooiing van de contralaterale iliacale poot ..161
148
148
149
149
149

NORSK
INNHOLDSFORTEGNELSE
1 BESKRIVELSE AV ANORDNINGEN ...................................162
1.1 Aortisk hoveddel og iliaca-benkomponenter .....................162
1.2 Hoveddelens innføringssystem ...................................162
1.3 Iliaca-ben innføringssystem ......................................162
1.4 Hjelpekomponenter for Zenith AAA endovaskulært implantat ....162
2 BRUKSOMRÅDER ..................................................162
3 KONTRAINDIKASJONER ............................................162
4 ADVARSLER OG FORHOLDSREGLER ................................162
4.1 Generelt .........................................................
4.2 Pasientutvelgelse, behandling og oppfølging ....................162
4.3 Målingsteknikker og avbilding før prosedyren. . . . . . . . . . . . . . . . . . . . 162
4.4 Valg av anordning ...............................................163
4.5 Implantasjon ....................................................163
4.6 Bruk av formingsballong ........................................163
4.7 MR-sikkerhet og kompatibilitet ...................................
5 UGUNSTIGE HENDELSER ...........................................163
5.1 Observerte ugunstige hendelser .................................163
Tabell 5.1.1 Død og ruptur fra klinisk studie ........................163
Tabell 5.1.2 Ugunstige hendelser i klinisk studie ....................164
5.2 Mulige ugunstige hendelser ......................................164
Rapportering av ugunstige hendelser, relatert til anordningen .....164
6 OVERSIKT AV KLINISKE STUDIER ...................................164
6.1 Målsetninger .....................................................164
6.2 Studiedesign .....................................................164
Tabell 6.2.1 Pasientoppfølging og ansvarlighet ..................... 165
6.3 Pasientbefolkningsstatistikk ..................................... 165
Tabell 6.3.1 Sammenligning av forsøkspersoners karakteristika .....165
Tabell 6.3.2 Distribusjon av aneurismens diameter ................. 165
6.4 Resultater ........................................................165
Tabell 6.4.1 Implanterte anordninger ..............................165
Tabell 6.4.2 Primære resultater .....................................166
Tabell 6.4.3 Suksessmål ............................................167
Tabell 6.4.4 Funn under abdominal røntgen –
anordningens integritet ........................................... 167
Tabell 6.4.5 CT-funn – implantatpatens .............................167
Tabell 6.4.6 CT-funn – implantat (hoveddel)-vandring .............. 167
Tabell 6.4.7 Funn under abdominal røntgen – løsning av lem ....... 168
6.5 Behandling av endolekkasje ......................................168
Tabell 6.5.1 Endolekkasjer (alle typer, nye og persistente) ...........168
Tabell 6.5.2 Første tilfelle av endolekkasje for
standardrisikopasienter ............................................168
Tabell 6.5.3 Første tilfelle av endolekkasje for høyrisikopasienter .... 168
Tabell 6.5.4 Første tilfelle av endolekkasje for innlæringspasienter .. 168
6.6 Aneurismeendringer .............................................169
Tabell 6.6.1 Aneurismens maksimale diameterendring pr. intervall ..169
Tabell 6.6.2 Endring i aneurismens størrelse og endolekkasje ved
12 måneder .......................................................169
Tabell 6.6.3 Endring i aneurismens størrelse og endolekkasje ved
24 måneder .......................................................169
6.7 AAA-relaterte sekundære intervensjoner .........................169
Tabell 6.7.1 Sekundære intervensjoner (til 12 måneder) ............ 169
Tabell 6.7.2 Sekundære intervensjoner (>12 til 24 måneder) ........169
6.8 Måltall for sekundære resultater ..................................170
Tabell 6.8.1 Sekundære resultater pr. behandlingsgruppe ..........170
7 PASIENTUTVALG OG BEHANDLING .................................170
Individualisering av behandlingen ...................................170
8 PASIENTRÅDFØRINGSINFORMASJON ...............................170
162
163
9 LEVERING .........................................................170
10 INFORMASJON OM KLINISK ANVENDELSE .......................170
10.1 Legeopplæring .................................................170
10.2 Inspeksjon før bruk .............................................170
10.3 Nødvendige materialer .........................................170
10.4 Anbefalte materialer ............................................170
10.5 Retningslinjer for størrelsesmåling av anordningens diameter ...170
Tabell 10.5.1 Diameterstørrelsesguide for hoveddel-implantat ...... 171
Tabell 10.5.2 Diameterstørrelsesguide for iliaca-ben implantat ......171
11 BRUKSVEILEDNING ...............................................171
Anatomiske krav .....................................................171
Allmenn bruksinformasjon ...........................................171
Avgjørende faktorer før implantasjonen ..............................171
Klargjøring av pasienten .............................................171
11.1 Bifurkert system ................................................171
11.1.1 Forberedelse/skylling av bifurkert hoveddel .................171
11.1.2 Forberedelse/skylling av kontralateral iliaca-ben .............171
11.1.3 Forberedelse/skylling av ipsilateralt iliaca-ben ...............171
11.1.4 Vaskulær tilgang og angiografi ..............................171
11.1.5 Plassering av hoveddel ......................................171
11.1.6 Plassering av kontralateral iliaca-ledevaier ...................171
11.1.7 Proksimal (topp) anleggelse av hoveddel ....................171
11.1.8 Plassering og anleggelse av
11.1.9 Distal (bunn) anleggelse av hoveddel ....................... 172
11.1.10 Sammenkobling av topphetten ............................172
11.1.11 Plassering og anleggelse av ipsilateralt iliaca-ben ..........172
11.1.12 Innføring av formingsballong ..............................172
Sluttangiogram ........................................... 172
12 RETNINGSLINJER FOR AVBILDING OG POSTOPERATIV
OPPFØLGING .................................................... 172
12.1 Generelt ........................................................172
Tabell 12.1 Anbefalt avbildingsprogram for pasienter
med endoimplantat ...............................................173
12.2 Anbefalinger for CT med og uten kontrast .......................173
Tabell 12.2 Godkjente avbildingsprotokoller ....................... 173
12.3 Abdominale røntgenbilder ......................................173
12.4 Ultralyd .........................................................173
12.5 MRI-sikkerhet og kompatibilitet .................................173
12.6 Ytterligere kontroll og behandling .............................. 173
13 SPORING AV PASIENTINFORMASJON ..............................173
14 FEILSØKING .........................................................173
14.1 Feilsøking for vaierutløsningsmekanisme ..........................
14.1.1 Proksimal (topp) anleggelse av hoveddel ....................173
14.1.2 Sammenkobling av topphetten ..............................
14.1.3 Plassering og anleggelse av ipsilateralt iliaca-ben ............
14.1.4 Plassering og anleggelse av kontralateralt iliaca-ben .........
14.2 Feilsøking for anleggelse av suprarenal stent .......................
14.2.1 Proksimal (topp) anleggelse av hoveddel ....................
14.2.2 Sammenkobling av topphetten ..............................
14.2.3 Plassering og anleggelse av ipsilateralt iliaca-ben ............
14.2.4 Plassering og anleggelse av kontralateralt iliaca-ben .........
kontralateralt
iliaca-ben. . . . . . . . . 172
173
173
174
174
174
174
174
174
174
POLSKI
SPIS TREŚCI
1 OPIS URZĄDZENIA ................................................175
1.1 Główny trzon aortalny i odnogi biodrowe ........................175
1.2 System podawania głównego trzonu ............................175
1.3 System podawania odnogi biodrowej ............................175
1.4 Elementy pomocnicze do stent-graftu wewnątrznaczyniowego
Zenith AAA .........................................................175
2 WSKAZANIA DO STOSOWANIA ....................................175
3 PRZECIWWSKAZANIA .............................................175
4 OSTRZEŻENIA I ŚRODKI OSTROŻNOŚCI ............................175
4.1 Ogólne ..........................................................
4.2 Wybór pacjentów, leczenie i kontrola po zabiegu .................175
4.3 Techniki pomiarów przedoperacyjnych i obrazowanie ............175
4.4 Wybór urządzenia ...............................................176
4.5 Procedura wszczepiania .........................................176
4.6 Użycie balonu kształtującego ....................................176
4.7 Bezpieczeństwo i zgodność z badaniem MRI ......................
5 ZDARZENIA NIEPOŻĄDANE .......................................176
5.1 Obserwowane zdarzenia niepożądane ...........................176
Tabela 5.1.1 Zgon i pęknięcie – dane z badania klinicznego .....176
Tabela 5.1.2 Zdarzenia niepożądane w badaniu klinicznym ......177
5.2 Potencjalne zdarzenia niepożądane .............................177
Zgłaszanie zdarzeń niepożądanych związanych z urządzeniem ..177
6 STRESZCZENIE BADAŃ KLINICZNYCH ..............................177
6.1 Cele .............................................................177
6.2 Schemat badania ................................................177
Tabela 6.2.1 Kontrola i ewidencja pacjentów ....................178
6.3 Dane demograficzne pacjentów .................................178
Tabela 6.3.1 Porównanie charakterystyki pacjentów .............178
Tabela 6.3.2 Rozkład średnic tętniaków .........................178
6.4 Wyniki ..........................................................178
Tabela 6.4.1 Wszczepione urządzenia ...........................179
Tabela 6.4.2 Wyniki pierwszorzędowe ...........................179
Tabela 6.4.3 Wyniki oceny skuteczności .........................180
Tabela 6.4.4 Wyniki badania radiologicznego jamy brzusznej –
integralność urządzenia ........................................180
Tabela 6.4.5 Wyniki badania TK – drożność stent-graftu .........180
Tabela 6.4.6 Wyniki badania TK – przemieszczenie stent-graftu
(głównego trzonu) .............................................180
Tabela 6.4.7 Wyniki badania rtg jamy brzusznej –
oddzielenie odgałęzienia .......................................181
6.5 Postępowanie w razie przecieku wewnętrznego. . . . . . . . . . . . . . . . . . 181
Tabela 6.5.1 Przecieki wewnętrzne (wszystkie typy, nowe
i przetrwałe) ...................................................181
Tabela 6.5.2 Pierwszorazowe wystąpienie przecieku
wewnętrznego u pacjentów ze standardowym ryzykiem ........181
Tabela 6.5.3 Pierwszorazowe wystąpienie przecieku
wewnętrznego u pacjentów z wysokim ryzykiem ...............181
Tabela 6.5.4 Pierwszorazowe wystąpienie przecieku
wewnętrznego u pacjentów z grupy próbnej ...................181
6.6 Zmiana w obrębie tętniaka ......................................182
Tabela 6.6.1 Zmiana maksymalnej średnicy tętniaka
w poszczególnych okresach badania ...........................182
Tabela 6.6.2 Zmiana średnicy tętniaka i przeciek wewnętrzny
w okresie 12 miesięcy ..........................................182
Tabela 6.6.3 Zmiana średnicy tętniaka i przeciek wewnętrzny
w okresie 24 miesięcy ..........................................182
6.7 Interwencje wtórne związane z AAA .............................182
Tabela 6.7.1 Interwencje wtórne (do 12 miesięcy) ...............182
Tabela 6.7.2 Interwencje wtórne (>12 do 24 miesięcy) ...........182
6.8 Wskaźniki wyników drugorzędowych ............................183
Tabela 6.8.1 Wyniki drugorzędowe w poszczególnych
grupach leczenia ...............................................183
7 DOBÓR I LECZENIE PACJENTÓW ...................................183
Indywidualizacja leczenia ...........................................183
8 INFORMACJE DLA PACJENTA ......................................183
9 RODZAJ OPAKOWANIA ...........................................183
10 INFORMACJE DOTYCZĄCE ZASTOSOWANIA KLINICZNEGO .........183
10.1 Szkolenie lekarza ...............................................183
10.2 Kontrola przed użyciem ........................................183
10.3 Wymagane materiały ...........................................183
10.4 Materiały zalecane .............................................183
10.5 Wskazówki dotyczące kalibrowania średnicy przyrządu .........184
Tabela 10.5.1 Wskazówki dotyczące kalibrowania średnicy
głównego trzonu stent-graftu ..................................184
Tabela 10.5.2 Wskazówki dotyczące kalibrowania średnicy
odnogi biodrowej stent-graftu .................................184
11 WSKAZANIA ....................................................184
Wymagania anatomiczne ............................................
Ogólne informacje o stosowaniu .....................................
Przedimplantacyjne czynniki determinujące ..........................
Przygotowanie pacjenta .............................................
11.1 System rozwidlony .............................................184
11.1.1 Przygotowanie/przepłukiwanie rozwidlonego trzonu
głównego .....................................................184
11.1.2 Przygotowanie/przepłukiwanie przeciwstronnej odnogi
biodrowej .....................................................184
11.1.3 Przygotowanie/przepłukiwanie tożsamostronnej odnogi
biodrowej .....................................................184
11.1.4 Dostęp naczyniowy i angiografia .........................184
11.1.5 Umieszczanie głównego trzonu ..........................184
11.1.6 Umieszczanie przeciwstronnego prowadnika
biodrowego ...................................................185
11.1.7 Rozprężanie proksymalnej (górnej) części głównego
trzonu .........................................................185
11.1.8 Umieszczanie i rozprężanie przeciwstronnej odnogi
biodrowej .....................................................185
11.1.9 Rozprężanie dystalnej (dolnej) części głównego trzonu ...185
11.1.10 Łączenie nasadki końcówki .............................185
11.1.11 Umieszczanie i rozprężanie tożsamostronnej odnogi
biodrowej .....................................................185
11.1.12 Wprowadzenie balonu kształtującego ...................185
Angiogram końcowy ...................................186
12 WSKAZÓWKI DOTYCZĄCE OBRAZOWANIA I KONTROLA PO ZABIEGU ..186
12.1 Ogólne ........................................................186
Tabela 12.1 Zalecany harmonogram badań dla pacjentów
ze stent-graftami wewnątrznaczyniowymi ......................186
12.2 Zalecenia dotyczące badania TK z kontrastem i bez kontrastu ...186
Tabela 12.2 Dopuszczalne protokoły obrazowania ..............186
12.3 Radiogramy brzuszne ..........................................186
12.4 Badanie USG ...................................................186
12.5 Bezpieczeństwo i zgodność z badaniem MRI ....................186
12.6 Dodatkowy nadzór i leczenie ...................................187
13 INFORMACJE DOTYCZĄCE OBSERWACJI PACJENTÓW ..............187
14 ROZWIĄZYWANIE PROBLEMÓW .....................................187
14.1 Rozwiązywanie problemów dotyczących uwalniania drutu
zwalniającego ..........................................................187
14.1.1 Rozprężanie proksymalnej (górnej) części głównego trzonu
14.1.2 Łączenie nasadki końcówki
14.1.3 Umieszczanie i rozprężanie tożsamostronnej odnogi biodrowej
14.1.4 Umieszczanie i rozprężanie przeciwstronnej odnogi biodrowej
14.2 Rozwiązywanie problemów dotyczących rozprężania stentu
nadnerkowego .........................................................187
14.2.1 Rozprężanie proksymalnej (górnej) części głównego trzonu
14.2.2 Łączenie nasadki końcówki
14.2.3 Umieszczanie i rozprężanie tożsamostronnej odnogi biodrowej
14.2.4 Umieszczanie i rozprężanie przeciwstronnej odnogi biodrowej
..................................187
..................................188
175
176
184
184
184
184
...187
. 187
..187
...187
. 188
..188

PORTUGUÊS
ÍNDICE
1 DESCRIÇÃO DO DISPOSITIVO ....................................... 189
1.1 Componentes do corpo aórtico principal e extremidades ilíacas ... 189
1.2 Sistema de colocação do corpo principal ......................... 189
1.3 Sistema de colocação da extremidade ilíaca ...................... 189
1.4 Componentes auxiliares da prótese endovascular AAA Zenith ..... 189
2 INDICAÇÕES DE UTILIZAÇÃO .......................................189
3 CONTRA-INDICAÇÕES ..............................................189
4 ADVERTÊNCIAS E PRECAUÇÕES .................................... 189
4.1 Geral ............................................................ 189
4.2 Selecção, tratamento e seguimento dos doentes .................
4.3 Técnicas de medição e imagiologia antes do procedimento ......
4.4 Escolha do dispositivo ...........................................190
4.5 Procedimento de implantação ...................................190
4.6 Utilização do balão de moldagem ...............................190
4.7 Segurança e compatibilidade com RMN ..........................
5 EFEITOS ADVERSOS ................................................190
5.1 Efeitos adversos observados ......................................
Tabela 5.1.1 Morte e rotura ocorridas no estudo clínico ..........
Tabela 5.1.2 Efeitos adversos no estudo clínico ...................191
5.2 Efeitos adversos potenciais ....................................... 191
Relato de efeitos adversos relacionados com o dispositivo ...........191
6 SÍNTESE DOS ESTUDOS CLÍNICOS ..................................191
6.1 Objectivos ....................................................... 191
6.2 Desenho do estudo .............................................. 191
Tabela 6.2.1 Seguimento e responsabilidade dos doentes ........192
6.3 Dados demográficos dos doentes ................................192
Tabela 6.3.1 Comparação das características dos sujeitos .........192
Tabela 6.3.2 Distribuição dos diâmetros dos aneurismas ......... 192
6.4 Resultados ....................................................... 192
Tabela 6.4.1 Dispositivos implantados ........................... 192
Tabela 6.4.2 Resultados primários ............................... 193
Tabela 6.4.3 Avaliações do sucesso .............................. 194
Tabela 6.4.4 Achados radiográficos abdominais - integridade
do dispositivo ..................................................194
Tabela 6.4.5 Achados da TAC - permeabilidade da prótese. . . . . . . . 194
Tabela 6.4.6 Achados da TAC - migração da prótese
(corpo principal) ................................................ 194
Tabela 6.4.7 Achados radiográficos abdominais -
separação dos ramos ........................................... 195
6.5 Tratamento de fugas intra-aneurismais ........................... 195
Tabela 6.5.1 Fugas intra-aneurismais (todos os tipos, novas
e persistentes) .................................................. 195
Tabela 6.5.2 Primeira ocorrência de fugas intra-aneurismais
para doentes de risco normal ................................... 195
Tabela 6.5.3 Primeira ocorrência de fugas intra-aneurismais
para doentes de alto risco ....................................... 195
Tabela 6.5.4 Primeira ocorrência de fugas intra-aneurismais
para doentes não permanentes ................................. 195
6.6 Alterações do aneurisma ......................................... 196
Tabela 6.6.1 Alterações nos diâmetros máximos dos
aneurismas por intervalo ........................................ 196
Tabela 6.6.2 Alterações no tamanho do aneurisma e fugas
intra-aneurismais aos 12 meses .................................196
Tabela 6.6.3 Alterações no tamanho do aneurisma e fugas
intra-aneurismais aos 24 meses .................................196
6.7 Intervenções secundárias relacionadas com AAA .................. 196
Tabela 6.7.1 Intervenções secundárias (aos 12 meses) ............ 196
Tabela 6.7.2 Intervenções secundárias (> 12 aos 24 meses) ....... 196
6.8 Avaliações dos resultados secundários ............................ 197
Tabela 6.8.1 Resultados secundários por grupo de tratamento ... 197
7 SELECÇÃO E TRATAMENTO DE DOENTES ............................197
Individualização do tratamento ...................................... 197
8 INFORMAÇÃO DE ACONSELHAMENTO AOS DOENTES ...............197
189
189
190
190
190
9 APRESENTAÇÃO ...................................................197
10 INFORMAÇÃO PARA UTILIZAÇÃO CLÍNICA ......................... 197
10.1 Formação de médicos ........................................... 197
10.2 Inspecção antes da utilização. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 197
10.3 Materiais necessários ............................................ 197
10.4 Materiais recomendados ........................................ 197
10.5 Orientações para escolha do diâmetro do dispositivo ............ 198
Tabela 10.5.1 Guia para escolha do diâmetro do corpo principal
da prótese ...................................................... 198
Tabela 10.5.2 Guia para escolha do diâmetro da extremidade
ilíaca da prótese ................................................ 198
11 INSTRUÇÕES DE UTILIZAÇÃO .....................................198
Requisitos anatómicos ............................................... 198
Informação geral sobre a utilização .................................. 198
Factores determinantes antes da implantação ........................ 198
Preparação do doente ............................................... 198
11.1 Sistema bifurcado ............................................... 198
11.1.1 Preparação/irrigação do corpo principal bifurcado ........ 198
11.1.2 Preparação/irrigação da extremidade ilíaca contralateral .. 198
11.1.3 Preparação/irrigação da extremidade ilíaca
11.1.4 Acesso vascular e angiografia ............................. 198
11.1.5 Colocação do corpo principal ............................. 198
11.1.6 Colocação do fio guia ilíaco contralateral .................. 199
11.1.7 Expansão da parte proximal (topo) do corpo principal ..... 199
11.1.8 Colocação e expansão da extremidade ilíaca contralateral ...199
11.1.9 Expansão da parte distal (inferior) do corpo principal ...... 199
11.1.10 Acoplagem da tampa superior. . . . . . . . . . . . . . . . . . . . . . . . . . . 199
11.1.11 Colocação e expansão da extremidade ilíaca ipsilateral .....
11.1.12 Inserção do balão de moldagem ......................... 199
Angiograma final ........................................ 199
12 ORIENTAÇÕES RELATIVAS À IMAGIOLOGIA E AO SEGUIMENTO
PÓS-OPERATÓRIO ................................................200
12.1 Geral ...........................................................200
Tabela 12.1 Plano de exames imagiológicos recomendado para
doentes com próteses endovasculares .......................... 200
12.2 Recomendações para TAC com e sem contraste .................. 200
Tabela 12.2 Protocolos de imagiologia aceitáveis ................200
12.3 Radiografias abdominais ........................................ 200
12.4 Ecografia .......................................................200
12.5 Segurança e compatibilidade com RMN ......................... 200
12.6 Vigilância e tratamento adicionais ...............................200
13 INFORMAÇÃO SOBRE A LOCALIZAÇÃO DE DOENTES ............... 200
14 RESOLUÇÃO DE PROBLEMAS ........................................200
14.1 Resoluções de problemas relativos ao mecanismo de libertação
com fio de comando ................................................... 201
14.1.1 Expansão da parte proximal (topo) do corpo principal. . . . . . . . . . 201
14.1.2 Acoplagem da tampa superior ..................................201
14.1.3 Colocação e expansão da extremidade ilíaca ipsilateral .........201
14.1.4 Colocação e expansão da extremidade ilíaca contralateral. . . . . . 201
14.2 Resolução de problemas relativos à expansão do stent supra-renal . 201
14.2.1 Expansão da parte proximal (topo) do corpo principal. . . . . . . . . . 201
14.2.2 Acoplagem da tampa superior ..................................201
14.2.3 Colocação e expansão da extremidade ilíaca ipsilateral .........201
14.2.4 Colocação e expansão da extremidade ilíaca contralateral. . . . . . 202
ipsilateral
. . . . . . 198
199
SVENSKA
INNEHÅLLSFÖRTECKNING
1 PRODUKTBESKRIVNING ............................................ 203
1.1 Aortahuvudstomme och komponenter för iliakaliskt graftben ..... 203
1.2 Införingssystem för huvudstommen .............................. 203
1.3 Det iliakaliska graftbenets införingssystem ........................ 203
1.4 Kompletterande komponenter för Zenith AAA endovaskulära
transplantat ......................................................... 203
2 AVSEDD ANVÄNDNING ............................................203
3 KONTRAINDIKATIONER ............................................ 203
4 VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ........................ 203
4.1 Allmänt .......................................................... 203
4.2 Patienturval, behandling och uppföljning ........................
4.3 Mättekniker och bildtagning före proceduren ....................
4.4 Val av anordning ................................................204
4.5 Implantationsförfarande .........................................204
4.6 Användning av formningsballong ...............................204
4.7 MRT-säkerhet och -kompabilitet ..................................
5 BIVERKNINGAR .................................................... 204
5.1 Observerade biverkningar ........................................
Tabell 5.1.1 Dödsfall och ruptur från den kliniska studien. . . . . . . . .
Tabell 5.1.2 Biverkningar i klinisk studie .......................... 205
5.2 Eventuella biverkningar .......................................... 205
Rapportering av biverkningar som har samband med anordningen ..205
6 SAMMANFATTNING AV KLINISKA STUDIER .......................... 205
6.1 Syfte ............................................................. 205
6.2 Studiedesign ..................................................... 205
Tabell 6.2.1 Patientuppföljning och redovisning ................. 206
6.3 Demografiska uppgifter om patienterna .......................... 206
Tabell 6.3.1 Jämförelse av patientkarakteristika .................. 206
Tabell 6.3.2 Distribution av aneurysmdiameter ................... 206
6.4 Resultat .......................................................... 206
Tabell 6.4.1 Implanterade anordningar .......................... 206
Tabell 6.4.2 Primära resultat ..................................... 207
Tabell 6.4.3 Mått på framgång ................................... 208
Tabell 6.4.4 Fynd vid radiografi av buken – anordningens
integritet ....................................................... 208
Tabell 6.4.5 DT-fynd – transplantatets öppenhet ................. 208
Tabell 6.4.6 DT-fynd – migration av transplantatet (huvudstommen) 208
Tabell 6.4.7 Fynd vid radiografi av buken – separation av lem ....209
6.5 Hantering av endoläckage ........................................ 209
Tabell 6.5.1 Endoläckage (alla typer, nya och ihållande) ..........209
Tabell 6.5.2 Första förekomst av endoläckage för standardrisk
patienter ....................................................... 209
Tabell 6.5.3 Första förekomst av endoläckage för högrisk patienter 209
Tabell 6.5.4 Första förekomst av endoläckage för prekliniska
patienter ....................................................... 209
6.6 Förändring av aneurysm. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 210
Tabell 6.6.1 Förändring av maximal aneurysmdiameter per
intervall ........................................................210
Tabell 6.6.2 Förändring av aneurysmstorlek och endoläckage
vid 12 månader ................................................. 210
Tabell 6.6.3 Förändring av aneurysmstorlek och endoläckage
vid 24 månader ................................................. 210
6.7 AAA-relaterade sekundära ingrepp ............................... 210
Tabell 6.7.1 Sekundära ingrepp (till 12 månader) ................. 210
Tabell 6.7.2 Sekundära ingrepp (> 12 till 24 månader) ............ 210
6.8 Sekundära resultatmått .......................................... 211
Tabell 6.8.1 Sekundära resultat enligt behandlingsgrupp .........211
7 PATIENTURVAL OCH BEHANDLING .................................. 211
Individualisering av behandlingen ................................... 211
8 PATIENTRÅDGIVNING .............................................. 211
9 LEVERANSFORM ................................................... 211
10 INFORMATION OM KLINISK ANVÄNDNING ......................... 211
10.1 Läkarens utbildning ............................................. 211
10.2 Besiktning före användning ..................................... 211
10.3 Material som behövs ............................................ 211
10.4 Rekommenderat material ....................................... 211
10.5 Riktlinjer för dimensionering av anordningens diameter ......... 212
Tabell 10.5.1 Dimensioneringsguide för
huvudstomtransplantatets diameter ............................ 212
Tabell 10.5.2 Dimensioneringsguide för det iliakaliska
graftbenets diameter ........................................... 212
11 BRUKSANVISNING ................................................ 212
Anatomiska krav ..................................................... 212
Allmän information .................................................. 212
Avgöranden före implantation ....................................... 212
Förberedelse av patienten ........................................... 212
11.1 Bifurkerat system ...............................................212
11.1.1 Förberedelse/spolning av bifurkerad huvudstomme .......212
11.1.2 Förberedning/spolning av kontralateralt iliakaliskt
graftben ........................................................ 212
11.1.3 Förberedning/spolning av ipsilateralt iliakaliskt graftben .. 212
11.1.4 Kärlåtkomst och angiografi ............................... 212
11.1.5 Placering av huvudstomme ............................... 212
11.1.6 Placering av den kontralaterala iliakaliska ledaren ......... 212
11.1.7
Proximal (hög) utplacering av huvudstomme
11.1.8
Placering och utplacering av det kontralaterala iliakaliska
graftbenet
.........................................................213
11.1.9 Distal (botten) insättning av huvudstomme ................
11.1.10 Inkoppling av det övre skyddet ...........................
11.1.11
Placering och utplacering av det ipsilaterala iliakaliska
graftbenet
.......................................................
11.1.12 Införande av formningsballong ...........................
Slutligt angiogram ........................................
12 RIKTLINJER FÖR BILDFRAMSTÄLLNING OCH POSTOPERATIV
UPPFÖLJNING. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213
12.1 Allmänt ..........................................................
Tabell 12.1 Rekommenderat bildtagningsschema för patienter
med endovaskulära implantat ................................... 214
12.2 Rekommendationer avseende DT med och utan kontrast ........ 214
Tabell 12.2 Godkända bildframställningsprotokoll ............... 214
12.3 Abdominala röntgenbilder ...................................... 214
12.4 Ultraljud ........................................................ 214
12.5 MRT-säkerhet och -kompabilitet ................................. 214
12.6 Ytterligare övervakning och behandling ......................... 214
13 PATIENTSPÅRNINGSINFORMATION ................................ 214
14 FELSÖKNING ........................................................214
14.1 Felsökning av frigöring av utlösningsstråden .......................
14.1.1 Proximal (hög) utplacering av huvudstomme ................
14.1.2 Inkoppling av det övre skyddet ..............................215
14.1.3 Placering och utplacering av det ipsilaterala iliakaliska
graftbenet .........................................................215
14.1.4 Placering och utplacering av det kontralaterala iliakaliska
graftbenet .........................................................215
14.2 Felsökning av utplacering av den suprarenala stenten. . . . . . . . . . . . . . 215
14.2.1 Proximal (hög) utplacering av huvudstomme ................215
14.2.2 Inkoppling av det övre skyddet ..............................215
14.2.3 Placering och utplacering av det ipsilaterala iliakaliska
graftbenet .........................................................215
14.2.4 Placering och utplacering av det kontralaterala iliakaliska
graftbenet .........................................................215
.............. 212
203
203
204
204
204
213
213
213
213
213
213
214
214
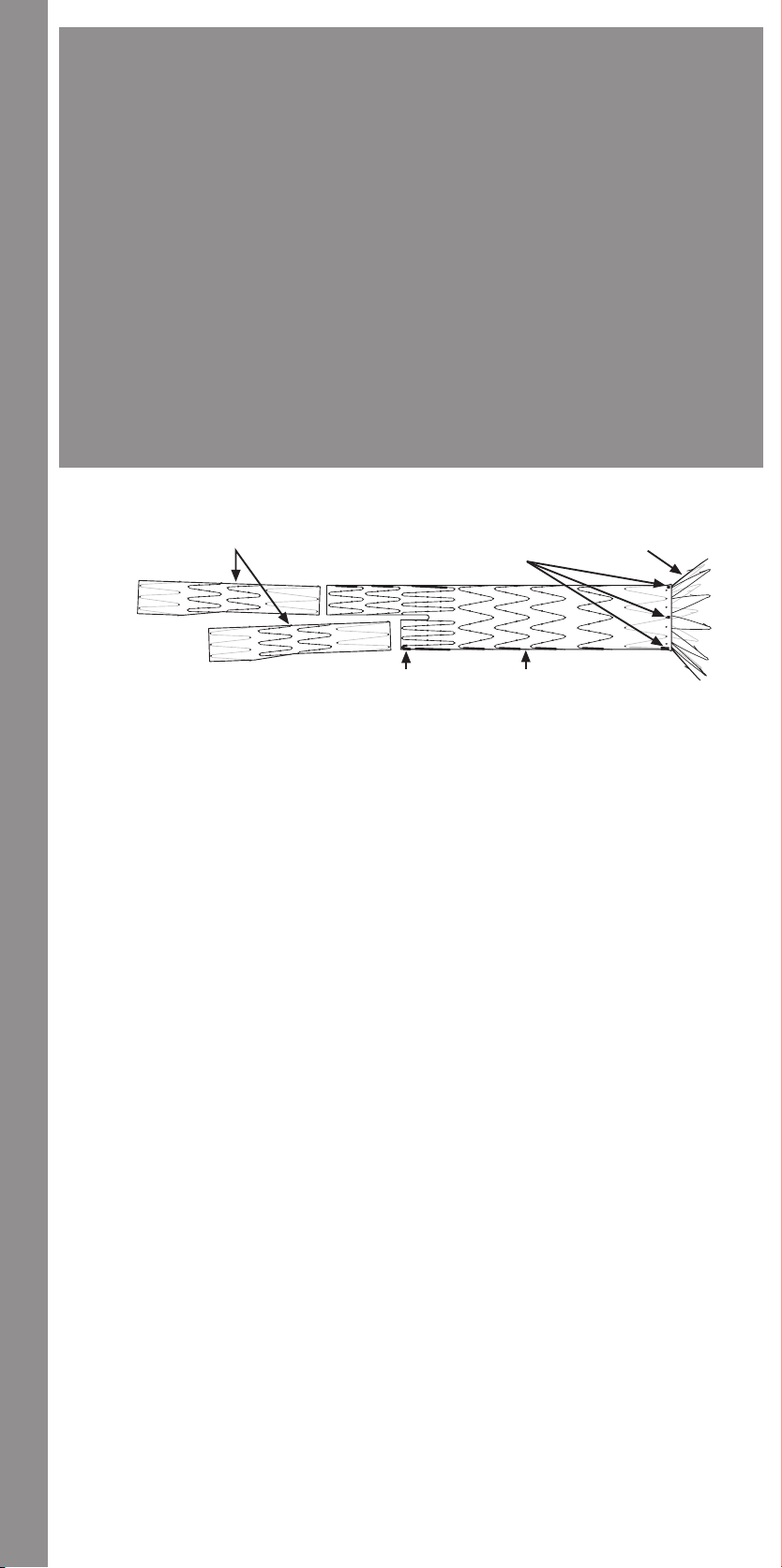
Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer
NOTE: The images in this section do not necessarily depict the actual sequence of procedures required to deploy this device. Please be
sure to reference the step-by-step instructions listed in Section 11 DIRECTIONS FOR USE and Section 14 TROUBLESHOOTING.
• POZNÁMKA:
• BEMÆRK:
af dette produkt. Sørg for at henvise til trin-for-trin anvisningerne angivet i Afsnit 11 VEJLEDNING og Afsnit 14 FEJLSØGNING.
• HINWEIS:
παρουσιάζονται σε αυτή την ενότητα δεν απεικονίζουν απαραίτητα την πραγματική αλληλουχία των διαδικασιών που απαιτούνται
ΧΡΗΣΗΣ» και στην ενότητα 14 «ΑΝΤΙΜΕΤΩΠΙΣΗ ΠΡΟΒΛΗΜΑΤΩΝ».
ilustran la secuencia real de los procedimientos requeridos para desplegar este dispositivo. Asegúrese de consultar las instrucciones
paso a paso indicadas en el apartado 11, MODO DE EMPLEO, y el apartado 14, SOLUCIÓN DE PROBLEMAS.
dispositif. Veiller à consulter les instructions étape par étape indiquées dans la Section 11 DIRECTIVES D’UTILISATION et la Section
14 DÉPANNAGE. • MEGJEGYZÉS:
tényleges sorrendjét ábrázolják. Mindenképpen tekintse át a „11., HASZNÁLATI UTASÍTÁS” és a „14., HIBAKERESÉS” c. szakasz
lépésenkénti utasításait.
richieste per il rilascio di questo dispositivo. Fare riferimento alle istruzioni passo-passo elencate nella Sezione 11 ISTRUZIONI PER
L’USO e nella Sezione 14 GUIDA ALLA RISOLUZIONE DEI PROBLEMI.
feitelijke volgorde van de procedures weer die nodig zijn om dit hulpmiddel te ontplooien. Raadpleeg de stapsgewijze instructies in
hoofdstuk 11, GEBRUIKSAANWIJZING, en hoofdstuk 14, OPLOSSEN VAN PROBLEMEN.
ikke nødvendigvis den faktiske sekvensen i prosedyrene som kreves for å anlegge denne anordningen. Sørg for å se trinn for trinn-
instruksjonene i avsnitt 11, BRUKSVEILEDNING, og avsnitt 14, FEILSØKING.
przedstawiają rzeczywistą kolejność procedur wymaganych do rozprężenia tego urządzenia. Należy koniecznie sprawdzić kolejne
instrukcje wyszczególnione w rozdziale 11, WSKAZANIA, i rozdziale 14, ROZWIĄZYWANIE PROBLEMÓW.
Certifique-se de que consulta as instruções passo a passo na Secção 11 INSTRUÇÕES DE UTILIZAÇÃO e na Secção 14 RESOLUÇÃO
DE PROBLEMAS.
anordningen. Använd de stegvisa anvisningarna i avsnitt 11, BRUKSANVISNING, och avsnitt 14, FELSÖKNING, som referens.
Obrázky v této části nemusí nezbytně zobrazovat skutečné pořadí postupů vyžadovaných k rozvinutí tohoto zařízení.
Prostudujte si laskavě podrobné pokyny uvedené v části 11 POKYNY K POUŽITÍ a v části 14 ŘEŠENÍ PROBLÉMŮ.
Billederne i dette afsnit afbilder ikke nødvendigvis den faktiske rækkefølge af de påkrævede procedurer til anlæggelse
Die Bilder in diesem Abschnitt stellen nicht notwendigerweise die tatsächliche Reihenfolge der Verfahrensschritte
dar, die zur Entfaltung dieser Vorrichtung erforderlich sind. Die in Abschnitt 11 „Gebrauchsanweisung“ und in Abschnitt
14 „Fehlerbehebung“ aufgeführten Schritt-für-Schritt-Anleitungen müssen beachtet werden.
για την απελευθέρωση αυτής της συσκευής. Φροντίστε να ανατρέχετε στις οδηγίες βήμα-βήμα στην ενότητα 11 «ΟΔΗΓΙΕΣ
• NOTA:
Las imágenes de este apartado no necesariamente
• ΣΗΜΕΙΩΣΗ:
Les images dans cette section ne représentent pas nécessairement la séquence réelle des procédures requises pour déployer ce
A jelen szakaszban szereplő képek nem feltétlenül az eszköz kinyitásához szükséges eljárások
• NOTA -
Le immagini in questa sezione non illustrano necessariamente la sequenza effettiva delle procedure
• NB:
De afbeeldingen in dit hoofdstuk geven niet per se de
• MERKNAD:
• UWAGA:
Ilustracje w tym rozdziale niekoniecznie
Bildene i dette avsnittet viser
desta secção não ilustram necessariamente a sequência real de procedimentos necessários para a expansão do dispositivo.
• OBS!
Bilderna i detta avsnitt återger inte nödvändigtvis den faktiska procedursekvens som krävs för att utplacera
Οι εικόνες που
• REMARQUE :
• NOTA:
As imagens
1
1. Iliac Legs
2. Gold Radiopaque Markers (4)
3. Suprarenal Stent
4. Main Body
5. Gold Radiopaque Marker
1. Iliakální ramena
2. Zlaté rentgenkontrastní značky (4)
3. Suprarenální stent
4. Hlavní tělo
5. Zlatá rentgenkontrastní značka
1. Iliaca-ben
2. Røntgenfaste guldmarkører (4)
3. Suprarenal stent
4. Hovedprotese
5. Røntgenfast guldmarkør
1. Iliakale Schenkel
2. Röntgendichte Goldmarkierungen (4)
3. Suprarenaler Stent
4. Hauptteil
5. Röntgendichte Goldmarkierung
1. Λαγόνια σκέλη
2. Χρυσοί ακτινοσκιεροί δείκτες (4)
3. Επινεφρική ενδοπρόσθεση
4. Κύριο σώμα
5. Χρυσός ακτινοσκιερός δείκτης
5
1. Ramas ilíacas
2. Marcadores radiopacos de oro (4)
3. Stent suprarrenal
4. Cuerpo principal
5. Marcador radiopaco de oro
1. Branches iliaques
2. Marqueurs radio-opaques en or (4)
3. Stent suprarénal
4. Corps principal
5. Marqueur radio-opaque en or
1. Iliaca-szárak
2. Arany sugárfogó markerek (4)
3. Suprarenális sztent
4. Fő graft-törzs
5. Arany sugárfogó marker
1. Branche iliache
2. Marker radiopachi d’oro (4)
3. Stent soprarenale
4. Corpo principale
5. Marker radiopaco d’oro
1. Iliacale poten
2. Gouden radiopake markeringen (4)
3. Suprarenale stent
4. Main body
5. Gouden radiopake markering
2
3
4
1. Iliaca-ben
2. Radioopake gullmarkører (4)
3. Suprarenal stent
4. Hoveddel
5. Radioopak gullmarkør
1. Odnogi biodrowe
2. Złote znaczniki cieniodajne (4)
3. Stent nadnerkowy
4. Główny trzon
5. Złoty znacznik cieniodajny
1. Extremidades ilíacas
2. Marcadores de ouro radiopacos (4)
3. Stent supra-renal
4. Corpo principal
5. Marcador de ouro radiopaco
1. Iliakaliskt graftben
2. Röntgentäta markeringar av guld (4)
3. Suprarenal stent
4. Huvudstomme
5. Röntgentät markering av guld
Fig. 1
10

5
6
3
1
2
4
7
14
1. Hub
2. Pin Vise
3. Safety Locks
4. Peel-Away® Sheath
5. Stopcock
6. Connecting Tube
7. Flexor® Introducer Sheath
8. Dilator Tip
9. Sheath Sideport
10. Captor® Hemostatic Valve
11. Gray Positioner
12. White Trigger-Wire Release Mechanism
13. Black Trigger-Wire Release Mechanism
14. Top Cap Inner Cannula
15. Endovascular Graft
1. Ústí
2. Svěrka
3. Pojistky
4. Sheath Peel-Away®
5. Uzavírací kohout
6. Spojovací hadička
7. Zaváděcí sheath Flexor®
8. Hrot dilatátoru
9. Boční port sheathu
10. Hemostatický ventil Captor®
11. Šedý polohovač
12. Bílá spoušť uvolňovacího drátu
13. Černá spoušť uvolňovacího drátu
14. Vnitřní kanyla horní čepičky
15. Endovaskulární graft
1. Muffe
2. Pin vise
3. Sikkerhedslåse
4. Peel-Away® sheath
5. Hane
6. Tilslutningsslange
7. Flexor® indføringssheath
8. Dilatatorspids
9. Sheath sideport
10. Captor® hæmostatisk ventil
11. Grå positioneringsanordning
12. Hvid udløsningsmekanisme for udløser-wire
13. Sort udløsningsmekanisme for udløser-wire
14. Tophættens indre kanyle
15. Endovaskulær protese
1. Ansatz
2. Klemmschraube
3. Sicherheitssperren
4. Peel-Away®-Schleuse
5. Absperrhahn
6. Verbindungsschlauch
7. Flexor®-Einführschleuse
8. Dilatatorspitze
9. Seitenport der Schleuse
10. Captor®-Hämostaseventil
11. Grauer Positionierer
12. Weißer Auslösedrahtmechanismus
13. Schwarzer Auslösedrahtmechanismus
14. Innere Kanüle der oberen Kappe
15. Endovaskuläre Prothese
1. Ομφαλός
2. Μέγγενη ακίδας
3. Ασφάλειες
4. Θηκάρι Peel-Away®
5. Στρόφιγγα
6. Σωλήνας σύνδεσης
7. Θηκάρι εισαγωγέα Flexor®
8. Άκρο διαστολέα
9. Πλευρική θύρα θηκαριού
10. Αιμοστατική βαλβίδα Captor®
11. Γκρι διάταξη τοποθέτησης
12. Λευκός μηχανισμός απελευθέρωσης
σύρματος ενεργοποίησης
13. Μαύρος μηχανισμός απελευθέρωσης
σύρματος ενεργοποίησης
14. Εσωτερική κάνουλα άνω πώματος
15. Ενδαγγειακό μόσχευμα
13
11
10
12
1. Conector
2. Manguito
3. Seguros
4. Vaina Peel-Away®
5. Llave de paso
6. Tubo conector
7. Vaina introductora Flexor®
8. Punta del dilatador
9. Orificio lateral de la vaina
10. Válvula hemostática Captor®
11. Posicionador gris
12. Mecanismo de liberación blanco mediante
alambre disparador
13. Mecanismo de liberación negro mediante
alambre disparador
14. Cánula interior de la cápsula superior
15. Endoprótesis vascular
1. Embase
2. Vis à broche
3. Verrous de sécurité
4. Gaine Peel-Away®
5. Robinet
6. Tube connecteur
7. Gaine d’introduction Flexor®
8. Extrémité du dilatateur
9. Orifice latéral de la gaine
10. Valve hémostatique Captor®
11. Positionneur gris
12. Mécanisme de largage blanc du guide de
déclenchement
13. Mécanisme de largage noir du guide de
déclenchement
14. Canule interne du capuchon supérieur
15. Endoprothèse vasculaire
1. Kónusz
2. Rögzítőelem
3. Biztosítózárak
4. Peel-Away® hüvely
5. Elzárócsap
6. Összekötőcső
7. Flexor® bevezetőhüvely
8. Dilatátor csúcsa
9. Hüvely oldalnyílása
10. Captor® vérzéscsillapító szelep
11. Szürke pozícionáló
12. Fehér elsütődrót-kioldószerkezet
13. Fekete elsütődrót-kioldószerkezet
14. Csúcsi sapka belső kanülje
15. Endovaszkuláris graft
1. Connettore
2. Morsetto
3. Blocchi di sicurezza
4. Guaina Peel-Away®
5. Rubinetto
6. Cannula di collegamento
7. Guaina di introduzione Flexor®
8. Punta del dilatatore
9. Foro laterale della guaina
10. Valvola emostatica Captor®
11. Posizionatore grigio
12. Meccanismo di rilascio bianco a filo di
sicurezza
13. Meccanismo di rilascio nero a filo di sicurezza
14. Cannula interna della calotta superiore
15. Endoprotesi addominale
1. Aanzetstuk
2. Borgschroef
3. Veiligheidsvergrendelingen
4. Peel-Away® sheath
5. Afsluitkraan
6. Verbindingsslang
7. Flexor® introducer sheath
8. Dilatatortip
9. Zijpoort voor sheath
10. Captor® hemostatische klep
11. Grijze pusher
12. Wit mechanisme met trigger wire
13. Zwart vrijgavemechanisme met trigger wire
14. Binnenste canule van topkap
15. Endovasculaire prothese
9
15
1. Kanylefeste
2. Klemmeskrue
3. Sikkerhetslåser
4. Peel-Away® hylse
5. Stoppekran
6. Tilkoblingsslange
7. Flexor® innføringshylse
8. Dilatorspiss
9. Hylsens sidepor t
10. Captor® hemostaseventil
11. Grå posisjoneringsenhet
12. Hvit vaierutløsermekanisme
13. Svart vaierutløsermekanisme
14. Topphettens indre kanyle
15. Endovaskulært implantat
1. Złączka
2. Imadło sztyftowe
3. Blokady
4. Koszulka Peel-Away®
5. Kranik odcinający
6. Rurka łącząca
7. Koszulka wprowadzająca Flexor®
8. Końcówka rozszerzadła
9. Otwór boczny koszulki
10. Zastawka hemostatyczna Captor®
11. Szary pozycjoner
12. Biały mechanizm zwalniający drut
13. Czarny mechanizm zwalniający drut
14. Nasadka końcówki kaniuli wewnętrznej
15. Stent-graft wewnątrznaczyniowy
1. Conector
2. Pino de fixação
3. Dispositivos de segurança
4. Bainha Peel-Away®
5. Torneira de passagem
6. Tubo de ligação
7. Bainha introdutora Flexor®
8. Ponta dilatadora
9. Orifício lateral da bainha
10. Válvula hemostática Captor®
11. Posicionador cinzento
12. Mecanismo de libertação branco com fio de
comando
13. Mecanismo de libertação preto com fio de
comando
14. Cânula interior da tampa superior
15. Prótese endovascular
1. Fattning
2. Pin vise
3. Säkerhetslås
4. Peel-Away®-hylsa
5. Kran
6. Kopplingsslang
7. Flexor® införarhylsa
8. Dilatatorspets
9. Hylsans sidopor t
10. Captor® hemostasventil
11. Grå lägeställare
12. Vit utlösningstråds utlösningsmekanism
13. Svart utlösningstråds utlösningsmekanism
14. Övre skyddets inre kanyl
15. Endovaskulärt transplantat
8
Fig. 2
11
Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer

Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer
4
5
1
2
11
1. Hub
2. Pin Vise
3. Peel-Away® Sheath
4. Stopcock
5. Connecting Tube
6. Sheath
7. Dilator Tip
8. Sheath Sideport
9. Hemostatic Valve
10. Gray Positioner
11. Inner Cannula
12. Contralateral/Ipsilateral Endovascular Graft
1. Ústí
2. Svěrka
3. Sheath Peel-Away®
4. Uzavírací kohout
5. Spojovací hadička
6. Sheath
7. Hrot dilatátoru
8. Boční port sheathu
9. Hemostatický ventil
10. Šedý polohovač
11. Vnitřní kanyla
12. Kontralaterální/ipsilaterální endovaskulární
graft
1. Muffe
2. Pin vise
3. Peel-Away® sheath
4. Hane
5. Tilslutningsslange
6. Sheath
7. Dilatatorspids
8. Sheath sideport
9. Hæmostatisk ventil
10. Grå positioneringsanordning
11. Indre kanyle
12. Kontralateral/ipsilateral endovaskulær
protese
1. Ansatz
2. Klemmschraube
3. Peel-Away®-Schleuse
4. Absperrhahn
5. Verbindungsschlauch
6. Schleuse
7. Dilatatorspitze
8. Seitenport der Schleuse
9. Hämostaseventil
10. Grauer Positionierer
11. Innere Kanüle
12. Kontralaterale/ipsilaterale endovaskuläre
Prothese
1. Ομφαλός
2. Μέγγενη ακίδας
3. Θηκάρι Peel-Away®
4. Στρόφιγγα
5. Σωλήνας σύνδεσης
6. Θηκάρι
7. Άκρο διαστολέα
8. Πλευρική θύρα θηκαριού
9. Αιμοστατική βαλβίδα
10. Γκρι διάταξη τοποθέτησης
11. Εσωτερική κάνουλα
12. Ετερόπλευρο/Σύστοιχο ενδαγγειακό
μόσχευμα
3
10
1. Conector
2. Manguito
3. Vaina Peel-Away®
4. Llave de paso
5. Tubo conector
6. Vaina
7. Punta del dilatador
8. Orificio lateral de la vaina
9. Válvula hemostática
10. Posicionador gris
11. Cánula interior
12. Endoprótesis vascular contralateral/ipsilateral
1. Embase
2. Vis à broche
3. Gaine Peel-Away®
4. Robinet
5. Tube connecteur
6. Gaine
7. Extrémité du dilatateur
8. Orifice latéral de la gaine
9. Valve hémostatique
10. Positionneur gris
11. Canule interne
12. Endoprothèse vasculaire controlatérale/
1. Kónusz
2. Rögzítőelem
3. Peel-Away® hüvely
4. Elzárócsap
5. Összekötőcső
6. Hüvely
7. Dilatátor csúcsa
8. Hüvely oldalnyílása
9. Vérzéscsillapító szelep
10. Szürke pozícionáló
11. Belső kanül
12. Kontralaterális/ipsilaterális endovaszkuláris
1. Connettore
2. Morsetto
3. Guaina Peel-Away®
4. Rubinetto
5. Cannula di collegamento
6. Guaina
7. Punta del dilatatore
8. Foro laterale della guaina
9. Valvola emostatica
10. Posizionatore grigio
11. Cannula interna
12. Branca iliaca controlaterale/ipsilaterale
1. Aanzetstuk
2. Borgschroef
3. Peel-Away® sheath
4. Afsluitkraan
5. Verbindingsslang
6. Sheath
7. Dilatatortip
8. Zijpoort voor sheath
9. Hemostatische klep
10. Grijze pusher
11. Binnenste canule
12. Contralaterale/ipsilaterale endovasculaire
9
homolatérale
graft
dell’endoprotesi addominale
prothese
6
7
8
12
1. Kanylefeste
2. Klemmeskrue
3. Peel-Away® hylse
4. Stoppekran
5. Tilkoblingsslange
6. Hylse
7. Dilatorspiss
8. Hylsens sideport
9. Hemostaseventil
10. Grå posisjoneringsenhet
11. Indre kanyle
12. Kontralateral/ipsilateral endovaskulært
1. Złączka
2. Imadło sztyftowe
3. Koszulka Peel-Away®
4. Kranik odcinający
5. Rurka łącząca
6. Koszulka
7. Końcówka rozszerzadła
8. Otwór boczny koszulki
9. Zastawka hemostatyczna
10. Szary pozycjoner
11. Kaniula wewnętrzna
12. Stent-graft wewnątrznaczyniowy
1. Conector
2. Pino de fixação
3. Bainha Peel-Away®
4. Torneira de passagem
5. Tubo de ligação
6. Bainha
7. Ponta dilatadora
8. Orifício lateral da bainha
9. Válvula hemostática
10. Posicionador cinzento
11. Cânula interna
12. Prótese endovascular contralateral/
1. Fattning
2. Pin vise
3. Peel-Away®-hylsa
4. Kran
5. Kopplingsslang
6. Hylsa
7. Dilatatorspets
8. Hylsans sidoport
9. Hemostasventil
10. Grå lägeställare
11. Inre kanyl
12. Kontralateralt/ipsilateralt endovaskulärt
Fig. 3
implantat
przeciwstronny/tożsamostronny
homolateral
transplantat
1. Converter
2. Iliac Plug
3. Iliac Leg Extension
4. Main Body Extension
1. Přechodový díl
2. Iliakální zátka
3. Extenze iliakálního ramena
4. Extenze hlavního těla
1. Konverteringsenhed
2. Iliaca-prop
3. Iliaca-benforlænger
4. Hovedproteseforlænger
1. Konverter
2. Iliakales Verschluss-Segment
3. Iliakale Schenkelverlängerung
4. Verlängerung des Hauptteils
1. Μετατροπέας
2. Λαγόνιο βύσμα
3. Προέκταση λαγόνιου σκέλους
4. Προέκταση κύριου σώματος
1
2
3
4
Fig. 4
1. Convertidor
2. Tapón ilíaco
3. Extensión de rama ilíaca
4. Extensión de cuerpo principal
1. Convertisseur
2. Obturateur iliaque
3. Extension de branche iliaque
4. Extension de corps principal
1. Konverter
2. Iliaca-dugó
3. Iliaca-szár toldaléka
4. Fő graft-törzs toldaléka
1. Convertitore
2. Dispositivo di occlusione iliaca
3. Estensione della branca iliaca
4. Estensione del corpo principale
1. Converteerder
2. Iliacale plug
3. Verlengstuk voor iliacale poot
4. Verlengstuk voor main body
12
1. Konverteringsenhet
2. Iliaca-propp
3. Iliaca-benforlengelse
4. Hoveddelforlengelse
1. Konwerter
2. Wtyk biodrowy
3. Przedłużenie odnogi biodrowej
4. Przedłużenie głównego trzonu
1. Conversor
2. Tampão ilíaco
3. Extensão da extremidade ilíaca
4. Extensão do corpo principal
1. Konverterare
2. Iliakalisk plugg
3. Förlängning för iliakaliskt graftben
4. Förlängning för huvudstomme
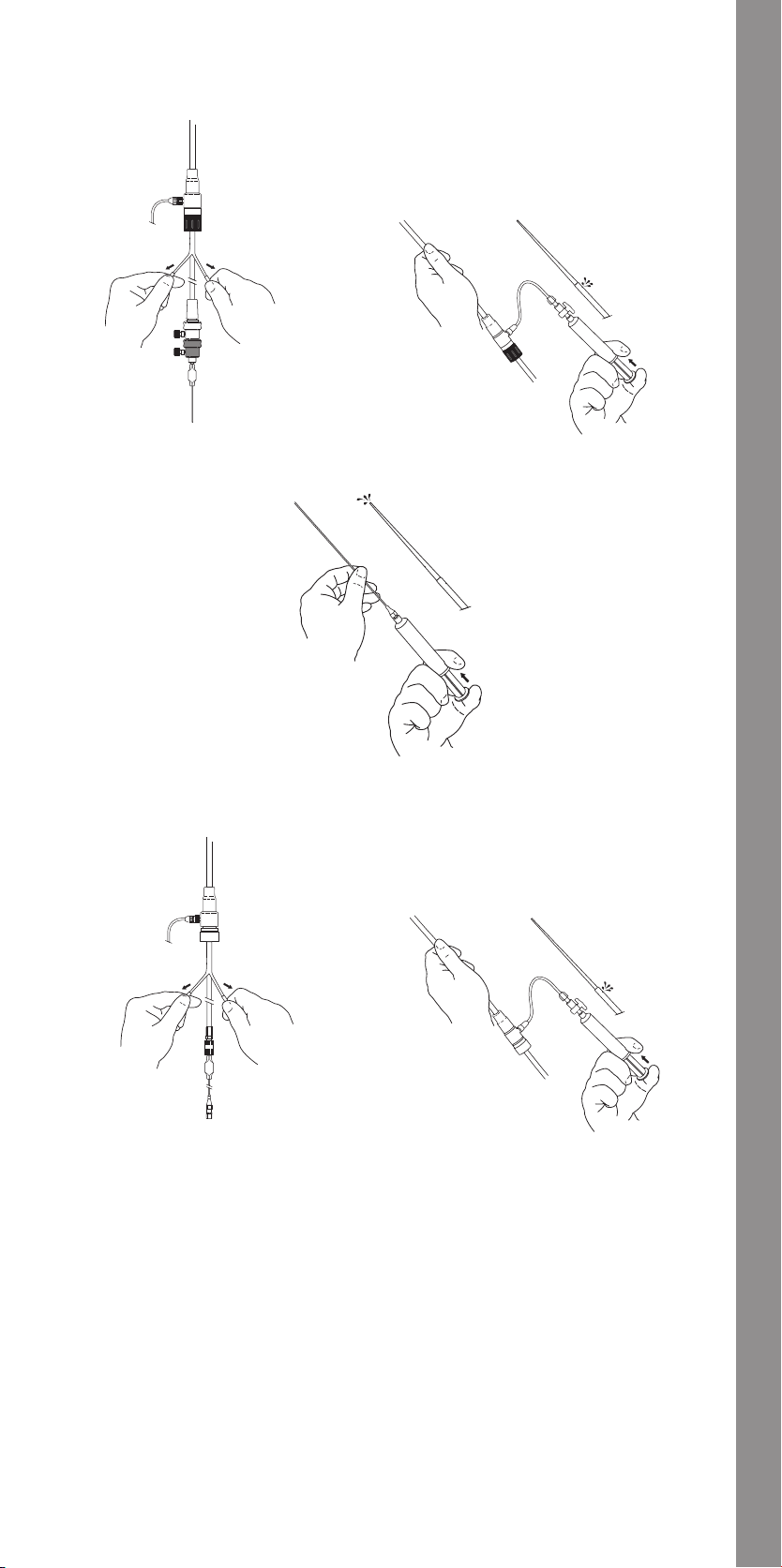
Fig. 5
Fig. 6
Fig. 7
Fig. 8
Fig. 9
13
Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer
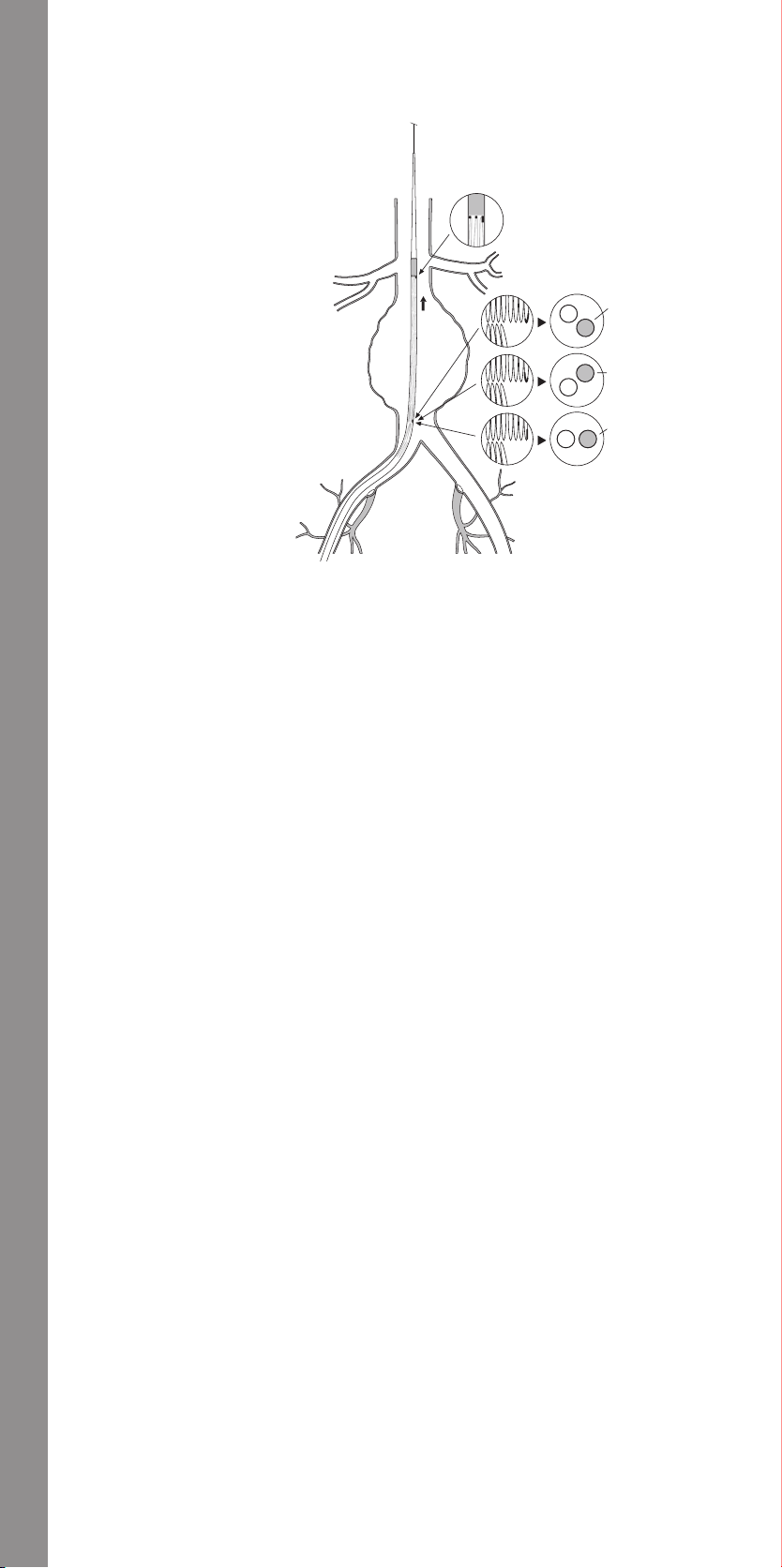
Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer
1
1. Small radiopaque markers provide top
(proximal) orientation of graft material. Long
radiopaque marker aligns with contralateral
limb.
2. Fluoroscopic Image
3. Top View Cross-Section
4. Anterior Contralateral Limb Orientation
5. Posterior Contralateral Limb Orientation
6. Lateral Contralateral Limb Orientation
7. Graft Positioning - Right Side Introduction
1. Malé rentgenkontrastní značky zajišťují
horní (proximální) orientaci materiálu graftu.
Dlouhé rentgenkontrastní značky jsou na
úrovni kontralaterální větve.
2. Rentgenový snímek
3. Průřez v pohledu shora
4. Přední orientace kontralaterální větve.
5. Zadní orientace kontralaterální větve.
6. Laterální orientace kontralaterální větve.
7. Polohování graftu – Zavedení z pravé strany
1. Små røntgenfaste markører giver top
(proksimal) orientering af protesemateriale.
Lange røntgenfaste markører tilpasser sig
med den kontralaterale kant.
2. Gennemlysningsbillede
3. Tværsnit set fra oven
4. Anterior orientering af kontralateral kant
5. Posterior orientering af kontralateral kant
6. Lateral orientering af kontralateral kant
7. Positionering af protese - Indføring i højre
side
1. Die kurzen röntgendichten Markierungen
geben die obere (proximale) Ausrichtung
des Prothesenmaterials an. Die lange
röntgendichte Markierung bildet eine Linie
mit dem kontralateralen Ansatz.
2. Fluoroskopische Ansicht
3. Aufsicht Querschnitt
4. Anteriore Ausrichtung des kontralateralen
Ansatzes
5. Posteriore Ausrichtung des kontralateralen
Ansatzes
6. Laterale Ausrichtung des kontralateralen
Ansatzes
7. Positionierung der Prothese - Einführung
von rechts
1. Μικροί ακτινοσκιεροί δείκτες παρέχουν
άνω (εγγύς) προσανατολισμό του υλικού
μοσχεύματος. Ο μακρός ακτινοσκιερός
δείκτης ευθυγραμμίζεται με το ετερόπλευρο
μέλος.
2. Ακτινοσκοπική εικόνα
3. Εγκάρσια διατομή άνω όψης
4. Πρόσθιος προσανατολισμός ετερόπλευρου
μέλους
5. Οπίσθιος προσανατολισμός ετερόπλευρου
μέλους
6. Πλευρικός προσανατολισμός ετερόπλευρου
μέλους
7. Τοποθέτηση μοσχεύματος - Εισαγωγή στη
δεξιά πλευρά
7
1. Los marcadores radiopacos pequeños
indican la orientación superior (proximal)
del material de la endoprótesis vascular. El
marcador radiopaco largo se alinea con la
ramificación contralateral.
2. Imagen fluoroscópica
3. Vista transversal superior
4. Orientación anterior de la ramificación
contralateral
5. Orientación posterior de la ramificación
contralateral
6. Orientación lateral de la ramificación
contralateral
7. Colocación de la endoprótesis vascular:
introducción por el lado derecho
1. Les petits marqueurs radio-opaques
indiquent l’orientation supérieure proximale
de l’endoprothèse. Le marqueur radio-opaque
long s’aligne avec le membre controlatéral.
2. Image radioscopique
3. Section transversale vue en plan
4. Orientation du membre controlatéral
antérieur
5. Orientation du membre controlatéral
postérieur
6. Orientation du membre controlatéral latéral
7. Positionnement de l’endoprothèse
-Introduction du côté droit
1. A kis sugárfogó markerek lehetővé teszik
a graft anyagának csúcsi (proximális)
orientációját. A hosszú sugárfogó marker a
kontralaterális ággal kerül egy vonalba.
2. Fluoroszkópos kép
3. Felülnézeti keresztmetszet
4. Kontralaterális ág anterior orientációja
5. Kontralaterális ág posterior orientációja
6. Kontralaterális ág lateralis orientációja
7. Graft pozícionálása – jobb oldali felvezetés
1. Piccoli marker radiopachi consentono di
rilevare l’orientamento della parte superiore
(prossimale) del materiale di rivestimento
dell’endoprotesi. Il marker radiopaco lungo si
allinea con l’estremità controlaterale.
2. Immagine fluoroscopica
3. Vista superiore, sezione trasversale
4. Orientamento anteriore dell’estremità
controlaterale
5. Orientamento posteriore dell’estremità
controlaterale
6. Orientamento laterale dell’estremità
controlaterale
7. Posizionamento dell’endoprotesi -
Inserimento dal lato destro
1. Kleine radiopake markeringen worden
gebruikt voor oriëntatie van de (proximale)
bovenkant van het prothesemateriaal. De
lange radiopake markering ligt in één lijn
met de contralaterale stomp.
2. Fluoroscopisch beeld
3. Bovenaanzicht dwarsdoorsnede
4. Anterieure oriëntatie contralaterale stomp
5. Posterieure oriëntatie contralaterale stomp
6. Laterale oriëntatie contralaterale stomp
7. Positioneren van de prothese - Introduceren
via de rechterzijde
2
3
4
5
6
Fig. 10
1. Små radioopake markører gir
toppdel (proksimal)-orientering av
implantatmaterialet. Lang radioopak markør
stilles på linje med kontralateralt lem.
2. Fluoroskopisk bilde
3. Tverrsnitt sett ovenifra
4. Anterior orientering av kontralateralt lem
5. Posterior orientering av kontralateralt lem
6. Lateral orientering av kontralateralt lem
7. Implantatplassering - Innføring høyre side
1. Małe znaczniki cieniodajne zapewniają
identyfikację górnej (proksymalnej) części
tworzywa stent-graftu. Długi znacznik
cieniodajny zrównuje się z odgałęzieniem
przeciwstronnym.
2. Obraz fluoroskopowy
3. Przekrój poprzeczny widziany od góry
4. Ustawienie przednie przeciwstronnego
odgałęzienia
5. Ustawienie tylne przeciwstronnego
odgałęzienia
6. Ustawienie boczne przeciwstronnego
odgałęzienia
7. Ustawianie stent-graftu – wprowadzenie
z prawej strony
1. Pequenos marcadores radiopacos fornecem
a orientação do topo (proximal) do material
da prótese. Marcador radiopaco longo
alinha-se com o ramo contralateral.
2. Imagem fluoroscópica
3. Secção transversal da vista superior
4. Orientação anterior do ramo contralateral
5. Orientação posterior do ramo contralateral
6. Orientação lateral do ramo contralateral
7. Posicionamento da prótese - Introdução do
lado direito
1. Små röntgentäta markeringar
anger toppläget (proximalt) för
transplantationsmaterialet. Den långa
röntgentäta markeringen riktar in sig i linje
med den kontralaterala lemmen.
2. Fluoroskopisk bild
3. Tvärsnitt uppifrån
4. Den främre kontralaterala lemmens
orientering
5. Den bakre kontralaterala lemmens
orientering
6. Den laterala kontralaterala lemmens
orientering
7. Transplantatplacering - Införing från högra
sidan
14
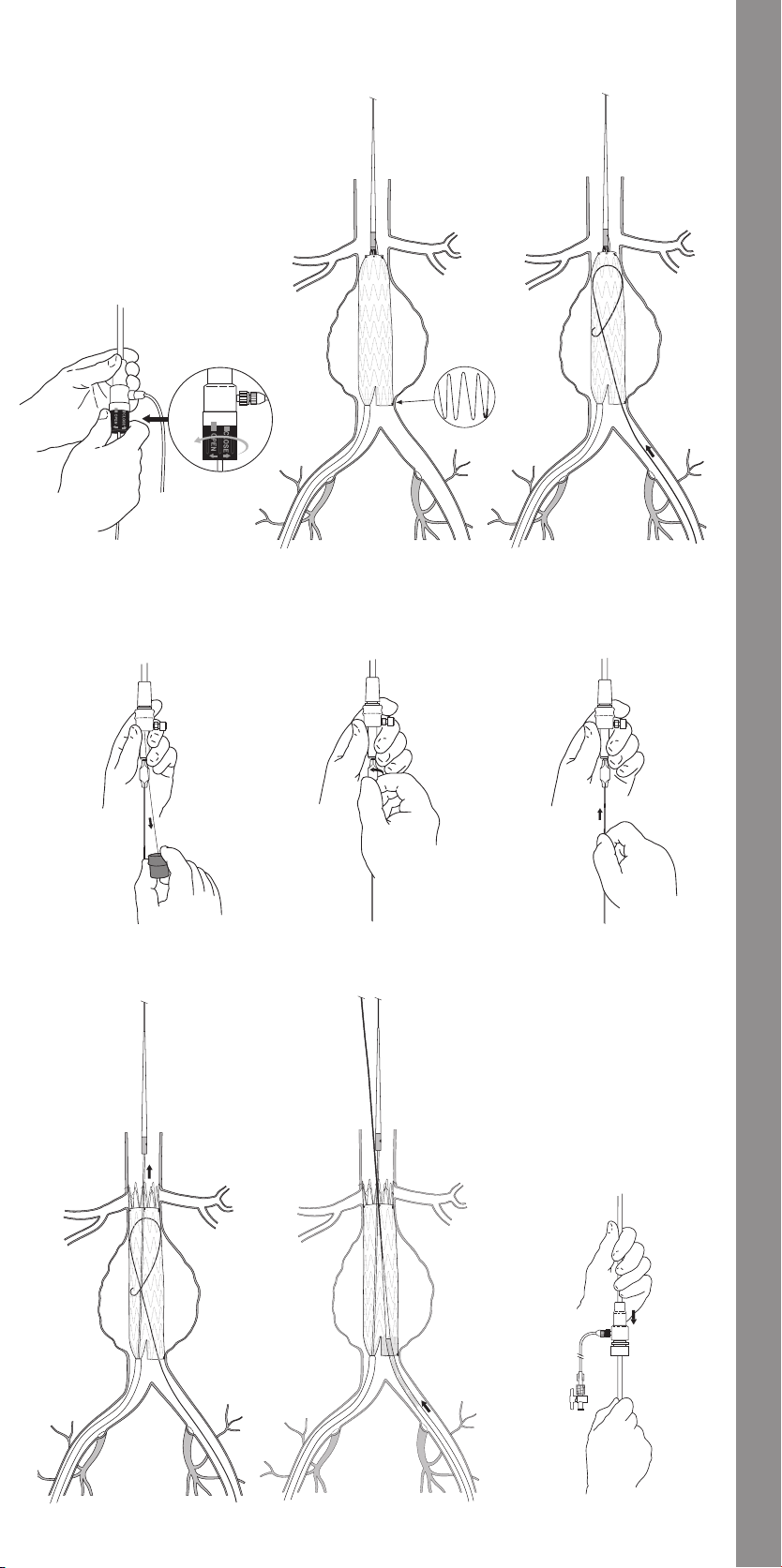
Fig. 11 Fig. 12
Fig. 14 Fig. 15 Fig. 16
Fig. 13
Fig. 17 Fig. 18 Fig. 19
15
Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer
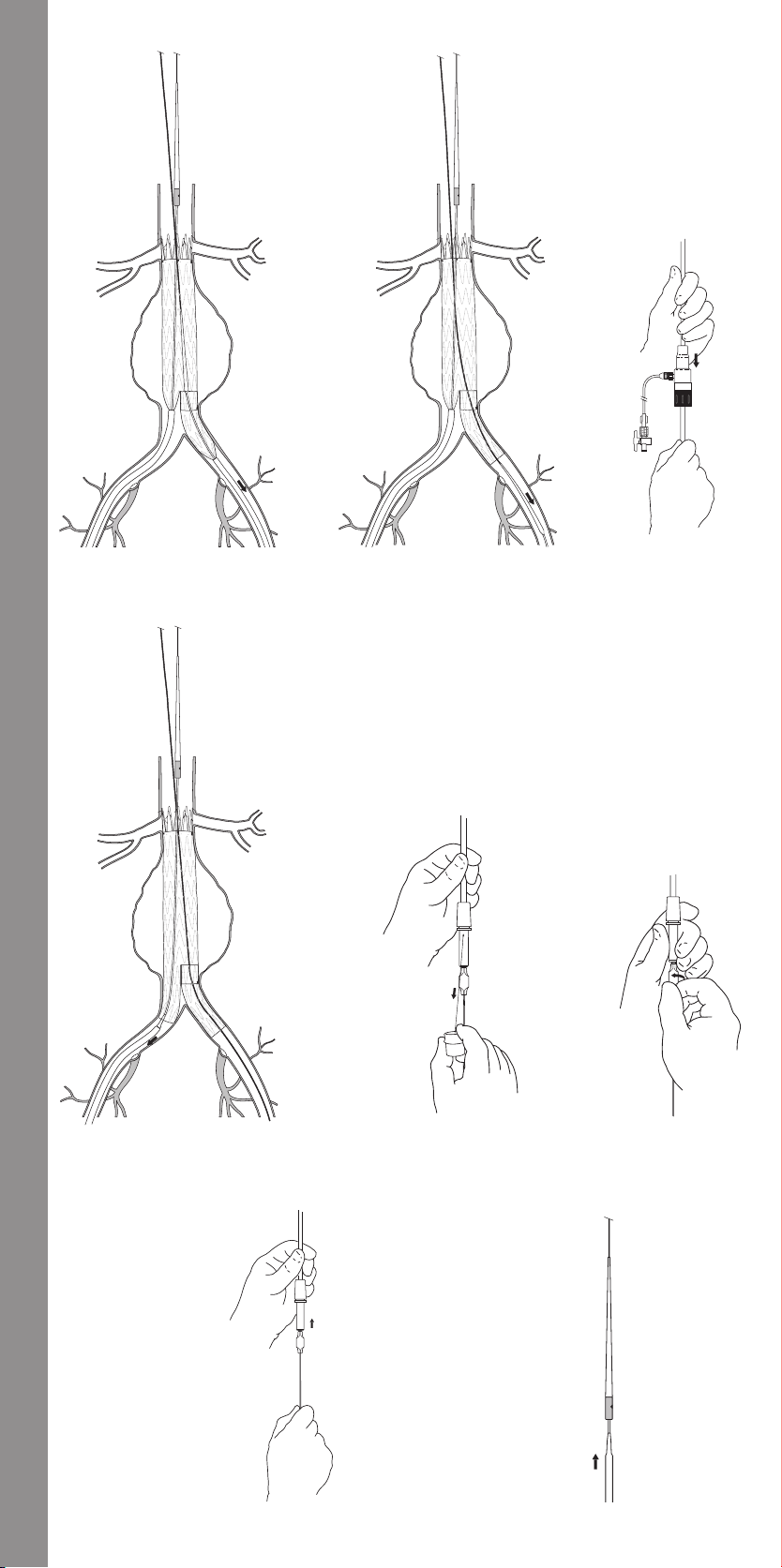
Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer
Fig. 20
Fig. 21 Fig. 22
Fig. 23 Fig. 24 Fig. 25
Fig. 26 Fig. 27
16
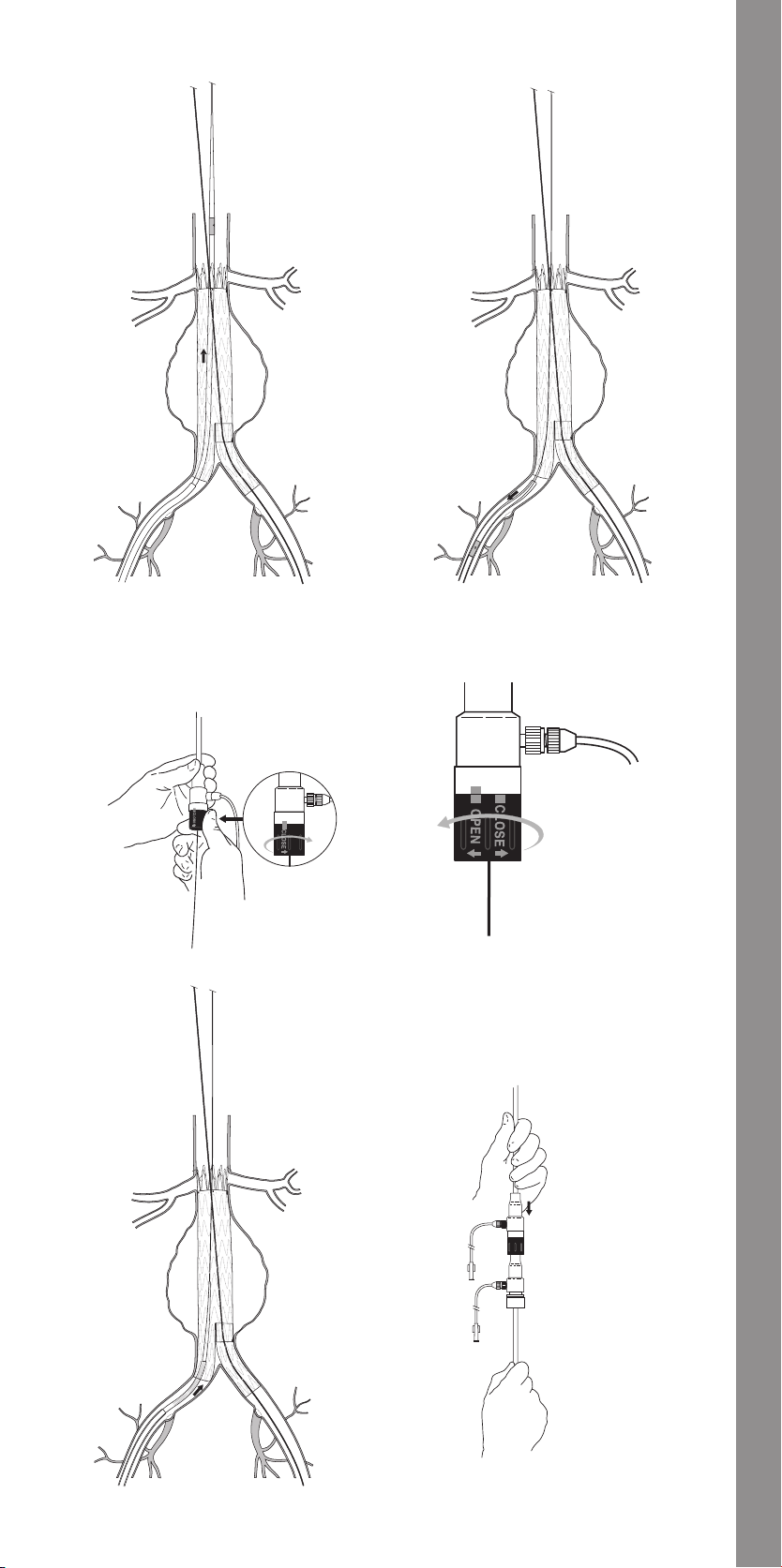
Fig. 28
Fig. 29
Fig. 30
Fig. 32 Fig. 33
Fig. 31
17
Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer
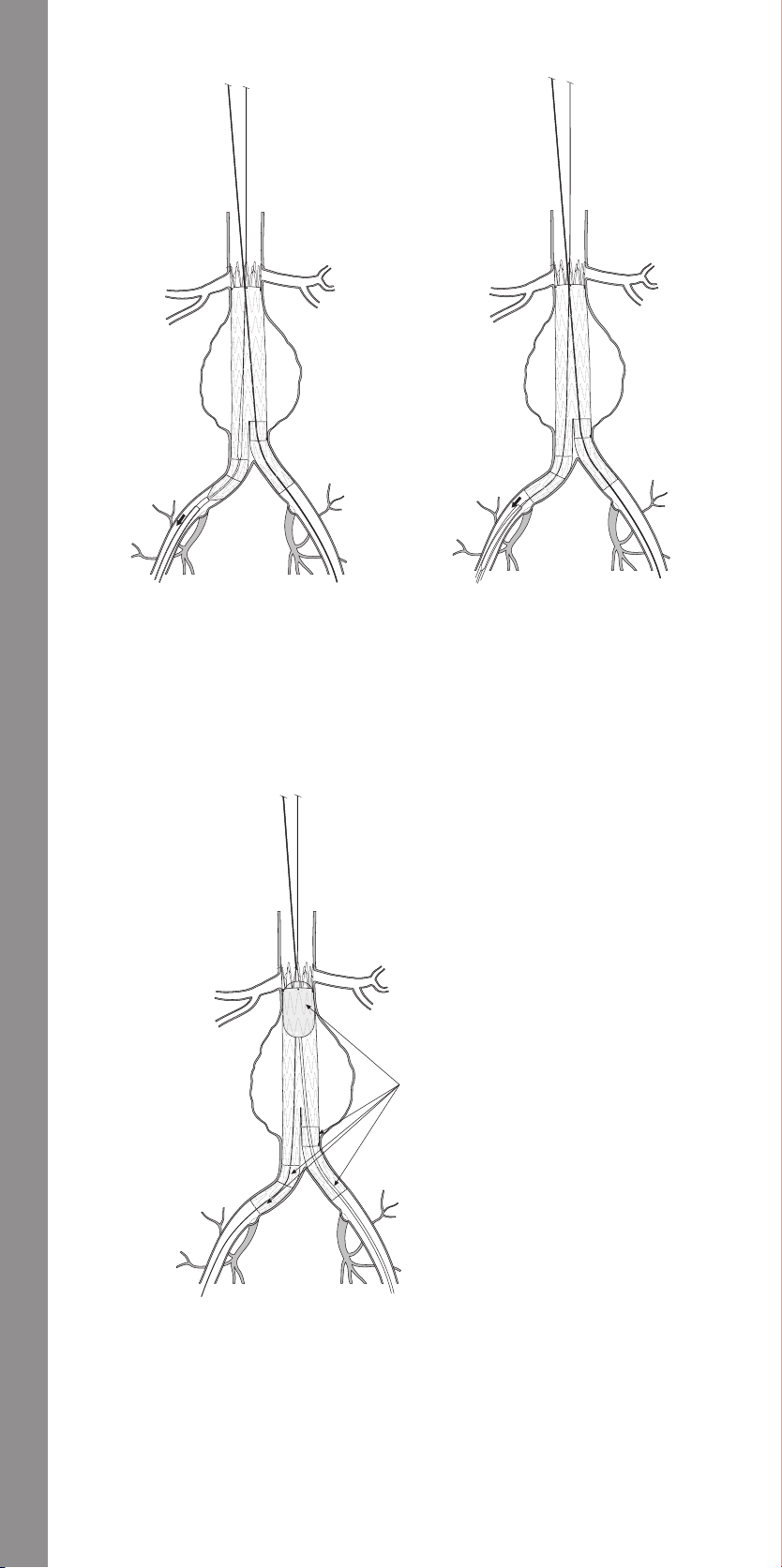
Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer
Fig. 36
Fig. 35Fig. 34
1. Balloon Expansion/Graft Sealing Sites
1
1. Expanze balónku/místa utěsnění graftu
1. Ballonekspansion/
proteseforseglingssteder
1. Ballonafweitungs-/
Prothesendichtungsstellen
1. Θέσεις διαστολής μπαλονιού/
στεγανοποίησης μοσχεύματος
1. Lugares de hinchado del balón y
sellado de la endoprótesis vascular
1. Sites de gonflage du ballonnet et
d’étanchéité de l’endoprothèse
1. Ballon feltöltés/graft tapadási helyei
1. Siti di dilatazione del palloncino/
fissazione dell’endoprotesi
1. Plaatsen voor ballonexpansie/
protheseafdichting
1. Ballongekspansjons-/
implantatforseglingssteder
1. Miejsca rozprężania balonu/
uszczelniania stent-graftu
1. Locais de expansão do balão/selagem
da prótese
1. Ballongexpansion/transplantatets
tätningsställen
18
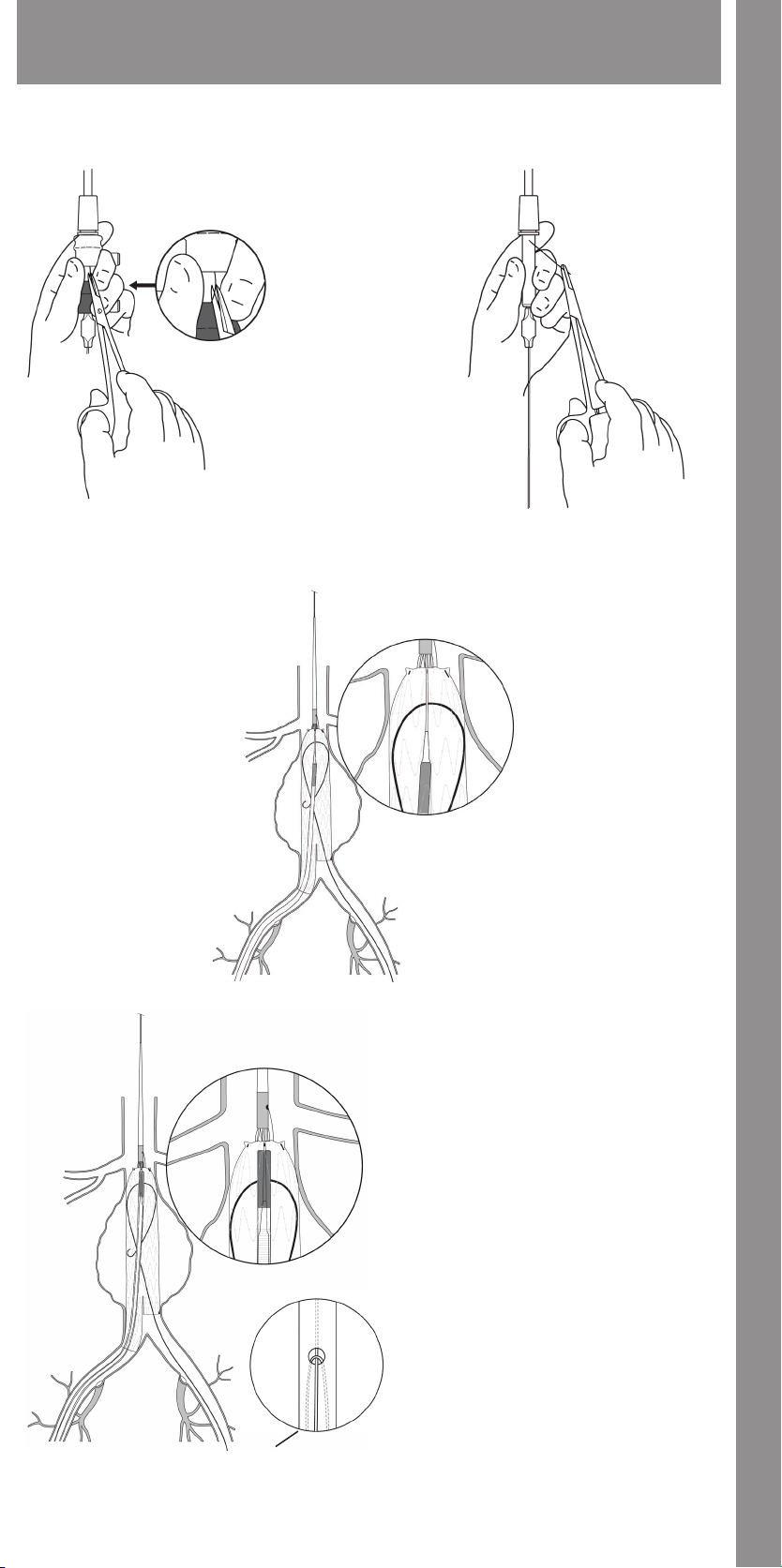
Troubleshooting (Section 14) • Řešení problémů (část 14) • Fejlsøgning (Afsnit 14) • Fehlerbehebung (Abschnitt 14)
• Αντιμετώπιση προβλημάτων (ενότητα 14) • Solución de problemas (apartado 14) • Dépannage (Section 14) • Hibakeresés (14. szakasz)
• Guida alla risoluzione dei problemi (Sezione 14) • Oplossen van problemen (hoofdstuk 14) • Feilsøking (avsnitt 14) • Rozwiązywanie
problemów (rozdział 14) • Resolução de problemas (secção 14) • Felsökning (avsnitt 14)
Fig. 37
Fig. 38
Fig. 39
Fig. 40
1. View of trigger-wire under tension
1. Pohled na napjatý uvolňovací drát
1. Visning af udløser-wiren under spænding
1. Ansicht des Auslösedrahts unter Zugspannung
1. Άποψη του σύρματος ενεργοποίησης υπό τάση
1. Vista del alambre disparador tenso
1. Vue du guide de déclenchement sous tension
1. A megfeszített elsütődrót képe
1. Vista del filo di sicurezza teso
1. Aanzicht van trigger wire onder spanning
1. Visning av stram utløservaier
1. Widok naprężonego drutu zwalniającego
1. Vista do fio de comando sob tensão
1. Bild av sträckt utlösningstråd
1
19
Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer
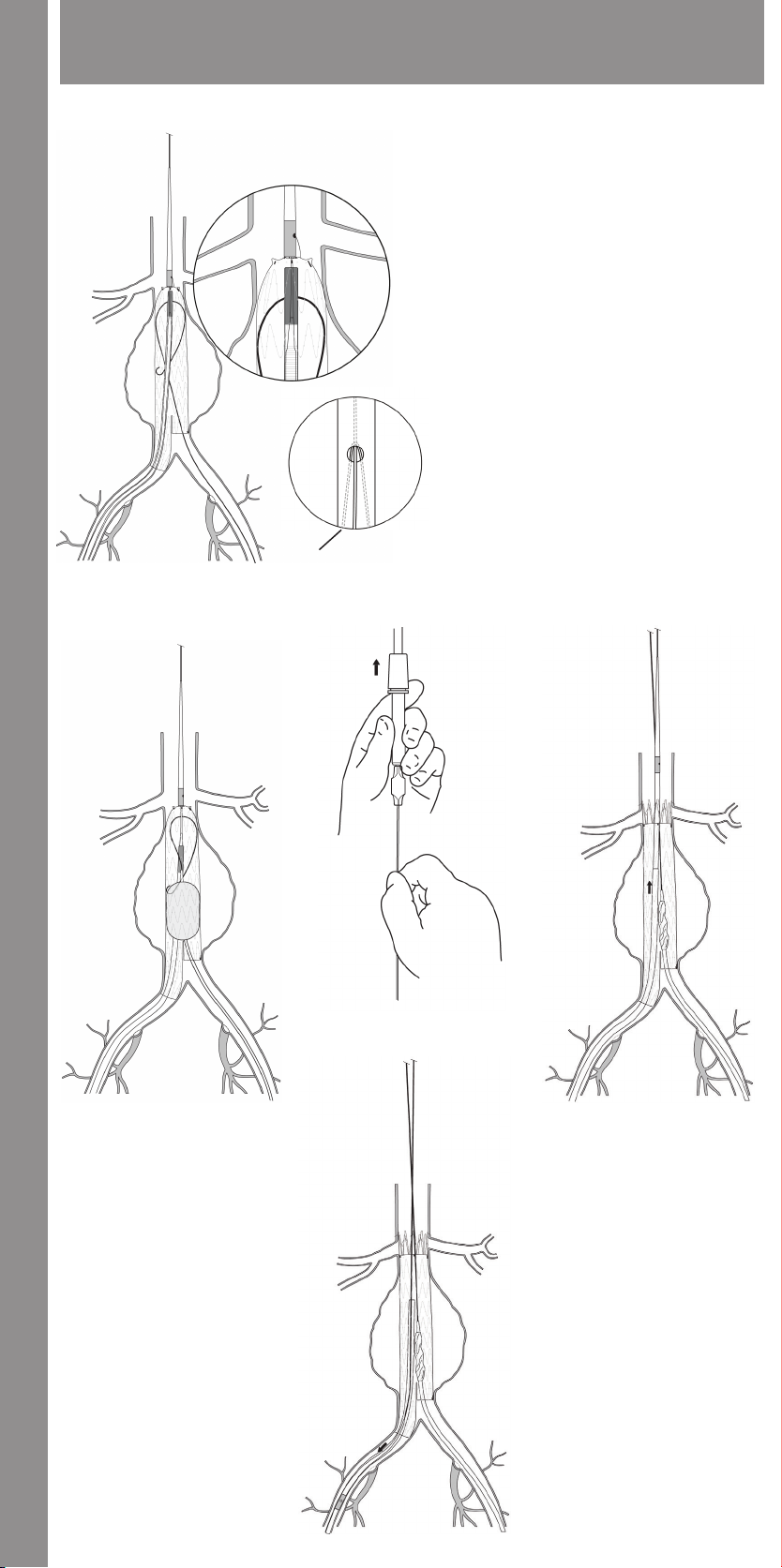
Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer
Troubleshooting (Section 14) • Řešení problémů (část 14) • Fejlsøgning (Afsnit 14) • Fehlerbehebung (Abschnitt 14)
• Αντιμετώπιση προβλημάτων (ενότητα 14) • Solución de problemas (apartado 14) • Dépannage (Section 14) • Hibakeresés (14. szakasz)
• Guida alla risoluzione dei problemi (Sezione 14) • Oplossen van problemen (hoofdstuk 14) • Feilsøking (avsnitt 14) • Rozwiązywanie
problemów (rozdział 14) • Resolução de problemas (secção 14) • Felsökning (avsnitt 14)
1. View of trigger-wire with inner cannula retracted,
releasing tension
1. Pohled na uvolňovací drát s vnitřní kanylou staženou
zpět, což uvolňuje napětí
1. Visning af udløser-wiren med den indre kanyle trukket
tilbage, mens den frigør spænding
1. Ansicht des entspannten Auslösedrahts bei
zurückgezogener innerer Kanüle
1. Άποψη του σύρματος ενεργοποίησης με αποσυρμένη
την εσωτερική κάνουλα, εκτόνωση τάσης
1. Vista del alambre disparador con la cánula interior
retraída, lo que destensa el alambre
1. Vue du guide de déclenchement avec la canule interne
tirée vers l’arrière pour relâcher la tension
1. Az elsütődrót képe a feszítést megszüntető visszahúzott
belső kanüllel
1. Vista del filo di sicurezza con la cannula interna retratta;
il filo è allentato
1. Aanzicht van trigger wire met binnenste canule
teruggetrokken om spanning te verminderen
1. Visning av slakk utløservaier med indre kanyle trukket
tilbake
1. Widok drutu zwalniającego z wycofaną kaniulą
wewnętrzną, przy zwolnionym naprężeniu
1. Vista do fio de comando com cânula interior recuada,
aliviando a tensão
1. Bild av utlösningstråd med tillbakadragen inre kanyl,
Fig. 41
1
vilket ger minskad sträckning
Fig. 42
Fig. 43
Fig. 44
Fig. 45
20
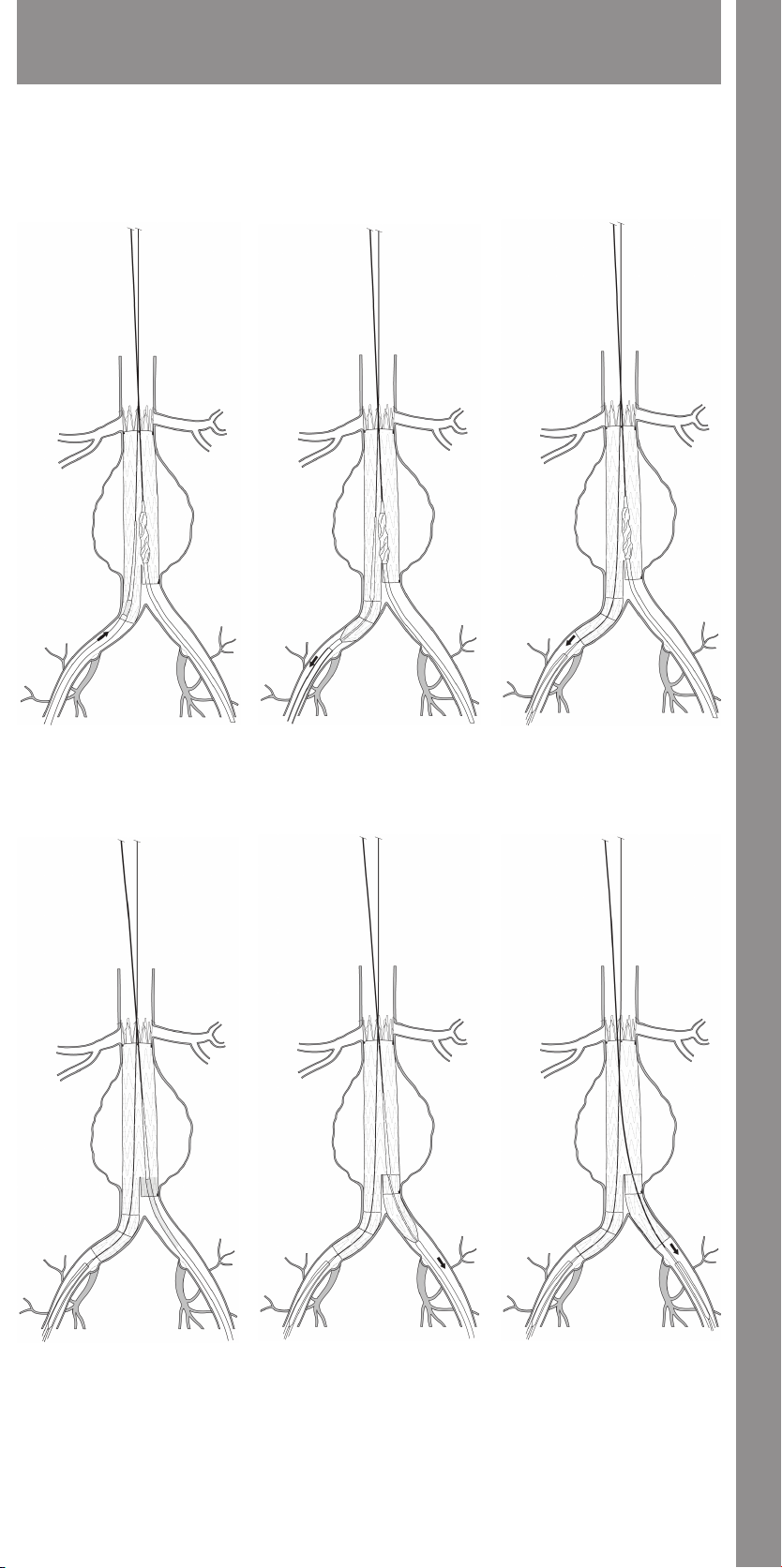
Troubleshooting (Section 14) • Řešení problémů (část 14) • Fejlsøgning (Afsnit 14) • Fehlerbehebung (Abschnitt 14)
• Αντιμετώπιση προβλημάτων (ενότητα 14) • Solución de problemas (apartado 14) • Dépannage (Section 14) • Hibakeresés (14. szakasz)
• Guida alla risoluzione dei problemi (Sezione 14) • Oplossen van problemen (hoofdstuk 14) • Feilsøking (avsnitt 14) • Rozwiązywanie
problemów (rozdział 14) • Resolução de problemas (secção 14) • Felsökning (avsnitt 14)
Fig. 46
Fig. 47
Fig. 48
Fig. 49
Fig. 50
21
Fig. 51
Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer

Illustrations • Ilustrace • Illustrationer • Abbildungen • Απεικονίσεις • Ilustraciones • Illustrations • Illusztrációk • Illustrazioni • Afbeeldingen • Illustrasjoner • Ilustracje • Ilustrações • Illustrationer
Troubleshooting (Section 14) • Řešení problémů (část 14) • Fejlsøgning (Afsnit 14) • Fehlerbehebung (Abschnitt 14)
• Αντιμετώπιση προβλημάτων (ενότητα 14) • Solución de problemas (apartado 14) • Dépannage (Section 14) • Hibakeresés (14. szakasz)
• Guida alla risoluzione dei problemi (Sezione 14) • Oplossen van problemen (hoofdstuk 14) • Feilsøking (avsnitt 14) • Rozwiązywanie
problemów (rozdział 14) • Resolução de problemas (secção 14) • Felsökning (avsnitt 14)
Fig. 52
22

ENGLISH
ZENITH FLEX® AAA ENDOVASCULAR GRAFT WITH THE
H&L-B ONE-SHOT™ INTRODUCTION SYSTEM
Read all instructions carefully. Failure to properly follow the instructions,
warnings and precautions may lead to serious surgical consequences or injury
to the patient.
CAUTION: Federal (U.S.A.) law restricts this device to sale by or on the
order of a physician.
CAUTION: All contents of the outer pouch (including the introduction
system and the endovascular grafts) are supplied sterile, for single use
only.
For the Zenith product line there are four applicable Suggested Instructions
for Use (IFU). This IFU describes the Suggested Instructions for Use for the
Zenith Flex AAA Endovascular Graft (main body and iliac legs). For information
on other Zenith components, please refer to the following Suggested
Instructions for Use:
• Zenith AAA Endovascular Graft (Zenith AAA Endovascular Graft main body
and iliac legs);
• Zenith AAA Endovascular Graft Ancillar y Components (main body
extension, iliac leg extension, converter and iliac plug);
• Zenith® Renu™ AAA Ancillary Graft (main body extension and converter
configurations); and
• CODA® Balloon Catheter.
1 DEVICE DESCRIPTION
1.1 Aortic Main Body and Iliac Leg Components
The Zenith Flex AAA Endovascular Graft is a modular system consisting of
three components, a bifurcated aortic main body and two iliac legs. (Fig. 1)
The graft modules are constructed of full-thickness woven polyester fabric
sewn to self-expanding stainless steel Cook-Z® stents with braided polyester
and monofilament polypropylene suture. The modules are fully stented to
provide stability and the expansile force necessary to open the lumen of
the graft during deployment. Additionally, the Cook-Z stents provide the
necessary attachment and seal of the graft to the vessel wall.
The bare suprarenal stent at the proximal end of the graft contains barbs
that are placed at 3 mm increments for additional fixation of the device. To
facilitate fluoroscopic visualization of the stent graft, gold radiopaque markers
are positioned as follows: one on the lateral aspect of the most distal stent on
the contralate ral limb of the bifurcated section of the main body and four in
a circumferential orientation within 2 mm of the most superior aspect of the
graft material.
1.2 Main Body Delivery System
The Zenith Flex AAA Endovascular Graft main body is shipped preloaded
onto the H&L-B One-Shot Introduction System. (Fig. 2) It has a sequential
deployment method with built-in features to provide continuous control of
the endovascular graft throughout the deployment procedure. The H&L-B
One-Shot Introduction System is designed for precise positioning and allows
readjustment of the final graft position before deployment of the barbed
suprarenal stent.
The main body graft delivery system uses an 18, 20 or 22 French H&L-B OneShot Introduction System. Dual trigger-wire release mechanisms lock the
endovascular graft onto the delivery system until released by the physician. All
systems are compatible with a .035 inch wire guide.
For added hemostasis, the Captor® Hemostatic Valve can be loosened or
tightened for the introduction and/or removal of ancillary devices into and
out of the sheath. The main body delivery systems feature a Flexor® introducer
sheath which resists kinking and is hydrophilically coated. Both features are
intended to enhance trackability in the iliac arteries and abdominal aorta.
1.3 Iliac Leg Delivery System
The Zenith AAA Endovascular Graft iliac legs are shipped preloaded onto the
H&L-B One-Shot Introduction System. (Fig. 3) The delivery system is designed
for ease of use with minimal preparation. The iliac leg delivery system uses
a 14 or 16 French H&L-B One-Shot Introduction System. All systems are
compatible with a .035 inch wire guide.
1.4 Zenith AAA Endovascular Graft Ancillary Components
Additional ancillary endovascular components (main body extensions, iliac
leg extensions, converters and iliac plugs) are available. (Fig. 4) Refer to the
Zenith AAA Endovascular Graft Ancillary Components Instructions for Use for
more information.
2 INDICATIONS FOR USE
The Zenith Flex AAA Endovascular Graft with the H&L-B One-Shot Introduction
System is indicated for the endovascular treatment of patients with abdominal
aortic or aorto-iliac aneurysms having morphology suitable for endovascular
repair, including:
• Adequate iliac/femoral access compatible with the required introduction
systems,
• Non-aneur ysmal infrarenal aortic segment (neck) proximal to the aneurysm:
• with a length of at least 15 mm,
• with a diameter measured outer wall to outer wall of no greater than
32 mm and no less than 18 mm,
• with an angle less than 60 degrees relative to the long axis of the
aneurysm, and
• with an angle less than 45 degrees relative to the axis of the suprarenal
aorta.
• I liac artery distal fixation site greater than 10 mm in length and 7.5-20 mm
in diameter (measured outer wall to outer wall).
3 CONTRAINDICATIONS
There are no known contraindications for these devices.
4 WARNINGS AND PRECAUTIONS
4.1 General
• Read all instructions carefully. Failure to properly follow the instructions,
warnings and precautions may lead to serious consequences or injury to the
patient.
• Always have a qualified surgery team available during implantation or
reintervention procedures in the event that conversion to open surgical
repair is necessary.
• The Zenith Flex AAA Endovascular Graft with the H&L-B One-Shot
Introduction System should only be used by physicians and teams trained in
vascular interventional techniques (catheter-based and surgical) and in the
use of this device. Specific training expectations are described in Section
10.1, Physician Training.
• Additional endovascular interventions or conversion to standard open
surgical repair following initial endovascular repair should be considered for
patients experiencing an enlarging aneurysm, unacceptable decrease in
fixation length (vessel and component overlap) and/or endoleak. An
increase in aneurysm size and/or persistent endoleak or migration may lead
to aneurysm rupture.
• Patients experiencing reduced blood flow through the graft limb and/or
leaks may be required to undergo secondary interventions or surgical
procedures.
4.2 Patient Selection, Treatment and Follow-Up
• The Zenith Flex AAA Endovascular Graft is designed to treat aortic neck
diameters no smaller than 18 mm and no larger than 32 mm. The Zenith
Flex AAA Endovascular Graft is designed to treat proximal aortic necks
(distal to the lowest renal artery) of at least 15 mm in length. Iliac artery
distal fixation site greater than 10 mm in length and 7.5 - 20 mm in diameter
(measured outer wall to outer wall) is required. These sizing measurements
are critical to the performance of the endovascular repair.
• Key anatomical elements that may affect successful exclusion of the
aneurysm include severe proximal neck angulation (>60 degrees for
infrarenal neck to axis of AAA or >45 degrees for suprarenal neck relative to
the immediate infrarenal neck); short proximal aortic neck (<15 mm); an
inverted funnel shape (greater than 10% increase in diameter over 15 mm of
proximal aortic neck length); and circumferential thrombus and/or
calcification at the arterial implantation sites, specifically the proximal aortic
neck and distal iliac artery interface. In the presence of anatomical
limitations, a longer neck may be required to obtain adequate sealing and
fixation. Irregular calcification and/or plaque may compromise the
attachment and sealing at the fixation sites. Necks exhibiting these key
anatomical elements may be more conducive to graft migration or
endoleak.
• Adequate iliac or femoral access is required to introduce the device into the
vasculature. Access vessel diameter (measured inner wall to inner wall) and
morphology (minimal tortuosity, occlusive disease and/or calcification)
should be compatible with vascular access techniques and delivery systems
of a 16 French to 22 French vascular introducer sheath. Vessels that are
significantly calcified, occlusive, tortuous or thrombus-lined may preclude
placement of the endovascular graft and/or may increase the risk of
embolization. A vascular conduit technique may be necessary to achieve
success in some patients.
• The Zenith Flex AAA Endovascular Graft with the H&L-B One-Shot
Introduction System is not recommended in patients who cannot tolerate
contrast agents necessary for intraoperative and postoperative follow-up
imaging. All patients should be monitored closely and checked periodically
for a change in the condition of their disease and the integrity of the
endoprosthesis.
• The Zenith Flex AAA Endovascular Graft with the H&L-B One-Shot
Introduction System is not recommended in patients exceeding weight
and/or size limits which compromise or prevent the necessary imaging
requirements.
• I nability to maintain patency of at least one internal iliac artery or occlusion
of an indispensable inferior mesenteric artery may increase the risk of
pelvic/bowel ischemia.
• Multiple large, patent lumbar arteries, mural thrombus and a patent inferior
mesenteric artery may all predispose a patient to Type II endoleaks. Patients
with uncorrectable coagulopathy may also have an increased risk of Type II
endoleak or bleeding complications.
• The safety and effectiveness of the Zenith Flex AAA Endovascular Graft with
the H&L-B One-Shot Introduction System has not been evaluated in the
following patient populations:
• traumatic aor tic injury
• leak ing, pending rupture or ruptured aneurysms
• mycotic aneurysms
• pseudoaneur ysms resulting from previous graft placement
• revision of previously placed endovascular grafts
• uncorrectable coagulopathy
• indispensable mesenteric ar tery
• genetic connec tive tissue disease (e.g., Marfans or Ehlers-Danlos
Syndromes)
• concomitant thoracic aortic or thoracoabdominal aneur ysms
• patients with active systemic infections
• pregnant or nursing females
• morbidly obese patients
• less than 18 years of age
• patients with less than 15 mm in length or greater than 60 degrees
angulation of the proximal aortic neck relative to the long axis of the
aneurysm.
• Successful patient selection requires specific imaging and accurate
measurements; please see Section 4.3 Pre-Procedure Measurement
Techniques and Imaging.
• All lengths and diameters of the devices necessar y to complete the
procedure should be available to the physician, especially when preoperative case planning measurements (treatment diameters/lengths) are
not certain. This approach allows for greater intraoperative flexibility to
achieve optimal procedural outcomes.
4.3 Pre-Procedure Measurement Techniques and Imaging
• Lack of non- contrast CT imaging may result in failure to appreciate iliac or
aortic calcification, which may preclude access or reliable device fixation
and seal.
• Pre-procedure imaging reconstruction thicknesses >3 mm may result in
sub-optimal device sizing, or in failure to appreciate focal stenoses from CT.
• Clinical experience indicates that contrast-enhanced spiral computed
tomographic angiography (CTA) with 3-D reconstruction is the strongly
recommended imaging modality to accurately assess patient anatomy prior
to treatment with the Zenith Flex AAA Endovascular Graft. If contrastenhanced spiral CTA with 3-D reconstruction is not available, the patient
should be referred to a facility with these capabilities.
• Clinicians recommend positioning the x-ray C-arm during procedural
angiography such that the origins of the renal arteries, and particularly the
lowest patent renal artery, are well demonstrated prior to deployment of
the proximal edge of the graft material (sealing stent) of the main body.
Additionally, angiography should demonstrate the iliac artery bifurcations
such that the distal common iliacs are well defined relative to the origin of
the internal iliac arteries bilaterally, prior to deployment of the iliac leg
components.
Diameters:
Utilizing CT, diameter measurements should be determined from the outer
wall to outer wall vessel diameter (not lumen measurement) to help with
proper device sizing and device selection. The contrast-enhanced spiral CT
scan must start 1 cm superior to the celiac axis and continue through the
femoral heads at an axial thickness slice of 3 mm or less.
Lengths:
Utilizing CT, length measurements should be determined to accurately
assess infrarenal proximal neck length as well as planning main body sizes
and leg components for the Zenith Flex AAA Endovascular Graft. These
reconstructions should be performed in sagittal, coronal, and 3-D.
• The long-term per formance of endovascular grafts has not yet been
established. All patients should be advised that endovascular
treatment requires life-long, regular follow-up to assess their health
and the performance of their endovascular graft. Patients with specific
clinical findings (e.g., endoleaks, enlarging aneurysm or changes in the
structure or position of the endovascular graft) should receive enhanced
follow-up. Specific follow-up guidelines are described in Section 12,
IMAGING GUIDELINES AND POSTOPERATIVE FOLLOW-UP.
23
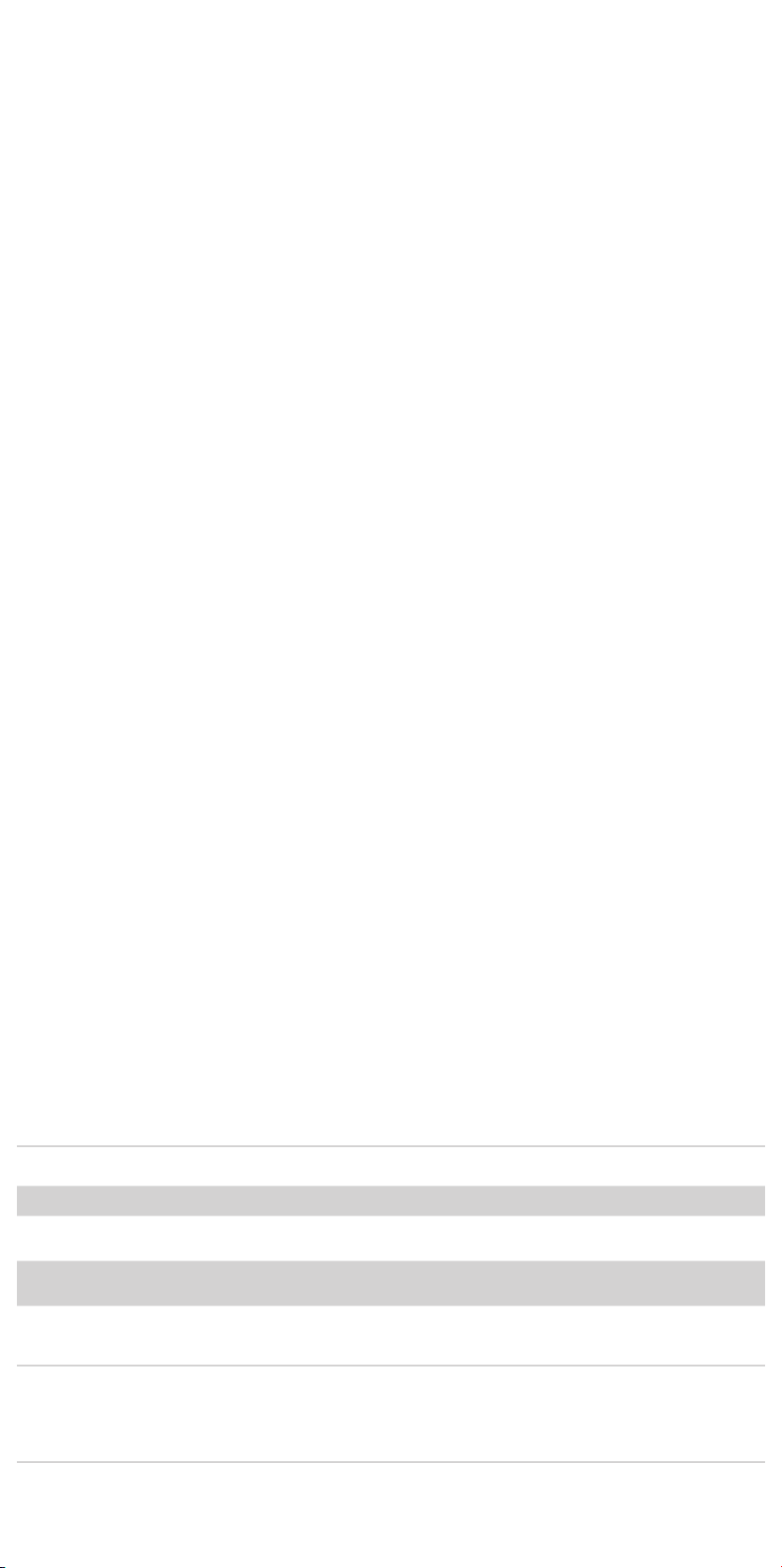
• The Zenith Flex AAA Endovascular Graft with the H&L-B One-Shot
Introduction System is not recommended in patients unable to undergo, or
who will not be compliant with, the necessary preoperative and
postoperative imaging and implantation studies as described in Section 12,
IMAGING GUIDELINES AND POSTOPERATIVE FOLLOW-UP.
• After endovascular graft placement, patients should be regularly monitored
for perigraft flow, aneurysm growth or changes in the structure or position
of the endovascular graft. At a minimum, annual imaging is required,
including: 1) abdominal radiographs to examine device integrity (separation
between components, stent fracture or barb separation) and 2) contrast and
non-contrast CT to examine aneurysm changes, perigraft flow, patency,
tortuosity and progressive disease. If renal complications or other factors
preclude the use of image contrast media, abdominal radiographs and
duplex ultrasound may provide similar information.
4.4 Device Selection
• Stric t adherence to the Zenith Flex AAA Endovascular Graft IFU sizing guide
is strongly recommended when selecting the appropriate device size
(Tables 10.5.1 and 10.5.2). Appropriate device oversizing has been
incorporated into the IFU sizing guide. Sizing outside of this range can
result in endoleak, fracture, migration, device infolding or compression.
4.5 Implant Procedure
(Refer to Section 11, DIRECTIONS FOR USE)
• Appropriate procedural imaging is required to successfully position the
Zenith Flex AAA Endovascular Graft and assure accurate apposition to the
aortic wall.
• Do not bend or k ink the delivery system. Doing so may cause damage to the
delivery system and the Zenith Flex AAA Endovascular Graft.
• To avoid any twist in the endovascular graft, during any rotation of the
delivery system, be careful to rotate all of the components of the system
together (from outer sheath to inner cannula).
• Do not continue advancing any portion of the deliver y system if resistance
is felt during advancement of the wire guide or delivery system. Stop and
assess the cause of resistance; vessel, catheter or graft damage may occur.
Exercise particular care in areas of stenosis, intravascular thrombosis or in
calcified or tortuous vessels.
• I nadvertent partial deployment or migration of the endoprosthesis may
require surgical removal.
• Unless medically indicated, do not deploy the Zenith Flex AAA Endovascular
Graft in a location that will occlude arteries necessary to supply blood flow
to organs or extremities. Do not cover significant renal or mesenteric
arteries (exception is the inferior mesenteric artery) with the endoprosthesis.
Vessel occlusion may occur. During the clinical study, this device was not
studied in patients with two occluded internal iliac arteries.
• Do not attempt to re -sheath the graft after partial or complete deployment.
• Repositioning the stent graft distally after par tial deployment of the covered
proximal stent may result in damage to the stent graft and/or vessel injury.
• I naccurate placement and/or incomplete sealing of the Zenith Flex AAA
Endovascular Graft within the vessel may result in increased risk of
endoleak, migration or inadvertent occlusion of the renal or internal iliac
arteries. Renal artery patency must be maintained to prevent/reduce the
risk of renal failure and subsequent complications.
• I nadequate fixation of the Zenith Flex AAA Endovascular Graft may result in
increased risk of migration of the stent graft. Incorrect deployment or
migration of the endoprosthesis may require surgical intervention.
• Systemic anticoagulation should be used during the implantation
procedure based on hospital and physician preferred protocol. If heparin is
contraindicated, an alternative anticoagulant should be considered.
• To activate the hydrophilic coating on the outside of the Flexor introducer
sheath, the surface must be wiped with sterile gauze pads soaked in saline
solution. Always keep the sheath hydrated for optimal performance.
• M inimize handling of the constrained endoprosthesis during preparation
and insertion to decrease the risk of endoprosthesis contamination and
infection.
• M aintain wire guide position during delivery system insertion.
• Fluoroscopy should be used during introduction and deployment to
confirm proper operation of the delivery system components, proper
placement of the graft, and desired procedural outcome.
• The use of the Zenith Flex AAA Endovascular Graft with the H&L-B One-Shot
Introduction System requires administration of intravascular contrast.
Patients with preexisting renal insufficiency may have an increased risk of
renal failure postoperatively. Care should be taken to limit the amount of
contrast media used during the procedure and to observe preventative
methods of treatment to decrease renal compromise (e.g., adequate
hydration).
• As the sheath and/or wire guide is withdrawn, anatomy and graft position
may change. Constantly monitor graft position and perform angiography to
check position as necessary.
• The Zenith Flex AAA Endovascular Graft incorporates a suprarenal stent with
fixation barbs. Exercise extreme caution when manipulating interventional
and angiographic devices in the region of the suprarenal stent.
• Use caution during manipulation of catheters, wires and sheaths within an
aneurysm. Significant disturbances may dislodge fragments of thrombus,
which can cause distal embolization, or rupture of the aneurysm.
• Avoid damaging the graft or disturbing graft positioning after placement in
the event reinstrumentation (secondary intervention) of the graft is
necessary.
• Before deployment of the suprarenal stent, verify that the position of the
access wire guide extends just distal to the aortic arch.
• Verify that the predetermined contralateral iliac leg is selected for insertion
on the contralateral side of the patient before implantation.
4.6 Molding Balloon Use
• Do not inflate the balloon in the vessel outside of the graft, as doing so may
cause damage to the vessel. Use the balloon in accordance with its labeling.
• Use care in inflating the balloon within the graft in the presence of
calcification, as excessive inflation may cause damage to the vessel.
• Confirm complete deflation of the balloon prior to repositioning.
• For added hemostasis, the Captor Hemostatic Valve can be loosened or
tightened to accommodate the insertion and subsequent withdrawal of a
molding balloon.
4.7 MRI Safety and Compatibility
Non-clinical testing has demonstrated that the Zenith AAA Endovascular Graft
is MR Conditional. It can be scanned safely under the following conditions:
1.5 Tesla Systems:
• Static magnetic field of 1.5 Tesla
• Spatial gradient field of 450 Gauss/cm
• M aximum whole-body-averaged specific absorption rate (SAR) of
2.0 W/kg for 15 minutes of scanning
In non-clinical testing, the Zenith AAA Endovascular Graft produced a
temperature rise of less than or equal to 1.4 °C at a maximum whole-bodyaveraged specific absorption rate (SAR) of 2.8 W/kg, as assessed by calorimetry
for 15 minutes of MR scanning in a 1.5 Tesla M agnetom, Siemens Medical
Magnetom, Numaris/4 Software, Version Syngo MR 2002B DHHS MR Scanner.
The maximum whole-body-averaged specific absorption rate (SAR) was 2.8 W/
kg, which corresponds to a calorimetry measured value of 1.5 W/kg.
3.0 Tesla Systems:
• Static magnetic field of 3.0 Tesla
• Spatial gradient field of 720 Gauss/cm
• M aximum whole-body-averaged specific absorption rate (SAR) of
2.0 W/kg for 15 minutes of scanning
In non-clinical testing, the Zenith AAA Endovascular Graft produced a
temperature rise of less than or equal to 1.9 °C at a maximum wholebody-averaged specific absorption rate (SAR) of 3.0 W/kg, as assessed by
calorimetry for 15 minutes of MR scanning in a 3.0 Tesla Excite, GE Electric
Healthcare, G3.0-052B Software, MR Scanner. The maximum whole-bodyaveraged specific absorption rate (SAR) was 3.0 W/kg, which corresponds to a
calorimetry measured value of 2.8 W/kg.
The image artifact extends throughout the anatomical region containing the
device, obscuring the view of immediately adjacent anatomical structures
within approximately 20 cm of the device, as well as the entire device and its
lumen, when scanned in non-clinical testing using the sequence: Fast spin
echo, in a 3.0 Tesla, Excite, GE Electric Healthcare, with G3.0-052B Software, MR
system with body radiofrequency coil.
For all scanners, the image artifact dissipates as the distance from the device
to the area of interest increases. MR scans of the head and neck and lower
extremities may be obtained without image artifact. Image artifact may be
present in scans of the abdominal region and upper extremities, depending
on distance from the device to the area of interest.
Clinical information is available for seventeen patients who received MRI scans
after stent-graft implantation. There have been no reported adverse events or
device problems in any of these patients as a result of having received an MRI.
Additionally, there have been well over 50,000 Zenith AAA Endovascular Grafts
implanted worldwide, in which there have been no reported adverse events or
device problems as a result of MRI.
Cook recommends that the patient register the MR conditions disclosed in
this IFU with the MedicAlert Foundation. The MedicAlert Foundation can be
contacted in the following manners:
Address: MedicAlert Foundation International
Fax: +1 209-669-2450
Web: www.medicalert.org
2323 Colorado Avenue
Turlock, CA 95382 USA
Phone: 888-633-4298 (toll free)
+1 209-668-3333 from outside the US
5 ADVERSE EVENTS
5.1 Observed Adverse Events
A U.S. multicenter, prospective study of a previous version of the device
(Zenith AAA Endovascular Graft) conducted at 15 centers which included
352 endovascular patients (200 standard risk, 100 high risk and 52 roll-in) and
80 control patients provides the basis of the observed adverse event rates
in Table 5.1.1. Patients were enrolled in the standard risk arm if they were
physiologically capable of withstanding an open or endovascular repair and
had anatomy suitable for treatment with the Zenith AAA Endovascular Graft
with the H&L-B One-Shot Introduction System. Patients with suitable anatomy,
but at higher risk of morbidity or mortality with open repair were enrolled
into the high risk arm. Initial patients treated in the study were enrolled in the
roll-in arm. The control group included patients whose vascular anatomy may
not have been suitable for endovascular AAA repair.
Table 5.1.1 Death and Rupture from Clinical Study
Death and Rupture Zenith Standard Risk
All death
2
(0-30 days)
3
(31-365 days)
AAA-related 0.0% (0/198) 1.3% (1/78) .29 3.1% (3/98) 0.0% (0/51)
Non-AAA-related 3.0% (6/198) 0.0% (0/78) .19 4.1% (4/98) 9.8% (5/51)
2,3,4
(0-365 days)
AAA-related 0.5% (1/199) 3.8% (3/80) .07 5.0% (5/100) 1.9% (1/52)
Non-AAA-related 3.0% (6/199) 0.0% (0/80) .19 4.0% (4/100) 9.6% (5/52)
Rupture
(0-30 days) 0.0% (0/199) n/a n/a 0.0% (0/100) 0.0% (0/52)
(31-365 days) 0.0% (0/198) n/a n/a 1.0% (1/98) 0.0% (0/51)
(0-365 days) 0.0% (0/199) n/a n/a 1.0% (1/100) 0.0% (0/52)
1
Denominator of 199 because one standard risk patient did not receive a device.
2
All deaths (0-30 days) were considered AAA and procedure related.
3
Of the deaths (31-365 days), four were considered AAA related: 1 surgical (septic shock from ischemic colitis) and 3 high risk (pancreatitis with renal failure and sepsis, hemorrhage
from upper abdominal aneurysm [not treated AAA] and multiple system failure).
4
Of the deaths (0-365 days), ten were considered AAA related: 1 standard risk (cardiac failure), 3 surgical (massive hemorrhage, mesenteric ischemia and septic shock from ischemic
colitis), 5 high risk (respiratory failure, cardiac failure with pulmonary embolism, pancreatitis with renal failure and sepsis, hemorrhage from upper abdominal aneurysm [not treated
AAA] and multiple system failure) and 1 roll-in (suspected cardiac failure).
0.5% (1/199) 2.5% (2/80) .20 2.0% (2/100) 1.9% (1/52)
3.0% (6/198) 1.3% (1/78) .68 7.1% (7/98) 9.8% (5/51)
3.5% (7/199) 3.8% (3/80) >.99 9.0% (9/100) 11.5% (6/52)
1
Surgical Standard Risk P value Zenith High Risk Zenith Roll-in
24
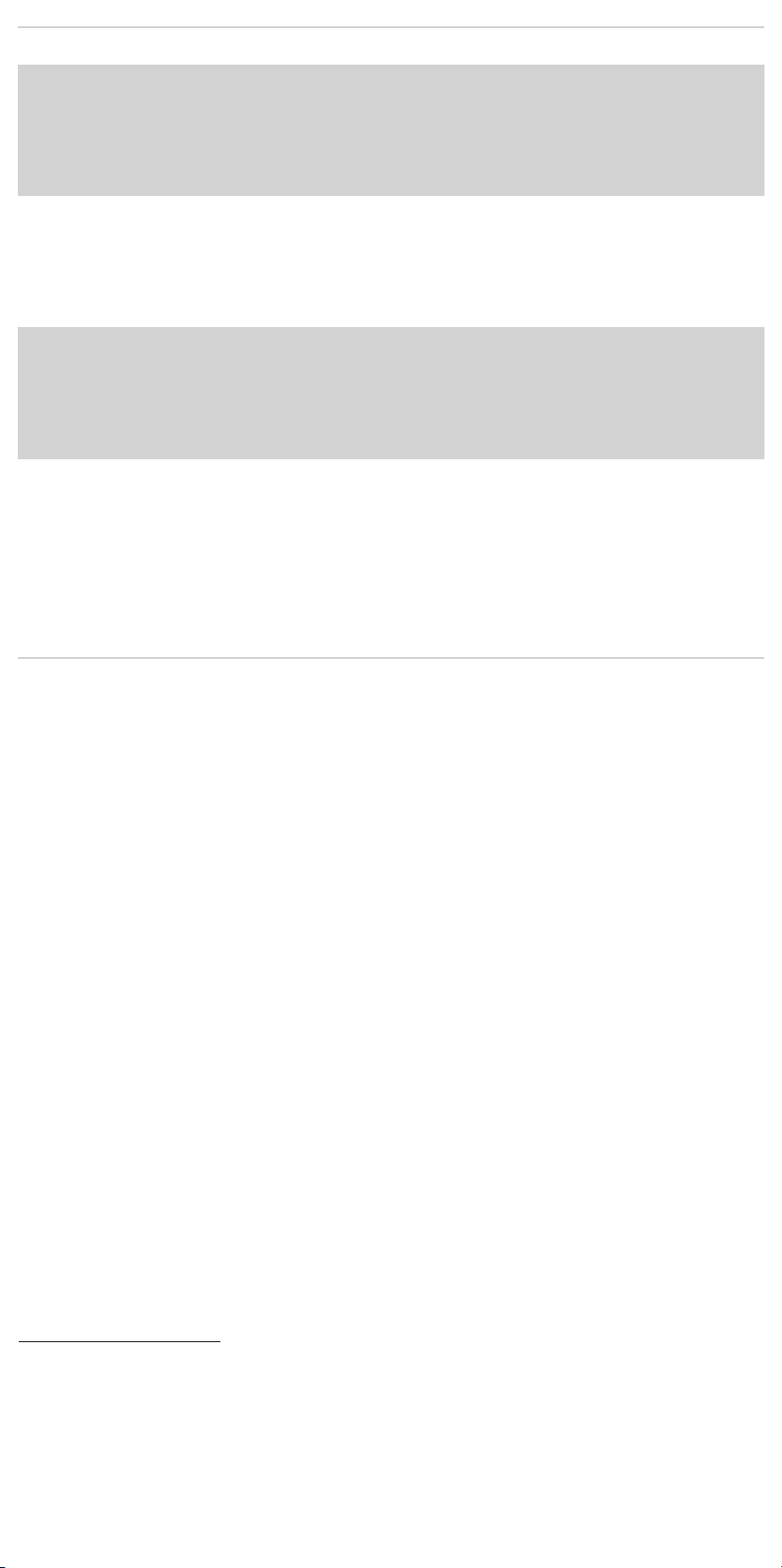
Zenith Standard Risk Surgical Standard Risk P value Zenith High Risk Zenith Roll-in
Table 5.1.2 Adverse Events1 in Clinical Study
Freedom from Morbidity
(0-30 days)
3
4, 9
5
6
7
8,11
(31-365 days)
3
4, 10
5
6
7
8
(0-365 days)
3
4, 9,10
5
6
7
8,11
2
2
2
Cardiovascular
Pulmonary
Renal
Bowel
Wound
Neurologic
Vascular
Freedom from Morbidity
Cardiovascular
Pulmonary
Renal
Bowel
Wound
Neurologic
Vascular
Freedom from Morbidity
Cardiovascular
Pulmonary
Renal
Bowel
Wound
Neurologic
Vascular
1
From the morbidity index.
2
Cardiovascular included: Q-wave and non-Q-wave myocardial infarctions, congestive heart failure, arrhythmias requiring new medication or treatment, cardiac ischemia requiring
intervention, inotropic support, medically intractable hypertension.
3
Pulmonary included: reintubation or ventilation >24 hours, pneumonia requiring antibiotics, supplemental oxygen at discharge.
4
Renal included: dialysis in patients with normal preoperative renal function, creatinine rise >30% from baseline on two or more follow-up tests.
5
Bowel included: bowel obstruction, bowel ischemia, aorto-enteric fistula, paralytic ileus >4 days.
6
Wound included: infection requiring antibiotic treatment, hernia, lymph fistula, dehiscence, necrosis requiring debridement.
7
Neurological included: stroke, TIA, spinal cord ischemia/paralysis.
8
Vascular included: limb thrombosis, distal embolization resulting in tissue loss or requiring intervention, transfusion postprocedure (resulting from pseudoaneurysm, vascular injury,
aneurysm leak or other procedure-related causes), pseudoaneurysm, vascular injury (such as inadvertent occlusion, dissection or other procedure related causes), aneurysm leak or
rupture, increase in aneurysm size by more than 0.5 cm relative to the smallest of any prior measurement.
9
Investigators reported one additional high risk patient to have occlusion of an accessory renal artery and one additional high risk patient to have chronic renal insufficiency as “other”
adverse events.
10
Investigators reported one additional roll-in patient to have renal insufficiency and one additional surgical patient to have renal insufficiency as “other” adverse events.
11
Investigators reported one additional roll-in patient to have experienced intraoperative aortic plaque rupture resulting in renal artery occlusion as an “other” adverse event.
80% (160/200) 58% (46/80) <.001 68% (68/100) 73% (38/52)
3.0% (6/200) 11% (9/80) .02 14% (14/100) 1.9% (1/52)
1.0% (2/200) 15% (12/80) <.001 2.0% (2/100) 0.0% (0/52)
2.5% (5/200) 10% (8/80) .01 6.0% (6/100) 5.8% (3/52)
1.0% (2/200) 3.8% (3/80) .14 1.0% (1/100) 1.9% (1/52)
4.5% (9/200) 7.5% (6/80) .38 2.0% (2/100) 3.8% (2/52)
0.0% (0/200) 2.5% (2/80) .08 0.0% (0/100) 0.0% (0/52)
11% (21/200) 31% (25/80) <.001 20% (20/100) 19% (10/52)
91% (181/198) 86% (67/78) .25 79% (77/98) 86% (44/51)
2.5% (5/198) 3.8% (3/78) .69 5.1% (5/98) 2.0% (1/51)
0.5% (1/198) 1.3% (1/78) .49 4.1% (4/98) 0.0% (0/51)
0.5% (1/198) 0.0% (0/78) >.99 3.1% (3/98) 0.0% (0/51)
0.5% (1/198) 1.3% (1/78) .49 0.0% (0/98) 0.0% (0/51)
2.0% (4/198) 5.1% (4/78) .23 3.1% (3/98) 2.0% (1/51)
1.0% (2/198) 0.0% (0/78) >.99 1.0% (1/98) 3.9% (2/51)
3.0% (6/198) 3.8% (3/78) .72 8.2% (8/98) 5.9% (3/51)
76% (151/200) 49% (39/80) <.001 55% (55/100) 62% (32/52)
5.0% (10/200) 14% (11/80) .02 19% (19/100) 3.8% (2/52)
1.5% (3/200) 16% (13/80) <.001 6.0% (6/100) 0.0% (0/52)
2.5% (5/200) 10% (8/80) .01 9.0% (9/100) 5.8% (3/52)
1.5% (3/200) 3.8% (3/80) .36 1.0% (1/100) 1.9% (1/52)
5.5% (11/200) 13% (10/80) .08 5.0% (5/100) 5.8% (3/52)
1.0% (2/200) 2.5% (2/80) .32 1.0% (1/100) 3.8% (2/52)
12% (24/200) 33% (26/80) <.001 25% (25/100) 23% (12/52)
5.2 Potential Adverse Events
Adverse events that may occur and/or require intervention include, but are
not limited to:
• Amputation
• Anesthetic complications and subsequent attendant problems (e.g.,
aspiration)
• Aneur ysm enlargement
• Aneur ysm rupture and death
• Aor tic damage, including perforation, dissection, bleeding, rupture and
death
• Ar terial or venous thrombosis and/or pseudoaneurysm
• Ar teriovenous fistula
• Bleeding, hematoma or coagulopathy
• Bowel complications (e.g., ileus, transient ischemia, infarction, necrosis)
• Cardiac complications and subsequent attendant problems (e.g.,
arrhythmia, myocardial infarction, congestive heart failure, hypotension,
hypertension)
• Claudication (e.g., buttock, lower limb)
• Death
• Edema
• Embolization (micro and macro) with transient or permanent ischemia or
infarction
• Endoleak
• Endoprosthesis: improper component placement; incomplete component
deployment; component migration; suture break; occlusion; infection; stent
fracture; graft material wear; dilatation; erosion; puncture; perigraft flow;
barb separation and corrosion
• Fever and localized inflammation
• Genitourinar y complications and subsequent attendant problems (e.g.,
ischemia, erosion, fistula, incontinence, hematuria, infection)
• Graf t or native vessel occlusion
• Hepatic failure
• Impotence
• I nfection of the aneurysm, device or access site, including abscess
formation, transient fever and pain
• Lymphatic complications and subsequent attendant problems (e.g., lymph
fistula)
• Neurologic local or systemic complications and subsequent attendant
problems (e.g., stroke, transient ischemic attack, paraplegia, paraparesis,
paralysis)
• Pulmonar y/respiratory complications and subsequent attendant problems
(e.g., pneumonia, respiratory failure, prolonged intubation)
• Renal complications and subsequent attendant problems (e.g., artery
occlusion, contrast toxicity, insufficiency, failure)
• Surgical conversion to open repair
• Vascular access site complications, including infection, pain, hematoma,
pseudoaneurysm, arteriovenous fistula
• Vascular spasm or vascular trauma (e.g., iliofemoral vessel dissection,
bleeding, rupture, death)
• Vessel damage
• Wound complications and subsequent attendant problems (e.g.,
dehiscence, infection)
Device Related Adverse Event Reporting
Any adverse event (clinical incident) involving the Zenith Flex AAA
Endovascular Graft should be reported to COOK immediately. For customers
in the United States, to report an incident, call the Customer Relations
Department at 1-800-457-4500 (24-hour) or 1-812-339-2235. For customers
outside the United States, please call your distributor.
6 SUMMARY OF CLINICAL STUDIES
6.1 Objectives
The primary objective of the clinical study was to evaluate the safety and
effectiveness of a previous version of the device (Zenith AAA Endovascular
Graft) as an alternative to open surgical repair in the primary treatment of
infrarenal abdominal aortic aneurysms. Study hypotheses examined whether
standard risk patients experienced less 30-day morbidity, non-inferior 30-day
survival r ates, non-inferior 12-mon th survival rates, non-inferior 12-month
treatment success and improved clinical utility measures compared to surgical
control patients. Safety was determined by evaluating whether the Zenith
AAA Endovascular Graft subjects would have reduced 30-day morbidity,
non-inferior 30-day and 12-month survival and non-inferior 12-month
treatment success compared to the subjects treated with open surgical
treatment. Effectiveness was based on exclusion of the aneurysm including
the absence of any endoleak, the absence of aneurysm enlargement (≥5 mm)
and the absence of major device adverse events evaluated through one year
follow-up. Secondary objectives included an assessment of clinical benefit
and quality-of-life measures.
6.2 Study Design
The U.S. clinical study was a multicenter, nonrandomized study comparing
standard medical risk patients who received an endovascular graft to an
open surgical control. There were two additional study arms for high medical
risk and roll-in treatment groups. Fifteen centers enrolled 200 standard risk,
80 surgical control, 100 high risk and 52 roll-in patients. The control group
included patients whose vascular anatomy may not have been suitable for
endovascular AAA repair. Follow-up evaluations were scheduled for
pre-discharge, 1 month, 6 months, 12 months and 24 months. Patient followup and accountability at 1 month, 12 months and 24 months are presented
in Table 6.2.1 as these were the primary data analysis time points. Imaging
data provided in this summary are based on findings from an independent
centralized image analysis laboratory (Core Lab) which reviewed CT scans and
abdominal X-rays to assess aneurysm diameter changes, device and relative
component migration, device integrity (wire and graft) and the presence and
type of endoleaks. Clinical events were adjudicated by an independent clinical
events committee and safety was monitored by a data safety monitoring
committee.
Surgical and Zenith standard risk patients met identical pathophysiologic risk
criteria. The endovascular groups excluded circumferential thrombus in the
proximal neck, proximal neck less than 15 mm in length, outer wall to outer
wall proximal neck diameter less than 18 mm or greater than 28 mm, severe
proximal neck angulation, outer wall to outer wall iliac artery diameter less
than 7.5 mm or greater than 20 mm at distal fixation site or iliac artery distal
fixation site less than 10 mm in length.
Patients were considered at higher risk for surgical repair if they had age
greater than 80, baseline creatinine >2.0 mg/dl, home oxygen therapy, FEV1
<1 liter, ejection fraction <25%, disabling COPD, New York Heart Classification
3 or 4, hostile abdomen, dialysis, MI within last 6 months, medically intractable
hypertension, previous stroke with residual deficit, cultural objection
to receipt of blood or blood products, previous renal bypass surgery or
inflammatory aneurysm.
Before enrolling patients into the pivotal trial, centers without Zenith AAA
Endovascular Graft experience were required to treat initial patients under the
supervision of a proctor. These roll-in patients were a combination of standard
and high risk patients and were followed according to the same schedule as
the patients in the pivotal trial.
25

Table 6.2.1 Patient Follow-Up and Accountability
1
Treatment Zenith Standard Risk Surgical Standard Risk
Interval
No device 1
Conversion to open repair 0
Expired 1 7 17 2 3 n/a
1 mo. 12 mo. 24 mo. 1 mo. 12 mo. 24 mo.
2
1
2
2
2
1
2
2
0 0 n/a
n/a n/a n/a
Withdrawn/lost to follow-up 0 0 2 0 4 n/a
Available 198 190 178 78 73 n/a
Site CT imaging 191 168 110 69 58 n/a
Core lab CT imaging 190 165 99 69 59 n/a
Site KUB imaging 179 153 108 n/a n/a n/a
Core lab KUB imaging 178 149 93 n/a n/a n/a
Site evaluated for endoleak 187 163 107 n/a n/a n/a
Core lab evaluated for endoleak 161 148 92 n/a n/a n/a
Site evaluated for aneurysm enlargement n/a 149 104 n/a n/a n/a
Core lab evaluated for aneurysm enlargement n/a 151 94 n/a n/a n/a
1
Data analysis sample size varies for each of the time points above and in the following tables. This variability is due to patient availability for follow-up, as well as quantity and quality
of images available from specific time points for evaluation. For example, the number and quality of images available for evaluation of endoleak at 12 months is different than the
number and quality of images available at 24 months due to variation in the number of image exams performed, the number of images provided from the clinical site to the Core Lab
and/or the number of images with acceptable evaluation quality. Totals at time points are not cumulative, unless otherwise noted.
2
Totals at time points are cumulative.
6.3 Patient Demographics
Tables 6.3.1 and 6.3.2 compare the subject characteristics and initial aneurysm diameter of the Zenith AAA Endovascular Graft and open surgical population,
respectively.
Table 6.3.1 Comparison of Subject Characteristics
Item Zenith Standard Risk Surgical Standard Risk P value
Zenith
High Risk
Zenith
Roll-in
Age (years) 71 ± 7 69 ± 7 .03 77 ± 7 74 ± 8
Gender male 94% (187/200) 89% (71/80) .22 92% (92/100) 90% (47/52)
Current medical conditions
Peripheral vascular disease
Hypertension 64% (127/200) 83% (65/78) .001 68% (67/99) 67% (35/52)
16% (31/195) 25% (19/76) .12 24% (23/96) 9.6% (5/52)
Renal failure 0.0% (0/197) 0.0% (0/79) >.99 5.2% (5/97) 1.9% (1/52)
COPD 20% (39/199) 18% (14/78) .87 34% (33/98) 22% (11/51)
Thromboembolic event 4.5% (9/199) 7.7% (6/78) .38 7.1% (7/99) 1.9% (1/52)
Liver disease 2.1% (4/192) 5.1% (4/79) .24 1.0% (1/99) 1.9% (1/52)
Diabetes mellitus 12% (24/199) 15% (12/79) .55 17% (17/99) 14% (7/51)
Insulin-dependent 17% (4/24) 8.3% (1/12) .65 24% (4/17) 43% (3/7)
Previous medical conditions
MI
39% (74/192) 29% (23/80) .13 35% (34/98) 35% (18/52)
Congestive heart failure 5.0% (10/199) 12% (9/78) .07 16% (16/100) 10% (5/50)
Angina 49% (98/198) 39% (31/79) .14 45% (44/98) 44% (23/52)
Arrhythmia 20% (40/197) 22% (17/78) .87 28% (27/98) 24% (12/51)
Cerebrovascular disease 9.5% (19/199) 16% (13/79) .14 20% (20/99) 9.8% (5/51)
Systemic infection 1.0% (2/196) 0.0% (0/78) >.99 3.1% (3/97) 0.0% (0/49)
Cancer 22% (43/200) 19% (15/80) .74 31% (31/99) 29% (15/51)
Family history of aneurysmal disease 16% (24/150) 27% (17/63) .09 14% (11/77) 26% (10/38)
Previous surgery at site 10% (20/200) 15% (12/79) .22 10% (10/99) 14% (7/51)
Previous radiation at site 0.5% (1/197) 0.0% (0/79) >.99 2.0% (2/100) 2.0% (1/51)
Excessive alcohol use 3.6% (7/193) 10% (8/77) .04 3.1% (3/96) 4.0% (2/50)
Tobacco use
Never smoked
Past smoker 69% (133/193) 60% (48/80) .03 69% (66/96) 57% (28/49)
Still smokes 21% (40/193) 35% (28/80) 18% (17/96) 18% (9/49)
Due to inclusion criteria, high risk patients were older (P<.001), had more renal failure (P=.004), COPD (P=.01), congestive heart failure (P=.004) and cerebrovascular disease (P=.02) than
standard risk patients.
10% (20/193) 5.0% (4/80)
14% (13/96) 24% (12/49)
Table 6.3.2 Aneurysm Diameter Distribution
Diameter Range Zenith Standard Risk Surgical Standard Risk Zenith High Risk Zenith Roll-in
<30 mm
30-39 mm
40-49 mm
50-59 mm
60-69 mm
70-79 mm
80-89 mm
≥90 mm
Aneurysm diameter distribution was not assessed in three high-risk and one roll-in patient.
0.0% (0/199) 0.0% (0/78) 0.0% (0/100) 0.0% (0/52)
0.5% (1/199) 0.0% (0/78) 1.0% (1/100) 0.0% (0/52)
23% (45/199) 7.7% (6/78) 15% (15/100) 13% (7/52)
48% (95/199) 33% (26/78) 47% (47/100) 40% (21/52)
24% (47/199) 29% (23/78) 27% (27/100) 42% (22/52)
3.0% (6/199) 21% (16/78) 5.0% (5/100) 1.9% (1/52)
2.5% (5/199) 6.4% (5/78) 1.0% (1/100) 0.0% (0/52)
0.0% (0/199) 2.6% (2/78) 1.0% (1/100) 0.0% (0/52)
6.4 Results
Data gathered in Tables 6.4.1 through 6.4.3 were collected by the clinical study sites and Core Lab. Where available, 24-month data are provided. Control
patients were not followed beyond 12 months and some data have not yet been adjudicated beyond 12 months. Therefore, some results are presented to
12 months while other results are presented to 24 months in this section. Table 6.4.1 describes the devices implanted in clinical study patients. Table 6.4.2
describes the primary results from the clinical study. Figures 6.4.1 and 6.4.2 are the Kaplan-Meier plots of all-cause and AAA-related survival to 24 months,
respectively. An independent clinical events committee adjudicated all deaths for possible relationship to aneurysm repair. All early deaths (0-30 days) were
considered AAA-related. Deaths after 30 days were considered AAA-related if AAA disease or device involvement was confirmed. Table 6.4.3 presents Success
Measures and Figure 6.4.3 is the Kaplan-Meier plot of Freedom from Morbidity.
Table 6.4.1 Devices Implanted
Item Zenith Standard Risk Zenith High Risk Zenith Roll-in
Main body and legs 99.5% (199/200)* 100%
Main body extension
Ipsilateral iliac leg extension
Contralateral iliac leg extension
Converter 0.5% (1/199)** 0.0% (0/100) 0.0% (0/52)
Occluder 0.0% (0/199) 0.0% (0/100) 0.0% (0/52)
*One standard risk patient did not receive a device due to tortuosity and calcification of the access vessel.
**Converter was used without occluder.
1
One device was custom.
2
Two standard risk and one high risk patient received main body extensions postprocedure; one standard risk patient received two main body extensions.
3
Two standard risk and one high risk patient received ipsilateral leg extensions postprocedure.
4
Four standard risk, two high risk and one roll-in patient received contralateral leg extensions postprocedure.
5
Three standard risk and three high risk patients received both ipsilateral and contralateral extensions during the procedure; one standard risk received both ipsilateral and
contralateral extensions postprocedure.
2
3,5
4,5
1.5% (3/199) 1.0% (1/100) 5.8% (3/52)
9.5% (19/199) 11% (11/100) 0.0% (0/52)
11% (21/199) 11% (11/100) 13.5% (7/52)
(100/100)
1
100% (52/52)
26
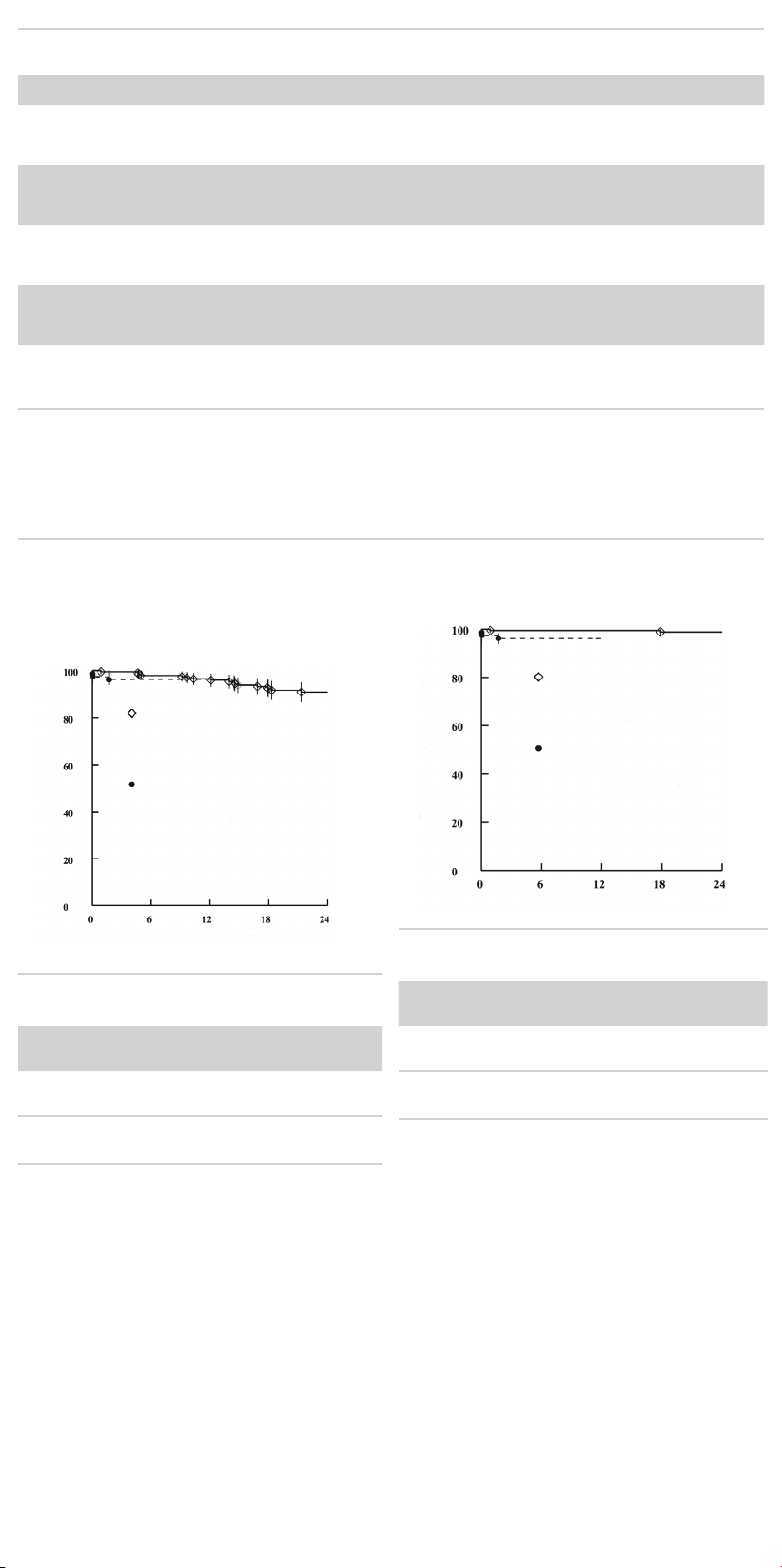
Table 6.4.2 Primary Results
Item Zenith Standard Risk
All death
All death
2
(0-30 days)
(31-365 days)
AAA-related 0.0% (0/199) 1.3% (1/80) .29 3.0% (3/100) 0.0% (0/52)
2, 3
0.5% (1/199) 2.5% (2/80) .20 2.0% (2/100) 1.9% (1/52)
3.0% (6/199) 1.3% (1/80) .68 7.0% (7/100) 9.6% (5/52)
1
Surgical Standard Risk P value Zenith High Risk Zenith Roll-in
Non-AAA-related 3.0% (6/199) 0.0% (0/80) .19 4.0% (4/100) 9.6% (5/52)
All death
2,3,4
(0-365 days)
AAA-related 0.5% (1/199) 3.8% (3/80) .07 5.0% (5/100) 1.9% (1/52)
3.5% (7/199) 3.8% (3/80) >.99 9.0% (9/100) 11.5% (6/52)
Non-AAA-related 3.0% (6/199) 0.0% (0/80) .19 4.0% (4/100) 9.6% (5/52)
Rupture
(0-30 days) 0.0% (0/199) n/a n/a 0.0% (0/100) 0.0% (0/52)
(31-365 days) 0.0% (0/199) n/a n/a 1.0% (1/100) 0.0% (0/52)
(0-365 days) 0.0% (0/199) n/a n/a 1.0% (1/100) 0.0% (0/52)
Conversion
(0-30 days) 0.0% (0/199) n/a n/a 0.0% (0/100) 0.0% (0/52)
(31-365 days)
(0-365 days)
Adverse events
(0-30 days)
(31-365 days)
(0-365 days)
1
Denominator of 199 because one standard risk patient did not receive a device.
2
All deaths (0-30 days) were considered AAA and procedure related.
3
Of the deaths (31-365 days), four were considered AAA related: 1 surgical (septic shock from ischemic colitis) and 3 high risk (pancreatitis with renal failure and sepsis, hemorrhage
from upper abdominal aneurysm [not treated AAA] and multiple system failure).
4
Of the deaths (0-365 days), ten were considered AAA related: 1 standard risk (cardiac failure), 3 surgical (massive hemorrhage, mesenteric ischemia and septic shock from ischemic
colitis), 5 high risk (respiratory failure, cardiac failure with pulmonary embolism, pancreatitis with renal failure and sepsis, hemorrhage from upper abdominal aneurysm [not treated
AAA] and multiple system failure) and 1 roll-in (suspected cardiac failure).
5
Standard risk patients underwent conversions due to a persistent, proximal Type I endoleak and a new suprarenal aortic aneurysm. Three surgical patients had massive
hemorrhages, of which 2 required re-operation and one died.
6
Adverse events included in the morbidity index.
5
5
6
6
6
1.0% (2/199) n/a n/a 1.0% (1/100) 0.0% (0/52)
1.0% (2/199) n/a n/a 1.0% (1/100) 0.0% (0/52)
20% (40/200) 43% (34/80) <.001 32% (32/100) 27% (14/52)
8.6% (17/199) 14% (11/78) .25 21% (21/98) 14% (7/51)
25% (49/200) 51% (41/80) <.001 45% (45/100) 38% (20/52)
Zenith AAA Endovascular Graft patients exhibited no significant differences
between males and females for survival and freedom from major adverse
events. (Error bars in Figures 6.4.1, 6.4.2 and 6.4.3 represent 95% confidence
limits.) Figure 6.4.1 presents all-cause survival to 24 months. The
accompanying table presents the Kaplan-Meier analysis at 1, 6, 12 and 24
months.
Figure 6.4.1 Survival at 24 Months
Zenith Standard Risk
(N=200, 1 death to 30 days
4 deaths to 6 months
7 deaths to 12 months
17 deaths to 24 months)
Surgical Standard Risk
(N=80, 2 deaths to 30 days
Percent Survival
3 deaths to 6 months
3 deaths to 12 months)
Months After Procedure
1 month 6 months 12 months 24 months
n % survival n % survival n % survival n % survival
Zenith
standard
198* 99.5 194 98.0 190 96.5 97 90.9
risk
Surgical
standard
risk
n= Patients alive and available for follow-up at the end of the interval
P = .81
*One patient expired before 1 month and one patient did not receive a device.
78 97.5 73 96.2 67 96.2 n/a n/a
Figure 6.4.2 presents AAA-related survival (determined by Clinical Events
Committee) to 24 months. The accompanying table presents the KaplanMeier analysis at 1, 6, 12 and 24 months.
Figure 6.4.2 AAA-Related Survival at 24 Months
Zenith Standard Risk
(N=200, 1 death to 30 days
1 death to 6 months
1 death to 12 months
2 deaths to 24 months)
Surgical Standard Risk
Relatd Death
(N=80, 2 deaths to 30 days
3 deaths to 6 months
3 deaths to 12 months)
Percent Freedom from Aneurysm
Event: Aneurysm Related Death
Months After Procedure
1 month 6 months 12 months 24 months
n % survival n % survival n % sur vival n % survival
Zenith
standard
198* 99.5 194 99.5 190 99.5 97 99.0
risk
Surgical
standard
risk
n= Patients alive and available for follow-up at the end of the interval
P = .04
*One patient expired before 1 month and one patient did not receive a device.
78 97.5 73 96.2 67 96.2 n/a n/a
27
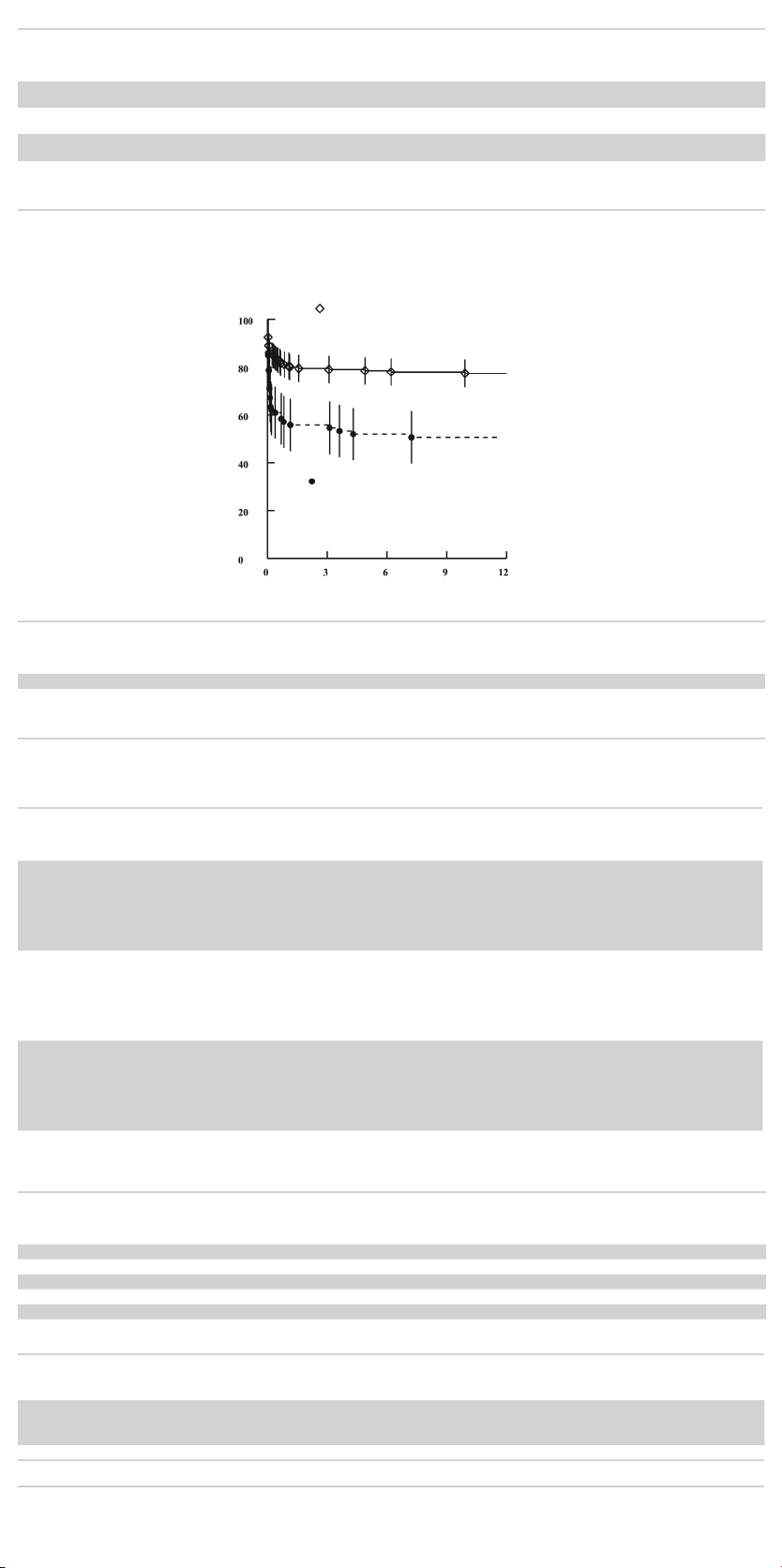
Table 6.4.3 Success Measures
Item Zenith Standard Risk Surgical Standard Risk Zenith High Risk Zenith Roll-in
Technical success
1
99.5% (199/200) 98.8% (79/80) 100% (100/100) 100% (52/52)
Procedural success at 30 days295.1% (155/163) 88% (60/68) 86% (70/81) 91% (30/33)
Treatment success at
3
12 months
1
Patent graft following deployment.
2
Technical success with no major complications, patent graft and no Type I or Type III endoleaks at 30 days.
3
Procedural success extended to 12 months with no aneurysm enlargement (>5 mm).
Figure 6.4.3 presents freedom from morbidity (events in the morbidity index) to 12 months. The accompanying table presents the Kaplan-Meier analysis at 1,
6 and 12 months.
89% (122/137) 85% (52/61) 70% (44/63) 87% (26/30)
Figure 6.4.3 Freedom from Morbidity (0-365 days)
Zenith Standard Risk
(N=200, 38 patients with events to 30 days
43 patients with events to 6 months
45 patients with events to 12 months)
Surgical Standard Risk
(N=80, 34 patients with events to 30 days
38 patients with events to 6 months
Percent Freedom from Morbidity
41 patients with events to 12 months)
Months After Procedure
1 month 6 months 12 months
n % n % n %
Zenith standard risk 162 81.0 154 78.5 133 77.4
Surgical standard risk 45 57.1 39 52.0 33 47.8
n= Patients alive and free of morbidity at the end of the interval
P = <.001
Tables 6.4.4 through 6.4.7 describe results of the Zenith AAA Endovascular Graft subjects as reported by the Core Lab. Device performance factors analyzed
by the Core Lab include device integrity (Table 6.4.4), device patency (Table 6.4.5), migration (Table 6.4.6) and limb separation (Table 6.4.7).
Table 6.4.4 Abdominal Radiographic Findings – Device Integrity
Item Zenith Standard Risk Zenith High Risk Zenith Roll-in
Stent Fractures
1
Pre-discharge 0.0% (0/172) 0.0% (0/81) 0.0% (0/39)
30 day 0.0% (0/172) 0.0% (0/83) 0.0% (0/43)
6 month 0.0% (0/166) 0.0% (0/78) 0.0% (0/35)
12 month 0.0% (0/148) 0.0% (0/60) 0.0% (0/28)
24 month 0.0% (0/93) 0.0% (0/42) 0.0% (0/19)
Barb Separation
2
Pre-discharge 0.0% (0/176) 0.0% (0/86) 0.0% (0/39)
30 day 0.0% (0/178) 0.0% (0/86) 0.0% (0/43)
6 month 1.2% (2/167) 2.5% (2/80) 0.0% (0/35)
12 month 2.0% (3/149) 1.7% (1/60) 0.0% (0/28)
24 month 1.1% (1/93) 0.0% (0/42) 0.0% (0/19)
Graft material rupture
Pre-discharge 0.0% (0/176) 0.0% (0/86) 0.0% (0/39)
30 day 0.0% (0/178) 0.0% (0/86) 0.0% (0/43)
6 month 0.0% (0/167) 0.0% (0/80) 0.0% (0/35)
12 month 0.0% (0/149) 0.0% (0/60) 0.0% (0/28)
24 month 0.0% (0/93) 0.0% (0/42) 0.0% (0/19)
1
Stent fracture percentages are for main body. There were also no right iliac leg, left iliac leg, occluder, converter, left iliac extension, right iliac extension or main body extension
fractures observed by the Core Lab.
2
Patients with separation of 1 or 2 barbs (of 10 or 12 total); no adverse clinical sequelae.
Table 6.4.5 CT Findings – Graft Patency
Item Zenith Standard Risk Zenith High Risk Zenith Roll-in
Graft patency
30 day 100% (185/185) 99% (85/86) 100% (47/47)
6 month 99% (183/184) 100% (74/74) 100% (39/39)
12 month 99% (153/155) 100% (62/62) 100% (30/30)
24 month 100% (96/96) 100% (33/33) 100% (25/25)
Table 6.4.6 CT Findings – Graft (Main Body) Migration
Item Zenith Standard Risk Zenith High Risk Zenith Roll-in
Graft migration (>5 mm) at 12 months
with clinical sequelae1 or intervention 0.0% (0/162) 0.0% (0/71) 0.0% (0/34)
without clinical sequelae1 or intervention 2.5% (4/162) 2.8% (2/71) 0.0% (0/34)
Graft migration (>10 mm) 0.0% (0/162) 0.0% (0/71) 0.0% (0/34)
1
Migration with clinical sequelae would include endoleak, conversion, rupture or AAA-related death.
28

Table 6.4.7 Abdominal Radiograph Findings – Limb Separation
Item Zenith Standard Risk Zenith High Risk Zenith Roll-in
Limb separation
Pre-discharge 0.0% (0/176) 0.0% (0/86) 0.0% (0/39)
30 day 0.0% (0/178) 0.0% (0/86) 0.0% (0/43)
6 month 0.0% (0/167) 0.0% (0/80) 0.0% (0/35)
12 month 0.0% (0/149) 0.0% (0/60) 0.0% (0/28)
24 month 0.0% (0/93) 0.0% (0/42) 0.0% (0/19)
6.5 Endoleak Management
During the clinical study Type I endoleaks were treated during the initial procedure by use of additional balloon seating or, if unsuccessful, additional
prostheses. Type II endoleaks were observed for a period of one to six months to determine if they would spontaneously thrombose, or in the absence of
enlarging aneurysms, they were treated with endovascular techniques at the discretion of the practicing physician. If the aneurysm enlarged, treatment by
embolization or ligation was considered and, in some cases, performed. Type III endoleaks caused by graft defects, inadequate seal or disconnection of the
modular components were treated with additional ballooning or prostheses. As reported by the angiographic Core Lab, there were no Type IV endoleaks
during the U.S. clinical study. The graft material used to manufacture the Zenith AAA Endovascular Graft is of standard thickness and is the same material used
in open surgical procedures. Table 6.5.1 presents the incidence of endoleaks by evaluation interval, as identified by the Core Lab for the standard risk, high
risk and roll-in patients, respectively.
Table 6.5.1 Endoleaks (All Types, New and Persistent)
Item Zenith Standard Risk Zenith High Risk Zenith Roll-in
Endoleaks
Pre-discharge 15% (23/153) 14% (11/78) 12% (3/26)
1
30-day
1
6-month
1
12-month
1
Includes both persistent endoleaks and new observations.
Tables 6.5.2 – 6.5.4 present the incidence of first occurrence of an endoleak according to evaluation interval, as identified by the Core Lab at or before the
30-day, 6-month and 12-month exams for the standard risk, high risk and roll-in patients, respectively. The number of patients who are leak-free thereafter is
also given.
9.9% (16/161) 12% (9/75) 6.3% (2/32)
8.7% (15/172) 11% (8/70) 8.6% (3/35)
7.4% (11/148) 8.8% (5/57) 3.4% (1/29)
Table 6.5.2 First Occurrence of Endoleak1 for Standard Risk Patients
Item
To One-Month Exam
N=179
% Endoleak
1
Leak-free
Thereafter
2
Six-Month Exam
N=172
% Endoleak
1
Leak-free
Thereafter
2
Twelve-Month Exam
N=148
% Endoleak
1
3
Leak-free
Thereafter
Endoleaks 17 31 17 2.3 4 3 3.4 5 2
Proximal Type I 2.8 5 4 0.0 0 0 0.0 0 0
Distal
Type I 1.7 3 1 0.0 0 0 0.7 1 1
Type II 9.5 17 9 2.3 4 3 1.4 2 1
Type III 1.1 2 2 0.0 0 0 0.7 1 0
Type IV 0.0 0 0 0.0 0 0 0.0 0 0
Multiple 1.1 2 1 0.0 0 0 0.0 0 0
Unknown 1.1 2 0 0.0 0 0 0.7 1 0
1
Identified by Core Lab.
2
Subsequent endoleaks may have been of different type than original.
3
Only 2 patients had new endoleaks after 12 months; follow-up after 24 months not available.
Table 6.5.3 First Occurrence of Endoleak1 for High Risk Patients
Item
To One-Month Exam
N=88
% Endoleak
1
Leak-free
Thereafter
2
Six-Month Exam
N=70
% Endoleak
1
Leak-free
Thereafter
2
Twelve-Month Exam3
N=57
% Endoleak
1
Thereafter
Endoleaks 18 16 6 2.9 2 0 3.5 2 1
Proximal Type I 2.3 2 1 0.0 0 0 0.0 0 0
Distal
Type I 1.1 1 1 0.0 0 0 0.0 0 0
Type II 9.1 8 3 1.4 1 0 1.8 1 0
Type III 0.0 0 0 1.4 1 0 1.8 1 1
Type IV 0.0 0 0 0.0 0 0 0.0 0 0
Multiple 4.5 4 0 0.0 0 0 0.0 0 0
Unknown 1.1 1 1 0.0 0 0 0.0 0 0
1
Identified by Core Lab.
2
Subsequent endoleaks may have been of different type than original.
3
No endoleaks after 12 months; follow-up after 24 months not available.
Leak-free
2
2
Table 6.5.4 First Occurrence of Endoleak1 for Roll-in Patients
Item
To One-Month Exam
N=36
% Endoleak
1
Leak-free
Thereafter
2
Six-Month Exam
N=35
% Endoleak
1
Leak-free
Thereafter
2
Twelve-Month Exam3
N=29
% Endoleak
1
Thereafter
Endoleaks 11 4 2 2.9 1 0 0.0 0 0
Proximal Type I 0.0 0 0 0.0 0 0 0.0 0 0
Distal
Type I 0.0 0 0 0.0 0 0 0.0 0 0
Type II 5.6 2 1 2.9 1 0 0.0 0 0
Type III 2.8 1 0 0.0 0 0 0.0 0 0
Type IV 0.0 0 0 0.0 0 0 0.0 0 0
Multiple 0.0 0 0 0.0 0 0 0.0 0 0
Unknown 2.8 1 1 0.0 0 0 0.0 0 0
1
Identified by Core Lab.
2
Subsequent endoleaks may have been of different type than original.
3
No endoleaks after 12 months; follow-up after 24 months not available.
29
Leak-free
2

6.6 Aneurysm Change
Tables 6.6.1 - 6.6.3 present the change in aneurysm diameter for the endovascular patients, as identified by the Core Lab. Table 6.6.1 presents maximum
aneurysm diameter change by interval. Tables 6.6.2 and 6.6.3 present aneurysm change and endoleak at 12 and 24 months, respectively.
Table 6.6.1 Change in Maximum Aneurysm Diameter by Interval*
Item Zenith Standard Risk Zenith High Risk Zenith Roll-in
From pre-discharge 30-day
Decrease >5 mm 1.7% (3/180) 4.8% (4/84) 0.0% (0/40)
Unchanged 97% (174/180) 94% (79/84) 97.5% (39/40)
Increase >5 mm 1.7% (3/180) 1.2% (1/84) 2.5% (1/40)
Change from pre-discharge 6-month
Decrease >5 mm 36% (63/173) 41% (30/73) 49% (18/37)
Unchanged 62% (108/173) 59% (43/73) 51% (19/37)
Increase >5 mm 1.2% (2/173) 0.0% (0/73) 0.0% (0/37)
Change from pre-discharge 12-month
Decrease >5 mm 68% (102/151) 63% (39/62) 67% (20/30)
Unchanged 31% (47/151) 35% (22/62) 33% (10/30)
Increase >5 mm 1.3% (2/151) 1.6% (1/62) 0.0% (0/30)
Change from pre-discharge 24-month
Decrease >5 mm 78% (74/94) 75% (27/36) 71% (17/24)
Unchanged 19% (18/94) 25% (9/36) 25% (6/24)
Increase >5 mm 2.1% (2/94) 0.0% (0/36) 4.2% (1/24)
*Only includes subjects with interpretable films and measurements of aneurysm change from 1 to 24 months.
Table 6.6.2 Change in Aneurysm Size and Endoleak at 12 Months
Zenith Standard Risk
N=140
Zenith High Risk
N=53
Zenith Roll-in
N=27
Endoleak Endoleak Endoleak
Item
Aneurysm size change from pre-discharge to 12 months
N n % N n % N n %
Decrease >5 mm 96 3 3.0 35 3 9.0 20 0 0.0
Unchanged 42 8 19 17 2 12 7 1 14
Increase >5 mm 2 0 0.0 1 0 0.0 0 0 0.0
Table 6.6.3 Change in Aneurysm Size and Endoleak at 24 Months
Zenith Standard Risk
N=90
Zenith High Risk
N=31
Zenith Roll-in
N=21
Endoleak Endoleak Endoleak
Item
Aneurysm size change from pre-discharge to 24 months
N n % N n % N n %
Decrease >5 mm 71 3 4.0 24 1 4.0 16 0 0.0
Unchanged 18 1 6.0 7 3 43 5 0 0.0
Increase >5 mm 1 1 100 1 0 0.0 0 0 0.0
6.7 AAA-Related Secondary Interventions
AAA-related secondary interventions within the first year were performed in 11% of the Zenith standard risk, 13% Zenith high risk and 5.8% Zenith roll-in
subjects as shown in Table 6.7.1. Greater than 50% of the secondary interventions involved catheterization to treat an endoleak. AAA-related secondary
interventions within the second year were performed in 4.2% of the Zenith standard risk, 2.2% Zenith high risk and 2.3% Zenith roll-in subjects as shown in
Table 6.7.2.
Table 6.7.1 Secondary Interventions (to 12 Months)
Intervention
Conversion to open repair 2 1.0 1 1.0 0 0.0
Zenith Standard Risk
N=199
Zenith High Risk
N=100
Zenith Roll-in
N=52
n % n % n %
Subjects with ≥1 intervention 21 11 13 13 3 5.8
Treat an endoleak
Embolization 5 2.5 3 3.0 1 2.0
Ancillary component 6 3.0 4 4.0 1 2.0
Stent 1 0.5 0 0.0 0 0.0
Angioplasty 0 0.0 1 1.0 0 0.0
Treat an aneurysm increase
Embolization 0 0.0 0 0.0 0 0.0
Treat a limb occlusion 1 0.5 3 3.0 1 2.0
Treat a limb stenosis 1 0.5 0 0.0 0 0.0
Treat a renal artery 5 2.5 0 0.0 0 0.0
Treat infra-inguinal ischemia 1 0.5 1 1.0 0 0.0
Treat multiple events 1 0.5 1 1.0 0 0.0
Table 6.7.2 Secondary Interventions (>12 to 24 Months)
Intervention
Conversion to open repair 1 0.5 1 1.1 0 0.0
Zenith Standard Risk
N=190
Zenith High Risk
N=90
Zenith Roll-in
N=44
n % n % n %
Subjects with ≥1 intervention 8 4.2 2 2.2 1 2.3
Treat an endoleak
Embolization
Ancillary component 1 0.5 0 0.0 0 0.0
1
3
1.6 0 0.0
2
1
Stent 0 0.0 1 1.1 0 0.0
Angioplasty 0 0.0 0 0.0 0 0.0
Treat an aneurysm increase
Embolization 1 0.5 0 0.0
Treat a limb occlusion 1 0.5 0 0.0 0 0.0
2
1
Treat a limb stenosis 0 0.0 0 0.0 0 0.0
Treat a renal artery 0 0.0 1 1.1 0 0.0
Treat infra-inguinal ischemia 1 0.5 0 0.0 0 0.0
Treat multiple events 1 0.5 0 0.0 0 0.0
1
Patient also received ancillary component.
2
Patient underwent intervention to treat aneurysm increase and endoleak.
30
2.3
2.3

6.8 Secondary Outcome Measures
As described in Table 6.8.1, treatment of AAA with the Zenith AAA Endovascular Graft compared to the surgical control group demonstrated significant
benefits in recovery and quality of life measures.
Table 6.8.1 Secondary Outcomes by Treatment Group
Item Zenith Standard Risk Surgical Standard Risk P value Zenith High Risk Zenith Roll-in
Anesthesia time (min) 221.6 ± 67.3 304.5 ± 102.7 <.001 218.9 ± 69.6 213.9 ± 57.7
Procedure time (min) 153.2 ± 56.3 238.7 ± 92.2 <.001 153.5 ± 58.6 155.9 ± 43.2
Blood bank products received 5.0% (10/200) 84% (67/80) <.001 12% (12/100) 3.8% (2/52)
Blood loss (cc) 299 ± 324 1676 ± 1676 <.001 356 ± 514 265 ± 226
Days in ICU 0.4 ± 0.9 3.4 ± 4.6 <.001 0.5 ± 1.2 0.5 ± 0.9
Days to discharge 2.6 ± 1.7 8.8 ± 5.6 <.001 3.0 ± 2.8 2.7 ± 1.5
Days to oral fluids 0.5 ± 0.8 3.9 ± 2.5 <.001 0.5 ± 0.6 0.7 ± 0.5
Days to normal diet 1.3 ± 1.2 6.6 ± 4.9 <.001 1.3 ± 0.8 1.1 ± 0.7
Days to normal bowel function 2.6 ± 1.4 4.2 ± 2.1 <.001 2.6 ± 1.5 2.0 ± 1.2
Days to ambulation 1.2 ± 0.7 3.5 ± 3.4 <.001 1.2 ± 0.7 1.2 ± 0.6
Hours of intubation 1.9 ± 2.2 11.7 ± 13.6 <.001 1.2 ± 1.7 2.6 ± 4.6
Maximum temperature 101.1 ± 1.3 100.7 ± 1.2 .06 100.8 ± 1.1 101.0 ± 1.2
7 PATIENT SELEC TION AND TREATMENT
See Section 4, WARNINGS AND PRECAUTIONS
Individualization of Treatment
Cook recommends that the Zenith Flex AAA Endovascular Graft component
diameters be selected as described in Tables 10.5.1 and 10.5.2. The length
of the Zenith Flex AAA Endovascular Graft should extend from the lowest
renal artery to just above the internal iliac (hypogastric) artery bifurcation. All
lengths and diameters of the devices necessary to complete the procedure
should be available to the physician, especially when preoperative case
planning measurements (treatment diameters/lengths) are not certain. This
approach allows for greater intraoperative flexibility to achieve optimal
procedural outcomes. The risks and benefits previously described in Section
6, SUMMARY OF CLINICAL STUDIES should be carefully considered for each
patient before use of the Zenith Flex AAA Endovascular Graft. Additional
considerations for patient selection include but are not limited to:
• Patient's age and life expectancy
• Co-mor bidities (e.g., cardiac, pulmonary or renal insufficiency prior to
surgery, morbid obesity)
• Patient's suitability for open surgical repair
• Patient's anatomical suitability for endovascular repair
• The risk of aneur ysm rupture compared to the risk of treatment with the
Zenith Flex AAA Endovascular Graft
• Abilit y to tolerate general, regional or local anesthesia
• I liofemoral access vessel size and morphology (minimal thrombus, calcium
and/or tortuosity) should be compatible with vascular access techniques
and accessories of the delivery profile of a 14 French to 22 French vascular
introducer sheath
• Non-aneur ysmal infrarenal aortic segment (neck) proximal to the aneurysm:
• with a length of at least 15 mm,
• with a diameter measured outer wall to outer wall of no greater than
32 mm and no less than 18 mm,
• with an angle less than 60 degrees relative to the long axis of the
aneurysm, and
• with an angle less than 45 degrees relative to the axis of the suprarenal
aorta
• Iliac artery distal fixation site greater than 10 mm in length and 7.5 to
20 mm in diameter (measured outer wall to outer wall)
• Freedom from significant femoral/iliac artery occlusive disease that would
impede flow through the endovascular graft
The final treatment decision is at the discretion of the physician and patient.
8 PATIENT COUNSELING INFORMATION
The physician and patient (and/or family members) should review the risks
and benefits when discussing this endovascular device and procedure,
including:
• R isks and differences between endovascular repair and surgical repair
• Potential advantages of traditional open surgical repair
• Potential advantages of endovascular repair
• The possibility that subsequent inter ventional or open surgical repair of the
aneurysm may be required after initial endovascular repair
In addition to the risks and benefits of an endovascular repair, the physician
should assess the patient's commitment and compliance to postoperative
follow-up as necessary to ensure continuing safe and effective results. Listed
below are additional topics to discuss with the patient as to expectations after
an endovascular repair:
• The long-term performance of endovascular grafts has not yet been
established. All patients should be advised that endovascular
treatment requires lifelong, regular follow-up to assess their health
and the performance of their endovascular graft. Patients with specific
clinical findings (e.g., endoleaks, enlarging aneurysms or changes in the
structure or position of the endovascular graft) should receive enhanced
follow-up. Specific follow-up guidelines are described in Section 12,
IMAGING GUIDELINES AND POSTOPERATIVE FOLLOW-UP.
• Patients should be counseled on the importance of adhering to the followup schedule, both during the first year and at yearly intervals thereafter.
Patients should be told that regular and consistent follow-up is a critical
part of ensuring the ongoing safety and effectiveness of endovascular
treatment of AAAs. At a minimum, annual imaging and adherence to
routine postoperative follow-up requirements is required and should be
considered a lifelong commitment to the patient’s health and well-being.
• The patient should be told that successful aneurysm repair does not arrest
the disease process. It is still possible to have associated degeneration of
vessels.
• Physicians must advise each patient that it is important to seek prompt
medical attention if he/she experiences signs of limb occlusion, aneurysm
enlargement or rupture. Signs of graft limb occlusion include pain in the
hip(s) or leg(s) during walking or at rest or discoloration or coolness of the
leg. Aneurysm rupture may be asymptomatic, but usually presents as: pain;
numbness; weakness in the legs; any back, chest, abdominal or groin pain;
dizziness; fainting; rapid heartbeat or sudden weakness.
• Due to the imaging required for successful placement and follow-up of
endovascular devices, the risks of radiation exposure to developing tissue
should be discussed with women who are or suspect they are pregnant.
Men who undergo endovascular or open surgical repair may experience
impotence.
Physicians should refer the patient to the Patient Guide regarding risks
occurring during or after implantation of the device. Procedure related risks
include cardiac, pulmonary, neurologic, bowel and bleeding complications.
Device related risks include occlusion, endoleak, aneurysm enlargement,
fracture, potential for reintervention and open surgical conversion, rupture
and death (See Section 5.1, Observed Adverse Events and Section 5.2,
Potential Adverse Events). The physician should complete the Patient ID Card
and give it to the patient so that he/she can carry it with him/her at all times.
The patient should refer to the card anytime he/she visits additional health
practitioners, particularly for any additional diagnostic procedures (e.g., MRI).
9 HOW SUPPLIED
• The Zenith Flex AAA Endovascular Graft is supplied sterile and preloaded in
peel-open packages.
• The device is intended for single use only. Do not re-sterilize the device.
• I nspect the device and packaging to verify that no damage has occurred as
a result of shipping. Do not use this device if damage has occurred or if the
sterilization barrier has been damaged or broken. If damage has occurred,
do not use the product and return to COOK.
• Prior to use, verify correct devices (quantity and size) have been supplied for
the patient by matching the device to the order prescribed by the physician
for that particular patient.
• The main body device is loaded onto an 18, 20 or 22 French Flexor®
introducer sheath. The sheath’s surface is treated with a hydrophilic coating
that, when hydrated, enhances trackability. To activate the hydrophilic
coating, the surface must be wiped with a sterile gauze pad soaked in saline
solution.
• Do not use af ter the "USE BY" (expiration) date printed on the label.
• Store in a cool, dry place.
10 CLINICAL USE INFORMATION
10.1 Physician Training
CAUTION: Always have a qualified surgery team available during
implantation or re-intervention procedures in the event that conversion
to open surgical repair is necessary.
CAUTION: The Zenith Flex AAA Endovascular Graft with the H&L-B
One-Shot Introduction System should only be used by physicians and
teams trained in vascular interventional techniques and in the use of this
device. The recommended skill/knowledge requirements for physicians
using the Zenith Flex AAA Endovascular Graft with the H&L-B One-Shot
Introduction System are outlined below:
Patient selection:
• K nowledge of the natural history of abdominal aortic aneurysms (AAA) and
co-morbidities associated with AAA repair.
• K nowledge of radiographic image interpretation, device selection, planning
and sizing.
A multidisciplinary team that has combined procedural experience with:
• Femoral cutdown, arteriotomy and repair
• Percutaneous access and closure techniques
• Non-selec tive and selective wire guide and catheter techniques
• Fluoroscopic and angiographic image interpretation
• Embolization
• Angioplasty
• Endovascular stent placement
• Snare techniques
• Appropriate use of radiographic contrast material
• Techniques to minimize radiation exposure
• Exper tise in necessary patient follow-up modalities
10.2 Inspection Prior to Use
Inspect the device and packaging to verify that no damage has occurred as
a result of shipping. Do not use this device if damage has occurred or if the
sterilization barrier has been damaged or broken. If damage has occurred, do
not use the product and return to COOK. Prior to use, verify correct devices
(quantity and size) have been supplied for the patient by matching the device
to the order prescribed by the physician for that particular patient.
10.3 Materials Required
(Not included in 3-piece modular system)
• Zenith AAA Endovascular Graft Ancillar y Kit
• Fluoroscope with digital angiography capabilities (C-arm or fixed unit)
• Contrast media
• Syringe
• Heparinized saline solution
• Sterile gauze pads
10.4 Materials Recommended
(Not included in 3-piece modular system)
The following products are recommended for implantation of any component
in the Zenith product line. For information on the use of these products, refer
to the individual product's Suggested Instructions for Use.
• .035 inch (0.89 mm) ex tra stiff wire guide, 260 cm; for example:
• Cook Lunderquist Extra Stiff Wire Guides (LES)
• .035 inch (0.89 mm) standard wire guide; for example:
• Cook .035 inch wire guides
• Cook Nimble™ Wire Guides
• M olding Balloons; for example:
• Cook CODA® Balloon Catheter
• Introducer sets; for example:
• Cook Check-Flo® Introducer Sets
• Cook Extra Large Check-Flo® Introducer Sets
• Cook Flexor® Balkin Up & Over® Contralateral Introducers
• Sizing catheter; for example:
• Cook Aurous® Centimeter Sizing Catheters
• Angiographic radiopaque tip catheters; for example:
• Cook Beacon® Tip Angiographic Catheters
• Cook Beacon® Tip Royal Flush Catheters
• Entr y needles; for example:
• Cook single wall entr y needles
31

10.5 Device Diameter Sizing Guidelines
The choice of diameter should be determined from the outer wall to outer
wall vessel diameter and not the lumen diameter. Undersizing or oversizing
may result in incomplete sealing or compromised flow.
Table 10.5.1 Main Body Graft Diameter Sizing Guide*
Intended Aortic
Vessel
Diameter
(mm)
18-19 22 82/112, 96/126, 111/141,
20-21 24 82/112, 96/126, 111/141,
23-24 28 82/112, 96/126, 111/141,
25-26 30 82/112, 96/126, 111/141,
27-28 32 82/112, 96/126, 111/141,
29-32 36 95/125, 113/143,
1
Maximum diameter along the proximal fixation site.
2
Round measured aortic diameter to nearest mm.
3
Additional considerations may affect choice of diameter.
*All dimensions are nominal.
Main Body
1,2
Diameter
(mm)
22 26 82/112, 96/126, 111/141,
Table 10.5.2 Iliac Leg Graft Diameter Sizing Guide
Intended
Iliac Vessel
Diameter
(mm)
<8 8 37, 54, 71, 88, 105, 122 14
8-9 10 37, 54, 71, 88, 105, 122 14
10-11 12 37, 54, 71, 88, 105, 122 14
12-13 14 37, 54, 71, 88 14
14-15 16 37, 54, 71, 88 14
16-17 18 37, 54, 71, 88 16
18 20 37, 54, 71, 88 16
19 22 37, 54, 71, 88 16
20 24 37, 54, 71, 88 16
1
Maximum diameter along the distal fixation site.
2
Round measured iliac diameter to nearest mm.
3
Additional considerations may affect choice of diameter.
4
Overall leg length = working length + 22 mm docking stent.
*All dimensions are nominal.
11 DIRECTIONS FOR USE
Anatomical Requirements
• I liofemoral access vessel size and morphology (minimal thrombus, calcium
and/or tortuosity) should be compatible with vascular access techniques
and accessories. Arterial conduit techniques may be required.
• Proximal aortic neck lengths should be a minimum of 15 mm with a
diameter measured outer wall to outer wall of 18 – 32 mm.
• I liac artery distal fixation site should be greater than 10 mm in length and
7.5 – 20 mm in diameter (measured outer wall to outer wall).
Prior to use of the Zenith Flex AAA Endovascular Graft with the H&L-B OneShot Introduction System, review this Suggested Instructions for Use booklet.
The following instructions embody a basic guideline for device placement.
Variations in the following procedures may be necessary. These instructions
are intended to help guide the physician and do not take the place of
physician judgment.
Iliac Leg
1,2
Diameter
(mm)
Overall Length to
Contralateral Limb/Overall
3
Length to Ipsilateral Limb
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
131/161, 149/179
Iliac Leg Working
3
(mm)
Length
(mm)
Introducer
Sheath
(Fr)
18
18
18
20
20
20
22
*
4
Introducer
Sheath
(Fr)
General Use Information
• Standard techniques for placement of arterial access sheaths, guiding
catheters, angiographic catheters and wire guides should be employed
during use of the Zenith Flex AAA Endovascular Graft with the H&L-B OneShot Introduction System. The Zenith Flex AAA Endovascular Graft with the
H&L-B One-Shot Introduction System is compatible with .035 inch diameter
wire guides.
• Endovascular stent grafting is a surgical procedure, and blood loss from
various causes may occur, infrequently requiring intervention (including
transfusion) to prevent adverse outcomes. It is important to monitor blood
loss from the hemostatic valve throughout the procedure, but is specifically
relevant during and after manipulation of the gray positioner. After the gray
positioner has been removed, if blood loss is excessive, consider placing an
uninflated molding balloon or an introduction system dilator within the
valve, restricting flow.
Pre-Implant Determinants
Verify from pre-implant planning that the correct device has been selected.
Determinants include:
1. Femoral artery selection for introduction of the main body system (i.e.,
define respective contralateral and ipsilateral iliac arteries).
2. Angulation of aortic neck, aneurysm and iliac ar teries.
3. Quality of the aortic neck.
4. Diameters of infrarenal aortic neck and distal iliac arteries.
5. Distance from renal arteries to the aortic bifurcation.
6. Length from the aortic bifurcation to the internal iliac arteries/attachment
site(s).
7. Aneurysm(s) extending into the iliac arteries may require special
consideration in selecting a suitable graft/artery interface site.
8. Consider the degree of vascular calcification.
Patient Preparation
1. Refer to institutional protocols relating to anesthesia, anticoagulation and
monitoring of vital signs.
2. Position patient on imaging table allowing fluoroscopic visualization from
the aortic arch to the femoral bifurcations.
3. Expose both common femoral arteries using standard surgical technique.
4. Establish adequate proximal and distal vascular control of both femoral
vessels.
11.1 Bifurcated System (Fig. 2)
11.1.1 Bifurcated Main Body Preparation/Flush
1. Remove black-hubbed shipping stylet (from the inner cannula), cannula
protector tube (from the inner cannula) and dilator tip protector (from
the dilator tip). Remove Peel-Away® sheath from back of hemostatic valve.
(Fig. 5) Elevate distal tip of system and flush through the stopcock on
the hemostatic valve until fluid emerges from the sideport near the tip of
the introducer sheath. (Fig. 6) Continue to inject a full 20 cc of flushing
solution through the device. Discontinue injection and close stopcock on
connecting tube.
NOTE: Graft flushing solution of heparinized saline is often used.
2. Attach syringe with heparinized saline to the hub on the inner cannula.
Flush until fluid exits the dilator tip. (Fig. 7)
NOTE: When flushing system, elevate distal end of system to facilitate
removal of air.
3. Soak sterile gauze pads in saline solution and use to wipe Flexor®
introducer sheath to activate the hydrophilic coating. Hydrate both
sheath and dilator liberally.
11.1.2 Contralateral Iliac Leg Preparation/Flush
1. Remove black-hubbed inner stylet (from the inner cannula), cannula
protector tube (from the inner cannula) and dilator tip protector (from
the dilator tip). Remove Peel-Away® sheath from back of the hemostatic
valve. (Fig. 8) Elevate distal tip of system and flush through the stopcock
on the hemostatic valve until fluid emerges from the sideport near the tip
of the introducer sheath. (Fig. 9) Continue to inject a full 20 cc of flushing
solution through the device. Discontinue injection and close stopcock on
connecting tube.
NOTE: Graft flushing solution of heparinized saline is often used.
2. Attach syringe with heparinized saline to the hub on the distal inner
cannula. Flush until fluid exits the distal dilator tip. (Fig. 7)
NOTE: When flushing system, elevate distal end of system to facilitate
removal of air.
11.1.3 Ipsilateral Iliac Leg Preparation/Flush
Follow the previous Section 11.1.2, Contralateral Iliac Leg Preparation/Flush
instructions to ensure proper flushing of the ipsilateral iliac leg graft.
11.1.4 Vascular Access and Angiography
1. Puncture the selected common femoral arteries using standard technique
with an 18 or 19 UT gage arterial needle. Upon vessel entry, insert:
• Wire guides — standard .035 inch diameter, 145 cm long, J tip or
Bentson Wire Guide
• Appropriate size sheaths (e.g., 6.0 or 8.0 French)
• Flush catheter (often radiopaque sizing catheters – e.g., Centimeter
Sizing Catheter or straight flush catheter)
2. Perform angiography to identify level(s) of renals, aortic bifurcation and
iliac bifurcations.
NOTE: If fluoroscope angulation is used with an angulated neck it may be
necessary to perform angiograms using various projections.
11.1.5 Main Body Placement
1. Ensure the delivery system has been flushed with heparinized saline and
that all air is removed from the system.
2. Give systemic heparin and check flushing solutions. Flush after each
catheter and/or wire guide exchange.
NOTE: Monitor the patient’s coagulation status throughout the procedure.
3. On ipsilateral side, replace J wire with stiff wire guide (LES) .035 inch,
260 cm long and advance through catheter and up to the thoracic aorta.
Remove flush catheter and sheath. Maintain wire guide position.
4. Before insertion, position main body delivery system on patient’s
abdomen under fluoroscopy to determine the orientation of the
contralateral limb radiopaque marker. The sidearm of the hemostatic
valve may serve as an external reference to the contralateral limb
radiopaque marker.
5. Insert main body delivery system over the wire, into the femoral artery
with attention to sidearm reference.
CAUTION: Maintain wire guide position during delivery system insertion.
CAUTION: To avoid any twist in the endovascular graft, during any
rotation of the delivery system, be careful to rotate all of the components
of the system together (from outer sheath to inner cannula).
6. Advance delivery system until the four gold radiopaque markers (which
are positioned 2 mm from the most proximal segment of the graft
material) (Fig. 10, Illustration 1) are just inferior to the most inferior renal
orifice.
7. Verify position of wire guide in the thoracic aorta. Ensure the graft system
is oriented such that the contralateral limb is positioned above and
anterior to the origin of the contralateral iliac. If the contralateral limb
radiopaque marker is not properly aligned, rotate the entire system until it
is correctly positioned half way between a lateral and an anterior position
on the contralateral side.
• A marker formation of a ✓ indicates an anterior position of the short
(contralateral) limb. (Fig. 10, Illustration 4)
• A marker formation of a indicates a posterior position of the short
(contralateral) limb. (Fig. 10, Illustration 5)
• A marker formation of a | indicates a lateral position of the short
(contralateral) limb. (Fig. 10, Illustration 6)
NOTE: Radiopaque cannula on each stent between the renal arteries and the
contralateral limb align with the contralateral limb radiopaque marker.
8. Repeat the angiogram to verify the four gold radiopaque markers are
2 mm or more below the most inferior renal orifice.
09. Ensure the Captor Hemostatic Valve on the Flexor® introducer sheath is
turned to the open position. (Fig. 11)
10. Stabilize the gray positioner (the shaft of the delivery system) while
withdrawing the sheath. Deploy the first two (2) covered stents by
withdrawing the sheath while monitoring device location.
11. Without moving the table, decrease magnification to check position of
the contralateral limb radiopaque marker and location of renal arteries.
Proceed with deployment until the contralateral limb is fully deployed.
(Fig. 12) Stop withdrawing sheath.
NOTE: Verify contralateral limb is at least 5 mm above the aortic bifurcation
and in desired location for cannulation.
11.1.6 Contralateral Iliac Wire Guide Placement
1. Manipulate catheter and wire guide through open end of contralateral
limb into body of the graft. Advance the wire guide until it curves inside
the body of the graft. AP and oblique fluoroscopic views can aid in
verification of device cannulation.
2. After cannulation, advance the angiographic catheter over the wire
into the body of the endovascular graft. Remove wire and perform
angiography to confirm position. Advance wire guide until it curves inside
the body of the graft. Remove angiographic catheter. (Fig. 13)
32

11.1.7 Main Body Proximal (Top) Deployment
1. Perform angiography through an angiographic catheter to verify position
of the endovascular graft with respect to the renal arteries. If necessary,
carefully reposition the covered portion of the endovascular graft with
respect to the renal arteries. (Repositioning can only take place over a
small range of distance at this stage.)
NOTE: Ensure patency of renal arteries by confirming that the proximal graft
markers are 2 mm or more below the lowest patent renal artery.
CAUTION: During proximal trigger-wire removal, top cap advancement,
and subsequent suprarenal stent deployment, verify that the position of
the main body wire guide extends just distal to the aortic arch and that
support of the system is maximized.
2. Remove the safety lock from the black trigger-wire release mechanism.
Under fluoroscopy, withdraw and remove the trigger-wire by sliding the
black trigger-wire release mechanism off the handle and then remove via
its slot over the inner cannula. (Fig. 14)
If resistance is felt or system bowing is noticed, the trigger-wire is under
tension. Excessive force may cause the graft position to be altered. If
excessive resistance or delivery system movement is noted, stop and assess
the situation. If unable to remove the black trigger-wire release mechanism
from the top cap, perform the following steps under fluoroscopy:
a. Remove tension on the trigger-wire by loosening the pin vise and
slightly pulling the inner cannula to move the top cap down over the
suprarenal stent. Avoid compressing the Zenith Flex main body.
b. Retighten the pin vise.
c. Remove the black trigger-wire release mechanism.
d. Continue with Step 3 in Section 11.1.7, Main Body Proximal (Top )
Deployment.
NOTE: If still unable to remove the top stent trigger-wire release mechanism
from the top cap, see Section 14.1, Trigger-Wire Release Troubleshooting.
3. Loosen the pin vise. (Fig. 15) Control the position of the graft by
stabilizing the gray positioner of the introducer.
4. Deploy the suprarenal stent by advancing the top cap inner cannula 1 to
2 mm at a time while controlling the position of the main body until the
top stent is fully deployed. (Fig. 16 and 17) Advance the top cap cannula
an additional 1 to 2 cm and then retighten the pin vise to avoid contact
with the deployed suprarenal stent.
NOTE: If unable to fully deploy suprarenal stent by advancing the top
cap inner cannula, see Section 14.2, Suprarenal Stent Deployment
Troubleshooting.
NOTE: Once the barbed suprarenal stent has been deployed, futher attempts
to reposition the graft are not recommended.
WARNING: The Zenith Flex AAA Endovascular Graft incorporates a
suprarenal stent with fixation barbs. Exercise extreme caution when
manipulating interventional devices in the region of the suprarenal
stent.
5. Advance the contralateral wire guide into the thoracic aorta.
11.1.8 Contralateral Iliac Leg Placement and Deployment
CAUTION: Verify that the predetermined contralateral iliac leg is selected
for insertion on the contralateral side of the patient before implantation.
1. Position the image intensifier to show both the contralateral internal iliac
artery and contralateral common iliac artery.
2. Prior to introduction of contralateral iliac leg delivery system, inject
contrast through the contralateral femoral sheath to locate the
contralateral internal iliac artery.
3. Introduce the contralateral iliac leg delivery system into the artery.
Advance slowly until the iliac leg graft overlaps at least one full iliac leg
stent (i.e., proximal stent of iliac leg graft) inside the contralateral limb of
the main body. (Fig. 18) If there is any tendency for the main body graft
to move during this maneuver, hold it in position by stabilizing the gray
positioner on the ipsilateral side.
NOTE: If difficulty is encountered advancing the iliac leg delivery system,
exchange to a more supportive wire guide. In tortuous vessels the anatomy may
alter significantly with the introduction of the rigid wires and sheath systems.
4. Confirm position of distal end of the iliac leg graft. Reposition the iliac leg
graft if necessary to ensure both internal iliac patency and a minimum
overlap of one full iliac leg stent (i.e., proximal stent of iliac leg graft,
maximum overlap of 1.5 stents) within the main body endovascular graft.
5. To deploy, hold the iliac leg graft in position with the gray positioner
while withdrawing the sheath. (Fig. 19 and 20) Ensure one stent overlap
is maintained.
6. Stop withdrawing the sheath as soon as the distal end of the iliac leg graft
is released.
7. Under fluoroscopy and after verification of iliac leg graft position, loosen
pin vise, retract inner cannula to dock tapered dilator to gray positioner.
Tighten pin vise. Maintain sheath position while withdrawing gray
positioner with secured inner cannula. (Fig. 21)
8. Re-check the position of the wire guide.
11.1.9 Main Body Distal (Bottom) Deployment
1. Return to the ipsilateral side.
2. Fully deploy the ipsilateral limb of the main body by withdrawing the sheath
until the most distal stent has expanded. (Fig. 22 and 23) Stop withdrawing
sheath.
NOTE: The distal stent is still secured by the trigger-wire.
3. Remove the safety lock from the white trigger-wire release mechanism.
Withdraw and remove the trigger-wire by sliding the white trigger-wire
release mechanism off the handle and then remove via its slot over the
device inner cannula. (Fig. 24)
11.1.10 Docking of Top Cap
1. Loosen the pin vise. (Fig. 25)
2. Secure sheath and inner cannula to avoid any movement of these
components.
3. Advance the gray positioner over the inner cannula until it docks with the
top cap. (Fig. 26, 27 and 28)
NOTE: If resistance occurs, slightly rotate gray positioner and continue to
gently advance.
4. Retighten the pin vise and withdraw the entire top cap and gray
positioner through the graft and through the sheath by pulling on the
inner cannula. (Fig. 29) Leave the sheath and wire guide in place.
NOTE: Maintain position of sheath and wire guide.
5. Close the Captor® Hemostatic Valve on the Flexor® introducer sheath by
turning it in a clockwise direction until it stops. (Fig. 30)
11.1.11 Ipsilateral Iliac Leg Placement and Deployment
NOTE: Ensure the Captor Hemostatic Valve on the introducer sheath is turned
to the open position. (Fig. 31)
1. Utilize the main body graft wire and sheath assembly to introduce the
ipsilateral iliac leg graft. Advance dilator and sheath assembly into the
main body sheath.
NOTE: In tortuous vessels, the position of the internal iliac arteries may alter
significantly with the introduction of the rigid wires and sheath systems.
2. Advance slowly until the ipsilateral iliac leg graft overlaps a minimum
of one full iliac leg stent (i.e., proximal stent of iliac leg graft) inside the
ipsilateral limb of the main body. (Fig. 32)
NOTE: If an overlap of greater than 3 iliac leg stents is required (greater than
2 iliac leg stents for 37 mm and 54 mm leg lengths), it may be necessary to
consider use of a leg extension in the bifurcation area of the opposite side.
3. Confirm position of distal end of the iliac leg graft. Reposition the iliac leg
graft if necessary to ensure internal iliac patency.
4. To deploy, stabilize the iliac leg graft with the gray positioner while
withdrawing the iliac leg sheath. (Fig. 33 and 34) If necessary, withdraw
the main body sheath.
5. Under fluoroscopy and after verification of iliac leg graft position, loosen
pin vise, retract inner cannula to dock tapered dilator to gray positioner.
Tighten pin vise. Maintain sheath position while withdrawing gray
positioner with secured inner cannula. (Fig. 35)
6. Close the Captor Hemostatic Valve on the Flexor® introducer sheath by
turning it in a clockwise direction until it stops.
7. Re-check the position of the wire guides. Leave sheath and wire guides in
place.
11.1.12 Molding Balloon Insertion
1. Prepare molding balloon as follows:
• Flush wire lumen with heparinized saline.
• Remove all air from balloon.
2. In preparation of the insertion of the molding balloon, open the Captor
Hemostatic Valve by turning counterclockwise.
3. Advance the molding balloon over the wire guide and through the Captor
Hemostatic Valve of the main body introduction system to the level of the
renal arteries. Maintain proper sheath position.
4. Tighten the Captor Hemostatic Valve around the molding balloon with
gentle pressure by turning it clockwise.
CAUTION: Do not inflate balloon in vessel outside of graft.
5. Expand the molding balloon with diluted contrast media (as directed by
the manufacturer) in the area of the most proximal covered stent and the
infrarenal neck, starting proximally and working in the distal direction.
(Fig. 36)
CAUTION: Confirm complete deflation of balloon prior to repositioning.
CAUTION: Captor Hemostatic Valve must be open prior to repositioning
of molding balloon.
6. Withdraw the molding balloon to the ipsilateral limb overlap and expand.
CAUTION: Captor Hemostatic Valve must be open prior to repositioning
of molding balloon.
7. Withdraw the molding balloon to the ipsilateral distal fixation site and
expand.
CAUTION: Do not inflate balloon in vessel outside of graft.
8. Deflate and remove molding balloon. Transfer the molding balloon
onto the contralateral wire guide and into the contralateral iliac leg
introduction system. Advance molding balloon to the contralateral limb
overlap and expand.
CAUTION: Confirm complete deflation of balloon prior to repositioning.
9. Withdraw the molding balloon to the contralateral iliac leg/vessel distal
fixation site and expand. (Fig. 36)
CAUTION: Do not inflate balloon in vessel outside of graft.
10. Remove molding balloon and replace it with an angiographic catheter to
perform completion angiograms.
11. Remove or replace all stiff wire guides to allow iliac arteries to resume
their natural position.
Final Angiogram
1. Position angiographic catheter just above the level of the renal arteries.
Perform angiography to verify that the renal arteries are patent and that
there are no endoleaks. Verify patency of internal iliac arteries.
2. Confirm there are no endoleaks or kinks and verify position of proximal
gold radiopaque markers. Remove the sheaths, wires and catheters.
NOTE: If endoleaks or other problems are observed, refer to the Suggested
Instructions for Use for the Zenith AAA Endovascular Graft Ancillary
Components.
3. Repair vessels and close in standard surgical fashion.
12 IMAGING GUIDELINES AND POSTOPERATIVE FOLLOW-UP
12.1 General
• The long-term performance of endovascular grafts has not yet been
established. All patients should be advised that endovascular
treatment requires lifelong, regular follow-up to assess their health
and performance of their endovascular graft. Patients with specific
clinical findings (e.g., endoleaks, enlarging aneurysms or changes in the
structure or position of the endovascular graft) should receive additional
follow-up.
• Patients should be counseled on the importance of adhering to the followup schedule, both during the first year and at yearly intervals thereafter.
Patients should be told that regular and consistent follow-up is a critical
part of ensuring the ongoing safety and effectiveness of endovascular
treatment of AAAs.
• Physicians should evaluate patients on an individual basis and prescribe
their follow-up relative to the needs and circumstances of each individual
patient. The recommended imaging schedule is presented in Table 12.1.
This schedule continues to be the minimum requirement for patient followup and should be maintained even in the absence of clinical symptoms
(e.g., pain, numbness, weakness). Patients with specific clinical findings (e.g.,
endoleaks, enlarging aneurysms or changes in the structure or position of
the stent graft) should receive follow-up at more frequent intervals.
• Annual imaging follow-up should include abdominal radiographs and both
contrast and non-contrast CT examinations. If renal complications or other
factors preclude the use of image contrast media, abdominal radiographs,
non-contrast CT and duplex ultrasound may be used.
• The combination of contrast and non-contrast CT imaging provides
information on aneurysm diameter change, endoleak, patency, tortuosity,
progressive disease, fixation length and other morphological changes.
• The abdominal radiographs provide information on device integrity
(separation between components, stent fracture and barb separation).
33

• Duplex ultrasound imaging may provide information on aneurysm diameter
change, endoleak, patency, tortuosity and progressive disease. In this
circumstance, a non-contrast CT should be performed to use in conjunction
with the ultrasound. Ultrasound may be a less reliable and sensitive
diagnostic method compared to CT.
Table 12.1 lists the minimum imaging follow-up for patients with the Zenith
Flex AAA Endovascular Graft. Patients requiring enhanced follow-up should
have interim evaluations.
Table 12.1 Recommended Imaging Schedule for Endograft Patients
Angiogram
Pre-procedure
Procedural X
Pre-discharge (within 7 days)
1 month
3 month
6 month
12 month (annually thereafter)
1
Imaging should be performed within 6 months before the procedure.
2
Duplex ultrasound may be used for those patients experiencing renal failure or who are
otherwise unable to undergo contrast enhanced CT scan. With ultrasound, non-contrast
CT is still recommended.
3
Either pre-discharge or 1 month CT recommended.
4
If Type I or III endoleak, prompt intervention and additional follow-up post-intervention
recommended, see Section 12.6, Additional Surveillance and Treatment.
5
Recommended if endoleak reported at pre-discharge or 1 month.
1
X
CT
(Contrast and
non-contrast)
1
X
2,3,4
X
2,3,4
X
2,4,5
X
2,4
X
2,4
X
Abdominal
Radiographs
X
X
X
X
12.2 Contrast and Non-Contrast CT Recommendations
• Film sets should include all sequential images at lowest possible slice
thickness (≤3 mm). DO NOT perform large slice thickness (>3 mm) and/or
omit consecutive CT images/film sets, as it prevents precise anatomical and
device comparisons over time.
• All images should include a scale for each film/image. Images should be
arranged no smaller than 20:1 images on 14 inch x 17 inch sheets if film is
used.
• Both non- contrast and contrast runs are required, with matching or
corresponding table positions.
• Pre- contrast and contrast run slice thickness and interval must match.
• DO NOT change patient orientation or re-landmark patient between noncontrast and contrast runs.
Non-contrast and contrast enhanced baseline and follow-up imaging are
important for optimal patient surveillance. It is important to follow acceptable
imaging protocols during the CT exam. Table 12.2 lists examples of
acceptable imaging protocols.
Table 12.2 Acceptable Imaging Protocols
Non-Contrast Contrast
IV contrast No Ye s
Acceptable machines
Injection volume n/a 150 cc
Injection rate n/a >2.5 cc/sec
Injection mode n/a Power
Bolus timing n/a
Coverage - start Diaphragm 1 cm superior to celiac axis
Coverage - finish Proximal femur Profunda femoris origin
Collimation <3 mm <3 mm
Reconstruction
Axial DFOV 32 cm 32 cm
Post-injection runs None None
Spiral capable of
>40 seconds
2.5 mm throughout –
soft algorithm
Spiral capable of >40 seconds
Test bolus: SmartPrep, C.A.R.E.
or equivalent
2.5 mm throughout – soft
algorithm
12.3 Abdominal Radiographs
The following views are required:
• Four films: supine-frontal (AP), cross-table lateral, 30 degree LPO and 30
degree RPO views centered on umbilicus.
• Record the table -to-film distance and use the same distance at each
subsequent examination.
Ensure entire device is captured on each single image format lengthwise.
If there is any concern about the device integrity (e.g., kinking,
stent breaks, barb separation, relative component migration), it is
recommended to use magnified views. The attending physician should
evaluate films for device integrity (entire device length including
components) using 2-4X magnification visual aid.
12.4 Ultrasound
Ultrasound imaging may be performed in place of contrast CT when patient
factors preclude the use of image contrast media. Ultrasound may be
paired with non-contrast CT. A complete aortic duplex is to be videotaped
for maximum aneurysm diameter, endoleaks, stent patency and stenosis.
Included on the videotape should be the following information as outlined
below:
• Transverse and longitudinal imaging should be obtained from the level of
the proximal aorta demonstrating mesenteric and renal arteries to the iliac
bifurcations to determine if endoleaks are present utilizing color flow and
color power angiography (if accessible).
• Spec tral analysis confirmation should be performed for any suspected
endoleaks.
• Transverse and longitudinal imaging of the maximum aneurysm should be
obtained.
12.5 MRI Safety and Compatibility
Non-clinical testing has demonstrated that the Zenith AAA Endovascular Graft
is MR Conditional. It can be scanned safely under the following conditions:
1.5 Tesla Systems:
• Static magnetic field of 1.5 Tesla
• Spatial gradient field of 450 Gauss/cm
• M aximum whole-body-averaged specific absorption rate (SAR) of
2.0 W/kg for 15 minutes of scanning
In non-clinical testing, the Zenith AAA Endovascular Graft produced a
temperature rise of less than or equal to 1.4 °C at a maximum whole-bodyaveraged specific absorption rate (SAR) of 2.8 W/kg, as assessed by calorimetry
for 15 minutes of MR scanning in a 1.5 Tesla M agnetom, Siemens Medical
Magnetom, Numaris/4 Software, Version Syngo MR 2002B DHHS MR Scanner.
The maximum whole-body-averaged specific absorption rate (SAR) was
2.8 W/kg, which corresponds to a calorimetry measured value of 1.5 W/kg.
3.0 Tesla Systems:
• Static magnetic field of 3.0 Tesla
• Spatial gradient field of 720 Gauss/cm
• M aximum whole-body-averaged specific absorption rate (SAR) of
2.0 W/kg for 15 minutes of scanning
In non-clinical testing, the Zenith AAA Endovascular Graft produced a
temperature rise of less than or equal to 1.9 °C at a maximum wholebody-averaged specific absorption rate (SAR) of 3.0 W/kg, as assessed by
calorimetry for 15 minutes of MR scanning in a 3.0 Tesla Excite, GE Electric
Healthcare, G3.0-052B Software, MR Scanner. The maximum whole-bodyaveraged specific absorption rate (SAR) was 3.0 W/kg, which corresponds to a
calorimetry measured value of 2.8 W/kg.
The image artifact extends throughout the anatomical region containing the
device, obscuring the view of immediately adjacent anatomical structures
within approximately 20 cm of the device, as well as the entire device and its
lumen, when scanned in non-clinical testing using the sequence: Fast spin
echo, in a 3.0 Tesla, Excite, GE Electric Healthcare, with G3.0-052B Software, MR
system with body radiofrequency coil.
For all scanners, the image artifact dissipates as the distance from the device
to the area of interest increases. MR scans of the head and neck and lower
extremities may be obtained without image artifact. Image artifact may be
present in scans of the abdominal region and upper extremities, depending
on distance from the device to the area of interest.
Clinical information is available for seventeen patients who received MRI scans
after stent-graft implantation. There have been no reported adverse events
or device problems in any of these patients as a result of having received an
MRI. Additionally, there have been well over 50,000 Zenith AAA Endovascular
Grafts implanted worldwide, in which there have been no reported adverse
events or device problems as a result of MRI.
Cook recommends that the patient register the MR conditions disclosed in
this IFU with the MedicAlert Foundation. The MedicAlert Foundation can be
contacted in the following manners:
M ail: M edicAlert Foundation International
Fax: +1 209-669-2450
Web: www.medicalert.org
2323 Colorado Avenue
Turlock, CA 95382 USA
Phone: 888-633-4298 (toll free)
+1 209-668-3333 from outside the US
12.6 Additional Surveillance and Treatment
Additional surveillance and possible treatment is recommended for:
• Aneur ysms with Type I endoleak
• Aneur ysms with Type III endoleak
• Aneur ysm enlargement, ≥5 mm of maximum diameter (regardless of
endoleak status)
• Migration
• I nadequate seal length
Consideration for reintervention or conversion to open repair should include
the attending physician's assessment of an individual patient's co-morbidities,
life expectancy and the patient's personal choices. Patients should be
counseled that subsequent reinterventions, including catheter based and
open surgical conversion, are possible following endograft placement.
13 PATIENT TRACKING INFORMATION
In addition to these Instructions for Use, the Zenith Flex AAA Endovascular
Graft with the H&L-B One-Shot Introduction System is packaged with a Device
Tracking Form which the hospital staff is required to complete and forward
to COOK for the purposes of tracking all patients who receive the Zenith Flex
AAA Endovascular Graft (as required by U. S. Federal Regulation).
14 TROUBLESHOOTING
NOTE: Technical assistance from a Cook product specialist may be obtained
by contacting your local Cook representative.
14.1 Trigger-Wire Release Troubleshooting
CAUTION: The following steps should be performed only if unable to
remove the proximal trigger-wire as described in section 11.1.7.
14.1.1 Main Body Proximal (Top ) Deployment
1. If the black (top cap) trigger-wire release mechanism cannot be removed
from the handle, cut the wire adjacent to the release mechanism (Fig. 37)
and remove the release mechanism from the handle.
2. Stabilize the gray positioner while withdrawing the sheath to fully deploy
the ipsilateral limb.
3. Remove the safety lock from the white trigger-wire release mechanism.
4. Withdraw and remove the trigger-wire by sliding the white trigger-wire
release mechanism off the handle, and then remove via its slot over the
device inner cannula.
5. Using locking forceps, clamp and secure the cut end of the top cap
trigger-wire. (Fig. 38)
6. Loosen the pin vise and, while maintaining inner cannula and trigger-wire
position, advance the gray positioner and sheath into the graft until the
tip of the gray positioner is approximately 2 cm from the gold markers.
(Fig. 39) The advanced gray positioner provides added support to the
inner cannula.
NOTE: Maintain gentle tension on the trigger-wire to remove any slack in the
wire as the gray positioner and sheath are being advanced.
NOTE: Ensure that the tip of the gray positioner is not advanced into the top
cap.
7. Lock the pin vise. Confirm that the trigger-wire is secured to the forceps.
8. Stabilize the gray positioner and slowly advance the sheath until the
sheath tip is 2 mm from the gold markers. (Fig. 40)
NOTE: Take care not to advance the graft itself during sheath advancement.
9. Stabilize the sheath and retract the gray positioner with inner cannula to
draw the top cap over the suprarenal stent. (Fig. 41)
10. Verify position of the gold markers inferior to the renal arteries.
11. Remove the trigger-wire.
12. Withdraw the sheath until the tapered tip of the gray positioner is
exposed.
13. Advance a molding balloon through the contralateral limb of the main
body and position it just superior to the graft bifurcation.
14. Inflate the balloon to the full diameter of the graft. (Fig. 42)
15. Loosen the pin vise.
16. Stabilize the gray positioner and balloon catheter, and advance the inner
cannula to deploy the top cap.
17. Tighten the pin vise.
18. Deflate the balloon and advance the contralateral wire guide into the
thoracic aorta.
NOTE: Due to early ipsilateral leg deployment and trigger-wire release, it is
suggested to leave the molding balloon in or just above the contralateral
limb for graft stabilization during placement of the ipsilateral limb.
34

14.1.2 Docking of Top Cap
1. Loosen the pin vise. (Fig. 25)
2. Secure sheath and inner cannula to avoid any movement of these
components.
3. Advance the gray positioner over the inner cannula until it docks with the
top cap. (Figs. 43, 27, 44)
NOTE: If resistance occurs, slightly rotate gray positioner and continue to
gently advance.
4. Retighten the pin vise and withdraw the entire top cap and gray
positioner through the graft and through the sheath by pulling on the
inner cannula. (Fig. 45) Leave the sheath and wire guide in place.
5. Close the Captor® Hemostatic Valve on the Flexor® introducer sheath by
turning it in a clockwise direction until it stops. (Fig. 30)
14.1.3 Ipsilateral Iliac Leg Placement and Deployment
NOTE: Ensure that the Captor Hemostatic Valve on the introducer sheath is
turned to the open position. (Fig. 31)
1. Utilize the main body graft wire and sheath assembly to introduce the
ipsilateral leg graft.
NOTE: Due to top cap release modification, the main body sheath assembly
must be withdrawn to a point 1-2 cm inside the proximal ipsilateral limb.
Advance the dilator sheath assembly of the ipsilateral limb into the main
body sheath.
NOTE: The molding balloon may be inflated in the contralateral limb of the
main body graft if additional graft stabilization is necessary.
CAUTION: Do not inflate the balloon outside of the graft.
NOTE: In tortuous vessels, the position of the internal iliac arteries may alter
significantly with the introduction of the rigid wires and sheath systems.
2. Advance slowly until the ipsilateral iliac leg graft overlaps a minimum
of one full iliac leg stent (i.e., proximal stent of iliac leg graft) inside the
ipsilateral limb of the main body. (Fig. 46)
NOTE: If an overlap of greater than three iliac leg stents is required (greater
than two iliac leg stents for 37, and 54 mm leg lengths), it may be necessary
to consider use of a leg extension in the bifurcation area of the opposite side.
3. Confirm position of distal end of the iliac leg graft. Reposition the iliac leg
graft if necessary to ensure internal iliac patency.
4. To deploy, stabilize the iliac leg graft with the gray positioner while
withdrawing the iliac leg sheath and main body sheath together. (Figs. 33
and 47) If necessary, withdraw the main body sheath.
5. Under fluoroscopy and after verification of iliac leg graft position, loosen
the pin vise, retract the inner cannula to dock the tapered dilator to gray
positioner. Tighten pin vise. Maintain sheath position while withdrawing
gray positioner with secured inner cannula. (Fig. 48)
6. Close the Captor Hemostatic Valve on the Flexor introducer sheath by
turning it in a clockwise direction until it stops.
7. Re-check the position of the wire guides. Leave sheath and wire guides in
place.
8. Remove deflated balloon from contralateral side.
14.1.4 Contralateral Iliac Leg Placement and Deployment
CAUTION: Verify the predetermined contralateral iliac leg is selected for
insertion on the contralateral side of the patient before implantation.
1. Position the image intensifier to show both the contralateral internal iliac
artery and contralateral common iliac artery.
2. Prior to introduction of contralateral iliac leg delivery system, inject
contrast through the contralateral femoral sheath to locate the
contralateral internal iliac artery.
3. Introduce the contralateral iliac leg delivery system into the artery.
Advance slowly until the iliac leg graft overlaps at least one full iliac leg
stent (i.e., proximal stent of iliac leg graft) inside the contralateral limb of
the main body. (Fig. 49)
NOTE: If difficulty is encountered advancing the iliac leg delivery system,
exchange to a more supportive wire guide. In tortuous vessels, the anatomy
may alter significantly with the introduction of the rigid wires and sheath
systems.
4. Confirm position of distal end of the iliac leg graft. Reposition the iliac leg
graft if necessary to ensure both internal iliac patency and a minimum
overlap of one full iliac leg stent (i.e., proximal stent of iliac leg graft,
maximum overlap of 1.5 stents) within the main body endovascular graft.
5. To deploy, hold the iliac leg graft in position with the gray positioner while
withdrawing the sheath. (Figs. 19 and 50) Ensure one stent overlap is
maintained.
6. Stop withdrawing the sheath as soon as the distal end of the iliac leg graft
is released.
7. Under fluoroscopy and after verification of the iliac leg graft position,
loosen the pin vise, retract the inner cannula to dock the tapered dilator
to the gray positioner. Tighten pin vise. Maintain sheath position while
withdrawing gray positioner with secured inner cannula. (Fig. 51)
8. Re-check the position of the wire guides.
9. Perform molding balloon insertion and final angiogram as described in
section 11.1.12 Molding Balloon Insertion.
14.2 Suprarenal Stent Deployment Troubleshooting
CAUTION: The following steps should be performed only if unable to
deploy the top cap as described in section 11.1.7.
14.2.1 Main Body Proximal (Top ) Deployment
1. If the suprarenal stent cannot be fully deployed by advancing the top cap
inner cannula, advance a molding balloon through the contralateral limb
of the main body and position it just superior to the bifurcation of the
stent graft.
2. To add support to the inner cannula, inflate the balloon to the full
diameter of the graft.
3. Loosen the pin vise. (Fig. 15)
4. Stabilize the gray positioner and balloon catheter, and advance the inner
cannula to deploy the suprarenal stent.
If the suprarenal stent is fully deployed:
5. Tighten the pin vise; deflate and withdraw the balloon.
6. Advance the contralateral wire guide into the thoracic aorta and return
to the appropriate step in the Instructions for Use to complete the
procedure.
If still unable to fully deploy the suprarenal stent:
5. Tighten the pin vise and deflate the balloon. While maintaining balloon
position, stabilize the gray positioner and withdraw the sheath to fully
deploy the ipsilateral limb.
6. Remove the safety lock from the ipsilateral limb trigger-wire release
mechanism.
7. Withdraw and remove the trigger-wire by sliding the ipsilateral limb
trigger-wire release mechanism off the handle, and remove via its slot
over the device inner cannula.
8. Loosen the pin vise (Fig. 25) and, while maintaining inner cannula
position, advance the gray positioner and sheath into the graft until the
tip of the gray positioner is approximately 2 cm inferior to the proximal
gold markers. (Fig. 52) The advanced gray positioner provides added
support to the inner cannula.
NOTE: Take care not to advance the graft during gray positioner and
sheath advancement.
NOTE: Ensure that the tip of the gray positioner is not advanced into the
top cap.
9. Lock the pin vise.
10. Verify position of the gold markers inferior to the renal arteries.
11. Reposition molding balloon so that it is seated against the bifurcation.
12. Inflate the balloon to the full diameter of the graft. (Fig. 42)
13. Loosen the pin vise.
14. Stabilize the gray positioner and balloon catheter, and advance the inner
cannula to deploy the suprarenal stent.
15. Tighten the pin vise.
16. Deflate the balloon and advance the contralateral wire guide into the
thoracic aorta.
NOTE: Due to early ipsilateral leg deployment and trigger-wire release,
it is suggested to leave the molding balloon in or just above the
contralateral limb for use in graft stabilization during placement of the
ipsilateral limb.
14.2.2 Docking of Top Cap
1. Loosen the pin vise. (Fig. 25)
2. Secure sheath and inner cannula to avoid any movement of these
components.
3. Advance the gray positioner over the inner cannula until it docks with the
top cap. (Figs. 43, 27 and 44)
NOTE: If resistance occurs, slightly rotate gray positioner and continue to
gently advance.
4. Retighten the pin vise and withdraw the entire top cap and gray
positioner through the graft and through the sheath by pulling on the
inner cannula. (Fig. 45) Leave the sheath and wire guide in place.
5. Close the Captor® Hemostatic Valve on the Flexor® introducer sheath by
turning it in a clockwise direction until it stops. (Fig. 30)
14.2.3 Ipsilateral Iliac Leg Placement and Deployment
NOTE: Ensure that the Captor Hemostatic Valve on the introducer sheath is
turned to the open position. (Fig. 31)
1. Position the image intensifier to show both the ipsilateral internal iliac
artery and ipsilateral common iliac artery.
2. After the introduction of the ipsilateral iliac leg delivery system, inject
contrast through the main body sheath to locate the ipsilateral internal
iliac artery.
3. Utilize the main body graft wire and sheath assembly to introduce the
ipsilateral leg graft.
NOTE: Due to top cap release modification, the main body sheath
assembly must be withdrawn to a point 1-2 cm inside the proximal
ipsilateral limb. Advance the dilator sheath assembly of the ipsilateral
limb into the main body sheath.
NOTE: The molding balloon may be inflated in the contralateral limb of
the main body graft if additional graft stabilization is necessary.
CAUTION: Do not inflate the balloon outside of the graft.
NOTE: In tortuous vessels, the position of the internal iliac arteries may
alter significantly with the introduction of the rigid wires and sheath
systems.
4. Advance the ipsilateral iliac leg delivery system slowly until the ipsilateral
iliac leg graft overlaps a minimum of one full iliac leg stent (i.e., proximal
stent of iliac leg graft) inside the ipsilateral limb of the main body. (Fig.
46)
NOTE: If an overlap of greater than three iliac leg stents is required
(greater than two iliac leg stents for 37, and 54 mm leg lengths), it may be
necessary to consider use of a leg extension into the bifurcation area of
the opposite side.
5. Confirm position of distal end of the iliac leg graft. Reposition the iliac leg
graft if necessary to ensure internal iliac patency.
6. To deploy, stabilize the iliac leg graft with the gray positioner while
withdrawing the iliac leg sheath and main body sheath together. (Figs.
33 and 47)
7. Under fluoroscopy and after verification of iliac leg graft position, loosen
the pin vise, retract the inner cannula to dock the tapered dilator to gray
positioner. Tighten pin vise. Maintain sheath position while withdrawing
gray positioner with secured inner cannula. (Fig. 48)
8. Close the Captor Hemostatic Valve on the Flexor introducer sheath by
turning it in a clockwise direction until it stops.
9. Re-check the position of the wire guides. Leave sheath and wire guides in
place.
10. Remove deflated balloon from contralateral side.
14.2.4 Contralateral Iliac Leg Placement and Deployment
CAUTION: Verify the predetermined contralateral iliac leg is selected for
insertion on the contralateral side of the patient before implantation.
1. Position the image intensifier to show both the contralateral internal iliac
artery and contralateral common iliac artery.
2. Prior to introduction of contralateral iliac leg delivery system, inject
contrast through the contralateral femoral sheath to locate the
contralateral internal iliac artery.
3. Introduce the contralateral iliac leg delivery system into the artery.
Advance slowly until the iliac leg graft overlaps at least one full iliac leg
stent (i.e., proximal stent of iliac leg graft) inside the contralateral limb of
the main body. (Fig. 49)
NOTE: If difficulty is encountered advancing the iliac leg delivery system,
exchange to a more supportive wire guide. In tortuous vessels, the
anatomy may alter significantly with the introduction of the rigid wires
and sheath systems.
4. Confirm position of distal end of the iliac leg graft. Reposition the iliac leg
graft if necessary to ensure both internal iliac patency and a minimum
overlap of one full iliac leg stent (i.e., proximal stent of iliac leg graft,
maximum overlap of 1.5 stents) within the main body endovascular graft.
5. To deploy, hold the iliac leg graft in position with the gray positioner
while withdrawing the sheath. (Figs. 19 and 50) Ensure one stent overlap
is maintained.
6. Stop withdrawing the sheath as soon as the distal end of the iliac leg graft
is released.
7. Under fluoroscopy and after verification of the iliac leg graft position,
loosen the pin vise, retract the inner cannula to dock the tapered dilator
to the gray positioner. Tighten pin vise. Maintain sheath position while
withdrawing gray positioner with secured inner cannula. (Fig. 51)
8. Re-check the position of the wire guides.
9. Perform molding balloon insertion and final angiogram as described in
the Molding Balloon Insertion section of the product‘s Instructions for
Use.
35

ČESKY
ENDOVASKULÁRNÍ GRAFT ZENITH FLEX® AAA SE
ZAVÁDĚCÍM SYSTÉMEM H&L-B ONE-SHOT™
Pečlivě si přečtěte všechny pokyny. Pokud nebudete postupovat přesně podle
pokynů, varování a upozornění, může to mít vážné chirurgické následky nebo
může dojít k poranění pacienta.
POZOR: Podle federálních zákonů USA je toto zařízení pouze na lékařský
předpis.
POZOR: Veškerý obsah vnějšího sáčku (včetně zaváděcího systému a endovaskulárních graftů) se dodává sterilní a je určen pouze na jedno použití.
K výrobkům řady Zenith se vztahují čtyři relevantní doporučené návody
k použití. Návod k použití obsahuje popis doporučeného použití
endovaskulárního graftu Zenith Flex AAA (hlavního těla a iliakálních ramen).
Informace o dalších komponentách systému Zenith najdete v doporučeném
příslušném návodu k použití:
• endovaskulárního graftu Zenith AAA (hlavního těla endovaskulárního graftu
Zenith AAA a iliakálních ramen);
• doplňkových pomocných zařízení endovaskulárního graftu Zenith AAA
(extenze hlavního těla graftu, extenze iliakálního ramena, přechodového
dílu a uzávěru iliakální větve);
• přídavného graftu Zenith® Renu™ AAA (konfigurace extenze hlavního těla
nebo přechodového dílu);
• balónkového katetru CODA®.
1 POPIS ZAŘÍZENÍ
1.1 Komponenty aortálního hlavního těla a iliakálního ramena
Endovaskulární graft Zenith Flex AAA je modulární systém skládající se ze
tří součástí: z hlavního aortálního těla graftu s bifurkací a ze dvou iliakálních
ramen. (Obr. 1) Moduly graftu jsou vyrobeny z plnostěnné polyesterové
tkaniny přišité k samoexpandujícím stentům z nerez oceli Cook-Z® nití
splétanou z polyesteru a jednovláknového polypropylénu. Moduly jsou plně
vyztuženy stenty, aby získaly stabilitu a expanzní sílu nezbytnou k otevření
lumen graftu při jeho rozvinutí. Navíc stenty Cook-Z zabezpečují nezbytné
přichycení a přilehnutí graftu ke stěně cévy.
Nezakrytý suprarenální stent na proximálním konci graftu je opatřen
kotvičkami umístěnými 3 mm od sebe, aby se zajistila další fixace zařízení. K
usnadnění skiaskopického sledování stentgraftu jsou na zařízení rozmístěny
zlaté rentgenokontrastní značky, a to následujícím způsobem: jedna na
laterální straně nejdistálnější části stentu na kontralaterální větvi bifurkované
části hlavního těla a čtyři rozmístěné po obvodu do 2 mm od nejvyšší strany
materiálu graftu.
1.2 Aplikační systém hlavního těla
Hlavní tělo endovaskulárního graftu Zenith Flex AAA se dodává
předinstalované v zaváděcím systému H&L-B One-Shot. (Obr. 2) Zavádí
se postupnou metodou a má integrované funkce k provádění nepřetržité
kontroly endovaskulárního graftu během celého procesu rozvinutí. Zaváděcí
systém H&L-B One-Shot je určen pro přesné umístění graftu a konečnou
úpravu polohy před rozvinutím suprarenálního stentu s kotvičkami.
Aplikační systém hlavního těla graftu používá zaváděcí systém H&L-B OneShot o velikosti 18, 20 nebo 22 French. Endovaskulární graft je aretován na
aplikačním systému dvojitým pojistným mechanismem spouštěcího drátu,
dokud jej lékař neuvolní. Všechny systémy jsou kompatibilní s vodicím drátem
o velikosti 0,035 palce (0,89 mm).
Krvácení lze omezovat utahováním a uvolňováním hemostatického ventilu
Captor® při zavádění pomocných zařízení do sheathu a/nebo vytahování z
něj. Aplikační systémy hlavního těla obsahují zaváděcí sheath Flexor®, který je
odolný proti zasmyčkování a má hydrofilní povlak. Obě tyto vlastnosti slouží k
zlepšení možnosti manévrování v iliakálních artériích a v břišní aortě.
1.3 Aplikační systém pro iliakální rameno
Iliakální ramena endovaskulárního graftu Zenith AAA se dodávají
předinstalovaná v zaváděcím systému H&L-B One-Shot. (Obr. 3) Aplikační
systém je konstruován k snadnému použití s minimální přípravou. Aplikační
systém iliakálního ramena používá zaváděcí systém H&L-B One-Shot o
velikosti 14 nebo 16 French. Všechny systémy jsou kompatibilní s vodicím
drátem o velikosti 0,035 palce (0,89 mm).
1.4 Doplňkové komponenty endovaskulárního graftu Zenith AAA
K dispozici jsou doplňková pomocná endovaskulární zařízení (extenze
hlavního těla graftu, extenze iliakálního ramena, přechodové díly a iliakální
zátky). (Obr. 4) Více informací najdete v návodu k použití pomocných
komponent endovaskulárního graftu Zenith AAA.
2 URČENÉ POUŽITÍ
Endovaskulární graft Zenith Flex AAA se zaváděcím systémem H&L-B
One-Shot je určen k endovaskulární léčbě pacientů s aneuryzmatem břišní
aorty nebo s aorto-iliakálními aneuryzmat y, jejichž mor fologie je vhodná
k endovaskulární reparaci, tj. která mají mimo jiné následující vlastnosti:
• adek vátní cévy pro iliakální nebo femorální přístup, kompatibilní s
potřebnými zaváděcími systémy;
• I nfrarenální segment aorty bez aneuryzmatu (krček) proximálně k
aneuryzmatu s následujícími parametry:
• délku nejméně 15 mm;
• průměr při měření od vnější stěny k vnější stěně v rozsahu max. 32 mm
do min. 18 mm;
• úhel vzhledem k dlouhé ose výdutě menší než 60 stupňů; a
• úhel vzhledem k ose suprarenální aorty menší než 45 stupňů.
• místo distální fixace v iliak ální artérii větší než 10 mm (délka) a o průměru
7,5-20 mm (měřeno od vnější stěny k vnější stěně).
3 KONTRAINDIKACE
Nejsou známy žádné kontraindikace použití těchto zařízení.
4 VAROVÁNÍ A UPOZORNĚNÍ
4.1 Obecně
• Pečlivě si přečtěte všechny pokyny. Pokud nebudete postupovat přesně
podle pokynů, varování a upozornění, může to mít vážné následky nebo
může dojít k poranění pacienta.
• Během implantace nebo reintervence musí být v pohotovosti kvalifikovaný
operační tým pro případ nutnosti přechodu na otevřenou chirurgickou
operaci.
• Endovaskulární graft Zenith Flex AAA se zaváděcím systémem H&L-B OneShot smí používat výhradně lékaři a operační týmy s odpovídající kvalifikací
pro cévní intervenční výkony (katetrizační i standardní chirurgické) a pro
použití tohoto zařízení. Specifické požadavky na školení jsou popsány v části
10.1 Školení lékařů.
• Možnost provedení další endovaskulární intervence nebo přechodu na
otevřenou chirurgickou operaci po primární endovaskulární operaci je třeba
zvážit u pacientů, u nichž se vyskytne zvětšující se aneuryzma,
neakceptovatelné zkrácení délky fixace (překrytí cévy nebo komponenty) a
(nebo) endoleak. Zvětšení aneuryzmatu a (nebo) přetrvávající endoleak
nebo migrace mohou být příčinou prasknutí aneuryzmatu.
• U pacientů s nižším průtokem krve větví graftu a/nebo s netěsnostmi může
být nezbytné provedení sekundární intervence nebo chirurgické operace.
4.2 Výběr, léčba a následné kontroly pacienta
• Endovaskulární graft Zenith Flex AAA je určen k léčbě při průměru
aortálního krčku min. 18 mm a max. 32 mm. Endovaskulární graft Zenith
Flex AAA je určen k léčbě při délce proximálního aortálního krčku (distálně k
nejnižší renální artérii) min. 15 mm. Je nutné místo distální fixace v iliakální
artérii větší než 10 mm (délka) a o průměru 7,5-20 mm (měřeno od vnější
stěny k vnější stěně). Výběr správné velikosti je kriticky důležitý pro
úspěšnost endovaskulární opravy.
• M ezi klíčové anatomické prvky, které mohou ovlivnit úspěšné ošetření
aneuryzmatu, patří závažná angulace proximálního krčku (>60 stupňů u
infrarenálního krčku vůči ose AAA nebo >45 stupňů u suprarenálního krčku
vzhledem k nejbližšímu infrarenálnímu krčku); krátký proximální krček aorty
(<15 mm); tvar obráceného trychtýře (zvětšení průměru proximálního krčku
aorty na délce 15 mm o více než 10 %); cirkumferenční trombus a/nebo
kalcifikace v místě arteriální implantace, zejména na rozhraní proximálního
krčku aorty a distální iliakální artérie. Při anatomickém omezení může být
nutný delší krček, aby se docílilo adekvátního přilehnutí a fixace.
Nepravidelná kalcifikace a (nebo) plak mohou ohrozit připojení a utěsnění v
místech fixace. U ohybů cév s těmito klíčovými anatomickými znaky lze také
očekávat větší náchylnost k migraci graftu nebo endoleaku.
• Pro zavedení prostředku do cévního systému je nutné zajistit adekvátní
iliakální nebo femorální přístup. Průměr přístupové cévy (měřený od vnitřní
stěny k vnitřní stěně) a její morfologie (minimální vinutost, okluzívní
onemocnění a/nebo kalcifikace) musí vyhovovat kritériím pro použití metod
cévního přístupu a zaváděcích systémů s cévním zaváděcím sheathem o
velikosti 16 French až 22 French. Cévy se závažným stupněm kalcifikace,
okludované, silně vinuté nebo pokryté tromby mohou znemožnit umístění
endovaskulárního graftu a (nebo) mohou zvyšovat riziko embolizace. U
některých pacientů může být úspěšnost výkonu podmíněna použitím
cévního bypassu.
• Endovaskulární graft Zenith Flex AAA se zaváděcím systémem H&L-B OneShot se nedoporučuje používat u pacientů, kteří nesnášejí kontrastní látky
nezbytné pro zobrazovací techniky při operaci a po ní. Všechny pacienty je
nutno pečlivě sledovat a pravidelně kontrolovat případné změny jejich
zdravotního stavu a integritu endoprotézy.
• Endovaskulární graft Zenith Flex AAA se zaváděcím systémem H&L-B OneShot se nedoporučuje používat u pacientů, jejichž hmotnost a (nebo)
tělesné rozměry překračují stanovené limity nezbytné pro realizaci nebo
dostatečnou kvalitu snímkování.
• Neschopnost udr žet průchodnost nejméně jedné vnitřní iliakální artérie
nebo uzávěr nepostradatelné a. mesenterica inferior může zvýšit riziko
ischémie pánve nebo střev.
• M nohočetné velké artérie o velkém průměru v bederní krajině, murální
trombus a průchozí a. mesenterica inferior mohou predisponovat pacienta k
endoleakům typu II. U pacientů s nekorigovatelnou koagulopatií může být
také zvýšené riziko endoleaku typu II nebo krvácivých komplikací.
• Bezpečnost a účinnost endovaskulárního graftu Zenith Flex AAA se
zaváděcím systémem H&L-B One-Shot nebyla dosud zjištěna u skupin
pacientů s následujícími stavy:
• trauma aor ty;
• aneur yzma s únikem, s hrozící rupturou nebo prasklé;
• mykotické aneur yzma;
• pseudoaneur yzma sekundárně po předchozí implantaci graftu;
• revize dříve implantovaného endovaskulárního graftu;
• nekorigovatelná koagulopatie;
• nepostradatelná a. mesenterica;
• dědičné onemocnění pojivové tkáně (např. Marfanův nebo EhlersDanlosův syndrom);
• současně se v yskytující aneuryzmata hrudní nebo thorakoabdominální
aorty;
• pacienti s aktivní celkovou infekcí;
• těhotné nebo kojící ženy;
• pacienti s morbidní obezitou;
• pacienti mladší 18 let;
• pacienti, u nichž je proximální krček aort y kratší než 15 mm nebo je pod
úhlem vzhledem k dlouhé ose aneuryzmatu větším než 60 stupňů.
• Úspěšný výběr pacienta vyžaduje specifické zobrazování a přesná měření;
viz část 4.3 Měřicí techniky a zobrazování před výkonem.
• Lékař má mít k dispozici všechny délky a průměr y součástí, nezbytných k
provedení výkonu, zejména pokud ještě nejsou k dispozici výsledky
předoperačních měření (hodnoty průměrů a délek). Tento přístup zaručuje
větší peroperační flexibilitu k dosažení optimálních výsledků.
4.3 Měřicí techniky a zobrazování před výkonem
• Pokud není k dispozici nekontrastní CT vyšetření, může dojít k nesprávnému
vyhodnocení kalcifikace v iliakální nebo aortální oblasti, která může
znemožnit přístup nebo spolehlivou fixaci a utěsnění zařízení.
• Rekonstrukční tloušťk a vrstvy před výkonem >3 mm může mít za následek
suboptimální volbu velikosti zařízení nebo neschopnost vyhodnotit fokální
stenózy z CT.
• Na zák ladě klinických zkušeností se k přesnému vyhodnocení anatomie
pacienta před ošetřením za použití endovaskulárního graftu Zenith Flex
AAA důrazně doporučuje použít zobrazovací metodu spirální CT angiografie
s kontrastní látkou a s 3D rekonstrukcí. Pokud kontrastní spirální CTA s 3D
rekonstrukcí není k dispozici, pacienta je nutno odeslat do zařízení, které
tyto možnosti má.
• Podle doporučení lékařů se má C rameno rentgenu během peroperační
angiografie umístit tak, aby byly před zahájením rozvinutí proximálního
okraje hlavního těla graftu (těsnicího stentu) dobře viditelné odstupy
renálních artérií a zejména nejnižší průchodná renální artérie. Kromě toho
musí být angiograficky potvrzeny bifurkace iliakální artérie tak, aby byly
před rozvinutím iliakálních ramen na obou stranách dobře definovány
distální společné iliakální artérie vzhledem k odstupům vnitřních iliakálních
artérií.
Průměry:
Pomocí CT je třeba změřit průměr od vnější stěny k vnější stěně cévy (nikoli
jako průměr lumen – světlost); tyto hodnoty jsou pomůckou při správné volbě
velikosti a typu prostředku. Kontrastní spirální CT musí začínat 1 cm nad a.
celiaca a pokračovat přes hlavice femurů při axiální tloušťce řezu 3 mm nebo
méně.
Délky:
Pomocí CT je nutno stanovit délky k přesnému vyhodnocení délky
infrarenálního proximálního krčku a k výběru velikosti hlavního těla a ramen
endovaskulárního graftu Zenith Flex AAA. Tyto rekonstrukce je nutno provést
v sagitální, koronální a 3D projekci.
• Funkčnost endovaskulárních graftů z dlouhodobého hlediska nebyla
zatím zjištěna. Všichni pacienti musí být poučeni, že endovaskulární
léčba vyžaduje celoživotní pravidelná kontrolní vyšetření ke zjištění
jejich zdravotního stavu a funkčnosti jejich endovaskulárního graftu.
Pacienti se specifickými klinickými nálezy (např. endoleaky, zvětšujícím se
aneuryzmatem nebo se změnami struktury nebo polohy endovaskulárního
graftu) se musí podrobit důkladnějším kontrolním vyšetřením. Konkrétní
pokyny ke kontrolním vyšetřením jsou uvedeny v části 12, POKYNY K
ZOBRAZOVÁNÍ A POOPERAČNÍ KONTROLE.
• Endovaskulární graft Zenith Flex AAA se zaváděcím systémem H&L-B One-
Shot není doporučeno používat u pacientů, kteří nejsou schopni podstoupit
nezbytné předoperační a pooperační snímkování a implantační studie nebo
36

kteří nebudou dodržovat jejich požadavky; viz popis v části 12, POKYNY K
ZOBRAZOVÁNÍ A POOPERAČNÍ KONTROLE.
• Po implantaci endovaskulárního graftu se u pacientů má pravidelně
kontrolovat prosakování kolem graftu, zvětšování aneuryzmatu a změny
struktury nebo polohy endovaskulárního graftu. Je nutné provést
snímkování nejméně jednou ročně, mimo jiné: 1) radiogram břicha ke
kontrole integrity implantátu (zda se neoddělily jeho komponenty nebo
kotvičky a zda nepraskl stent) a 2) kontrastní a nekontrastní CT k vyšetření
změn aneuryzmatu, prosakování kolem graftu, průchodnosti, tortuozity a
vývoje onemocnění. Pokud není možné použít kontrastní látku vzhledem k
renálním komplikacím nebo jiným faktorům, mohou podobné informace
poskytnout radiogramy břicha a duplexní ultrasonografie.
4.4 Volba zařízení
• Při výběru správné velikosti prostředku se důrazně doporučuje přísně
dodržovat instrukce uvedené v pokynech pro výběr velikosti
endovaskulárního graftu Zenith Flex AAA v návodu k jeho použití (tabulky
10.5.1 a 10.5.2). Pokyny pro výběr velikosti v návodu k použití nyní obsahují
informace o výběru příslušné větší velikosti zařízení. Výběr velikosti mimo
tento rozsah může způsobit endoleak, prasknutí, migraci a kolaps nebo
kompresi zařízení.
4.5 Postup implantace
(Viz část 11, POKYNY K POUŽITÍ)
• K úspěšnému umístění endovaskulárního graftu Zenith Flex AAA a k
dosažení jeho přesného přilehnutí na aortální stěnu je nutné vhodné
peroperační zobrazování.
• Aplik ační systém neohýbejte ani nezasmyčkujte. Mohlo by dojít k poškození
aplikačního systému a endovaskulárního graftu Zenith Flex AAA.
• Pro vyloučení zkroucení endovaskulárního graftu při rotaci aplikačního
systému otáčejte všemi komponentami systému najednou (od vnějšího
sheathu po vnitřní kanylu).
• Pokud ucítíte odpor při posouvání vodicího drátu nebo aplikačního
systému, nikdy nepokračujte v zasouvání jakékoli části aplikačního systému.
Zastavte operaci a zjistěte příčinu odporu; mohlo by dojít k poranění cévy a
poškození katetru nebo graftu. Zvlášť opatrně postupujte v oblastech se
stenózami a s intravaskulárními tromby a v kalcifikovaných a silně vinutých
cévách.
• Neúmyslné částečné rozvinutí endoprotézy nebo její migrace může
vyžadovat chirurgické odstranění.
• Pokud to není indikováno z lékařských důvodů, neimplantujte endovaskulární
graft Zenith Flex AAA v místě, kde by došlo k okluzi artérií zásobujících orgány
nebo končetiny. Endoprotézou nezakrývejte důležité renální nebo
mesenterické artérie (výjimkou může být a. mesenterica inferior). Může dojít k
okluzi cévy. Během klinické studie nebylo toto zařízení testováno u pacientů
se dvěma okludovanými vnitřními iliakálními artériemi.
• Nepokoušejte se graft po částečném nebo úplném rozvinutí zatáhnout zpět
do sheathu.
• Repozice stentgraftu distálně po částečném rozvinutí kr ytého proximálního
stentu může způsobit poškození stentgraftu a (nebo) poranění cévy.
• Nepřesné umístění a/nebo neúplné přilehnutí endovaskulárního graftu
Zenith Flex AAA v cévě může vést ke zvýšenému riziku vzniku endoleaku,
migrace nebo nežádoucí okluze renální nebo vnitřní iliakální artérie.
Průchodnost renální artérie musí být zachována, aby se omezilo nebo
eliminovalo riziko selhání ledvin a následných komplikací.
• Neadek vátní fixace endovaskulárního graftu Zenith Flex AAA může mít za
následek zvýšené riziko migrace stentgraftu. Nesprávné rozvinutí
endoprotézy nebo její migrace může vyžadovat chirurgický zásah.
• V průběhu implantace je třeba používat celkovou antikoagulační léčbu
podle protokolů předepsaných zdravotnickým zařízením nebo zvolených
lékařem. Je-li kontraindikován heparin, je nutno zvážit možnost použití
jiného antikoagulačního přípravku.
• K aktivaci hydrofilního povlaku na vnějším povrchu zaváděcího sheathu
Flexor je třeba povrch otřít sterilními gázovými polštářky, navlhčenými
fyziologickým roztokem. Sheath udržujte v hydratovaném stavu, abyste
dosáhli jeho optimální funkce.
• Během př ípravy a zavádění omezte manipulaci se složenou endoprotézou,
aby se snížilo riziko kontaminace a infekce endoprotézy.
• Při vkládání aplik ačního systému udržujte polohu vodicího drátu.
• Při zavádění a rozvinutí se k potvrzení správné funkce komponent
aplikačního systému, správného umístění graftu a požadovaného výsledku
zákroku musí používat skiaskopie.
• Použití endovaskulárního graftu Zenith Flex AAA se zaváděcím systémem
H&L-B One-Shot vyžaduje podání intravaskulární kontrastní látky. U
pacientů s preexistující renální nedostatečností může existovat zvýšené
riziko pooperačního selhání ledvin. Kvůli snížení rizika poškození ledvin je
třeba pečlivě minimalizovat množství kontrastní látky použité při výkonu a
dodržovat preventivní metody léčby (např. dostatečnou hydrataci).
• Jak mile je sheath a/nebo vodicí drát stažen zpět, může dojít ke změně
anatomických poměrů a polohy graftu. Neustále kontrolujte polohu graftu a
podle potřeby proveďte kontrolní angiografii.
• Endovaskulární graft Zenith Flex AAA obsahuje suprarenální stent s
fixačními kotvičkami. Při manipulaci s intervenčními a angiografickými
zařízeními v blízkosti suprarenálního stentu postupujte s extrémní
opatrností.
• Při manipulaci s k atetry, dráty a sheathy uvnitř aneuryzmatu postupujte
opatrně. Velké narušení může uvolnit fragmenty trombu, které mohou
způsobit embolizaci v distálních částech těla nebo rupturu aneuryzmatu.
• Pokud je nezbytné znovuzavedení nástrojů (sekundární intervence), nesmí
dojít k poškození graftu nebo ke změně jeho polohy.
• Před rozvinutím suprarenálního stentu ověřte, že přístupový drát zasahuje
bezprostředně distálně za aortální oblouk.
• Zkontrolujte, zda je k zavedení na kontralaterální straně pacienta vybráno
kontralaterální iliakální rameno určené před implantací.
4.6 Použití tvarovacího balónku
• Nenaplňujte balónek v cévě, pokud není uvnitř graftu, neboť takový postup
může způsobit poškození cévy. Balónek používejte v souladu s informacemi
uvedenými na štítku a v jakékoliv další doprovodné dokumentaci.
• Při plnění balónku v graftu za přítomnosti k alcifikace postupujte opatrně,
neboť nadměrné naplnění může způsobit poškození cévy.
• Před repozicí zkontrolujte, zda je balónek úplně vyprázdněný.
• K rvácení lze dále eliminovat utahováním a uvolňováním hemostatického
ventilu Captor tak, aby se přizpůsobil zavádění a následnému vytahování
tvarovacího balónku.
4.7 Bezpečnost a kompatibilita vyšetření MRI
Neklinické testy prokázaly, že endovaskulární graft Zenith AAA je podmínečně
bezpečný při vyšetření MRI (MR Conditional). Může být bezpečně snímán za
následujících podmínek:
Systémy 1,5 tesla:
• Statické magnetické pole 1,5 tesla
• Prostorový gradient pole 450 gaussů/cm
• M aximální průměrná hodnota měrného absorbovaného výkonu
přepočteného na celé tělo (SAR) 2,0 W/kg pro 15 minut snímkování
Při neklinickém testování došlo u endovaskulárního graftu Zenith AAA ke
zvýšení teploty maximálně o 1,4 °C při maximálním měrném absorbovaném
výkonu přepočteném na celé tělo (SAR) 2,8 W/kg při kalorimetrickém měření
po dobu 15 minut snímkování MR scannerem Magnetom (Siemens Medical
Magnetom, software Numaris/4, verze Syngo MR 2002B DHHS) s magnetickým
polem 1,5 tesla. Maximální průměrná hodnota měrného absorbovaného
výkonu přepočteného na celé tělo (SAR) byla 2,8 W/kg, což odpovídá
kalorimetricky změřené hodnotě 1,5 W/kg.
Systémy 3,0 tesla:
• Statické magnetické pole 3,0 tesla
• Prostorový gradient pole 720 gaussů/cm
• M aximální průměrná hodnota měrného absorbovaného výkonu
přepočteného na celé tělo (SAR) 2,0 W/kg pro 15 minut snímkování
Při neklinickém testování došlo u endovaskulárního graftu Zenith AAA ke
zvýšení teploty maximálně o 1,9 °C při maximálním měrném absorbovaném
výkonu přepočteném na celé tělo (SAR) 3,0 W/kg při kalorimetrickém měření
po dobu 15 minut snímkování MR scannerem Excite (GE Electric Healthcare,
software G3.0-052B) s magnetickým polem 3,0 tesla. Maximální průměrná
hodnota měrného absorbovaného výkonu přepočteného na celé tělo (SAR)
byla 3,0 W/kg, což odpovídá kalorimetricky změřené hodnotě 2,8 W/kg.
Při neklinickém skenování zasahoval obrazový artefakt celou anatomickou
oblast obsahující implantát, přičemž byl zacloněn pohled na bezprostředně
sousedící anatomické struktury do vzdálenosti přibližně 20 cm od implantátu
stejně jako implantát a jeho lumen; použitá sekvence byla následující: fast
spin echo, MR systém Excite (GE Electric Healthcare, se softwarem G3.0-052B);
3,0 tesla, s tělovou radiofrekvenční cívkou.
U všech skenerů se obrazové artefakty rozptýlily při zvětšující se vzdálenosti
oblasti zájmu od implantátu. Snímky MR hlavy a krku a dolních končetin
lze pořídit bez obrazových artefaktů. Obrazový artefakt může být přítomen
u skenů břišní oblasti a horních končetin, v závislosti na vzdálenosti mezi
implantátem a oblastí zájmu.
Jsou k dispozici klinické údaje o sedmnácti pacientech, kteří podstoupili
MR vyšetření po implantaci stentgraftu. U žádného z těchto pacientů
nebyla hlášena žádná nežádoucí příhoda ani problémy s implantátem
vzniklé v souvislosti s podstoupením MRI. Kromě toho bylo na celém světě
implantováno více než 50 000 endovaskulárních graftů Zenith AAA, u nichž
nebyly hlášeny žádné nežádoucí příhody ani problémy s implantátem
způsobené MRI.
Společnost Cook doporučuje, aby pacienti zaregistrovali podmínky pro MRI
snímkování popsané v tomto návodu u nadace MedicAlert. Nadaci MedicAlert
lze kontaktovat následujícími způsoby:
Poštou: MedicAlert Foundation International
Telefon: +1 888-633-4298 (bezplatná linka)
Fax: +1 209-669-2450
Web: www.medicalert.org
2323 Colorado Avenue
Turlock, CA 95382 USA
+1 209-668-3333 mimo USA
5 NEŽÁDOUCÍ PŘÍHODY
5.1 Zaznamenané nežádoucí příhody
Zaznamenané nežádoucí příhody, uvedené v Tabulce 5.1.1. byly založeny na
datech získaných z multicentrické prospektivní studie předchozí verze zařízení
(endovaskulárního graftu Zenith AAA), provedené na 15 pracovištích v USA, do
níž bylo zahrnuto 352 pacientů po endovaskulárním výkonu (200 se
standardním rizikem, 100 vysoce rizikových a 52 roll-in pacientů) a 80
kontrolních pacientů. Pacienti byli zařazeni do větve se standardním rizikem,
pokud byli fyziologicky schopni podstoupit otevřenou nebo endovaskulární
reparaci a pokud byly jejich anatomické poměry vhodné pro léčbu pomocí
endovaskulárního graftu Zenith AAA se zaváděcím systémem H&L-B One-Shot.
Do vysoce rizikové větve byli zařazeni pacienti s vhodnými anatomickými
poměry, ale s vyšším rizikem morbidity nebo mortality u otevřené reparace.
Pacienti léčení na začátku studie byli zařazeni do roll-in větve. Skupina
kontrolních pacientů zahrnovala pacienty, jejichž cévní anatomie nemusela být
vhodná pro endovaskulární reparaci AAA.
Tabulka 5.1.1 Úmrtí a ruptura v klinické studii
Úmrtí a ruptura
Všechna úmrtí
2
(0-30 dnů)
3
(31-365 dnů)
Související s AAA 0,0 % (0/198) 1,3 % (1/78) 0,29 3,1 % (3/98) 0,0 % (0/51)
Nesouvisející s AAA 3,0 % (6/198) 0,0 % (0/78) 0,19 4,1 % (4/98) 9,8 % (5/51)
2,3,4
(0-365 dnů)
Související s AAA 0,5 % (1/199) 3,8 % (3/80) 0,07 5,0 % (5/100) 1,9 % (1/52)
Nesouvisející s AAA 3,0 % (6/199) 0,0 % (0/80) 0,19 4,0 % (4/100) 9,6 % (5/52)
Ruptura
(0-30 dnů) 0,0 % (0/199) Nevztahuje se Nevztahuje se 0,0 % (0/100) 0,0 % (0/52)
(31-365 dnů) 0,0 % (0/198) Nevztahuje se Nevztahuje se 1,0 % (1/98) 0,0 % (0/51)
(0-365 dnů) 0,0 % (0/199) Nevztahuje se Nevztahuje se 1,0 % (1/100) 0,0 % (0/52)
1
Denominátor je 199, protože jednomu pacientovi se standardním rizikem nebylo zařízení implantováno.
2
Všechna úmrtí (0-30 dnů) byla shledána jako související s AAA a s výkonem.
3
Čtyři z úmrtí (31-365 dnů) byla shledána jako související s AAA: 1 chirurgicky léčený pacient (septický šok z ischemické kolitidy) a 3 vysoce rizikoví pacienti (pankreatitida se selháním
ledvin a sepsí, krvácení z aneuryzmatu v horní břišní aortě [neléčené AAA] a multiorgánové selhání).
4
Deset z úmrtí (0-365 dnů) bylo shledáno jako související s AAA: 1 pacient se standardním rizikem (srdeční selhání) a 3 chirurgicky léčení pacienti (masivní krvácení, mesenterická
ischémie a septický šok z ischemické kolitidy), 5 vysoce rizikových pacientů (selhání dýchacího systému, srdeční selhání s plicní embolií, pankreatitida se selháním ledvin a sepsí,
krvácení z aneuryzmatu v horní břišní aortě [neléčené AAA] a multiorgánové selhání) a 1 roll-in pacient (podezření na srdeční selhání).
Zenith – větev se
standardním rizikem
0,5 % (1/199) 2,5 % (2/80) 0,20 2,0 % (2/100) 1,9 % (1/52)
3,0 % (6/198) 1,3 % (1/78) 0,68 7,1 % (7/98) 9,8 % (5/51)
3,5 % (7/199) 3,8 % (3/80) >0,99 9,0 % (9/100) 11,5 % (6/52)
Chirurgicky – větev se
1
standardním rizikem Hodnota P Zenith – vysoce riziková větev Zenith – roll-in větev
37

Tabulka 5.1.2 Nežádoucí příhody1 v klinické studii
Zenith – větev se
Bez onemocnění
(0-30 dnů)
Kardiovaskulární
Pulmonální
Renální
Střevní
Rána
Neurologické
Cévní
Bez onemocnění
(31-365 dnů)
Kardiovaskulární
Pulmonální
Renální
Střevní
Rána
Neurologické
Cévní
Bez onemocnění
(0-365 dnů)
Kardiovaskulární
Pulmonální
Renální
Střevní
Rána
Neurologické
Cévní
1
Z indexu nemocnosti.
2
Položka „kardiovaskulární“ obsahovala následující: infarkt myokardu s Q vlnou a bez Q vlny, městnavé srdeční selhání, arytmie vyžadující novou medikaci nebo léčbu, srdeční ischémie
vyžadující intervenci, inotropická léčba, medikamentózně nezvladatelná hypertenze.
3
Položka „pulmonální“ obsahovala následující: reintubace nebo ventilace >24 hodin, pneumonie vyžadující antibiotickou léčbu, kyslíková suplementace při propuštění.
4
Položka „renální“ obsahovala následující: dialýza u pacientů s normální funkcí ledvin před operací, vzestup hladiny kreatininu o více než 30 % oproti počáteční hodnotě při dvou nebo
více kontrolních testech.
5
Položka „střevní“ obsahovala následující: obstrukce střeva, ischémie střeva, aortoenterická píštěl, paralytický ileus >4 dny.
6
Položka „rána“ obsahovala následující: infekce vyžadující antibiotickou léčbu, hernie, mízní píštěl, dehiscence, nekróza vyžadující seškrabání.
7
Položka „neurologické“ obsahovala následující: mrtvice, TIA, ischémie nebo paralýza míchy.
8
Položka „cévní“ obsahovala následující: trombóza větve, distální embolizace s následnou ztrátou tkáně nebo nutnou intervencí, pooperační transfuze (z důvodu pseudoaneuryzmatu,
poranění cévy, prosakování z aneuryzmatu nebo z jiných příčin souvisejících s výkonem), pseudoaneuryzma, poranění cévy (neúmyslná okluze, disekce nebo jiné příčiny související se
výkonem), prosakování nebo prasknutí aneuryzmatu, zvětšení aneuryzmatu o více než 0,5 cm oproti nejmenší dříve naměřené velikosti.
9
Zkoušející zaznamenali okluzi renální artérie u jednoho dalšího vysoce rizikového pacienta a chronickou renální insuficienci u jednoho dalšího vysoce rizikového pacienta do kategorie
nežádoucích příhod „jiné“.
10
Zkoušející zaznamenali renální insuficienci u jednoho dalšího roll-in pacienta a renální insuficienci u jednoho dalšího pacienta podstupujícího chirurgický zásah do kategorie
nežádoucích příhod „jiné“.
11
Zkoušející zaznamenali prasknutí plátu v aortě při operaci s následnou okluzí renální artérie u jednoho dalšího roll-in pacienta do kategorie nežádoucích příhod „jiné“.
5.2 Potenciální nežádoucí příhody
Mezi další nežádoucí příhody, ke kterým může dojít a které mohou vyžadovat
intervenci, mimo jiné patří:
• Amputace
• Arteriovenózní píštěl
• Edém
• Embolizace (mikro a makro) s transientní nebo trvalou ischémií nebo
infarktem
• Endoleak
• Endoprotéza: nesprávné umístění komponent; neúplné rozvinutí komponent;
migrace komponent; prasknutí stehů; okluze; infekce; prasknutí stentu;
opotřebení materiálu graftu; dilatace; eroze; propíchnutí; prosakování kolem
graftu; separace a koroze kotviček
• Horečka a lokalizovaný zánět
• Impotence
• Infekce v místě aneuryzmatu, implantátu nebo místa vstupu, včetně abscesu,
transientní horečky a bolesti
• Klaudikace (např. v hýždi nebo v lýtku)
• Komplikace močového a pohlavního systému s následnými doprovodnými
potížemi (např. ischémie, eroze, píštěl, inkontinence, hematurie, infekce)
• Komplikace související s anestézií a následné doprovodné problémy
(například aspirace)
• Komplikace v lymfatickém systému a následné doprovodné problémy
(například mízní píštěl)
• Komplikace v místě cévního vstupu včetně infekce, bolesti, hematomu,
pseudoaneuryzmatu a arteriovenózní píštěle
• Komplikace v oblasti ledvin s následnými doprovodnými potížemi (např.
okluze artérie, toxicita kontrastní látky, renální nedostatečnost nebo selhání)
• Komplikace v oblasti plic a dýchacích cest s následnými doprovodnými
potížemi (např. pneumonie, selhání respiračního systému, prolongovaná
intubace)
• Komplikace v ráně a následné doprovodné problémy (například dehiscence,
infekce)
• Krvácení, hematom nebo koagulopatie
• Neurologické lokální nebo celkové komplikace s následnými doprovodnými
potížemi (např. mrtvice, transientní ischemická ataka, paraplegie, paraparéza,
paralýza)
• Okluze graftu nebo nativní cévy
• Poranění aorty včetně perforace, disekce, krvácení, ruptury a úmrtí
• Poranění cévy
• Prasknutí aneuryzmatu a úmrtí
• Přechod na otevřenou chirurgickou operaci
• Selhání jater
• Spasmus nebo trauma cévy (např. disekce iliofemorální cévy, krvácení,
ruptura, úmrtí)
• Srdeční komplikace s následnými doprovodnými potížemi (např. arytmie,
infarkt myokardu, městnavé srdeční selhání, hypotenze, hypertenze)
• Střevní komplikace (například ileus, transientní ischémie, infarkt, nekróza)
• Trombóza tepny nebo žíly nebo pseudoaneuryzma
• Úmrtí
• Zvětšení aneuryzmatu
Hlášení nežádoucích příhod souvisejících se zařízením
Všechny nežádoucí příhody (klinické události) související s endovaskulárním
graftem Zenith Flex AAA se musí neodkladně hlásit společnosti COOK. Pro
zákazníky v USA: Chcete-li nahlásit klinickou událost, volejte oddělení vztahů
se zákazníky 1-800-457-4500 (nepřetržitá služba) nebo 1-812-339-2235. Pro
zákazníky mimo USA: Obraťte se prosím na svého distributora.
2
3
4, 9
5
6
7
8,11
2
3
4, 10
5
6
7
8
2
3
4, 9,10
5
6
7
8,11
standardním rizikem
80 % (160/200) 58 % (46/80) <0,001 68 % (68/100) 73 % (38/52)
3,0 % (6/200) 11 % (9/80) 0,02 14 % (14/100) 1,9 % (1/52)
1,0 % (2/200) 15 % (12/80) <0,001 2,0 % (2/100) 0,0 % (0/52)
2,5 % (5/200) 10 % (8/80) 0,01 6,0 % (6/100) 5,8 % (3/52)
1,0 % (2/200) 3,8 % (3/80) 0,14 1,0 % (1/100) 1,9 % (1/52)
4,5 % (9/200) 7,5 % (6/80) 0,38 2,0 % (2/100) 3,8 % (2/52)
0,0 % (0/200) 2,5 % (2/80) 0,08 0,0 % (0/100) 0,0 % (0/52)
11 % (21/200) 31 % (25/80) <0,001 20 % (20/100) 19 % (10/52)
91 % (181/198) 86 % (67/78) 0,25 79 % (77/98) 86 % (44/51)
2,5 % (5/198) 3,8 % (3/78) 0,69 5,1 % (5/98) 2,0 % (1/51)
0,5 % (1/198) 1,3 % (1/78) 0,49 4,1 % (4/98) 0,0 % (0/51)
0,5 % (1/198) 0,0 % (0/78) >0,99 3,1 % (3/98) 0,0 % (0/51)
0,5 % (1/198) 1,3 % (1/78) 0,49 0,0 % (0/98) 0,0 % (0/51)
2,0 % (4/198) 5,1 % (4/78) 0,23 3,1 % (3/98) 2,0 % (1/51)
1,0 % (2/198) 0,0 % (0/78) >0,99 1,0 % (1/98) 3,9 % (2/51)
3,0 % (6/198) 3,8 % (3/78) 0,72 8,2 % (8/98) 5,9 % (3/51)
76 % (151/200) 49 % (39/80) <0,001 55 % (55/100) 62 % (32/52)
5,0 % (10/200) 14 % (11/80) 0,02 19 % (19/100) 3,8 % (2/52)
1,5 % (3/200) 16 % (13/80) <0,001 6,0 % (6/100) 0,0 % (0/52)
2,5 % (5/200) 10 % (8/80) 0,01 9,0 % (9/100) 5,8 % (3/52)
1,5 % (3/200) 3,8 % (3/80) 0,36 1,0 % (1/100) 1,9 % (1/52)
5,5 % (11/200) 13 % (10/80) 0,08 5,0 % (5/100) 5,8 % (3/52)
1,0 % (2/200) 2,5 % (2/80) 0,32 1,0 % (1/100) 3,8 % (2/52)
12 % (24/200) 33 % (26/80) <0,001 25 % (25/100) 23 % (12/52)
Chirurgicky – větev se
standardním rizikem Hodnota P Zenith – vysoce riziková větev Zenith – roll-in větev
6 SOUHRN KLINICKÝCH STUDIÍ
6.1 Cíle
Primárním cílem klinické studie bylo vyhodnotit bezpečnost a účinnost
předchozí verze zařízení (endovaskulárního graftu Zenith AAA) jako alternativy
otevřeného chirurgického zásahu při primární léčbě infrarenálních aneuryzmat
břišní aorty. Hypotéza studie zkoumala, zda u skupiny pacientů se standardní
mírou rizika došlo ke snížení nemocnosti do 30 dnů, non-inferiorní 30denní míry
přežití, non-inferiorní 12měsíční míry přežití, non-inferiorní 12měsíční úspěšnosti
léčby a ke zvýšení míry klinické úspěšnosti v porovnání s kontrolní skupinou
pacientů, která podstoupila chirurgický zásah. Bezpečnost byla zjišťována
hodnocením, zda se u pacientů s endovaskulárním graftem Zenith AAA
vyskytuje snížená míra 30denní nemocnosti, non-inferiorní 30denní a 12měsíční
míra přežití a non-inferiorní 12měsíční úspěšnost léčby v porovnání s léčbou
otevřeným chirurgickým zásahem. Hodnocení účinnosti bylo založeno na
vyloučení aneuryzmatu s absencí veškerých endoleaků, bez jakéhokoli zvětšení
aneuryzmatu (≥5 mm) a bez závažnějších nežádoucích příhod souvisejících se
zařízením po dobu následujícího jednoho roku. Mezi sekundární cíle patřilo
hodnocení klinického přínosu a přispění ke kvalitě života.
6.2 Uspořádání studie
V USA byla provedena multicentrická, nerandomizovaná studie porovnávající
pacienty se standardním medicínským rizikem, kterým byl implantován
endovaskulární graft, s kontrolní skupinou pacientů léčenou otevřeným
chirurgickým zásahem. Studie obsahovala dvě další větve, a sice skupinu
vysoce rizikových pacientů a skupinu roll-in pacientů z počáteční fáze použití
léčby. Na patnácti pracovištích bylo do studie zahrnuto 200 pacientů se
standardní mírou rizika, 80 kontrolních pacientů, kteří podstoupili chirurgickou
operaci, 100 pacientů s vysokou mírou rizika a 52 roll-in pacientů. Skupina
kontrolních pacientů zahrnovala pacienty, jejichž cévní anatomie nemusela být
vhodná pro endovaskulární reparaci AAA. Kontrolní vyhodnocení byla
naplánována takto: před propuštěním, po 1 měsíci, po 6 měsících, po
12 měsících a po 24 měsících. Kontrolní vyšetření a dostupnost pacientů po
1 měsíci, po 12 měsících a po 24 měsících ukazuje Tabulka 6.2.1, jelikož se
jednalo o primární časové body datové analýzy. Údaje ze zobrazovacích
vyšetření, uvedené v tomto souhrnu, jsou založeny na nálezech nezávislé
laboratoře pro centralizované analýzy snímků (centrální laboratoře), která
prověřovala CT snímky a rentgeny břicha a tak zjišťovala změny průměru
aneuryzmatu, migraci zařízení a relativní pohyb komponent, integritu zařízení
(drátu a graftu) a přítomnost a typ endoleaku. Klinické události byly
posuzovány nezávislým výborem pro klinické události a bezpečnost
monitoroval nezávislý výbor pro bezpečnost dat.
Pacienti se standardní mírou rizika, podstupující léčbu pomocí zařízení Zenith,
a pacienti léčení pomocí standardní chirurgické operace splňovali identická
kritéria patofyziologických rizik. U skupin pacientů léčených endovaskulárně
byl vyloučen cirkumferenční trombus proximálního krčku, proximální krček
kratší než 15 mm, průměr proximálního krčku od vnější stěny k vnější stěně
menší než 18 mm nebo větší než 28 mm, závažný stupeň angulace
proximálního krčku, průměr iliakální artérie od vnější stěny k vnější stěně
menší než 7,5 mm nebo větší než 20 mm v místě distální fixace nebo délka
místa distální fixace v iliakální artérii menší než 10 mm.
Riziko při podstoupení chirurgické operace bylo považováno za zvýšené u
pacientů starších 80 let, u pacientů s počáteční hladinou kreatininu >2,0 mg/dl,
s domácí kyslíkovou suplementací, s FEV1 (usilovná sekundová vitální kapacita)
<1 litr, s ejekční frakcí <25 %, s invalidizující CHOPN, s klasifikací NYHA 3 nebo 4,
s nepříznivým stavem břišní krajiny („hostile abdomen“), u pacientů
podstupujících dialýzu, u pacientů s IM v posledních 6 měsících, u pacientů s
medikamentózně nezvladatelnou hypertenzí, u pacientů s mrtvicí v anamnéze
s reziduálním deficitem, u pacientů, jejichž kulturní přesvědčení zakazuje příjem
krve nebo krevních produktů, u pacientů s operací bypassu renální cévy v
anamnéze a u pacientů se zánětlivým aneuryzmatem.
Před zahrnutím pacientů do pilotní studie byla pracoviště bez předchozí praxe
v implantaci endovaskulárního graftu Zenith AAA požádána, aby provedla
léčbu prvních pacientů pod supervizí dohlížejícího. Tito tzv. roll-in pacienti
měli jak standardní, tak vysokou míru rizika a byli sledování podle stejného
plánu kontrol jako pacienti zahrnutí do pilotní studie.
38

Tabulka 6.2.1 Kontrolní vyšetření a dostupnost pacienta
1
Léčba Zenith – větev se standardním rizikem Chirurgicky – větev se standardním rizikem
Interval
Bez zařízení 1 1
Přechod na otevřenou chirurgickou operaci 0 2
1 měsíc 12 měsíce 24 měsíce 1 měsíc 12 měsíce 24 měsíce
2
2
2
1
2
2
0 0 Nevztahuje se
Nevztahuje se Nevztahuje se Nevztahuje se
Zemřel/a 1 7 17 2 3 Nevztahuje se
Zrušil/a účast nebo se nedostavil/a na kontrolu 0 0 2 0 4 Nevztahuje se
K dispozici 198 190 178 78 73 Nevztahuje se
CT snímek na místě 191 168 110 69 58 Nevztahuje se
CT snímek v centrální laboratoři 190 165 99 69 59 Nevztahuje se
Zobrazovací vyšetření močového systému na místě 179 153 108 Nevztahuje se Nevztahuje se Nevztahuje se
Zobrazovací vyšetření močového systému v centrální laboratoři 178 149 93 Nevztahuje se Nevztahuje se Nevztahuje se
Vyhodnocení endoleaku na místě 187 163 107 Nevztahuje se Nevztahuje se Nevztahuje se
Vyhodnocení endoleaku v centrální laboratoři 161 148 92 Nevztahuje se Nevztahuje se Nevztahuje se
Vyhodnocení zvětšení aneuryzmatu na místě Nevztahuje se 149 104 Nevztahuje se Nevztahuje se Nevztahuje se
Vyhodnocení zvětšení aneuryzmatu v centrální laboratoři Nevztahuje se 151 94 Nevztahuje se Nevztahuje se Nevztahuje se
1
Velikost vzorku pro datovou analýzu byla různá u jednotlivých výše uvedených časových bodů a u níže uvedených tabulek. Tato variabilita je způsobena nedostupností pacienta
pro následné kontroly, a také množstvím a kvalitou snímků dostupných v určitých časových bodech pro vyhodnocení. Například počet a kvalita snímků dostupných k vyhodnocení
endoleaku po 12 měsících je jiná než počet a kvalita snímků po 24 měsících kvůli různému počtu provedených zobrazovacích vyšetření, počtu snímků poskytnutému klinickým
pracovištěm centrální laboratoři a/nebo počtu snímků s kvalitou akceptovatelnou pro vyhodnocení. Celkové počty v časových bodech nejsou kumulativní, pokud není uvedeno jinak.
2
Celkové počty v časových bodech jsou kumulativní.
6.3 Data o pacientech
Tabulka 6.3.1 a Tabulka 6.3.2 porovnávají charakteristiky subjektů a počáteční průměr aneuryzmatu u pacientů léčených pomocí endovaskulárního graftu
Zenith AAA a u pacientů léčených standardní chirurgickou operací.
Tabulka 6.3.1 Porovnání charakteristik subjektů
Položka
Zenith – větev se
standardním rizikem
Chirurgicky – větev se
standardním rizikem Hodnota P
Zenith – vysoce
riziková větev Zenith – roll-in větev
Věk (let) 71 ± 7 69 ± 7 0,03 77 ± 7 74 ± 8
Pohlaví mužské 94 % (187/200) 89 % (71/80) 0,22 92 % (92/100) 90 % (47/52)
Aktuální zdravotní stav
Onemocnění periferních cév
Hypertenze 64 % (127/200) 83 % (65/78) 0,001 68 % (67/99) 67 % (35/52)
16 % (31/195) 25 % (19/76) 0,12
24 % (23/96) 9,6 % (5/52)
Selhání ledvin 0,0 % (0/197) 0,0 % (0/79) >0,99 5,2 % (5/97) 1,9 % (1/52)
CHOPN 20 % (39/199) 18 % (14/78) 0,87 34 % (33/98) 22 % (11/51)
Tromboembolická příhoda 4,5 % (9/199) 7,7 % (6/78) 0,38 7,1 % (7/99) 1,9 % (1/52)
Onemocnění jater 2,1 % (4/192) 5,1 % (4/79) 0,24 1,0 % (1/99) 1,9 % (1/52)
Diabetes mellitus 12 % (24/199) 15 % (12/79) 0,55 17 % (17/99) 14 % (7/51)
Závislost na inzulinu 17 % (4/24) 8,3 % (1/12) 0,65 24 % (4/17) 43 % (3/7)
Předchozí zdravotní stav
IM
39 % (74/192) 29 % (23/80) 0,13
35 % (34/98) 35 % (18/52)
Městnavé srdeční selhání 5,0 % (10/199) 12 % (9/78) 0,07 16 % (16/100) 10 % (5/50)
Angina pectoris 49 % (98/198) 39 % (31/79) 0,14 45 % (44/98) 44 % (23/52)
Arytmie 20 % (40/197) 22 % (17/78) 0,87 28 % (27/98) 24 % (12/51)
Cerebrovaskulární onemocnění 9,5 % (19/199) 16 % (13/79) 0,14 20 % (20/99) 9,8 % (5/51)
Celková infekce 1,0 % (2/196) 0,0 % (0/78) >0,99 3,1 % (3/97) 0,0 % (0/49)
Rakovina 22 % (43/200) 19 % (15/80) 0,74 31 % (31/99) 29 % (15/51)
Aneuryzma v rodinné anamnéze 16 % (24/150) 27 % (17/63) 0,09 14 % (11/77) 26 % (10/38)
Chirurgický zásah v místě onemocnění
v anamnéze
10 % (20/200) 15 % (12/79) 0,22 10 % (10/99) 14 % (7/51)
Ozařování v místě onemocnění v anamnéze 0,5 % (1/197) 0,0 % (0/79) >0,99 2,0 % (2/100) 2,0 % (1/51)
Nadměrné požívání alkoholu 3,6 % (7/193) 10 % (8/77) 0,04 3,1 % (3/96) 4,0 % (2/50)
Kuřáctví
Nikdy nekouřil/a
Kouřil/a v minulosti 69 % (133/193) 60 % (48/80) 0,03 69 % (66/96) 57 % (28/49)
Stále kouří 21 % (40/193) 35 % (28/80) 18 % (17/96) 18 % (9/49)
V důsledku vstupních kritérií byli vysoce rizikoví pacienti starší (P<0,001), s vyšším výskytem selhání ledvin (P=0,004), CHOPN (P=0,01), městnavého srdečního selhání (P=0,004) a
cerebrovaskulárních onemocnění (P=0,02) oproti pacientům se standardním rizikem.
10 % (20/193) 5,0 % (4/80)
14 % (13/96) 24 % (12/49)
Rozsah průměru
<30 mm
30-39 mm
40-49 mm
50-59 mm
60-69 mm
70-79 mm
80-89 mm
≥90 mm
Do distribuce průměru aneuryzmatu nebyli zahrnuti tři vysoce rizikoví pacienti a jeden roll-in pacient.
6.4 Výsledky
Data shromážděná v Tabulkách 6.4.1 až 6.4.3 byla shromážděna na pracovištích klinické studie a v centrální laboratoři. Jsou uvedena data za 24 měsíců, pokud
byla k dispozici. Kontrolní pacienti nebyli sledováni později než po 12 měsících a některá data nebyla posuzována později než po 12 měsících. Proto jsou v této
části uvedeny některé výsledky po 12 měsících a některé po 24 měsících. Tabulka 6.4.1 popisuje zařízení implantovaná pacientům zahrnutým do klinické studie.
Tabulka 6.4.2 popisuje primární výsledky klinické studie. Obrázky 6.4.1 a 6.4.2 jsou Kaplan-Meierovy grafy přežití ze všech příčin a v souvislosti s AAA po dobu
24 měsíců. U všech úmrtí posuzoval nezávislý výbor pro klinické události případnou souvislost s reparací aneuryzmatu. Všechna časná úmrtí (0-30 dnů) byla
shledána jako související s AAA. Úmrtí po 30 dnech byla shledána jako související s AAA, pokud bylo potvrzeno onemocnění AAA nebo vztah k zařízení.
Tabulka 6.4.3 uvádí měřítka úspěšnosti a Obrázek 6.4.3 je Kaplan-Meierův graf nepřítomnosti onemocnění.
Zenith – větev se
standardním rizikem
0,0 % (0/199) 0,0 % (0/78) 0,0 % (0/100) 0,0 % (0/52)
0,5 % (1/199) 0,0 % (0/78) 1,0 % (1/100) 0,0 % (0/52)
23 % (45/199) 7,7 % (6/78) 15 % (15/100) 13 % (7/52)
48 % (95/199) 33 % (26/78) 47 % (47/100) 40 % (21/52)
24 % (47/199) 29 % (23/78) 27 % (27/100) 42 % (22/52)
3,0 % (6/199) 21 % (16/78) 5,0 % (5/100) 1,9 % (1/52)
2,5 % (5/199) 6,4 % (5/78) 1,0 % (1/100) 0,0 % (0/52)
0,0 % (0/199) 2,6 % (2/78) 1,0 % (1/100) 0,0 % (0/52)
Chirurgicky – větev se
standardním rizikem Zenith – vysoce riziková větev Zenith – roll-in větev
Tabulka 6.4.1 Implantované zařízení
Položka Zenith – větev se standardním rizikem Zenith – vysoce riziková větev Zenith – roll-in větev
Tabulka 6.3.2 Distribuce průměru aneuryzmatu
Hlavní tělo a ramena 99,5 % (199/200)* 100 % (100/100)
Extenze hlavního těla
Extenze ipsilaterálního iliakálního ramena
Extenze kontralaterálního iliakálního ramena
Přechodový díl 0,5 % (1/199)** 0,0 % (0/100) 0,0 % (0/52)
Okluzor 0,0 % (0/199) 0,0 % (0/100) 0,0 % (0/52)
*Jeden pacient se standardním rizikem neobdržel zařízení kvůli tortuozitě a kalcifikaci přístupové cévy.
**Přechodový díl byl použit bez okluzoru.
1
Jedno zařízení bylo individuálně upravené.
2
Dvěma pacientům se standardním rizikem a jednomu vysoce rizikovému pacientovi byla implantována extenze hlavního těla sekundárně; jednomu pacientovi se standardním rizikem
byly implantovány dvě extenze hlavního těla.
3
Dvěma pacientům se standardním rizikem a jednomu vysoce rizikovému pacientovi byly sekundárně implantovány extenze ipsilaterálního ramena.
4
Čtyřem pacientům se standardním rizikem, dvěma vysoce rizikovým pacientům a jednomu roll-in pacientovi byly sekundárně implantovány extenze kontralaterálního ramena.
5
Tři pacienti se standardním rizikem a tři vysoce rizikoví pacienti obdrželi primárně ipsilateriální i kontralaterální extenzi; jeden pacient se standardním rizikem obdržel ipsilateriální i
kontralaterální extenzi sekundárně.
2
3,5
4,5
1,5 % (3/199) 1,0 % (1/100) 5,8 % (3/52)
9,5 % (19/199) 11 % (11/100) 0,0 % (0/52)
11 % (21/199) 11 % (11/100) 13,5 % (7/52)
1
100 % (52/52)
39

Tabulka 6.4.2 Primární výsledky
Položka
Všechna úmrtí
(0-30 dnů)
Všechna úmrtí
(31-365 dnů)
Zenith – větev se
standardním rizikem
2
2, 3
0,5 % (1/199) 2,5 % (2/80) 0,20 2,0 % (2/100) 1,9 % (1/52)
3,0 % (6/199) 1,3 % (1/80) 0,68 7,0 % (7/100) 9,6 % (5/52)
Chirurgicky – větev se
1
standardním rizikem Hodnota P
Zenith – vysoce
riziková větev Zenith – roll-in větev
Související s AAA 0,0 % (0/199) 1,3 % (1/80) 0,29 3,0 % (3/100) 0,0 % (0/52)
Nesouvisející s AAA 3,0 % (6/199) 0,0 % (0/80) 0,19 4,0 % (4/100) 9,6 % (5/52)
Všechna úmrtí
(0-365 dnů)
2,3,4
3,5 % (7/199) 3,8 % (3/80) >0,99 9,0 % (9/100) 11,5 % (6/52)
Související s AAA 0,5 % (1/199) 3,8 % (3/80) 0,07 5,0 % (5/100) 1,9 % (1/52)
Nesouvisející s AAA 3,0 % (6/199) 0,0 % (0/80) 0,19 4,0 % (4/100) 9,6 % (5/52)
Ruptura
(0-30 dnů) 0,0 % (0/199) Nevztahuje se Nevztahuje se 0,0 % (0/100) 0,0 % (0/52)
(31-365 dnů) 0,0 % (0/199) Nevztahuje se Nevztahuje se 1,0 % (1/100) 0,0 % (0/52)
(0-365 dnů) 0,0 % (0/199) Nevztahuje se Nevztahuje se 1,0 % (1/100) 0,0 % (0/52)
Konverze
(0-30 dnů) 0,0 % (0/199) Nevztahuje se Nevztahuje se 0,0 % (0/100) 0,0 % (0/52)
5
(31-365 dnů)
5
(0-365 dnů)
Nežádoucí příhody
6
(0-30 dnů)
6
(31-365 dnů)
6
(0-365 dnů)
1
Denominátor je 199, protože jednomu pacientovi se standardním rizikem nebylo zařízení implantováno.
2
Všechna úmrtí (0-30 dnů) byla shledána jako související s AAA a s výkonem.
3
Čtyři z úmrtí (31-365 dnů) byla shledána jako související s AAA: 1 chirurgicky léčený pacient (septický šok z ischemické kolitidy) a 3 vysoce rizikoví pacienti (pankreatitida se
selháním ledvin a sepsí, krvácení z aneuryzmatu v horní břišní aortě [neléčené AAA] a multiorgánové selhání).
4
Deset z úmrtí (0-365 dnů) bylo shledáno jako související s AAA: 1 pacient se standardním rizikem (srdeční selhání) a 3 chirurgicky léčení pacienti (masivní krvácení, mesenterická
ischémie a septický šok z ischemické kolitidy), 5 vysoce rizikových pacientů (selhání dýchacího systému, srdeční selhání s plicní embolií, pankreatitida se selháním ledvin a sepsí,
krvácení z aneuryzmatu v horní břišní aortě [neléčené AAA] a multiorgánové selhání) a 1 roll-in pacient (podezření na srdeční selhání).
5
Příčinou konverze u pacientů se standardním rizikem byl perzistující proximální endoleak typu 1 a nové suprarenální aortální aneuryzma. U třech pacientů, jimž byl proveden
chirurgický zásah, došlo k masivnímu krvácení; u 2 byla nutná revize a jeden zemřel.
6
Nežádoucí příhody zahrnuté do indexu morbidity.
1,0 % (2/199) Nevztahuje se Nevztahuje se 1,0 % (1/100) 0,0 % (0/52)
1,0 % (2/199) Nevztahuje se Nevztahuje se 1,0 % (1/100) 0,0 % (0/52)
20 % (40/200) 43 % (34/80) <0,001 32 % (32/100) 27 % (14/52)
8,6 % (17/199) 14 % (11/78) 0,25 21 % (21/98) 14 % (7/51)
25 % (49/200) 51 % (41/80) <0,001 45 % (45/100) 38 % (20/52)
U pacientů s endovaskulárním graftem Zenith AAA nebyly z hlediska přežití a
nepřítomnosti závažnějších nežádoucích příhod žádné signifikantní rozdíly
mezi muži a ženami. (Chybové pruhy, které ukazují Obrázky 6.4.1, 6.4.2 a
6.4.3, reprezentují intervaly 95 % spolehlivosti.) Obrázek 6.4.1 představuje
24měsíční přežití ze všech příčin. Doprovodná tabulka představuje KaplanMeierovu analýzu po 1, 6, 12 a 24 měsících.
Obrázek 6.4.1 24 měsíční přežití
Zenith – větev se standardním rizikem
(N=200, 1 úmr tí do 30 dnů
4 úmrtí do 6 měsíců
7 úmrtí do 12 měsíců
17 úmrtí do 24 měsíců)
Chirurgicky – větev se standardním rizikem
(N=80, 2 úmrtí do 30 dnů
Procento přežití
3 úmrtí do 6 měsíců
3 úmrtí do 12 měsíců)
Měsíců po výkonu
Po 1 měsíci Po 6 měsících Po 12 měsících Po 24 měsících
n % přežití n % přežití n % přežití n % přežití
Zenith –
větev se
standardním
rizikem
Chirurgicky –
větev se
standardním
rizikem
n = Živí pacienti schopní kontrolního vyšetření na konci intervalu
P = 0,81
*Jeden pacient zemřel před uplynutím 1 měsíce a jednomu pacientovi nebylo zařízení
implantováno.
198* 99,5 194 98,0 190 96,5 97 90,9
Nevzta-
78 97,5 73 96,2 67 96,2
huje se
Nevzta-
huje se
Obrázek 6.4.2 představuje 24měsíční přežití související s AAA (určeno
výborem pro klinické události). Doprovodná tabulka představuje KaplanMeierovu analýzu po 1, 6, 12 a 24 měsících.
Obrázek 6.4.2 24 měsíční přežití související s AAA
Zenith – větev se standardním rizikem
(N=200, 1 úmr tí do 30 dnů
1 úmrtí do 6 měsíců
1 úmrtí do 12 měsíců
2 úmrtí do 24 měsíců)
Chirurgicky – větev se standardním rizikem
(N=80, 2 úmrtí do 30 dnů
3 úmrtí do 6 měsíců
3 úmrtí do 12 měsíců)
Procento nepřítomnosti úmrtí
souvisejícího s aneuryzmatem
Událost: úmrtí související s aneuryzmatem
Měsíců po výkonu
Po 1 měsíci Po 6 měsících Po 12 měsících Po 24 měsících
n % přežití n % přežití n % přežití n % přežití
Zenith –
větev se
standardním
rizikem
Chirurgicky –
větev se
standardním
rizikem
n = Živí pacienti schopní kontrolního vyšetření na konci intervalu
P = 0,04
*Jeden pacient zemřel před uplynutím 1 měsíce a jednomu pacientovi nebylo zařízení
implantováno.
198* 99,5 194 99,5 190 99,5 97 99,0
Nevzta-
78 97,5 73 96,2 67 96,2
huje se
Nevzta-
huje se
40

Tabulka 6.4.3 Měřítka úspěšnosti
Položka
Technická úspěšnost
Úspěšnost výkonu po
30 dnech
Úspěšnost léčby po
12 měsících
1
Průchodný graft po rozvinutí.
2
Technická úspěšnost bez závažnějších komplikací, průchodný graft a nepřítomnost endoleaku I nebo III typu po 30 dnech.
3
Úspěšnost výkonu po 12 měsících bez zvětšení aneuryzmatu (>5 mm).
Obrázek 6.4.3 představuje 12měsíční nepřítomnost onemocnění (událostí v indexu morbidity). Doprovodná tabulka představuje Kaplan-Meierovu analýzu po 1, 6 a
12 měsících.
1
2
3
Zenith – větev se
standardním rizikem
99,5 % (199/200) 98,8 % (79/80) 100 % (100/100) 100 % (52/52)
95,1 % (155/163) 88 % (60/68) 86 % (70/81) 91 % (30/33)
89 % (122/137) 85 % (52/61) 70 % (44/63) 87 % (26/30)
Chirurgicky – větev se
standardním rizikem Zenith – vysoce riziková větev Zenith – roll-in větev
Obrázek 6.4.3 Bez onemocnění (0-365 dnů)
Zenith – větev se standardním rizikem
(N=200, 38 pacientů s příhodami do 30 dnů
43 pacientů s příhodami do 6 měsíců
45 pacientů s příhodami do 12 měsíců)
Chirurgicky – větev se standardním rizikem
(N=80, 34 pacientů s příhodami do 30 dnů
38 pacientů s příhodami do 6 měsíců
Procento nepřítomnosti onemocnění
41 pacientů s příhodami do 12 měsíců)
Měsíců po výkonu
Po 1 měsíci Po 6 měsících Po 12 měsících
n % n % n %
Zenith – větev se standardním rizikem 162 81,0 154 78,5 133 77,4
Chirurgicky – větev se standardním rizikem 45 57,1 39 52,0 33 47,8
n = Živí pacienti bez onemocnění na konci intervalu
P = <0,001
Tabulky 6.4.4 až 6.4.7 popisují výsledky léčby subjektů pomocí endovaskulárního graftu Zenith AAA podle hlášení centrální laboratoře. Mezi výkonnostní
faktory zařízení, analyzované centrální laboratoří, patřila integrita zařízení (Tabulka 6.4.4), průchodnost zařízení (Tabulka 6.4.5), migrace (Tabulka 6.4.6) a
separace větve (Tabulka 6.4.7).
Tabulka 6.4.4 Radiografické nálezy v břišní oblasti – integrita zařízení
Položka Zenith – větev se standardním rizikem Zenith – vysoce riziková větev Zenith – roll-in větev
Prasknutí stentu
1
Před propuštěním 0,0 % (0/172) 0,0 % (0/81) 0,0 % (0/39)
Po 30 dnech 0,0 % (0/172) 0,0 % (0/83) 0,0 % (0/43)
Po 6 měsících 0,0 % (0/166) 0,0 % (0/78) 0,0 % (0/35)
Po 12 měsících 0,0 % (0/148) 0,0 % (0/60) 0,0 % (0/28)
Po 24 měsících 0,0 % (0/93) 0,0 % (0/42) 0,0 % (0/19)
Separace kotviček
2
Před propuštěním 0,0 % (0/176) 0,0 % (0/86) 0,0 % (0/39)
Po 30 dnech 0,0 % (0/178) 0,0 % (0/86) 0,0 % (0/43)
Po 6 měsících 1,2 % (2/167) 2,5 % (2/80) 0,0 % (0/35)
Po 12 měsících 2,0 % (3/149) 1,7 % (1/60) 0,0 % (0/28)
Po 24 měsících 1,1 % (1/93) 0,0 % (0/42) 0,0 % (0/19)
Prasknutí materiálu graftu
Před propuštěním 0,0 % (0/176) 0,0 % (0/86) 0,0 % (0/39)
Po 30 dnech 0,0 % (0/178) 0,0 % (0/86) 0,0 % (0/43)
Po 6 měsících 0,0 % (0/167) 0,0 % (0/80) 0,0 % (0/35)
Po 12 měsících 0,0 % (0/149) 0,0 % (0/60) 0,0 % (0/28)
Po 24 měsících 0,0 % (0/93) 0,0 % (0/42) 0,0 % (0/19)
1
Procento prasknutí stentu je uvedeno pro hlavní tělo Centrální laboratoří také nebyly pozorovány žádné případy prasknutí pravého iliakálního ramena, levého iliakálního ramena,
okluzoru, přechodového dílu, levé iliakální extenze, pravé iliakální extenze nebo extenze hlavního těla.
2
Pacienti se separací 1 nebo 2 kotviček (z celkového počtu 10 nebo 12); bez klinických následků.
Tabulka 6.4.5 Nálezy na CT – průchodnost graftu
Položka Zenith – větev se standardním rizikem Zenith – vysoce riziková větev Zenith – roll-in větev
Průchodnost graftu
Po 30 dnech 100 % (185/185) 99 % (85/86) 100 % (47/47)
Po 6 měsících 99 % (183/184) 100 % (74/74) 100 % (39/39)
Po 12 měsících 99 % (153/155) 100 % (62/62) 100 % (30/30)
Po 24 měsících 100 % (96/96) 100 % (33/33) 100 % (25/25)
Tabulka 6.4.6 Nálezy na CT – migrace graftu (hlavního těla)
Položka Zenith – větev se standardním rizikem Zenith – vysoce riziková větev Zenith – roll-in větev
Migrace graftu (>5 mm) po 12 měsících
s klinickými následky1 nebo intervencí 0,0 % (0/162) 0,0 % (0/71) 0,0 % (0/34)
bez klinických následků1 nebo intervence 2,5 % (4/162) 2,8 % (2/71) 0,0 % (0/34)
Migrace graftu (>10 mm) 0,0 % (0/162) 0,0 % (0/71) 0,0 % (0/34)
1
Migrace s klinickými následky by zahrnovala endoleak, konverzi, rupturu nebo úmrtí související s AAA.
41

Tabulka 6.4.7 Radiografické nálezy v břišní oblasti – separace větve
Položka Zenith – větev se standardním rizikem Zenith – vysoce riziková větev Zenith – roll-in větev
Separace větve
Před propuštěním 0,0 % (0/176) 0,0 % (0/86) 0,0 % (0/39)
Po 30 dnech 0,0 % (0/178) 0,0 % (0/86) 0,0 % (0/43)
Po 6 měsících 0,0 % (0/167) 0,0 % (0/80) 0,0 % (0/35)
Po 12 měsících 0,0 % (0/149) 0,0 % (0/60) 0,0 % (0/28)
Po 24 měsících 0,0 % (0/93) 0,0 % (0/42) 0,0 % (0/19)
6.5 Léčba endoleaku
V klinické studii byly endoleaky typu I léčeny při primárním výkonu pomocí dalšího balónku nebo usazením dalších protéz, pokud reparace balónkem nebyla
úspěšná. Endoleaky typu II byly pozorovány po dobu jednoho až šesti měsíců, aby se zjistilo, zda se spontánně tromboticky uzavřou; v případě, že se
aneuryzmata nezvětšovala, byly endoleaky léčeny endovaskulárními technikami podle rozhodnutí ošetřujícího lékaře. Pokud se aneuryzma zvětšovalo, byla
zvážena léčba embolizací nebo ligací a v některých případech byla i realizována. Endoleaky typu III, způsobené defekty graftu, neadekvátním utěsněním nebo
odpojením modulárních komponent, byly léčeny dalšími balónky nebo protézami. Podle hlášení centrální angiografické laboratoře se ve studii v USA
nevyskytovaly endoleaky typu IV. Materiál, použitý k výrobě endovaskulárního graftu Zenith AAA, má standardní tloušťku a je stejný jako materiál používaný při
otevřených chirurgických postupech. Tabulka 6.5.1 ukazuje výskyt endoleaků podle intervalu hodnocení, tak, jak byl zjištěn centrální laboratoří u pacientů se
standardním rizikem, u pacientů s vysokým rizikem a u roll-in pacientů.
Tabulka 6.5.1 Endoleaky (všechny typy, nové a perzistující)
Položka Zenith – větev se standardním rizikem Zenith – vysoce riziková větev Zenith – roll-in větev
Endoleaky
Před propuštěním 15 % (23/153) 14 % (11/78) 12 % (3/26)
1
Po 30 dnech
Po 6 měsících
Po 12 měsících
1
Zahrnuje jak přetrvávající endoleaky, tak nové nálezy.
Tabulka 6.5.2 – 6.5.4 ukazuje incidenci prvního výskytu endoleaku podle intervalu hodnocení tak, jak byl zjištěn centrální laboratoří při kontrolním vyšetření po 30
dnech, 6 měsících nebo 12 měsících (nebo před tímto kontrolním vyšetřením), u pacientů se standardním rizikem, u pacientů s vysokým rizikem a u roll-in pacientů. Je
také uveden počet pacientů bez endoleaku po této době.
1
1
9,9 % (16/161) 12 % (9/75) 6,3 % (2/32)
8,7 % (15/172) 11 % (8/70) 8,6 % (3/35)
7,4 % (11/148) 8,8 % (5/57) 3,4 % (1/29)
Tabulka 6.5.2 První výskyt endoleaku1 u pacientů se standardním rizikem
Položka
Do vyšetření po 1 měsíci
N=179
% Endoleak
Bez endoleaku
1
později
2
Vyšetření po 6 měsících
N=172
% Endoleak
Bez endoleaku
1
později
Vyšetření po 12 měsících
N=148
2
% Endoleak
Bez endoleaku
1
později
Endoleaky 17 31 17 2,3 4 3 3,4 5 2
Proximální, typ I 2,8 5 4 0,0 0 0 0,0 0 0
Distální
Typ I 1,7 3 1 0,0 0 0 0,7 1 1
Typ II 9,5 17 9 2,3 4 3 1,4 2 1
Typ III 1,1 2 2 0,0 0 0 0,7 1 0
Typ IV 0,0 0 0 0,0 0 0 0,0 0 0
Vícečetný 1,1 2 1 0,0 0 0 0,0 0 0
Neznámý 1,1 2 0 0,0 0 0 0,7 1 0
1
Zjištěno centrální laboratoří.
2
Následné endoleaky mohou být odlišného typu než původní.
3
Pouze 2 pacienti měli nové endoleaky po 12 měsících; kontrola po 24 měsících nebyla k dispozici.
Tabulka 6.5.3 První výskyt endoleaku1 u pacientů s vysokým rizikem
Položka
Do vyšetření po 1 měsíci
N=88
% Endoleak
Bez endoleaku
1
později
2
Vyšetření po 6 měsících
N=70
% Endoleak
Bez endoleaku
1
později
2
Vyšetření po 12 měsících
N=57
% Endoleak
Bez endoleaku
1
Endoleaky 18 16 6 2,9 2 0 3,5 2 1
Proximální, typ I 2,3 2 1 0,0 0 0 0,0 0 0
Distální
Typ I 1,1 1 1 0,0 0 0 0,0 0 0
Typ II 9,1 8 3 1,4 1 0 1,8 1 0
Typ III 0,0 0 0 1,4 1 0 1,8 1 1
Typ IV 0,0 0 0 0,0 0 0 0,0 0 0
Vícečetný 4,5 4 0 0,0 0 0 0,0 0 0
Neznámý 1,1 1 1 0,0 0 0 0,0 0 0
1
Zjištěno centrální laboratoří.
2
Následné endoleaky mohou být odlišného typu než původní.
3
Bez endoleaků po 12 měsících; kontrola po 24 měsících nebyla k dispozici.
3
3
později
2
2
Tabulka 6.5.4 První výskyt endoleaku1 u roll-in pacientů
Položka
Do vyšetření po 1 měsíci
N=36
% Endoleak
Bez endoleaku
1
později
2
Vyšetření po 6 měsících
N=35
% Endoleak
Bez endoleaku
1
později
2
Vyšetření po 12 měsících
N=29
% Endoleak
Bez endoleaku
1
Endoleaky 11 4 2 2,9 1 0 0,0 0 0
Proximální, typ I 0,0 0 0 0,0 0 0 0,0 0 0
Distální
Typ I 0,0 0 0 0,0 0 0 0,0 0 0
Typ II 5,6 2 1 2,9 1 0 0,0 0 0
Typ III 2,8 1 0 0,0 0 0 0,0 0 0
Typ IV 0,0 0 0 0,0 0 0 0,0 0 0
Vícečetný 0,0 0 0 0,0 0 0 0,0 0 0
Neznámý 2,8 1 1 0,0 0 0 0,0 0 0
1
Zjištěno centrální laboratoří.
2
Následné endoleaky mohou být odlišného typu než původní.
3
Bez endoleaků po 12 měsících; kontrola po 24 měsících nebyla k dispozici.
42
3
později
2

6.6 Změna aneuryzmatu
Tabulky 6.6.1 až 6.6.3 uvádějí změny průměru aneuryzmat u pacientů léčených pomocí endovaskulárního graftu podle zjištění centrální laboratoře. Tabulka
6.6.1 uvádí maximální změnu průměru aneuryzmatu v časovém intervalu. Tabulky 6.6.2 a 6.6.3 uvádí změnu aneuryzmatu a endoleak po 12 a 24 měsících.
Tabulka 6.6.1 Změna maximálního průměru aneuryzmatu podle časového intervalu*
Položka Zenith – větev se standardním rizikem Zenith – vysoce riziková větev Zenith – roll-in větev
Změna po 30 dnech oproti stavu před propuštěním
Zmenšení >5 mm 1,7 % (3/180) 4,8 % (4/84) 0,0 % (0/40)
Beze změny 97 % (174/180) 94 % (79/84) 97,5 % (39/40)
Zvětšení >5 mm 1,7 % (3/180) 1,2 % (1/84) 2,5 % (1/40)
Změna po 6 měsících oproti stavu před propuštěním
Zmenšení >5 mm 36 % (63/173) 41 % (30/73) 49 % (18/37)
Beze změny 62 % (108/173) 59 % (43/73) 51 % (19/37)
Zvětšení >5 mm 1,2 % (2/173) 0,0 % (0/73) 0,0 % (0/37)
Změna po 12 měsících oproti stavu před propuštěním
Zmenšení >5 mm 68 % (102/151) 63 % (39/62) 67 % (20/30)
Beze změny 31 % (47/151) 35 % (22/62) 33 % (10/30)
Zvětšení >5 mm 1,3 % (2/151) 1,6 % (1/62) 0,0 % (0/30)
Změna po 24 měsících oproti stavu před propuštěním
Zmenšení >5 mm 78 % (74/94) 75 % (27/36) 71 % (17/24)
Beze změny 19 % (18/94) 25 % (9/36) 25 % (6/24)
Zvětšení >5 mm 2,1 % (2/94) 0,0 % (0/36) 4,2 % (1/24)
*Jsou zahrnuty pouze subjekty s interpretovatelnými snímky a měřeními změn aneuryzmatu v době od 1 do 24 měsíců.
Tabulka 6.6.2 Změna velikosti aneuryzmatu a endoleaku po 12 měsících
Zenith – větev se standardním rizikem
N=140
Zenith – vysoce riziková větev
N=53
Zenith – roll-in větev
N=27
Endoleak Endoleak Endoleak
Položka
Změna velikosti aneuryzmatu po 12 měsících oproti stavu před propuštěním
N n % N n % N n %
Zmenšení >5 mm 96 3 3,0 35 3 9,0 20 0 0,0
Beze změny 42 8 19 17 2 12 7 1 14
Zvětšení >5 mm 2 0 0,0 1 0 0,0 0 0 0,0
Tabulka 6.6.3 Změna velikosti aneuryzmatu a endoleaku po 24 měsících
Zenith – větev se standardním rizikem
N=90
Zenith – vysoce riziková větev
N=31
Zenith – roll-in větev
N=21
Endoleak Endoleak Endoleak
Položka
Změna velikosti aneuryzmatu po 24 měsících oproti stavu před propuštěním
N n % N n % N n %
Zmenšení >5 mm 71 3 4,0 24 1 4,0 16 0 0,0
Beze změny 18 1 6,0 7 3 43 5 0 0,0
Zvětšení >5 mm 1 1 100 1 0 0,0 0 0 0,0
6.7 Sekundární intervence související s AAA
Sekundární intervence související s AAA v prvním roce byly provedeny u 11 % pacientů se standardním rizikem léčených pomocí zařízení Zenith, u 13 %
pacientů s vysokým rizikem léčených pomocí zařízení Zenith, a u 5,8 % roll-in subjektů, jak uvádí Tabulka 6.7.1. Více než 50 % sekundárních intervencí
zahrnovalo katetrizaci k léčbě endoleaku. Sekundární intervence související s AAA v druhém roce byly provedeny u 4,2 % pacientů se standardním rizikem
léčených pomocí zařízení Zenith, u 2,2 % pacientů s vysokým rizikem léčených pomocí zařízení Zenith, a u 2,3 % roll-in subjektů, jak uvádí Tabulka 6.7.2.
Tabulka 6.7.1 Sekundární intervence (do 12 měsíců)
Zenith – větev se standardním rizikem
Intervence
Přechod na otevřenou chirurgickou operaci 2 1,0 1 1,0 0 0,0
N=199
n % n % n %
Zenith – vysoce riziková větev
N=100
Zenith – roll-in větev
N=52
Subjekty s ≥1 intervencí 21 11 13 13 3 5,8
Léčba endoleaku
Embolizace 5 2,5 3 3,0 1 2,0
Pomocná komponenta 6 3,0 4 4,0 1 2,0
Stent 1 0,5 0 0,0 0 0,0
Angioplastika 0 0,0 1 1,0 0 0,0
Léčba zvětšujícího se aneuryzmatu
Embolizace 0 0,0 0 0,0 0 0,0
Léčba okluze větve 1 0,5 3 3,0 1 2,0
Léčba stenózy větve 1 0,5 0 0,0 0 0,0
Léčba renální artérie 5 2,5 0 0,0 0 0,0
Léčba infrainguinální ischémie 1 0,5 1 1,0 0 0,0
Léčba více příhod 1 0,5 1 1,0 0 0,0
Tabulka 6.7.2 Sekundární intervence (>12 až 24 měsíců)
Zenith – větev se standardním rizikem
Intervence
Přechod na otevřenou chirurgickou operaci 1 0,5 1 1,1 0 0,0
N=190
n % n % n %
Zenith – vysoce riziková větev
N=90
Zenith – roll-in větev
N=44
Subjekty s ≥1 intervencí 8 4,2 2 2,2 1 2,3
Léčba endoleaku
Embolizace 3
1
1,6 0 0,0 1
2
Pomocná komponenta 1 0,5 0 0,0 0 0,0
Stent 0 0,0 1 1,1 0 0,0
Angioplastika 0 0,0 0 0,0 0 0,0
Léčba zvětšujícího se aneuryzmatu
Embolizace 1 0,5 0 0,0 1
2
Léčba okluze větve 1 0,5 0 0,0 0 0,0
Léčba stenózy větve 0 0,0 0 0,0 0 0,0
Léčba renální artérie 0 0,0 1 1,1 0 0,0
Léčba infrainguinální ischémie 1 0,5 0 0,0 0 0,0
Léčba více příhod 1 0,5 0 0,0 0 0,0
1
Pacient obdržel také pomocnou komponentu.
2
Pacient podstoupil intervenci k léčbě zvětšení aneuryzmatu a endoleaku.
43
2,3
2,3

6.8 Měření sekundárních výsledků
Jak ukazuje Tabulka 6.8.1, léčba AAA pomocí endovaskulárního graftu Zenith AAA v porovnání s kontrolní skupinou léčenou chirurgicky prokázala značně lepší
výsledky v ukazatelích sledujících rekonvalescenci a kvalitu života.
Tabulka 6.8.1 Sekundární výsledky podle léčených skupin
Položka
Trvání anestézie (min) 221,6 ± 67,3 304,5 ± 102,7 <0,001 218,9 ± 69,6 213,9 ± 57,7
Trvání výkonu (min) 153,2 ± 56,3 238,7 ± 92,2 <0,001 153,5 ± 58,6 155,9 ± 43,2
Podané produkty z krevní banky 5,0 % (10/200) 84 % (67/80) <0,001 12 % (12/100) 3,8 % (2/52)
Ztráta krve (ml) 299 ± 324 1676 ± 1676 <0,001 356 ± 514 265 ± 226
Počet dnů na JIP 0,4 ± 0,9 3,4 ± 4,6 <0,001 0,5 ± 1,2 0,5 ± 0,9
Počet dnů do propuštění 2,6 ± 1,7 8,8 ± 5,6 <0,001 3,0 ± 2,8 2,7 ± 1,5
Počet dnů do příjmu tekutin ústy 0,5 ± 0,8 3,9 ± 2,5 <0,001 0,5 ± 0,6 0,7 ± 0,5
Počet dnů do normální stravy 1,3 ± 1,2 6,6 ± 4,9 <0,001 1,3 ± 0,8 1,1 ± 0,7
Počet dnů do normálního vyprazdňování
střev
Počet dnů do chůze 1,2 ± 0,7 3,5 ± 3,4 <0,001 1,2 ± 0,7 1,2 ± 0,6
Trvání intubace (h) 1,9 ± 2,2 11,7 ± 13,6 <0,001 1,2 ± 1,7 2,6 ± 4,6
Maximální teplota (°F) 101,1 ± 1,3 100,7 ± 1,2 0,06 100,8 ± 1,1 101,0 ± 1,2
Zenith – větev se
standardním rizikem
2,6 ± 1,4 4,2 ± 2,1 <0,001 2,6 ± 1,5 2,0 ± 1,2
Chirurgicky – větev se
standardním rizikem Hodnota P Zenith – vysoce riziková větev Zenith – roll-in větev
7 VÝBĚR A LÉČBA PACIENTA
Viz kapitola 4 Varování a upozornění
Individualizace léčby
Společnost Cook doporučuje provést výběr průměrů komponent
endovaskulárního graftu Zenith Flex AAA tak, jak uvádí Tabulky 10.5.1 a
10.5.2. Délka endovaskulárního graftu Zenith Flex AAA musí být taková, aby
graft zasahoval od nejnižší renální artérie přesně nad bifurkaci vnitřní iliakální
(hypograstrické) artérie. Lékař má mít k dispozici všechny délky a průměry
součástí, nezbytných k provedení výkonu, zejména pokud ještě nejsou k
dispozici výsledky předoperačních měření (hodnoty průměrů a délek). Tento
přístup zaručuje větší operační flexibilitu k dosažení optimálních výsledků. U
každého pacienta je nutno před použitím endovaskulárního graftu Zenith Flex
AAA pečlivě posoudit rizika a přínosy popsané v kapitole 6 SOUHRN
KLINICKÝCH STUDIÍ. K dalším aspektům pro posouzení vhodnosti pacienta
mimo jiné patří:
• Pacientův věk a očekávaná doba života
• Doprovodná onemocnění (např. srdeční, plicní nebo renální nedostatečnost
před operací, morbidní obezita)
• Vhodnost pacienta pro otevřenou chirurgickou operaci
• Vhodnost anatomických poměrů pacienta pro endovaskulární reparaci
• Riziko prasknutí aneuryzmatu v porovnání s riziky vyplývajícími z léčby za
použití endovaskulárního graftu Zenith Flex AAA
• Schopnost tolerovat celkovou, svodovou nebo lokální anestézii
• Velikost a morfologie iliofemorální přístupové cévy (minimální trombus,
kalcifikace a [nebo] tortuozita) mají vyhovovat kritériím pro použití metod
cévního přístupu a zaváděcích systémů profilu cévního zaváděcího sheathu
o velikosti 14 French až 22 French
• Infrarenální segment aorty bez aneuryzmatu (krček) proximálně k
aneuryzmatu s následujícími parametry:
• o délce nejméně 15 mm;
• s průměrem při měření od vnější stěny k vnější stěně v rozsahu max.
32 mm do min. 18 mm;
• s úhlem vzhledem k dlouhé ose výdutě menším než 60 stupňů; a
• s úhlem vzhledem k ose suprarenální aorty menším než 45 stupňů.
• Místo distální fixace v iliakální artérii delší než 10 mm a s průměrem
7,5-20 mm (měřeno od vnější stěny k vnější stěně)
• Nepřítomnost závažných forem okluzívních onemocnění femorálních/
iliakálních artérií, které by omezily průtok endovaskulárním graftem
Konečné rozhodnutí o léčbě záleží na lékaři a pacientovi.
8 PORADENSTVÍ PRO PACIENTY
Při diskusi o tomto endovaskulárním zařízení a příslušném zákroku musí lékař
a pacient (popřípadě také rodinní příslušníci) zvážit rizika a přínosy, zejména:
• Rizika a rozdíly mezi endovaskulární operací a otevřenou chirurgickou operací
• Potenciální výhody tradiční otevřené chirurgické operace
• Potenciální výhody endovaskulární reparace
• Možnost, že po primární endovaskulární reparaci může být nutné provést
další intervenci nebo standardní chirurgickou operaci aneuryzmatu
Vedle posouzení rizik a přínosů endovaskulární reparace musí lékař zhodnotit
pacientův postoj k následným kontrolám a k dodržování pooperační péče
potřebné k zajištění dlouhodobých bezpečných a efektivních výsledků. Níže
jsou uvedena další témata, která je vhodné prodiskutovat s pacientem a která
se týkají jeho očekávání, co se týče výsledků endovaskulární opravy:
• Funkčnost endovaskulárních graftů z dlouhodobého hlediska nebyla
zatím zjištěna. Všichni pacienti mají být poučeni, že endovaskulární léčba
vyžaduje celoživotní pravidelná kontrolní vyšetření ke zjištění jejich
zdravotního stavu a funkčnosti jejich endovaskulárního graftu. Pacienti se
specifickými klinickými nálezy (např. endoleaky, zvětšujícími se aneuryzmaty
nebo se změnami struktury nebo polohy endovaskulárního graftu) se musí
podrobit důkladnějším kontrolním vyšetřením. Konkrétní pokyny ke
kontrolním vyšetřením jsou uvedeny v kapitole 12 POKYNY K
ZOBRAZOVÁNÍ A POOPERAČNÍ KONTROLE.
• Pacienti mají být poučeni o důležitosti dodržování plánu kontrolních
prohlídek, a to jak v prvním roce, tak později jednou ročně. Pacienti mají být
informováni, že pravidelné a konzistentní kontroly jsou kriticky důležitou
součástí zajištění trvale bezpečné a účinné endovaskulární léčby AAA. Jako
minimum se vyžaduje podstoupení zobrazovacího vyšetření jednou ročně a
docházení na pravidelné pooperační kontroly jako celoživotní závazek k
zajištění pacientova dobrého zdravotního stavu.
• Pacient musí být poučen, že úspěšná oprava aneuryzmatu nezastavuje postup
choroby. Stále trvá nebezpečí přidružené degenerace cév.
• Lékař musí každému pacientovi sdělit, že je důležité vyhledat rychlou
lékařskou pomoc, pokud pocítí příznaky okluze větve a zvětšení nebo
prasknutí aneuryzmatu. Mezi příznaky okluze větve graftu patří mimo jiné
bolest jedné nebo obou kyčlí nebo nohou při chůzi nebo v klidu a zblednutí
nebo chlad nohy. Prasknutí aneuryzmatu může být asymptomatické,
obvykle se však prezentuje následujícími příznaky: bolest; znecitlivění;
slabost nohou; bolest v zádech, hrudníku, břicha nebo třísel; závratě;
mdloby; rychlý srdeční tep nebo náhlá slabost.
•
Vzhledem k tomu, že pro úspěšnou implantaci a kontrolu endovaskulárních
zařízení je nutno použít zobrazovací metody, je nutno s pacientkami ženského
pohlaví, které jsou těhotné nebo mohou otěhotnět, prodiskutovat rizika spojená
s expozicí vyvíjejících se tkání rentgenovému záření. U mužů, kteří podstoupí
endovaskulární nebo standardní chirurgický výkon, může dojít k impotenci.
Lékař má poučit pacienta, aby si přečetl Příručku pacienta a informoval se o
rizicích objevujících se během implantace zařízení nebo po ní. Mezi rizika
související s výkonem patří kardiologické, pulmonální, neurologické, střevní a
krvácivé komplikace. Mezi rizika související ze zařízením patří okluze, endoleak,
zvětšení aneuryzmatu, prasknutí zařízení, riziko opakované intervence nebo
konverze na otevřenou chirurgickou operaci, ruptura a úmrtí (viz část 5.1
Zaznamenané nežádoucí příhody a část 5.2 Potenciální nežádoucí příhody).
Lékař má vyplnit Identifikační kartu pacienta a předat ji pacientovi, aby ji pacient
mohl neustále nosit u sebe. Pacient má kartu předložit při každé návštěvě jiného
lékaře, zejména při jakýchkoli dalších diagnostických výkonech (např. MRI).
9 STAV PŘI DODÁNÍ
• Endovaskulární graft Zenith Flex AAA se dodává sterilní a předinstalovaný v
odtrhovacích obalech.
• Toto zařízení je určeno pouze pro jednorázové použití. Zařízení neresterilizujte.
• Před použitím prohlédněte zařízení i jeho obal a zkontrolujte, zda při
transportu nedošlo k jejich poškození. Nepoužívejte toto zařízení, pokud je
poškozené nebo pokud je poškozena nebo zničena sterilní bariéra. Došlo-li k
poškození, výrobek nepoužívejte a vraťte jej společnosti COOK.
• Před použitím zkontrolujte, zda bylo dodáno správné zařízení (množství a
velikost) pro daného pacienta, a to srovnáním dodaného zařízení s předpisem
lékaře pro konkrétního pacienta.
• Hlavní tělo prostředku je předinstalováno na zaváděcím sheathu Flexor® o
velikosti 18, 20 nebo 22 French. Povrch sheathu je ošetřen hydrofilním
povlakem, který po hydratování zvyšuje jeho schopnost manévrování. K
aktivaci hydrofilního povlaku je třeba otřít povrch sterilním gázovým
polštářkem navlhčeným fyziologickým roztokem.
• Nepoužívejte po uplynutí data „ POUŽÍT DO“ (data exspirace), vytištěného na
štítku.
• Skladujte v suchu a chladu.
10 INFORMACE O KLINICKÉM POUŽITÍ
10.1 Školení lékařů
POZOR: Během implantace nebo reintervence musí být v pohotovosti
kvalifikovaný operační tým pro případ nutnosti přechodu na otevřenou
chirurgickou operaci.
POZOR: Endovaskulární graft Zenith Flex AAA se zaváděcím systémem
H&L-B One-Shot smí používat výhradně lékaři a týmy s kvalifikací pro
vaskulární intervenční výkony a pro použití tohoto zařízení. Doporučené
znalosti a zkušenosti lékařů používajících endovaskulární graft Zenith
Flex AAA se zaváděcím systémem H&L-B One-Shot jsou uvedeny níže:
Výběr pacientů:
• Znalosti vzniku a vývoje aneuryzmatu abdominální aorty (AAA) a
doprovodných onemocnění souvisejících s reparací AAA.
• Znalost interpretace radiologických snímků, výběru a volby velikosti zařízení.
Souhrn operatérských zkušeností multidisciplinárního týmu:
• Femorální přístup, arteriotomie a reparace
• Techniky perkutánního přístupu a uzavření
• Ovládání neselektivních a selektivních vodicích drátů a katetrů
• Interpretace skiaskopických a angiografických snímků
• Embolizace
• Angioplastika
• Umístění endovaskulárního stentu
• Metody odstraňování cizího tělesa z cév
• Správné použití radiografické kontrastní látky
• Metody omezení expozice záření
• Zkušenost s variantami potřebné následné péče o pacienta
10.2 Kontrola před použitím
Před použitím prohlédněte zařízení i jeho obal a zkontrolujte, zda při
transportu nedošlo k jejich poškození. Nepoužívejte toto zařízení, pokud je
poškozené nebo pokud je poškozena nebo zničena sterilní bariéra. Došlo-li k
poškození, výrobek nepoužívejte a vraťte jej společnosti COOK. Před použitím
zkontrolujte, zda bylo dodáno správné zařízení (množství a velikost) pro
daného pacienta, a to srovnáním dodaného zařízení s předpisem lékaře pro
konkrétního pacienta.
10.3 Požadovaný materiál
(Není obsažen v 3kusovém modulárním systému.)
• Pomocná souprava endovaskulárního graftu Zenith AAA
• Skiaskop s funkcemi pro digitální angiografii (rameno C nebo pevná
jednotka)
• Kontrastní látka
• Stříkačka
• Heparinizovaný fyziologický roztok
• Sterilní gázové polštářky
10.4 Doporučený materiál
(Není obsažen v 3kusovém modulárním systému.)
K implantaci jakékoli komponenty řady výrobků Zenith se doporučují
následující výrobky. Informace o použití těchto výrobků najdete v příslušném
návodu k použití příslušného výrobku.
• Extra tuhý vodicí drát 0,035 palce (0,89 mm) o délce 260 cm; například:
• Ultra tuhé vodicí dráty Cook Lunderquist (LES)
• Standardní vodicí drát 0,035 palce (0,89 mm); například:
• Vodicí dráty Cook 0,035 palce (0,89 mm)
• Vodicí dráty Cook Nimble™
• Tvarovatelné balónky; například:
• Balónkový katetr Cook CODA®
• Zaváděcí sady; například:
• Zaváděcí sady Cook Check-Flo®
• Extra velké zaváděcí sady Cook Check-Flo®
• Kontralaterální zavaděče Cook Flexor® Balkin Up & Over®
• Měřicí katetr; například:
• Centimetrové měřicí katetry Cook Aurous®
• Angiografické katetry s rentgenkontrastním hrotem; například:
• Angiografické katetry Cook s hrotem Beacon®
• Katetry Cook Royal Flush s hrotem Beacon®
• Přístupové jehly; například:
• Jehly Cook pro punkci jedné stěny cévy
44

10.5 Pokyny k určení průměru zařízení
Průměr je třeba měřit od vnější stěny k vnější stěně cévy, nikoli jako průměr
lumen (světlost). Volba příliš velké nebo naopak příliš malé velikosti může vést
k netěsnostem nebo k omezení průtoku.
Tabulka 10.5.1 Návod k měření průměru hlavního těla graftu*
Průměr
aorty v místě
implantace
(mm)
18-19 22 82/112, 96/126, 111/141,
20-21 24 82/112, 96/126, 111/141,
22 26 82/112, 96/126, 111/141,
23-24 28 82/112, 96/126, 111/141,
25-26 30 82/112, 96/126, 111/141,
27-28 32 82/112, 96/126, 111/141,
29-32 36 95/125, 113/143,
1
Maximální průměr podél místa proximální fixace.
2
Změřený průměr aorty zaokrouhlete na nejbližší mm.
3
Volbu průměru mohou ovlivňovat také další aspekty.
*Všechny rozměry jsou jmenovité.
Průměr
hlavního
1,2
3
těla
(mm)
Celková délka ke
kontralaterální větvi/celková
délka k ipsilaterální větvi
(mm)
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
131/161, 149/179
Zaváděcí
sheath
(French)
18
18
18
20
20
20
22
Tabulka 10.5.2 Návod k měření průměru iliakálního ramena graftu*
Průměr
iliakální cévy
v místě
implantace
(mm)
<8 8 37, 54, 71, 88, 105, 122 14
8-9 10 37, 54, 71, 88, 105, 122 14
10-11 12 37, 54, 71, 88, 105, 122 14
12-13 14 37, 54, 71, 88 14
14-15 16 37, 54, 71, 88 14
16-17 18 37, 54, 71, 88 16
18 20 37, 54, 71, 88 16
19 22 37, 54, 71, 88 16
20 24 37, 54, 71, 88 16
1
Maximální průměr podél místa distální fixace.
2
Změřený průměr iliakální artérie zaokrouhlete na nejbližší mm.
3
Volbu průměru mohou ovlivňovat také další aspekty.
4
Celková délka ramena = pracovní délka + 22 mm pro aretaci stentu.
*Všechny rozměry jsou jmenovité.
Průměr
iliakálního
1,2
ramena
(mm)
3
Pracovní délka
iliakálního ramena
(mm)
Zaváděcí
4
sheath
(French)
11 POKYNY K POUŽITÍ
Anatomické požadavky
• Velikost a morfologie iliofemorální přístupové cévy (minimální trombus,
kalcifikace a/nebo vinutost) musí vyhovovat kritériím pro použití metod
cévního přístupu a zaváděcích systémů. Je možné, že bude nutno použít
techniky arteriálního bypassu.
• Délk a proximálního aortálního krčku musí být minimálně 15 mm při
průměru měřeném od vnější stěny k vnější stěně 18-32 mm.
• M ísto distální fixace v iliakální artérii musí být delší než 10 mm a s
průměrem 7,5-20 mm (měřeno od vnější stěny k vnější stěně).
Před použitím endovaskulárního graftu Zenith Flex AAA se zaváděcím
systémem H&L-B One-Shot si přečtěte tuto příručku Doporučený návod k
použití. Následující instrukce obsahují základní návod k umístění zařízení. Je
možné, že v praxi bude nutno popsaný postup upravit. Tyto instrukce mají
lékaři sloužit jako vodítko a nemají nahrazovat jeho úsudek.
Obecné informace o použití
• Při použití endovaskulárního graftu Zenith Flex AAA se zaváděcím
systémem H&L-B One-Shot se uplatňují standardní metody zavádění
sheathů pro arteriální přístup, vodicích katetrů, angiografických katetrů a
vodicích drátů. Endovaskulární graft Zenith Flex AAA se zaváděcím
systémem H&L-B One-Shot je kompatibilní s vodicími dráty o průměru
0,035 palce (0,89 mm).
• Zavedení endovaskulárního stentgraftu je chirurgický zákrok, při kterém
může z různých důvodů dojít ke ztrátě krve, což v nepříliš častých případech
vyžaduje další intervenci (včetně transfuze) k zamezení nežádoucích
následků. Je důležité monitorovat, zda při zákroku nedochází ke ztrátě krve
přes hemostatický ventil. Toto je obz vláště relevantní při manipulaci se
šedým polohovačem a po ní. Pokud po vyjmutí šedého polohovače dochází
k nadměrné ztrátě krve, zvažte možnost zavedení nenaplněného
tvarovacího balónku nebo dilatátoru zaváděcího systému do ventilu, což
omezí průtok.
Rozhodující činitele před implantací
Podle předoperačního plánu zkontrolujte, zda bylo vybráno správné zařízení.
Mezi rozhodující činitele patří:
1. Výběr femorální artérie k zavedení systému hlavního těla graftu (tj. určení
příslušných kontralaterálních a ipsilaterálních iliakálních artérií).
2. Angulace krčku aorty, aneuryzmatu a iliakálních artérií.
3. Kvalita krčku aorty.
4. Průměry infrarenálního krčku aorty a distálních iliakálních ar térií.
5. Vzdálenost od renálních artérií k bifurkaci aorty.
6. Vzdálenost od bifurkace aorty k vnitřním iliakálním ar tériím nebo k místu/
místům jejich odstupu.
7. Výdutě zasahující do iliakálních artérií mohou vyžadovat zvláštní
pozornost při výběru vhodného místa styku graftu a artérie.
8. Posuďte stupeň kalcifikace cévy.
Příprava pacienta
1. Při anestézii, antikoagulaci a monitorování vitálních známek postupujte
podle protokolů zdravotnického zařízení.
2. Položte pacienta na snímkovací stůl umožňující skiaskopickou vizualizaci
od aortálního oblouku po femorální bifurkaci.
3. Standardní chirurgickou technikou obnažte obě společné femorální artérie.
4. Zaveďte adekvátní proximální a distální cévní kontrolu obou femorálních
artérií.
11.1 Systém s bifurkací (Obr. 2)
11.1.1 Příprava a propláchnutí hlavního těla s bifurkací
1. Odstraňte z hrotu ochranný transportní stylet s černým ústím (z vnitřní
kanyly) a sejměte ochrannou trubičku kanyly (z vnitřní kanyly) a chránič
hrotu dilatátoru (z hrotu dilatátoru). Ze zadní strany hemostatického
ventilu odstraňte odlepovací sheath Peel-Away®. (Obr. 5) Zvedněte
distální hrot systému a proplachujte systém přes uzavírací kohout na
hemostatickém ventilu, až začne kapalina vytékat z bočního portu v
blízkosti hrotu zaváděcího sheathu. (Obr. 6) Pokračujte v nástřiku celé
dávky 20 ml proplachovacího roztoku do zařízení. Nástřik ukončete a
zavřete uzavírací kohout na přípojné hadičce.
POZNÁMKA: Často používaným roztokem k proplachování graftu je
heparinizovaný fyziologický roztok.
2. K ústí vnitřní kanyly připojte stříkačku s heparinizovaným fyziologickým
roztokem. Proplachujte tak dlouho, až začne roztok vytékat z hrotu
dilatátoru. (Obr. 7)
POZNÁMKA: Při proplachování systému zvedněte distální konec systému, aby
se usnadnilo odstranění vzduchu.
3. Sterilní gázové polštářky navlhčete fyziologickým roztokem a otřete
jimi zaváděcí sheath Flexor®, aby se aktivoval hydrofilní povlak. Hojně
hydratujte sheath i dilatátor.
11.1.2 Příprava a propláchnutí kontralaterálního iliakálního ramena
1. Odstraňte z hrotu vnitřní stylet s černým ústím (z vnitřní kanyly) a sejměte
ochrannou trubičku kanyly (z vnitřní kanyly) a chránič hrotu dilatátoru
(z hrotu dilatátoru). Ze zadní strany hemostatického ventilu odstraňte
odlepovací sheath Peel-Away®. (Obr. 8) Zvedněte distální hrot systému
a proplachujte systém přes uzavírací kohout na hemostatickém ventilu,
až začne kapalina vytékat z bočního portu v blízkosti hrotu zaváděcího
sheathu. (Obr. 9) Pokračujte v nástřiku celé dávky 20 ml proplachovacího
roztoku do zařízení. Nástřik ukončete a zavřete uzavírací kohout na
přípojné hadičce.
POZNÁMKA: Často používaným roztokem k proplachování graftu je
heparinizovaný fyziologický roztok.
2. K ústí na distálním konci vnitřní kanyly připojte stříkačku s
heparinizovaným fyziologickým roztokem. Proplachujte tak dlouho, až
začne roztok vytékat z distálního hrotu dilatátoru. (Obr. 7)
POZNÁMKA: Při proplachování systému zvedněte distální konec systému, aby
se usnadnilo odstranění vzduchu.
11.1.3 Příprava a propláchnutí ipsilaterálního iliakálního ramena
K zajištění správného propláchnutí ipsilaterálního iliakálního ramena graftu se
řiďte podle výše uvedené části 11.1.2. Příprava/proplach kontralaterálního
iliakálního ramena.
11.1.4 Cévní přístup a angiografie
1. Standardní metodou napíchněte vybranou společnou femorální artérii za
použití arteriální jehly s ultratenkou stěnou 18UT gauge nebo 19UT gauge.
Po vytvoření přístupu do cévy zaveďte:
• vodicí dráty – standardní o průměru 0,035 palce (0,89 mm) a délce 145 cm,
s hrotem J, nebo vodicí drát Bentson;
• sheathy vhodné velikosti (např. 6,0 nebo 8,0 French);
• proplachovací katetr (obvykle rentgenkontrastní měřicí katetr, např. katetr
s centimetrovými značkami nebo přímý proplachovací katetr).
2. Proveďte angiografii k identifikaci renálních artérií, bifurkace aorty a
iliakálních bifurkací.
POZNÁMKA: Používá-li se u zešikmeného krčku angulace skiaskopu, může být
nutné provést angiogramy v různých projekcích.
11.1.5 Umístění hlavního těla
1. Zajistěte propláchnutí aplikačního systému heparinizovaným
fyziologickým roztokem a odstranění veškerého vzduchu ze systému.
2. Podávejte celkově heparin a kontrolujte proplachovací roztoky. Proplach
provádějte po každé výměně katetru a (nebo) vodicího drátu.
POZNÁMKA: Po celou dobu výkonu sledujte stav koagulace pacienta.
3. Na ipsilaterální straně nahraďte drát s hrotem J tuhým vodicím drátem
(LES) o průměru 0,035 palce (0,89 mm) a délce 260 cm a posuňte jej přes
katetr do hrudní aorty. Vyjměte proplachovací katetr a sheath. Udržujte
polohu vodicího drátu.
4. Před zavedením umístěte aplikační systém hlavního těla graftu na
břicho pacienta pod skiaskopickým naváděním, abyste si pomohli s
orientací rentgenkontrastní značky kontralaterální větve. Postranní
rameno hemostatického ventilu může sloužit jako externí reference pro
rentgenkontrastní značku kontralaterální větve.
5. Zaveďte aplikační systém hlavního těla přes drát do femorální artérie a
přitom věnujte pozornost postranním referenčním značkám.
POZOR: Při vkládání aplikačního systému udržujte polohu vodicího drátu.
POZOR: Pro vyloučení zkroucení endovaskulárního graftu při rotaci
aplikačního systému otáčejte všemi komponentami systému najednou
(od vnějšího sheathu po vnitřní kanylu).
6. Posunujte aplikační systém tak, až se čt yři zlaté rentgenkontrastní značky
(umístěné 2 mm od nejproximálnějšího segmentu materiálu graftu)
(Obr. 10, Ilustrace 1) dostanou přesně pod nejspodnější ústí renální
artérie.
7. Zkontrolujte polohu vodicího drátu v hrudní aortě. Systém graftu musí
být orientován tak, aby byla kontralaterální větev umístěna nad a před
počátkem kontralaterální iliakální tepny. Není-li rentgenkontrastní značka
kontralaterální větve správně sesazena, otočte celý systém tak, aby byla
značka správně umístěna v poloviční vzdálenosti mezi laterální a přední
polohou na kontralaterální straně.
• Značka tvaru ✓ indikuje přední pozici krátké (kontralaterální) větve.
(Obr. 10, Ilustrace 4)
• Značka tvaru indikuje zadní pozici krátké (kontralaterální) větve.
(Obr. 10, Ilustrace 5)
• Značka tvaru | indikuje boční pozici krátké (kontralaterální) větve.
(Obr. 10, Ilustrace 6)
POZNÁMKA: Rentgenkontrastní kanyla na každém stentu mezi renálními
artériemi a kontralaterální větví se srovná s rentgenkontrastní značkou na
kontralaterální větvi.
8. Opakovanou angiografií potvrďte polohu čtyřech zlatých
rentgenkontrastních značek ve vzdálenosti 2 mm nebo více od
nejspodnějšího ústí renální artérie.
9. Zkontrolujte, zda je hemostatický ventil Captor na zaváděcím sheathu
Flexor® v otevřené poloze. (Obr. 11)
10. Stabilizujte šedý polohovač (tubus aplikačního systému) a přitom vytahujte
sheath. Rozviňte první dva (2) zakryté stenty vytažením sheathu; přitom
monitorujte polohu zařízení.
11. Bez pohybu stolem zmenšete zvětšení, abyste mohli zkontrolovat polohu
rentgenkontrastní značky kontralaterální větve a polohu renálních artérií.
Pokračujte v rozvíjení, dokud není kontralaterální větev zcela rozvinuta.
(Obr. 12) Zastavte vytahování sheathu.
POZNÁMKA: Zkontrolujte, zda je kontralaterální větev nejméně 5 mm nad
bifurkací aorty a v požadované pozici pro kanylaci.
45

11.1.6 Umístění vodicího drátu kontralaterální iliakální větve
1. Naveďte katetr a vodicí drát skrz otevřený konec kontralaterální větve do
těla graftu. Posunujte vodicí drát tak, až se zakřiví do těla graftu. K ověření
kanylace zařízení napomohou AP a šikmé skiaskopické projekce.
2. Po dokončení kanylace posunujte angiografický katetr přes vodicí drát
do těla endovaskulárního graftu. Vyjměte drát a proveďte angiografické
potvrzení polohy. Posunujte vodicí drát tak, až se zakřiví do těla graftu.
Vyjměte angiografický katetr. (Obr. 13)
11.1.7 Rozvinutí proximální (horní) části hlavního těla
1. Pomocí angiografického katetru proveďte angiografické potvrzení polohy
endovaskulárního graftu vůči renálním artériím. Je-li třeba, opatrně
posuňte krytou část endovaskulárního graftu vůči renálním artériím.
(Posunutí se v této fázi může provádět pouze po malých vzdálenostech.)
POZNÁMKA: Zajistěte průchodnost renálních artérií potvrzením, že
proximální značky graftu jsou nejméně 2 mm pod nejnižší průchodnou
renální artérií.
POZOR: Při vyjímání proximálního uvolňovacího drátu, posunu horní
čepičky vpřed a následném rozvinutí suprarenálního stentu ověřte, že
vodicí drát hlavního těla zasahuje bezprostředně distálně za aortální
oblouk, a že se systému dostává maximální podpory.
2. Odstraňte bezpečnostní západku z černé spouště uvolňovacího drátu. Pod
skiaskopickou kontrolou vytáhněte a odstraňte uvolňovací drát vysunutím
černé spouště uvolňovacího drátu z rukojeti a následným vyjmutím přes
příslušnou štěrbinu nad vnitřní kanylou. (Obr. 14)
Pokud pociťujete odpor nebo pokud se systém ohýbá, uvolňovací drát je
příliš napjatý. Použití nadměrné síly může změnit polohu graftu. Pokud zjistíte
nadměrný odpor nebo pohyb aplikačního systému, zastavte zákrok a
zhodnoťte situaci. Pokud nelze černou spoušť uvolňovacího drátu vyjmout z
horní čepičky, pod skiaskopickou kontrolou proveďte následující kroky:
a. Uvolněte napětí uvolňovacího drátu uvolněním svěrky a lehkým
povytáhnutím vnitřní kanyly, aby se horní čepička posunula dolů po
suprarenálním stentu. Nedovolte, aby došlo ke stlačení hlavního těla
Zenith Flex.
b. Znova svěrku utáhněte.
c. Odstraňte černou spoušť uvolňovacího drátu.
d. Pokračujte krokem 3 v části 11.1.7, Rozvinutí proximální (horní) části
hlavního těla.
POZNÁMKA: Pokud nadále nelze vyjmout spoušť uvolňovacího drátu
horního stentu z horní čepičky, prostudujte část 14.1, Řešení problémů s
uvolňovacím drátem.
3. Uvolněte svěrku. (Obr. 15) Pozici graftu kontrolujte stabilizováním šedého
polohovače zavaděče.
4. Rozviňte suprarenální stent posouváním vnitřní kanyly horní čepičky o 1
až 2 mm najednou a přitom kontrolujte polohu hlavního těla, dokud horní
stent není zcela rozvinutý. (Obr. 16 a 17) Posuňte kanylu horní čepičky o
další 1 až 2 cm a pak znovu utáhněte svěrku, aby nedošlo ke kontaktu s
rozvinutým suprarenálním stentem.
POZNÁMKA: Pokud suprarenální stent nelze plně rozvinout posunováním
vnitřní kanyly horní čepičky, viz část 14.2, Řešení problémů s rozvinováním
suprarenálního stentu.
POZNÁMKA: Po rozvinutí suprarenálního stentu s kotvičkami se již
nedoporučuje provádět další pokusy o repozici graftu.
VAROVÁNÍ: Endovaskulární graft Zenith Flex AAA obsahuje suprarenální
stent s fixačními kotvičkami. Při manipulaci s intervenčními zařízeními v
blízkosti suprarenálního stentu postupujte s extrémní opatrností.
5. Kontralaterální vodicí drát posuňte do hrudní aorty.
11.1.8 Umístění a rozvinutí kontralaterálního iliakálního ramena
POZOR: Zkontrolujte, zda je k zavedení na kontralaterální straně pacienta
vybráno kontralaterální iliakální rameno určené před implantací.
1. Umístěte zesilovač obrazu tak, aby ukazoval kontralaterální vnitřní iliakální
artérii a kontralaterální společnou iliakální artérii.
2. Před zavedením aplikačního systému kontralaterálního iliakálního ramena
injikujte kontrastní látku kontralaterálním femorálním sheathem a
lokalizujte kontralaterální vnitřní iliakální artérii.
3. Zaveďte aplikační systém kontralaterálního iliakálního ramena do artérie.
Posunujte pomalu, až graft iliakálního ramena překryje nejméně jeden
celý stent iliakálního ramena (tj. proximální stent iliakálního ramena graftu)
uvnitř kontralaterální větve hlavního těla. (Obr. 18) Pokud má hlavní tělo
graftu během tohoto manévru tendenci k pohybu, přidržte je v jeho pozici
stabilizací šedého polohovače na ipsilaterální straně.
POZNÁMKA: Pokud narazíte na obtíže při posouvání aplikačního systému
iliakálního ramena, použijte tužší vodicí drát. V silně vinutých cévách se mohou
anatomické poměry po zavedení tuhých drátů a systémů sheathů podstatně
změnit.
4. Potvrďte polohu distálního konce iliakálního ramena graftu. Podle
potřeby upravte polohu iliakálního ramena graftu tak, aby byla zajištěna
průchodnost vnitřní iliakální artérie a zároveň minimální překrytí o
jeden úplný stent iliakálního ramena (tj. proximální stent iliakálního
ramena graftu, maximální překrytí 1,5 stentu) v rámci hlavního těla
endovaskulárního graftu.
5. Při rozvinování držte iliakální rameno graftu na místě šedým polohovačem
a současně vytahujte sheath. (Obr. 19 a 20) Zajistěte, aby bylo zachováno
překrytí o jeden stent.
6. Zastavte vytahování sheathu, jakmile je uvolněn distální konec iliakálního
ramena graftu.
7. Pod rentgenovým naváděním a po ověření polohy iliakálního ramena
graftu uvolněte svěrku a zatáhněte vnitřní kanylu tak, aby se zkosený
dilatátor aretoval k šedému polohovači. Utáhněte svěrku. Udržujte sheath
ve stejné poloze a současně vytahujte šedý polohovač se zajištěnou vnitřní
kanylou. (Obr. 21)
8. Zopakujte ověření polohy vodicího drátu.
11.1.9 Rozvinutí distální (dolní) části hlavního těla
1. Vraťte se k ipsilaterální straně.
2. Úplně rozviňte ipsilaterální větev hlavního těla vytažením sheathu tak, až
se rozvine nejdistálnější stent. (Obr. 22 a 23) Zastavte vytahování sheathu.
POZNÁMKA: Distální stent je stále zajištěn uvolňovacím drátem.
3. Odstraňte pojistku bílé spouště uvolňovacího drátu. Vytáhněte a odstraňte
uvolňovací drát vysunutím bílé spouště uvolňovacího drátu z rukojeti a pak
vyjmutím přes štěrbinu nad vnitřní kanylou zařízení. (Obr. 24)
11.1.10 Aretace horní čepičky
1. Uvolněte svěrku. (Obr. 25)
2. Zajistěte sheath a vnitřní kanylu, aby se vyloučil jakýkoli pohyb těchto
komponent.
3. Posuňte šedý polohovač přes vnitřní kanylu tak, až se aretuje k horní
čepičce. (Obr. 26, 27 a 28)
POZNÁMKA: Narazíte-li na odpor, lehce pootočte šedým polohovačem a
pokračujte v šetrném zavádění.
4. Opět utáhněte svěrku a vytáhněte celou horní čepičku a šedý polohovač
přes graft a přes sheath tažením za vnitřní kanylu. (Obr. 29) Sheath a vodicí
drát ponechte na místě.
POZNÁMKA: Udržujte polohu sheathu a vodicího drátu.
5. Zavřete hemostatický ventil Captor® na zaváděcím sheathu Flexor®
otočením po směru hodinových ručiček až na doraz. (Obr. 30)
11.1.11 Umístění a rozvinutí ipsilaterálního iliakálního ramena
POZNÁMKA: Zkontrolujte, zda je hemostatický ventil Captor na zaváděcím
sheathu v otevřené poloze. (Obr. 31)
1. Za použití drátu hlavního těla graftu a sestavy sheathu zaveďte ipsilaterální
iliakální rameno graftu. Sestavu dilatátoru a sheathu posuňte do sheathu
hlavního těla.
POZNÁMKA: V silně vinutých cévách se může po zavedení tuhých drátů a
systémů sheathů podstatně změnit poloha vnitřních iliakálních artérií.
2. Posunujte pomalu, až graft ipsilaterálního iliakálního ramena graftu
překryje nejméně jeden celý stent iliakálního ramena (tj. proximální stent
iliakálního ramena graftu) uvnitř ipsilaterální větve hlavního těla. (Obr. 32)
POZNÁMKA: Je-li zapotřebí překrytí o více než 3 stenty iliakálního ramena
(více než 2 stenty iliakálního ramena při délce ramena 37 mm a 54 mm), může
být nezbytné zvážit použití extenze ramena v místě bifurkace na protější
straně.
3. Potvrďte polohu distálního konce iliakálního ramena graftu. Podle potřeby
reponujte iliakální rameno graftu, abyste zajistili průchodnost vnitřní
iliakální artérie.
4. Při rozvinování držte iliakální rameno graftu na místě šedým polohovačem
a současně vytahujte sheath iliakálního ramena. (Obr. 33 a 34) Je-li to
nutné, vytáhněte sheath hlavního těla.
5. Pod rentgenovým naváděním a po ověření polohy iliakálního ramena
graftu uvolněte svěrku a zatáhněte vnitřní kanylu tak, aby se zkosený
dilatátor aretoval k šedému polohovači. Utáhněte svěrku. Udržujte sheath
ve stejné poloze a současně vytahujte šedý polohovač se zajištěnou vnitřní
kanylou. (Obr. 35)
6. Zavřete hemostatický ventil Captor na zaváděcím sheathu Flexor®
otočením po směru hodinových ručiček až na doraz.
7. Zopakujte ověření polohy vodicích drátů. Sheath a vodicí dráty ponechte
na místě.
11.1.12 Zavedení tvarovacího balónku
1. Připravte tvarovací balónek podle následujícího popisu:
• Lumen vodicího drátu propláchněte heparinizovaným fyziologickým
roztokem.
• Odstraňte z balónku všechen vzduch.
2. Během přípravy k zavedení tvarovacího balónku otevřete hemostatický
ventil Captor otočením proti směru hodinových ručiček.
3. Tvarovací balónek posuňte po vodicím drátu přes hemostatický ventil
Captor zaváděcího systému hlavního těla do úrovně renálních artérií.
Udržujte správnou polohu sheathu.
4. Utáhněte hemostatický ventil Captor kolem tvarovacího balónku
otočením s jemným tlakem po směru hodinových ručiček.
POZOR: Balónek nenaplňujte v cévě, pokud není uvnitř graftu.
5. Expandujte tvarovací balónek zředěnou kontrastní látkou (podle pokynů
výrobce) v oblasti nejproximálnějšího krytého stentu a infrarenálního
krčku; začněte proximálně a pracujte v distálním směru. (Obr. 36)
POZOR: Před repozicí zkontrolujte, zda je balónek úplně vyprázdněný.
POZOR: Hemostatický ventil Captor se musí před repozicí tvarovacího
balónku otevřít.
6. Vytáhněte tvarovací balónek k místu překrytí ipsilaterální větve a
expandujte jej.
POZOR: Hemostatický ventil Captor se musí před repozicí tvarovacího
balónku otevřít.
7. Vytáhněte tvarovací balónek k místu distální fixace ipsilaterální větve a
expandujte jej.
POZOR: Balónek nenaplňujte v cévě, pokud není uvnitř graftu.
8. Vyprázdněte tvarovací balónek a vyjměte jej. Tvarovací balónek přeneste
na kontralaterální vodicí drát a do zaváděcího systému kontralaterálního
iliakálního ramena. Posuňte tvarovací balónek k místu překrytí
kontralaterální větve a expandujte jej.
POZOR: Před repozicí zkontrolujte, zda je balónek úplně vyprázdněný.
9. Vytáhněte tvarovací balónek k místu distální fixace kontralaterálního
iliakálního ramena k cévě a expandujte jej. (Obr. 36)
POZOR: Balónek nenaplňujte v cévě, pokud není uvnitř graftu.
10. Vyjměte tvarovací balónek a nahraďte jej angiografickým katetrem k
provedení závěrečných angiogramů.
11. Vyjměte všechny tuhé vodicí dráty nebo je nahraďte tak, aby se iliakální
artérie mohly vrátit do přirozené polohy.
Finální angiogram
1. Angiografický katetr umístěte přímo nad úroveň renálních artérií.
Angiograficky potvrďte průchodnost renálních artérií a nepřítomnost
endoleaků. Ověřte průchodnost vnitřních iliakálních artérií.
2. Zkontrolujte, zda se nevytvořily endoleaky nebo smyčky, a ověřte polohu
proximálních zlatých rentgenkontrastních značek. Vyjměte sheathy, dráty a
katetry.
POZNÁMKA: Pokud zjistíte endoleaky nebo jiné problémy, viz Doporučený
návod k použití pomocných komponent endovaskulárního graftu Zenith AAA.
3. Sešijte cévy a uzavřete pole standardními chirurgickými technikami.
12 POKYNY K ZOBRAZOVÁNÍ A POOPERAČNÍ KONTROLE
12.1 Obecně
• Funkčnost endovaskulárních graftů z dlouhodobého hlediska nebyla
zatím zjištěna. Všichni pacienti mají být poučeni, že endovaskulární
léčba vyžaduje celoživotní pravidelná kontrolní vyšetření ke zjištění
jejich zdravotního stavu a funkčnosti jejich endovaskulárního graftu.
Pacienti se specifickými klinickými nálezy (např. endoleaky, zvětšujícími se
aneuryzmaty nebo se změnami struktury nebo polohy endovaskulárního
graftu) se musí podrobit dalším kontrolním vyšetřením.
• Pacienti mají být poučeni o důležitosti dodržování plánu kontrolních
prohlídek, a to jak v prvním roce, tak později jednou ročně. Pacienti mají být
informováni, že pravidelné a konzistentní kontroly jsou kriticky důležitou
součástí zajištění trvale bezpečné a účinné endovaskulární léčby AAA.
• Lékař má vyhodnotit každého pacienta individuálně a předepsat následné
kontroly podle potřeb a okolností konkrétního pacienta. Doporučený plán
kontrolních zobrazovacích vyšetření uvádí Tabulka 12.1. Tento plán
představuje minimální požadavky na následná kontrolní vyšetření pacienta a
má se dodržovat i v případě asymptomatického průběhu (např. při
nepřítomnosti bolesti, znecitlivění a slabosti). Pacienti se specifickými
klinickými nálezy (např. endoleaky, zvětšujícími se aneuryzmaty nebo se
změnami struktury nebo polohy endovaskulárního graftu) se musí podrobit
kontrolním vyšetřením v kratších intervalech.
46

• Každoroční snímkovací vyšetření má zahrnovat radiogram břicha a kontrastní
i nekontrastní CT vyšetření. Pokud není možné použít kontrastní látku
vzhledem k renálním komplikacím nebo jiným faktorům, lze použít
radiogramy břicha, nekontrastní CT a duplexní ultrasonografii.
• Kombinace kontrastního a nekontrastního CT poskytuje informace o
změnách průměru aneuryzmatu, o endoleaku, průchodnosti, tortuozitě,
postupu choroby, délce fixace a dalších morfologických změnách.
• Radiogramy břicha poskytují informace o integritě zařízení (separaci
komponent, prasknutí stentu a separaci kotviček).
• Zobrazení pomocí duplexní ultrasonografie může poskytnout informace o
změnách průměru aneuryzmatu, o endoleaku, průchodnosti, tortuozitě a
postupu choroby. Za těchto okolností je třeba spolu s ultrazvukem provést
nekontrastní CT. Ultrazvuk může být méně spolehlivou a citlivou
diagnostickou metodou ve srovnání s CT.
Tabulka 12.1 uvádí seznam minimálních požadavků na kontrolní zobrazovací
vyšetření pacientů s endovaskulárním graftem Zenith Flex AAA. U pacientů, u
kterých je nutná důkladnější kontrola, se má provádět častější vyhodnocení.
Tabulka 12.1 Doporučený plán zobrazovacích vyšetření
Před výkonem X
V průběhu výkonu X
Před propuštěním (do 7 dnů) X
Po 1 měsíci X
Po 3 měsících X
Po 6 měsících X
Po 12 měsících (později
jednou ročně)
1
Snímkování je nutno provést do 6 měsíců před výkonem.
2
Duplexní ultrasonografii lze použít u pacientů se selháváním ledvin nebo u těch, kteří
nejsou schopni podstoupit kontrastní vyšetření CT. I při použití ultrazvuku se doporučuje
použít nekontrastní CT.
3
Doporučuje se CT před propuštěním nebo po 1 měsíci.
4
V případě endoleaku typu I nebo III se doporučuje promptní intervence a po ní další
kontrola; viz část 12.6 Další sledování a léčba.
5
Doporučeno při hlášeném endoleaku před propuštěním nebo po 1 měsíci.
pro pacienty s endograftem
1
(kontrastní a
nekontrastní)
Angiogram
CT
Abdominální
radiogramy
1
X
2,3,4
2,3,4
2,4,5
2,4
2,4
X
X
X
X
X
12.2 Doporučení pro kontrastní a nekontrastní CT
• Sady filmů mají zahrnovat všechny sekvenční snímky s nejmenší možnou
tloušťkou vrstvy (≤3 mm). NEPOŘIZUJTE snímky s velkou tloušťkou vrstvy
(>3 mm) ani nevynechávejte následné CT snímky nebo sady filmů, protože
pak není možné přesné pozdější srovnání anatomických struktur a polohy
implantátu.
• Všechny snímky mají obsahovat měřítko pro každý film/snímek. Snímky mají
být v měřítku min. 20:1 na arších 35,5 x 43,2 cm (14 palců x 17 palců), pokud
se používá film.
• Je zapotřebí pořídit snímky s použitím kontrastní látky i bez použití kontrastní
látky, se shodnou nebo odpovídající polohou stolu.
• Tloušťka vrstvy a interval snímků bez kontrastní látky a po jejím podání musí
být stejné.
• NEMĚŇTE orientaci pacienta ani referenčních bodů mezi nekontrastním a
kontrastním vyšetřením.
Snímky s použitím kontrastní látky a bez použití kontrastní látky z počátku
léčby a při následných kontrolách jsou důležité pro optimální sledování
pacientova stavu. Při vyšetření CT je nutné postupovat podle příslušných
protokolů pro snímkování. Tabulka 12.2 uvádí seznam akceptovatelných
protokolů pro snímkování.
Tabulka 12.2 Akceptovatelné zobrazovací protokoly
Bez kontrastní látky S kontrastní látkou
IV kontrastní látka Ne Ano
Akceptovatelné přístroje Spirální s možností >40 s Spirální s možností >40 s
Objem injekce Nevztahuje se 150 ml
Rychlost injekce Nevztahuje se >2,5 ml/s
Režim injekce Nevztahuje se Výkon
Načasování bolusu Nevztahuje se
Pokrytí – začátek Bránice 1 cm nad břišní osou
Pokrytí – konec Proximální femur Odstup a. profunda femoris
Kolimace <3 mm <3 mm
Rekonstrukce
Axiální DFOV (duální
zorné pole)
Série po injekci Není Není
2,5 mm v celém rozsahu –
„měkký algoritmus“
32 cm 32 cm
Testovací bolus: SmartPrep,
C.A.R.E. nebo ekvivalent
2,5 mm v celém rozsahu –
„měkký algoritmus“
12.3 Abdominální radiogramy
Potřebné jsou následující snímky:
• Čtyři filmy: projekce supinačně – frontální (AP), příčná laterální, LPO (levá
zadní šikmá) 30 stupňů a RPO (pravá zadní šikmá) 30 stupňů, vycentrované
na pupek.
• Zaznamenejte vzdálenost od stolu k filmu a použijte stejnou vzdálenost při
každém dalším vyšetření.
Zajistěte, aby byl celý implantát zachycen na každém jednotlivém snímku v
podélné orientaci.
Existují-li jakékoli pochybnosti ohledně integrity zařízení (např.
zasmyčkování, zlomení stentu, oddělení kotviček, vzájemný pohyb
komponent), doporučuje se použít zvětšené obrazy. Ošetřující lékař má
na snímcích vyhodnotit integritu implantátu (celou délku zařízení včetně
komponent) za použití zvětšovacího skla s 2-4násobným zvětšením.
12.4 Ultrazvuk
Zobrazení ultrazvukem je možno provést místo kontrastního CT, pokud stav
pacienta vylučuje použití kontrastní látky. Ultrazvukové vyšetření je možno
doplnit nekontrastním CT. Je třeba pořídit videozáznam kompletního
duplexního ultrasonografického vyšetření aorty, ze kterého lze určit maximální
průměr výdutě, endoleaky, průchodnost stentu a stenózu. Videozáznam má
obsahovat následující informace:
• Příčný a podélný obraz má být pořízen z úrovně proximální aorty a má
demonstrovat mesenterické a renální artérie až k iliakální bifurkaci, aby bylo
možné zjistit přítomnost endoleaků, a to pomocí color flow a color power
angiografie (je-li tato technologie dostupná).
• V případě podezření na endoleak je nutno provést potvrzení spektrální
analýzou.
• Je nutno pořídit příčný a podélný snímek maximální výdutě.
12.5 Bezpečnost a kompatibilita vyšetření MRI
Neklinické testy prokázaly, že endovaskulární graft Zenith AAA je podmínečně
bezpečný při vyšetření MRI (MR Conditional). Může být bezpečně snímán za
následujících podmínek:
Systémy 1,5 tesla:
• Statické magnetické pole 1,5 tesla
• Prostorový gradient pole 450 gaussů/cm
• M aximální průměrná hodnota měrného absorbovaného výkonu
přepočteného na celé tělo (SAR) 2,0 W/kg pro 15 minut snímkování
Při neklinickém testování došlo u endovaskulárního graftu Zenith AAA ke
zvýšení teploty maximálně o 1,4 °C při maximálním měrném absorbovaném
výkonu přepočteném na celé tělo (SAR) 2,8 W/kg při kalorimetrickém měření
po dobu 15 minut snímkování MR scannerem Magnetom (Siemens Medical
Magnetom, software Numaris/4, verze Syngo MR 2002B DHHS) s magnetickým
polem 1,5 tesla. Maximální průměrná hodnota měrného absorbovaného
výkonu přepočteného na celé tělo (SAR) byla 2,8 W/kg, což odpovídá
kalorimetricky změřené hodnotě 1,5 W/kg.
Systémy 3,0 tesla:
• Statické magnetické pole 3,0 tesla
• Prostorový gradient pole 720 gaussů/cm
• M aximální průměrná hodnota měrného absorbovaného výkonu
přepočteného na celé tělo (SAR) 2,0 W/kg pro 15 minut snímkování.
Při neklinickém testování došlo u endovaskulárního graftu Zenith AAA ke
zvýšení teploty maximálně o 1,9 °C při maximálním měrném absorbovaném
výkonu přepočteném na celé tělo (SAR) 3,0 W/kg při kalorimetrickém měření
po dobu 15 minut snímkování MR scannerem Excite (GE Electric Healthcare,
software G3.0-052B) s magnetickým polem 3,0 tesla. Maximální průměrná
hodnota měrného absorbovaného výkonu přepočteného na celé tělo (SAR)
byla 3,0 W/kg, což odpovídá kalorimetricky změřené hodnotě 2,8 W/kg.
Při neklinickém skenování zasahoval obrazový artefakt celou anatomickou
oblast obsahující implantát, přičemž byl zacloněn pohled na bezprostředně
sousedící anatomické struktury do vzdálenosti přibližně 20 cm od implantátu
stejně jako implantát a jeho lumen; použitá sekvence byla následující: Fast spin
echo, MR systém Excite (GE Electric Healthcare, se softwarem G3.0-052B);
3,0 tesla, s tělovou radiofrekvenční cívkou.
U všech skenerů se obrazové artefakty rozptýlily při zvětšující se vzdálenosti
oblasti zájmu od implantátu. Snímky MR hlavy a krku a dolních končetin lze
pořídit bez obrazových artefaktů. Obrazový artefakt může být přítomen u
skenů břišní oblasti a horních končetin, v závislosti na vzdálenosti mezi
implantátem a oblastí zájmu.
Jsou k dispozici klinické údaje o sedmnácti pacientech, kteří podstoupili MR
vyšetření po implantaci stentgraftu. U žádného z těchto pacientů nebyla
hlášena žádná nežádoucí příhoda ani problémy s implantátem vzniklé v
souvislosti s podstoupením MRI. Kromě toho bylo na celém světě
implantováno více než 50 000 endovaskulárních graftů Zenith AAA, u nichž
nebyly hlášeny žádné nežádoucí příhody ani problémy s implantátem
způsobené MRI.
Společnost Cook doporučuje, aby pacienti zaregistrovali podmínky pro MRI
snímkování popsané v tomto návodu u nadace MedicAlert. Nadaci MedicAlert
lze kontaktovat následujícími způsoby:
Poštou: MedicAlert Foundation International
Telefon: +1 888-633-4298 (bezplatná linka)
Fax: +1 209-669-2450
2323 Colorado Avenue
Turlock, CA 95382 USA
+1 209-668-3333 mimo USA
Web: w ww.medicalert.org
12.6 Další sledování a léčba
Další sledování a potenciálně další léčba se doporučuje v následujících
případech:
• Aneuryzma s endoleakem typu I
• Aneuryzma s endoleakem typu III
• Zvětšení aneuryzmatu, maximální průměr ≥5 mm (bez ohledu na stav
endoleaku)
• Migrace
• Nedostatečná délka přilehnutí
Při zvažování opakované intervence nebo přechodu na otevřenou
chirurgickou reparaci musí lékař posoudit doprovodná onemocnění
konkrétního pacienta, předpokládanou dobu jeho života a pacientovu osobní
volbu. Pacienti mají být poučeni, že po implantaci endograftu může být nutný
další intervenční zásah, a to buď endovaskulární, nebo chirurgický.
13 INFORMACE PRO SLEDOVÁNÍ PACIENTŮ
Kromě tohoto Návodu k použití je k endovaskulárnímu graftu Zenith Flex AAA
se zaváděcím systémem H&L-B One-Shot přibalen Formulář ke sledování
zařízení, který má nemocniční personál vyplnit a odeslat společnosti COOK za
účelem sledování všech pacientů s implantovaným endovaskulárním graftem
Zenith Flex AAA (v souladu s požadavky federálních předpisů USA).
14 ŘEŠENÍ PROBLÉMŮ
POZNÁMKA: Kontaktujte místního zástupce společnosti Cook a požádejte o
technickou pomoc, kterou může poskytnout specializovaný technik.
14.1 Řešení problémů s uvolňovacím drátem
POZOR: Následující kroky použijte, pouze pokud nelze proximální
uvolňovací drát vyjmout postupem popsaným v části 11.1.7.
14.1.1 Rozvinutí proximální (horní) části hlavního těla
1. Pokud nelze černou spoušť uvolňovacího drátu (horní čepičky) sejmout
z rukojeti, ustřihněte drát u spouště (Obr. 37) a poté spoušť z rukojeti
odstraňte.
2. Stabilizujte šedý polohovač a zároveň stahujte sheath zpět, a tak zcela
rozviňte ipsilaterální větev.
3. Odstraňte pojistku bílé spouště uvolňovacího drátu.
4. Vytáhněte a odstraňte uvolňovací drát následovně: stáhněte bílou spoušť
uvolňovacího drátu z rukojeti a pak drát vytáhněte po vnitřní kanyle
příslušným otvorem.
5. Fixačním peánem zajistěte zastřižený konec uvolňovacího drátu horní
čepičky. (Obr. 38)
6. Uvolněte svěrku, udržujte vnitřní kanylu a uvolňovací drát ve stabilní
poloze a zaveďte šedý polohovač a sheath do graftu, až hrot šedého
polohovače dosáhne vzdálenosti přibližně 2 cm od zlatých značek.
(Obr. 39) Zavedený šedý polohovač poskytuje dodatečnou podporu
vnitřní kanyle.
POZNÁMKA: Uvolňovací drát udržujte lehce napnutý, aby se odstranil jeho
případný průvěs a aby nebyl při zavádění šedého polohovače a sheathu
volný.
POZNÁMKA: Zajistěte, aby hrot šedého polohovače nebyl zaveden do horní
čepičky.
7. Zamkněte svěrku. Zkontrolujte, že je uvolňovací drát zajištěn peánem.
8. Stabilizujte šedý polohovač a pomalu posunujte sheath vpřed, až hrot
sheathu dosáhne vzdálenosti 2 mm od zlatých značek. (Obr. 40)
POZNÁMKA: Dávejte pozor, aby při posouvání sheathu nedošlo k posunutí
samotného graftu.
9. Stabilizujte sheath a stáhněte zpět šedý polohovač s vnitřní kanylou, aby
se horní čepička přetáhla po suprarenálním stentu. (Obr. 41)
10. Ověřte polohu zlatých značek pod renálními artériemi.
47

11. Odstraňte uvolňovací drát.
12. Stahujte sheath zpět, až se obnaží zkosený hrot šedého polohovače.
13. Zaveďte tvarovací balónek kontralaterální větví hlavního těla a umístěte
jej těsně nad bifurkací graftu.
14. Naplňte balónek na celý průměr graftu. (Obr. 42)
15. Uvolněte svěrku.
16. Stabilizujte šedý polohovač a balónkový katetr a posuňte vpřed vnitřní
kanylu, aby se rozvinula horní čepička.
17. Utáhněte svěrku.
18. Vyprázdněte balónek a zaveďte kontralaterální vodicí drát do hrudní aorty.
POZNÁMKA: Vzhledem k riziku předčasného rozvinutí ipsilaterálního ramene
a spuštění uvolňovacího drátu doporučujeme ponechat při umisťování
ipsilaterální větve tvarovací balónek v kontralaterální větvi nebo těsně nad ní,
aby se stabilizoval graft.
14.1.2 Aretace horní čepičky
1. Uvolněte svěrku. (Obr. 25)
2. Zajistěte sheath a vnitřní kanylu, aby se vyloučil jakýkoli pohyb těchto
komponent.
3. Posuňte šedý polohovač po vnitřní kanyle, až se aretuje k horní čepičce.
(Obr. 43, 27 a 44)
POZNÁMKA: Narazíte-li na odpor, lehce pootočte šedým polohovačem a
pokračujte v šetrném zavádění.
4. Opět utáhněte svěrku a vytáhněte celou horní čepičku a šedý polohovač
skrz graft a skrz sheath tažením za vnitřní kanylu. (Obr. 45) Sheath a vodicí
drát ponechte na místě.
5. Zavřete hemostatický ventil Captor® na zaváděcím sheathu Flexor®
otočením po směru hodinových ručiček až na doraz. (Obr. 30)
14.1.3 Umístění a rozvinutí ipsilaterálního iliakálního ramena
POZNÁMKA: Zkontrolujte, zda je hemostatický ventil Captor na zaváděcím
sheathu v otevřené poloze. (Obr. 31)
1. Pomocí sestavy drátu hlavního těla graftu a sheathu zaveďte ipsilaterální
rameno graftu.
POZNÁMKA: Vzhledem ke změně v postupu uvolňování horní čepičky musí
být sestava sheathu hlavního těla stažena zpět 1-2 cm proximálně dovnitř
ipsilaterální větve. Zaveďte sestavu dilatátoru a sheathu ipsilaterální větve do
sheathu hlavního těla.
POZNÁMKA: Tvarovací balónek lze naplnit v kontralaterální větvi hlavního
těla graftu, pokud bude nutná dodatečná stabilizace graftu.
POZOR: Balónek nenaplňujte, pokud není uvnitř graftu.
POZNÁMKA: V silně vinutých cévách se může po zavedení tuhých drátů a
systémů sheathů podstatně změnit poloha vnitřních iliakálních artérií.
2. Posunujte pomalu, až ipsilaterální iliakální rameno graftu překryje
nejméně jeden celý stent iliakálního ramena (tj. proximální stent
iliakálního ramena graftu) uvnitř ipsilaterální větve hlavního těla. (Obr. 46)
POZNÁMKA: Pokud bude nutné překrytí delší než tři délky stentu iliakálního
ramena (delší než dvě délky stentu iliakálního ramena pro 37 a 54mm stenty),
může být nutné uvážit použití extenze ramena do oblasti bifurkace na
protilehlé straně.
3. Potvrďte polohu distálního konce iliakálního ramena graftu. Podle potřeby
reponujte iliakální rameno graftu, abyste zajistili průchodnost vnitřní
iliakální artérie.
4. Při rozvinování držte iliakální rameno graftu na místě šedým polohovačem
a současně společně vytahujte sheath iliakálního ramena a sheath
hlavního těla. (Obr. 33 a 47) Je-li to nutné, vytáhněte sheath hlavního těla.
5. Pod skiaskopickým naváděním a po ověření polohy iliakálního ramena
graftu uvolněte svěrku a stáhněte zpět vnitřní kanylu tak, aby se zkosený
dilatátor aretoval k šedému polohovači. Utáhněte svěrku. Udržujte sheath
ve stejné poloze a současně vytahujte šedý polohovač se zajištěnou
vnitřní kanylou. (Obr. 48)
6. Zavřete hemostatický ventil Captor na zaváděcím sheathu Flexor
otočením po směru hodinových ručiček až na doraz.
7. Znovu ověřte polohu vodicích drátů. Sheath a vodicí dráty ponechte na
místě.
8. Vyjměte vyprázdněný balónek z kontralaterální strany.
14.1.4 Umístění a rozvinutí kontralaterálního iliakálního ramena
POZOR: Před implantací zkontrolujte, zda je k zavedení na
kontralaterální straně pacienta vybráno kontralaterální iliakální rameno
určené dříve.
1. Umístěte zesilovač obrazu tak, aby ukazoval kontralaterální vnitřní iliakální
artérii a kontralaterální společnou iliakální artérii.
2. Před zavedením aplikačního systému kontralaterálního iliakálního ramena
injikujte kontrastní látku kontralaterálním femorálním sheathem a
lokalizujte kontralaterální vnitřní iliakální artérii.
3. Zaveďte aplikační systém kontralaterálního iliakálního ramena do artérie.
Posunujte pomalu, až iliakální rameno graftu překryje nejméně jeden celý
stent iliakálního ramena (tj. proximální stent iliakálního ramena graftu)
uvnitř kontralaterální větve hlavního těla. (Obr. 49)
POZNÁMKA: Pokud narazíte na obtíže při posouvání aplikačního systému
iliakálního ramena, použijte tužší vodicí drát. V silně vinutých cévách se
mohou anatomické poměry po zavedení tuhých drátů a systémů sheathů
podstatně změnit.
4. Potvrďte polohu distálního konce iliakálního ramena graftu. Podle
potřeby upravte polohu iliakálního ramena graftu tak, aby byla zajištěna
průchodnost vnitřní iliakální artérie a zároveň minimální překrytí o
jeden úplný stent iliakálního ramena (tj. proximální stent iliakálního
ramena graftu, maximální překrytí 1,5 stentu) uvnitř hlavního těla
endovaskulárního graftu.
5. Při rozvinování držte iliakální rameno graftu na místě šedým polohovačem
a současně vytahujte sheath. (Obr. 19 a 50) Zajistěte, aby bylo zachováno
překrytí o jeden stent.
6. Zastavte vytahování sheathu, jakmile je uvolněn distální konec iliakálního
ramena graftu.
7. Pod skiaskopickou kontrolou a po ověření polohy iliakálního ramena
graftu uvolněte svěrku a stáhněte zpět vnitřní kanylu tak, aby se zkosený
dilatátor aretoval k šedému polohovači. Utáhněte svěrku. Udržujte sheath
ve stejné poloze a současně vytahujte šedý polohovač se zajištěnou
vnitřní kanylou. (Obr. 51)
8. Znovu ověřte polohu vodicích drátů.
9. Zaveďte tvarovací balónek a proveďte závěrečný angiogram, jak je
popsáno v části 11.1.12 Zavedení tvarovacího balónku.
14.2 Řešení problémů s rozvinováním suprarenálního stentu
POZOR: Níže uvedené kroky je třeba provádět pouze tehdy, když není
možné horní čepičku rozvinout, jak je popsáno v části 11.1.7.
14.2.1 Rozvinutí proximální (horní) části hlavního těla
1. Pokud nelze suprarenální stent zcela rozvinout posunutím vnitřní kanyly
horní čepičky vpřed, zaveďte kontralaterální větví hlavního těla tvarovací
balónek a umístěte jej těsně nad bifurkaci stentgraftu.
2. K zajištění dodatečné podpory vnitřní kanyly naplňte balónek na rozměr
celého průměru graftu.
3. Uvolněte svěrku. (Obr. 15)
4. Stabilizujte šedý polohovač a balónkový katetr a posuňte vnitřní kanylu
vpřed, aby se rozvinul suprarenální stent.
Pokud je suprarenální stent zcela rozvinut:
5. Utáhněte svěrku a pak vyprázdněte a vytáhněte balónek.
6. Posuňte kontralaterální vodicí drát do hrudní aorty, vraťte se k
příslušnému kroku v návodu k použití a zákrok dokončete.
Pokud nadále není možné suprarenální stent zcela rozvinout:
5. Utáhněte svěrku a vyprázdněte balónek. Udržujte balónek ve stabilní
poloze, stabilizujte šedý polohovač a stáhněte zpět sheath, aby se zcela
rozvinula ipsilaterální větev.
6. Odstraňte bezpečnostní západku ze spouště uvolňovacího drátu
ipsilaterální větve.
7. Vytáhněte a odstraňte uvolňovací drát tak, že vysunete spoušť
uvolňovacího drátu ipsilaterální větve z rukojeti a pak drát vyjmete skrz
příslušnou štěrbinu po vnitřní kanyle zařízení.
8. Uvolněte svěrku (Obr. 25), udržujte vnitřní kanylu ve stabilní poloze a
přitom posunujte šedý polohovač a sheath do graftu, až hrot šedého
polohovače dostoupí přibližně 2 cm pod proximální zlaté značky.
(Obr. 52) Zavedený šedý polohovač poskytuje dodatečnou podporu
vnitřní kanyle.
POZNÁMKA: Postupujte opatrně, aby se při zavádění šedého polohovače
a sheathu graft neposunul vpřed.
POZNÁMKA: Zajistěte, aby hrot šedého polohovače nebyl zaveden do
horní čepičky.
9. Zamkněte svěrku.
10. Ověřte polohu zlatých značek pod renálními artériemi.
11. Upravte polohu tvarovacího balónku tak, aby přisedl na bifurkaci.
12. Naplňte balónek na celý průměr graftu. (Obr. 42)
13. Uvolněte svěrku.
14. Stabilizujte šedý polohovač a balónkový katetr a posuňte vnitřní kanylu
vpřed, aby se rozvinul suprarenální stent.
15. Utáhněte svěrku.
16. Vyprázdněte balónek a zaveďte kontralaterální vodicí drát do hrudní
aorty.
POZNÁMKA: Vzhledem k časnému rozvinutí ipsilaterálního ramena a
spuštění uvolňovacího drátu se doporučuje ponechat tvarovací balónek
v kontralaterální větvi nebo těsně nad ní, za účelem použití pro stabilizaci
graftu při umisťování ipsilaterální větve.
14.2.2 Aretace horní čepičky
1. Uvolněte svěrku. (Obr. 25)
2. Zajistěte sheath a vnitřní kanylu, aby se vyloučil jakýkoli pohyb těchto
komponent.
3. Posuňte šedý polohovač po vnitřní kanyle, až se aretuje k horní čepičce.
(Obr. 43, 27 a 44)
POZNÁMKA: Narazíte-li na odpor, lehce pootočte šedým polohovačem a
pokračujte v šetrném zavádění.
4. Opět utáhněte svěrku a vytáhněte celou horní čepičku a šedý polohovač
skrz graft a skrz sheath tažením za vnitřní kanylu. (Obr. 45) Sheath a
vodicí drát ponechte na místě.
5. Zavřete hemostatický ventil Captor® na zaváděcím sheathu Flexor®
otočením po směru hodinových ručiček až na doraz. (Obr. 30)
14.2.3 Umístění a rozvinutí ipsilaterálního iliakálního ramena
POZNÁMKA: Zkontrolujte, zda je hemostatický ventil Captor na zaváděcím
sheathu v otevřené poloze. (Obr. 31)
1. Umístěte zesilovač obrazu tak, aby zobrazoval ipsilaterální vnitřní iliakální
artérii a ipsilaterální společnou iliakální artérii.
2. Po zavedení aplikačního systému ipsilaterálního iliakálního ramena
injikujte kontrastní látku skrz sheath hlavního těla, abyste lokalizovali
ipsilaterální vnitřní iliakální artérii.
3. Pomocí sestavy drátu hlavního těla graftu a sheathu zaveďte ipsilaterální
rameno graftu.
POZNÁMKA: Vzhledem ke změně v postupu uvolňování horní čepičky
musí být sestava sheathu hlavního těla stažena zpět 1-2 cm dovnitř
proximální ipsilaterální větve. Zaveďte sestavu dilatátoru a sheathu
ipsilaterální větve do sheathu hlavního těla.
POZNÁMKA: Tvarovací balónek lze naplnit v kontralaterální větvi
hlavního těla graftu, pokud bude nutná dodatečná stabilizace graftu.
POZOR: Balónek nenaplňujte, pokud není uvnitř graftu.
POZNÁMKA: V silně vinutých cévách se může po zavedení tuhých drátů a
systémů sheathů podstatně změnit poloha vnitřních iliakálních artérií.
4. Pomalu zavádějte aplikační systém ipsilaterálního iliakálního ramena, až
graft ipsilaterálního iliakálního ramena překryje nejméně jeden celý stent
iliakálního ramena (tj. proximální stent iliakálního ramena graftu) uvnitř
ipsilaterální větve hlavního těla. (Obr. 46)
POZNÁMKA: Pokud bude nutné překrytí delší než tři délky stentu
iliakálního ramena (delší než dvě délky stentu iliakálního ramena pro 37 a
54mm stenty), může být nutné uvážit použití extenze ramena do oblasti
bifurkace na protilehlé straně.
5. Potvrďte polohu distálního konce iliakálního ramena graftu. Podle
potřeby reponujte iliakální rameno graftu, abyste zajistili průchodnost
vnitřní iliakální artérie.
6. Při rozvinování držte iliakální rameno graftu na místě šedým
polohovačem a současně společně vytahujte sheath iliakálního ramena a
sheath hlavního těla. (Obr. 33 a 47)
7. Pod skiaskopickým naváděním a po ověření polohy iliakálního ramena
graftu uvolněte svěrku a stáhněte zpět vnitřní kanylu tak, aby se zkosený
dilatátor aretoval k šedému polohovači. Utáhněte svěrku. Udržujte sheath
ve stejné poloze a současně vytahujte šedý polohovač se zajištěnou
vnitřní kanylou. (Obr. 48)
8. Zavřete hemostatický ventil Captor na zaváděcím sheathu Flexor
otočením po směru hodinových ručiček až na doraz.
9. Znovu ověřte polohu vodicích drátů. Sheath a vodicí dráty ponechte na
místě.
10. Vyjměte vyprázdněný balónek z kontralaterální strany.
14.2.4 Umístění a rozvinutí kontralaterálního iliakálního ramena
POZOR: Před implantací zkontrolujte, zda je k zavedení na kontralaterální
straně pacienta vybráno kontralaterální iliakální rameno určené dříve.
1. Umístěte zesilovač obrazu tak, aby ukazoval kontralaterální vnitřní
iliakální artérii a kontralaterální společnou iliakální artérii.
2. Před zavedením aplikačního systému kontralaterálního iliakálního ramena
injikujte kontrastní látku kontralaterálním femorálním sheathem a
lokalizujte kontralaterální vnitřní iliakální artérii.
3. Zaveďte aplikační systém kontralaterálního iliakálního ramena do artérie.
Posunujte pomalu, až iliakální rameno graftu překryje nejméně jeden celý
stent iliakálního ramena (tj. proximální stent iliakálního ramena graftu)
uvnitř kontralaterální větve hlavního těla. (Obr. 49)
POZNÁMKA: Pokud narazíte na obtíže při posouvání aplikačního systému
iliakálního ramena, použijte tužší vodicí drát. V nadměrně vinutých cévách
se mohou anatomické poměry po zavedení tuhých drátů a systémů
sheathů podstatně změnit.
4. Potvrďte polohu distálního konce iliakálního ramena graftu. Podle
potřeby upravte polohu iliakálního ramena graftu tak, aby byla zajištěna
průchodnost vnitřní iliakální artérie a zároveň minimální překrytí o
jeden úplný stent iliakálního ramena (tj. proximální stent iliakálního
ramena graftu, maximální překrytí 1,5 stentu) v rámci hlavního těla
endovaskulárního graftu.
48

5. Při rozvinování držte iliakální rameno graftu na místě šedým polohovačem
a současně vytahujte sheath. (Obr. 19 a 50) Zajistěte, aby bylo zachováno
překrytí o jeden stent.
6. Zastavte vytahování sheathu, jakmile je uvolněn distální konec iliakálního
ramena graftu.
7. Pod skiaskopickou kontrolou a po ověření polohy iliakálního ramena
graftu uvolněte svěrku a stáhněte zpět vnitřní kanylu tak, aby se zkosený
dilatátor aretoval k šedému polohovači. Utáhněte svěrku. Udržujte sheath
ve stejné poloze a současně vytahujte šedý polohovač se zajištěnou
vnitřní kanylou. (Obr. 51)
8. Znovu ověřte polohu vodicích drátů.
9. Zaveďte tvarovací balónek a proveďte závěrečný angiogram podle popisu
v části Zavedení tvarovacího balónku v návodu k použití produktu.
49

DANSK
ZENITH FLEX® AAA ENDOVASKULÆR PROTESE MED H&L-B
ONE-SHOT™ INDFØRINGSSYSTEM
Læs alle instruktioner omhyggeligt. Hvis instruktionerne, advarslerne og
forholdsreglerne ikke følges nøje, kan det få alvorlige kirurgiske konsekvenser
eller medføre personskade på patienten.
FORSIGTIG: I henhold til amerikansk lovgivning må dette produkt kun
sælges af en læge eller efter dennes anvisning.
FORSIGTIG: Hele indholdet af den ydre pose (inklusive
indføringssystemet og de endovaskulære proteser) leveres sterile, kun
til engangsbrug.
Der findes 4 gældende udgaver af Anbefalet brugsanvisning for Zenith
produktgruppen. Denne brugsanvisning beskriver Anbefalet brugsanvisning
for Zenith Flex AAA endovaskulær protese (hovedprotese og iliaca-ben).
Vedrørende oplysninger om andre Zenith komponenter henvises der til
Anbefalet brugsanvisning nedenfor:
• Zenith AAA endovaskulær protese (Zenith AAA endovaskulær hovedprotese
og iliaca-ben);
• Zenith hjælpekomponenter til AAA endovaskulær protese
(hovedproteseforlænger, iliaca-benforlænger, konverteringsenhed og iliacaprop);
• Zenith® Renu™ AAA hjælpeprotese (konfigurationer for
hovedproteseforlænger og konverteringsenhed); og
• CODA® ballonkateter.
1 BESKRIVELSE AF PRODUKTET
1.1 Komponenter til aortisk hovedprotese og til iliaca-ben
Zenith Flex AAA endovaskulær protese er et modulært system, der består
af tre komponenter: en aortisk bifurkationshovedprotese og to iliaca-ben.
(Fig. 1) Protesemodulerne er konstrueret af et vævet polyesterstof i fuld
tykkelse, der er syet på de selvekspanderende Cook-Z® stents i rustfrit stål
med sutur af flettet polyester og monofilament polypropylen. Modulerne er
fuldt stentede for at give stabilitet og den nødvendige ekspansionskraft til at
åbne proteselumen under anlæggelsen. Derudover giver Cook-Z stents den
nødvendige fastgørelse og forsegling af protesen til karvæggen.
Den bare suprarenale stent ved protesens proksimale ende indeholder
modhager, der sidder med 3 mm mellemrum for yderligere fiksering af
anordningen. Til lettelse af visualisering af den stentede protese under
gennemlysning er der placeret røntgenfaste guldmarkører som følger: En
på det laterale aspekt i den mest distale stent på det kontralaterale lem i
bifurkationshovedprotesen og fire i en perifer retning inden for 2 mm af det
mest superiore aspekt i protesematerialet.
1.2 Hovedprotesens fremføringssystem
Zenith Flex AAA endovaskulær hovedprotese forsendes præmonteret
på H&L-B One-Shot indføringssystemet. (Fig. 2) Den er udstyret med
en sekventiel anlæggelsesmetode med indbyggede funktioner til at
give kontinuerlig kontrol over den endovaskulære protese under hele
anlæggelsesproceduren. H&L-B One-Shot indføringssystemet muliggør præcis
positionering og tillader omjustering af den endelige proteseposition inden
anlæggelse af den suprarenale stent med modhager.
Fremføringssystemet for hovedprotesen bruger et 18, 20 eller 22 French H&L-B
One-Shot indføringssystem. En udløsningsmekanisme med dobbelt udløserwire fastlåser den endovaskulære protese på fremføringssystemet, indtil
lægen frakobler protesen. Alle systemer er kompatible med en 0,035 tomme
(0,89 mm) kateterleder.
Captor® hæmostatisk ventil kan løsnes eller strammes for at opnå yderligere
hæmostase ved indføringen og/eller ernelsen af hjælpeprodukter ind i og ud
af sheathen. Fremføringssystemerne for hovedprotesen er udstyret med en
Flexor® indføringssheath, der modstår knæk, og som har hydrofil belægning.
Begge funktioner har til hensigt at øge sporbarheden i aa. iliacae og aorta
abdominalis.
1.3 Fremføringssystem for iliaca-ben
Iliaca-ben til Zenith AAA endovaskulær protese forsendes præmonteret på
H&L-B One-Shot indføringssystemet. (Fig. 3) Fremføringssystemet er designet
til at være let at bruge med minimal klargøring. Fremføringssystemet for iliacabenet bruger et 14 French eller 16 French H&L-B One-Shot indføringssystem.
Alle systemer er kompatible med en 0,035 tomme (0,89 mm) kateterleder.
1.4 Hjælpekomponenter til Zenith AAA endovaskulær protese
Der findes yderligere endovaskulære hjælpekomponenter
(hovedproteseforlængere, iliaca-benforlængere, konverteringsenheder
og iliaca-propper). (Fig. 4) For yderligere oplysninger skal der refereres til
brugsanvisningen til Zenith hjælpekomponenter til AAA endovaskulær
protese.
2 TILSIGTET ANVENDELSE
Zenith Flex AAA endovaskulær protese med H&L-B One-Shot
indføringssystemet er indiceret til endovaskulær behandling af patienter med
abdominal aortisk eller aorta-iliaca aneurisme med en morfologi egnet til
endovaskulær reparation, inklusive:
• Tilstrækkelig iliaca/femoral adgang, kompatibel med de krævede
indføringssystemer,
• Non-aneurismalt infrarenalt aor tasegment (hals) proksimalt for aneurismet:
• med en længde på mindst 15 mm,
• med en diameter, der er målt ydre væg til ydre væg, på højst 32 mm og
ikke mindre end 18 mm,
• med en vinkel, der er mindre end 60 grader i forhold til aneurismets
længdeakse, og
• med en vinkel, der er mindre end 45 grader i forhold til den suprarenale
aortaakse.
• Et distalt fikseringssted i iliaca-ar terien, der er mere end 10 mm langt og
7,5-20 mm i diameter (målt fra ydre væg til ydre væg).
3 KONTRAINDIKATIONER
Der er ingen kendte kontraindikationer for disse produkter.
4 ADVARSLER OG FORHOLDSREGLER
4.1 Generelle
• Læs alle instruktioner omhyggeligt. Hvis instruktionerne, advarslerne og
forholdsreglerne ikke følges nøje, kan det få alvorlige konsekvenser eller
medføre personskade på patienten.
• Der skal altid være et kvalificeret kirurgisk team til rådighed under
implantations- eller reinterventionsprocedurer i tilfælde af, at det bliver
nødvendigt at konvertere til åben kirurgisk reparation.
• Zenith Flex AAA endovaskulær protese med H&L-B One-Shot
indføringssystem må kun anvendes af læger og teams, der er uddannet i
vaskulære interventionsteknikker (kateterbaserede og kirurgiske) og i
brugen af denne anordning. Specifikke forventninger til uddannelse
beskrives i Afsnit 10.1, Lægeuddannelse.
• Yderligere endovaskulære interventioner eller konvertering til almindelig
åben kirurgisk reparation efter initial endovaskulær reparation bør overvejes
for patienter med aneurismer, der forstørres, uacceptabelt fald i
fikseringslængde (overlapning af kar og komponent) og/eller endolækage.
En stigning i aneurismestørrelse og/eller en persisterende endolækage eller
migration kan medføre ruptur af aneurismet.
• Det kan være nødvendigt for patienter, der oplever reduceret blodflow
gennem proteselemmet og/eller lækager, at gennemgå sekundære
interventioner eller kirurgiske procedurer.
4.2 Patientudvælgelse, behandling og opfølgning
• Zenith Flex AAA endovaskulær protese er designet til behandling af
aortahalsdiametre på mindst 18 mm og højst 32 mm. Zenith Flex AAA
endovaskulær protese er designet til at behandle proksimale aortahalse
(distalt for den nedre nyrearterie) på mindst 15 mm i længde. Det er
påkrævet med et distalt fikseringssted i iliaca-arterien, der er mere end
10 mm langt og 7,5-20 mm i diameter (målt fra ydre væg til ydre væg). Disse
størrelsesmålinger er yderst vigtige for udførelsen af den endovaskulære
reparation.
• Anatomiske nøgleelementer, der kan påvirke vellykket eksklusion af
aneurismet, inkluderer svær proksimal halsvinkling (>60 grader for
infrarenal hals til akse af AAA eller >45 grader for suprarenal hals i forhold til
den umiddelbare infrarenale hals); kort proksimal aortahals (<15 mm); en
inverteret tragtform (større end 10 % stigning i diameter over 15 mm af den
proksimale aortahalslængde); og periferisk trombe og/eller forkalkning ved
implantationsstederne i arterien, især grænsefladen for den proksimale
aortahals og den distale iliaca-arterie. Hvis der findes anatomiske
begrænsninger, kan det være nødvendigt med en længere hals for at opnå
adækvat forsegling og fiksering. Uregelmæssig forkalkning og/eller plaque
kan kompromittere fastgørelsen og forseglingen ved fikseringsstederne.
Halse med disse vigtige anatomiske elementer kan have større tendens til
protesemigration eller endolækage.
• Adækvat iliaca- eller femoral adgang er nødvendig for at indføre protesen i
vaskulaturen. Adgangskarrets diameter (målt fra indre væg til indre væg) og
morfologi (minimal snirklethed, okklusiv sygdom og/eller forkalkning) bør
være kompatibel med vaskulære adgangsteknikker og fremføringssystemer
til en 16 French til 22 French vaskulær indføringssheath. Kar, der er
signifikant forkalkede, okklusive, snirklede eller med vægtromber, kan
udelukke placering af den endovaskulære protese og/eller kan øge risikoen
for embolisering. En vaskulær kanalteknik kan være nødvendig for at opnå
succes hos nogle patienter.
• Zenith Flex AAA endovaskulær protese med H&L-B One-Shot
indføringssystem kan ikke anbefales til patienter, der ikke tåler de
kontraststoffer, der er nødvendige ved intraoperativ og postoperativ
opfølgningsbilleddiagnostik. Alle patienter skal monitoreres nøje og
kontrolleres regelmæssigt med henblik på ændring i deres sygdomstilstand
og integriteten af endoprotesen.
• Zenith Flex AAA endovaskulær protese med H&L-B One-Shot
indføringssystem kan ikke anbefales til patienter, der overstiger grænserne
for vægt og/eller størrelse, hvilket kompromitterer eller forhindrer de
nødvendige billeddiagnostiske krav.
• M anglende evne til at opretholde åbenhed i mindst en a. iliaca interna eller
okklusion af en uundværlig a. mesenterica inferior kan øge risikoen for
pelvis-/tarmiskæmi.
• M ange store, åbne lumbalarterier, vægtromber og en åben a. mesenterica
inferior kan alle prædisponere en patient for type II endolækager. Patienter
med koagulopati, der ikke kan korrigeres, kan også have en øget risiko for
type II endolækage eller blødningskomplikationer.
• Sik kerheden og effektiviteten af Zenith Flex AAA envaskulære protese med
H&L-B One-Shot indføringssystemet i følgende patientgrupper er ikke
blevet evalueret:
• traumatisk sk ade på aorta
• læk age fra foreliggende ruptur eller ruptur af aneurismer
• mykotiske aneurismer
• pseudoaneurismer, opstået fra tidligere placering af protese
• revision af tidligere placerede endovaskulære proteser
• uoprettelig koagulationsdefekt
• uundværlig mesenterialar terie
• genetisk bindevævssygdom (f.eks. Marfans eller Ehlers-Danlos syndrom)
• ledsagende thorak al aortaaneurisme eller thorakal abdominal aneurisme
• patienter med aktive systemiske infektioner
• gravide eller ammende kvinder
• morbidt fede patienter
• yngre end 18 år
• patienter, hvis proksimale aortahals er mindre end 15 mm i længde eller
større end 60 grader i vinkel i forhold til aneurismets lange akse.
• Vellykket patientudvælgelse kræver specifik billeddiagnostik og nøjagtige
målinger; se Afsnit 4.3 Måleteknik og billeddiagnostik før proceduren.
• Alle de nødvendige længder og diametre af produktet til at gennemføre
proceduren skal være tilgængelige for lægen, især når de præ-operative
planlægningsmål til tilfældet (behandlingsdiametre/længder) ikke er sikre.
Denne fremgangsmåde tillader større intraoperativ fleksibilitet til at opnå
optimale resultater af proceduren.
4.3 Måleteknik og billeddiagnostik før proceduren
• M anglende CT-scanning uden kontrast kan resultere i, at forkalkning af
iliaca eller aorta ikke kan vurderes, hvilket kan udelukke adgang eller
pålidelig fiksering og forsegling af produktet.
• En rekonstruktionstyk kelse ved gennemlysning før proceduren på >3 mm
kan resultere i suboptimal størrelsesbestemmelse af produktet eller i
manglende vurdering af fokal stenose fra CT-scanning.
• K linisk erfaring angiver, at kontrastforstærket spiralcomputer-tomografisk
angiografi (CTA) med 3-D rekonstruktion er den stærkt anbefalede
billeddiagnostiske modalitet til korrekt bedømmelse af patientens anatomi
inden behandling med Zenith Flex AAA endovaskulær protese. Hvis
kontrastforstærket spiral-CTA med 3-D rekonstruktion ikke er tilgængelig,
bør patienten henvises til et hospital, der råder over disse muligheder.
• K linikere anbefaler at placere C-buen på gennemlysningen under
proceduremæssig angiografi således, at nyrearteriernes udspring, og især
den nedre, åbne nyrearterie, ses tydeligt inden ekspansion af protesens
proksimale gren (forseglende stent) på hovedprotesen. Desuden skal
angiografien vise iliaca-arteriebifurkaturerne således, at de distale aa. iliacae
communes er veldefinerede i forhold til udspringet for a. iliaca interna
bilateralt, inden ekspansion af iliaca-benene.
Diametre:
Under anvendelse af CT skal diametermålinger bestemmes ud fra
kardiameteren mellem de udvendige vægge (ikke lumenmåling) for at
hjælpe med korrekt størrelsesbestemmelse og udvælgelse af produkt. Den
kontrastforstærkede spiral CT-scanning skal starte 1 cm superiort for axis
celiacus og fortsætte gennem femurhovederne ved en aksial skivetykkelse på
3 mm eller derunder.
Længder:
Under anvendelse af CT skal længdemålinger bestemmes for at vurdere
infrarenal proksimal halslængde nøjagtigt, såvel som planlægning af
hovedprotesestørrelse og benkomponenter for Zenith Flex AAA endovaskulær
protese. Disse rekonstruktioner skal udføres i sagittal-, frontal- og 3-D-plan.
• Endovaskulære protesers ydeevne på langt sigt er endnu ikke fastlagt.
Alle patienter skal gøres opmærksom på, at endovaskulær behandling
kræver regelmæssig opfølgning resten af deres liv til bedømmelse af
deres helbredstilstand og ydeevnen af deres endovaskulære protese.
Patienter med særlige kliniske fund (f.eks. endolækager, aneurismer, der
forstørres, eller ændringer i den endovaskulære proteses struktur eller
50

position) bør følges nøjere. Specifikke retningslinjer for opfølgning beskrives
i Afsnit 12, RETNINGSLINJER FOR BILLEDDIAGNOSTIK OG
POSTOPERATIV OPFØLGNING.
• Zenith Flex AAA endovaskulær protese med H&L-B One-Shot
indføringssystemet anbefales ikke til patienter, der ikke er i stand til eller
ikke har samarbejdsvilje til at få foretaget de nødvendige præoperative og
postoperative billeddiagnostiske og implantationsundersøgelser, der er
beskrevet i Afsnit 12, RETNINGSLINJER FOR BILLEDDIAGNOSTIK OG
POSTOPERATIV OPFØLGNING.
• Efter placering af en endovaskulær protese bør patienter monitoreres
regelmæssigt for periprotese flow, aneurismevækst eller ændringer i den
endovaskulære proteses struktur eller position. Som et minimum kræves der
årlig billeddiagnostik, herunder: 1) abdominale røntgenbilleder for at
undersøge produktets integritet (adskillelse mellem komponenter, stentbrud
eller modhageadskillelse) og 2) CT-scanning med og uden kontrast for at
undersøge aneurismeændringer, periprotese flow, åbenhed, snirklethed og
fremskreden sygdom. Hvis nyrekomplikationer eller andre faktorer udelukker
brugen af billedkontraststoffer, kan røntgenbilleder af abdomen og duplexultralydsscanning give lignende information.
4.4 Udvælgelse af produkt
• Det anbefales kraftigt at følge størrelsesguiden i brugsanvisningen til Zenith
Flex AAA endovaskulær protese, når den rette størrelse protese skal vælges
(tabel 10.5.1 og 10.5.2). Der er inkorporeret passende overstørrelse af
protesen i brugsanvisningens størrelsesguide. Størrelsesbestemmelse uden for
dette område kan resultere i endolækage, fraktur, migration, at produktet
folder ind eller bliver komprimeret.
4.5 Implantationsprocedure
(Se Afsnit 11, VEJLEDNING)
• Passende billeddiagnostik under proceduren er nødvendig for at sikre vellykket
placering af Zenith Flex AAA endovaskulær protese og sikre nøjagtig
apposition i forhold til aortavæggen.
• Fremføringssystemet må ikke bøjes eller knækkes. Dette vil kunne beskadige
fremføringssystemet og Zenith Flex AAA endovaskulær protese.
• Alle komponenter i systemet skal drejes samtidig (fra ydre sheath til indre
kanyle) for at undgå snoninger i den endovaskulære protese under drejning af
fremføringssystemet.
• Fortsæt ikke med at fremføre nogen del af fremføringssystemet, hvis der
mærkes modstand under fremføringen af kateterlederen eller
fremføringssystemet. Stop og vurdér årsagen til modstanden; der kan ske
skade på karret, kateteret eller protesen. Udvis særlig forsigtighed i områder
med stenoser, intravaskulær trombose eller i forkalkede eller snirklede kar.
• Utilsigtet delvis ekspansion eller migration af endoprotesen kan kræve kirurgisk
fjernelse.
• Medmindre det er medicinsk indiceret, må Zenith Flex AAA endovaskulær
protese ikke anlægges på et sted, der vil okkludere arterier, der er nødvendige
til forsyning af blodflow til organer eller ekstremiteter. Dæk ikke signifikante
nyrearterier eller mesenterialarterier (a. mesenterica inferior er en undtagelse)
med endoprotesen. Der kan ske karokklusion. Under den kliniske undersøgelse
blev denne anordning ikke undersøgt i patienter med to okkluderede a. iliaca
interna.
• Forsøg ikke at føre protesen tilbage i sheathen efter delvis eller fuldstændig
ekspansion.
• Omplacering af stentprotesen distalt efter delvis ekspansion af den dækkede
proksimale stent kan resultere i beskadigelse af stentprotesen
og/eller karlæsion.
• Unøjagtig placering og/eller ufuldstændig forsegling af Zenith Flex AAA
endovaskulær protese i karret kan resultere i øget risiko for endolækage,
migration eller utilsigtet okklusion af aa. renales eller aa. iliacae internae.
Nyrearteriens åbenhed skal opretholdes for at forhindre/reducere risikoen for
nyresvigt og efterfølgende komplikationer.
• Inadækvat fiksering af Zenith Flex AAA endovaskulær protese kan resultere i
øget risiko for migration af stentprotesen. Forkert anlæggelse eller migration af
endoprotesen kan kræve kirurgisk intervention.
• Der skal anvendes systemisk antikoagulation under implantationsproceduren
på grundlag af hospitalets og lægens foretrukne protokol. Hvis heparin er
kontraindiceret, bør det overvejes at bruge en alternativ antikoagulans.
• For at aktivere den hydrofile coating på ydersiden af Flexor indføringssheathen
skal overfladen aftørres med sterile gazekompresser, der er gennemvædet med
saltvandsopløsning. Hold altid sheathen hydreret for optimal funktion.
• Minimér håndteringen af den tvungne endoprotese under klargøringen og
indføringen for at nedsætte risikoen for kontaminering og infektion af
endoprotesen.
• Oprethold kateterlederens position under indføring af fremføringssystemet.
• Gennemlysning bør anvendes under indføring og anlæggelse for at bekræfte
korrekt funktion af indføringssystemets komponenter, korrekt placering af
protesen og det ønskede proceduremæssige resultat.
• Det er nødvendigt at administrere intravaskulær kontrast ved brugen af Zenith
Flex AAA endovaskulær protese med H&L-B One-Shot indføringssystemet.
Patienter med allerede eksisterende nyreinsufficiens kan have øget risiko for
nyresvigt efter operationen. Vær omhyggelig med at begrænse mængden af
anvendt kontraststof under proceduren og overholde forebyggende
behandlingsmetoder for at nedsætte nyresvigt (f.eks. adækvat hydrering).
• Når sheathen og/eller kateterlederen trækkes tilbage, kan anatomien og
protesens position blive ændret. Monitorér protesens position konstant og
udfør angiografi for at kontrollere positionen efter behov.
• Zenith Flex AAA endovaskulær protese omfatter en suprarenal stent med
fikseringsmodhager. Der skal udvises meget stor forsigtighed ved manipulering
af interventionelle og angiografiske produkter i området med den suprarenale
stent.
• Vær forsigtig ved manipulering af katetre, kateterledere og sheaths i et
aneurisme. Signifikante forstyrrelser kan rive trombefragmenter løs, hvilket kan
forårsage distal embolisering eller ruptur af aneurismet.
• Undgå at beskadige protesen eller at forstyrre positioneringen af protesen efter
placering i tilfælde af, at yderligere brug af kirurgiske instrumenter (sekundær
intervention) nær protesen er nødvendig.
• Det skal verificeres, at adgangskateterlederens position går ud lige distalt for
aortabuen, inden den suprarenale stent anlægges.
• Verificér inden implantationen, at det forudfastlagte kontralaterale iliaca-ben er
valgt til indføring på patientens kontralaterale side.
4.6 Brug af formningsballon
• Undgå at inflatere ballonen i kar uden for protesen, da det kan resultere i
skader på karret. Brug ballonen i overensstemmelse med den medfølgende
dokumentation.
• Vær forsigtig ved inflation af ballonen i protesen, hvis der foregår en
forkalkning, da overdreven inflation kan beskadige karret.
• Kontrollér fuld deflation af ballonen inden omplacering.
• For yderligere hæmostasekontrol kan Captor hæmostatiske ventil løsnes eller
strammes for at imødekomme indføring og senere tilbagetrækning af en
formningsballon.
4.7 Sikkerhed og kompatibilitet ved MR-scanning
Ikke-klinisk testning har påvist, at Zenith AAA endovaskulær protese er MR
conditional. Den er ufarlig at scanne på følgende betingelser:
1,5 Tesla systemer:
• Statisk magnetisk feltstyrke på 1,5 Tesla
• Rumlig feltstyrke for gradient på 450 Gauss/cm
• Maksimal helkropsgennemsnitlig specifik absorptionsrate (SAR) på
2,0 W/kg i 15 minutters scanning
I ikke-klinisk testning frembragte Zenith AAA endovaskulær protese en
temperaturstigning på mindre end eller lig med 1,4 °C ved en maksimal
helkropsgennemsnitlig specifik absorptionsrate (SAR) på 2,8 W/kg, bedømt ved
kalorimetri i 15 minutters MR-scanning i en 1,5 Tesla Magnetom, Siemens Medical
Magnetom, Numaris/4 Software, Version Syngo MR 2002B DHHS MR-scanner. D en
maksimale helkropsgennemsnitlige specifikke absorptionsrate (SAR) var 2,8 W/kg,
hvilket svarer til en kalorimetrisk målt værdi på 1,5 W/kg.
3,0 Tesla systemer:
• Statisk magnetisk feltstyrke på 3,0 Tesla
• Rumlig feltstyrke for gradient på 720 Gauss/cm
• Maksimal helkropsgennemsnitlig specifik absorptionsrate (SAR) på
2,0 W/kg i 15 minutters scanning
I ikke-klinisk testning frembragte Zenith AAA endovaskulær protese en
temperaturstigning på mindre end eller lig med 1,9 °C ved en maksimal
helkropsgennemsnitlig specifik absorptionsrate (SAR) på 3,0 W/kg, bedømt ved
kalorimetri i 15 minutters MR-scanning i en 3,0 Tesla Excite, GE Electric Healthcare,
G3.0-052B Software, MR-scanner. Den maksimale helkropsgennemsnitlige
specifikke absorptionsrate (SAR) var 3,0 W/kg, hvilket svarer til en kalorimetrisk
målt værdi på 2,8 W/kg.
Billedartefaktet strækker sig gennem hele den anatomiske region, der indeholder
implantatet, og slører billedet af umiddelbart nærliggende anatomiske strukturer
inden for ca. 20 cm fra implantatet, såvel som hele implantatet og dets lumen, når
der scannes i ikke-klinisk testning ved hjælp af sekvensen: Hurtigt spinekko i en
3,0 Tesla, Excite, GE Electric Healthcare, med G3.0-052B software, MR-system med
kropsradiofrekvensspiral.
For alle scannere spredes billedartefaktet, når afstanden fra implantatet til
området af interesse øges. MR-scanninger af hoved og hals og underekstremiteter
kan tages uden billedartefakt. Billedartefakt kan findes i scanninger af
abdominalregionen og overekstremiteterne, afhængigt af afstanden fra
implantatet til området af interesse.
Der findes klinisk information om sytten patienter, som fik MR-scanninger efter
implantation af stent-protese. Der har ikke været nogen rapporterede uønskede
hændelser eller implantatproblemer hos nogen af disse patienter som et resultat
af, at de er blevet MR-scannet. Desuden er der blevet implanteret langt over
50.000 Zenith AAA endovaskulære proteser globalt, hvor der ikke har været
nogen rapporterede uønskede hændelser eller implantatproblemer som et
resultat af MR-scanning.
Cook anbefaler, at patienten registrerer betingelserne for MR-scanning, der
er beskrevet i denne brugsanvisning, hos MedicAlert Foundation. MedicAlert
Foundation kan kontaktes på følgende måder:
Adresse: MedicAlert Foundation International
Telefon: +1 888-633-4298 frikaldsnummer
FAX: +1 209-669-2450
2323 Colorado Avenue
Turlock, CA 95382 USA
+1 209-668-3333 fra uden for USA
Web: www.medicalert.org
5 UØNSKEDE HÆNDELSER
5.1 Observerede uønskede hændelser
En amerikansk prospektiv multicenterundersøgelse af en tidligere version af
anordningen (Zenith AAA endovaskulær protese), gennemført på 15 centre, der
inkluderede 352 endovaskulære patienter (200 standardrisiko-, 100 højrisiko- og
52 indlæringspatienter) og 80 kontrolpatienter danner basis for de observerede
uønskede hændelser i Tabel 5.1.1. Patienter blev tilmeldt standardrisikogrenen,
hvis de var fysiologisk i stand til at tåle en åben eller endovaskulær reparation, og
havde en anatomi, der var egnet til behandling med Zenith AAA endovaskulær
protese med H&L-B One-Shot indføringssystemet. Patienter med en egnet
anatomi, men med en højere risiko for morbiditet eller dødelighed ved åben
reparation, blev tilmeldt højrisikogrenen. Initiale patienter, der blev behandlet i
undersøgelsen, blev tilmeldt indlæringspatientgrenen. Kontrolgruppen
inkluderede patienter, hvis vaskulære anatomi muligvis ikke var egnet til
endovaskulær AAA reparation.
Tabel 5.1.1 Død og ruptur fra klinisk undersøgelse
Død og ruptur Zenith standardrisiko
Alle dødsfald
2
(0-30 dage)
(31-365 dage)
(0-365 dage)
Ruptur
1
Nævner på 199, da én standardrisikopatient ikke modtog en anordning.
2
Alle dødsfald (0-30 dage) blev regnet som relateret til AAA og proceduren.
3
Af dødsfaldene (31-365 dage) blev fire regnet som AAA-relaterede: 1 kirurgisk (septisk shock fra iskæmisk colitis) og 3 højrisiko (pancreatitis med nyresvigt og sepsis, hæmorragi fra
øvre abdominal aneurisme [ubehandlet AAA] og flere systemsvigt).
4
Af dødsfaldene (0-365 dage) blev ti regnet som AAA-relaterede: 1 standardrisiko (hjertesvigt), 3 kirurgiske (alvorlig hæmorragi, mesenterisk iskæmi og septisk shock fra iskæmisk
colitis), 5 højrisiko (respirationsinsufficiens, hjertesvigt med pulmonær emboli, pancreatitis med nyresvigt og sepsis, hæmorragi fra øvre abdominal aneurisme [ikke AAA-behandlet] og
flere systemsvigt) og 1 indlæring (mistanke om hjertesvigt).
3
AAA-relaterede 0,0 % (0/198) 1,3 % (1/78) 0,29 3,1 % (3/98) 0,0 % (0/51)
Ikke-AAA-relaterede 3,0 % (6/198) 0,0 % (0/78) 0,19 4,1 % (4/98) 9,8 % (5/51)
2,3,4
AAA-relaterede 0,5 % (1/199) 3,8 % (3/80) 0,07 5,0 % (5/100) 1,9 % (1/52)
Ikke-AAA-relaterede 3,0 % (6/199) 0,0 % (0/80) 0,19 4,0 % (4/100) 9,6 % (5/52)
(0-30 dage) 0,0 % (0/199) Ikke relevant Ikke relevant 0,0 % (0/100) 0,0 % (0/52)
(31-365 dage) 0,0 % (0/198) Ikke relevant Ikke relevant 1,0 % (1/98) 0,0 % (0/51)
(0-365 dage) 0,0 % (0/199) Ikke relevant Ikke relevant 1,0 % (1/100) 0,0 % (0/52)
0,5 % (1/199) 2,5 % (2/80) 0,20 2,0 % (2/100) 1,9 % (1/52)
3,0 % (6/198) 1,3 % (1/78) 0,68 7,1 % (7/98) 9,8 % (5/51)
3,5 % (7/199) 3,8 % (3/80) > 0,99 9,0 % (9/100) 11,5 % (6/52)
1
Kirurgisk standardrisiko P værdi Zenith højrisiko Zenith indlæring
51

Zenith standardrisiko Kirurgisk standardrisiko P værdi Zenith højrisiko Zenith indlæring
Tabel 5.1.2 Uønskede hændelser1 i klinisk undersøgelse
Fravær af morbiditet
(0-30 dage)
3
4, 9
5
6
7
8,11
(31-365 dage)
3
4, 10
5
6
7
8
(0-365 dage)
3
4, 9,10
5
6
7
8,11
2
2
2
Kardiovaskulær
Pulmonær
Renal
Tar m
Sår
Neurologisk
Vaskulær
Fravær af morbiditet
Kardiovaskulær
Pulmonær
Renal
Tar m
Sår
Neurologisk
Vaskulær
Fravær af morbiditet
Kardiovaskulær
Pulmonær
Renal
Tar m
Sår
Neurologisk
Vaskulær
1
Fra morbiditetsindekset.
2
Kategorien kardiovaskulær inkluderede: Q-tak og ikke-Q-tak myokardieinfarkter, kongestiv hjerteinsufficiens, arytmier, der kræver ny medicin eller behandling, hjerteiskæmi der
kræver behandling, inotropisk støtte, hypertension der er vanskelig at behandle medicinsk.
3
Kategorien pulmonær inkluderede: reintubation eller ventilation i > 24 timer, lungebetændelse der kræver antibiotikum, supplerende ilt ved udskrivning.
4
Kategorien renal inkluderede: dialyse hos patienter med normal, præoperativ renal funktion, en stigning i kreatinin > 30 % fra basislinjen på to eller flere efterfølgende tests.
5
Kategorien tarm inkluderede: tarmobstruktion, tarmiskæmi, aortointestinal fistel, paralytisk ileus > 4 dage.
6
Kategorien sår inkluderede: infektion der krævede antibiotisk behandling, hernia, lymfefistel, dehiscens, nekrose der krævede debridement.
7
Kategorien neurologi inkluderede: slagtilfælde, TIA, rygmarvsiskæmi/paralyse.
8
Kategorien vaskulær inkluderede: trombose i lem, distal emboli med resulterende vævstab eller behov for behandling, transfusion efter proceduren (som resultat af pseudoaneurisme,
vaskulær skade, lækage fra aneurisme eller andre procedurerelaterede årsager), pseudoaneurisme, vaskulær skade (som f.eks. utilsigtet okklusion, dissektion eller andre
procedurerelaterede årsager), lækage fra aneurisme eller ruptur, øgning af aneurismets størrelse med mere end 0,5 cm, relativt til det mindste af de tidligere mål.
9
Forskere rapporterede, at én højrisikopatient yderligere havde okklusion af en ekstra nyrearterie, og én højrisikopatient yderligere havde kronisk nyreinsufficiens som “anden” uønsket
hændelse.
10
Forskere rapporterede, at én indlæringspatient yderligere havde nyreinsufficiens og én kirurgisk patient yderligere havde nyreinsufficiens som “anden” uønsket hændelse.
11
Forskere rapporterede, at én indlæringspatient yderligere havde oplevet intraoperativ aortisk plaque ruptur, der medførte arterieokklussion som en “anden” uønsket hændelse.
80 % (160/200) 58 % (46/80) < 0,001 68 % (68/100) 73 % (38/52)
3,0 % (6/200) 11 % (9/80) 0,02 14 % (14/100) 1,9 % (1/52)
1,0 % (2/200) 15 % (12/80) < 0,001 2,0 % (2/100) 0,0 % (0/52)
2,5 % (5/200) 10 % (8/80) 0,01 6,0 % (6/100) 5,8 % (3/52)
1,0 % (2/200) 3,8 % (3/80) 0,14 1,0 % (1/100) 1,9 % (1/52)
4,5 % (9/200) 7,5 % (6/80) 0,38 2,0 % (2/100) 3,8 % (2/52)
0,0 % (0/200) 2,5 % (2/80) 0,08 0,0 % (0/100) 0,0 % (0/52)
11 % (21/200) 31 % (25/80) < 0,001 20 % (20/100) 19 % (10/52)
91 % (181/198) 86 % (67/78) 0,25 79 % (77/98) 86 % (44/51)
2,5 % (5/198) 3,8 % (3/78) 0,69 5,1 % (5/98) 2,0 % (1/51)
0,5 % (1/198) 1,3 % (1/78) 0,49 4,1 % (4/98) 0,0 % (0/51)
0,5 % (1/198) 0,0 % (0/78) > 0,99 3,1 % (3/98) 0,0 % (0/51)
0,5 % (1/198) 1,3 % (1/78) 0,49 0,0 % (0/98) 0,0 % (0/51)
2,0 % (4/198) 5,1 % (4/78) 0,23 3,1 % (3/98) 2,0 % (1/51)
1,0 % (2/198) 0,0 % (0/78) > 0,99 1,0 % (1/98) 3,9 % (2/51)
3,0 % (6/198) 3,8 % (3/78) 0,72 8,2 % (8/98) 5,9 % (3/51)
76 % (151/200) 49 % (39/80) < 0,001 55 % (55/100) 62 % (32/52)
5,0 % (10/200) 14 % (11/80) 0,02 19 % (19/100) 3,8 % (2/52)
1,5 % (3/200) 16 % (13/80) < 0,001 6,0 % (6/100) 0,0 % (0/52)
2,5 % (5/200) 10 % (8/80) 0,01 9,0 % (9/100) 5,8 % (3/52)
1,5 % (3/200) 3,8 % (3/80) 0,36 1,0 % (1/100) 1,9 % (1/52)
5,5 % (11/200) 13 % (10/80) 0,08 5,0 % (5/100) 5,8 % (3/52)
1,0 % (2/200) 2,5 % (2/80) 0,32 1,0 % (1/100) 3,8 % (2/52)
12 % (24/200) 33 % (26/80) < 0,001 25 % (25/100) 23 % (12/52)
5.2 Mulige uønskede hændelser
Uønskede hændelser, der kan forekomme og/eller kræve indgriben
inkluderer, men er ikke begrænset til:
• Amputation
• Aneurismeforstørrelse
• Aneurismeruptur og død
• Anæstetiske komplikationer og efterfølgende ledsagende problemer (f.eks.
aspiration)
• Aortaskade, herunder perforation, dissektion, blødning, ruptur og død
• Arterie- eller venetrombose og/eller pseudoaneurisme
• Arteriovenøs fistel
• Blødning, hæmatom eller koagulopati
• Claudicatio (f.eks. balde, underekstremitet)
• Død
• Emboli (mikro og makro) med transitorisk eller permanent iskæmi eller infarkt
• Endolækage
• Endoprotese: ukorrekt placering af komponent, ufuldstændig ekspansion
af komponent, migration af komponent, brud på sutur, okklusion, infektion,
stentfraktur, slittage af protesematerialet, dilatation, erosion, punktur, flow
omkring protesen, løsrevet modhage og korrosion
• Feber og lokaliseret inflammation
• Impotens
• Infektion af aneurismet, produktet eller adgangsstedet, herunder
abscesdannelse, transitorisk feber og smerte
• Kardielle komplikationer og efterfølgende ledsagende problemer (f.eks.
arytmi, myokardieinfarkt, hjerteinsufficiens, hypotension, hypertension)
• Karskade
• Karspasme eller kartraume (f.eks. iliofemoral kardissektion, blødning, ruptur,
død)
• Kirurgisk konvertering til åben reparation
• Komplikationer ved vaskulært adgangssted, herunder infektion, smerte,
hæmatom, pseudoaneurisme, arteriovenøs fistel
• Leversvigt
• Lymfatiske komplikationer og efterfølgende ledsagende problemer (f.eks.
lymfefistel)
• Neurologiske lokale eller systemiske komplikationer og efterfølgende
ledsagende problemer (f.eks. slagtilfælde, transitorisk iskæmisk attak (TIA),
paraplegi, paraparese, paralyse)
• Nyrekomplikationer og efterfølgende ledsagende problemer (f.eks.
arterieokklusion, kontrasttoksicitet, insufficiens, svigt)
• Okklusion af protese eller nativt kar
• Pulmonale/respirationskomplikationer og efterfølgende ledsagende
problemer (f.eks. pneumoni, respirationsinsufficiens, langvarig intubation)
• Sårkomplikationer og efterfølgende ledsagende problemer (f.eks. sårruptur,
infektion)
• Tarmkomplikationer (f.eks. ileus, transitorisk iskæmi, infarkt, nekrose)
• Urogenitale komplikationer og efterfølgende ledsagende problemer (f.eks.
iskæmi, erosion, fistel, inkontinens, hæmaturi, infektion)
• Ødem.
Rapportering af uønskede hændelser, relateret til anordning
Alle uønskede hændelser (kliniske tilfælde), der involverer Zenith Flex AAA
endovaskulær protese, skal straks rapporteres til COOK. Kunder i USA skal
rapportere tilfælde ved at ringe til Customer Relations Department på
1-800-457-4500 (24 timer) eller 1-812-339-2235. Kunder udenfor USA bedes
ringe til forhandleren.
6 RESUME OVER KLINISKE UNDERSØGELSER
6.1 Målsætning
Den kliniske undersøgelses primære målsætning var at evaluere sikkerheden og
effektiviteten af en tidligere version af anordningen (Zenith AAA endovaskulær
protese) som et alternativ til åben kirurgisk reparation i den primære behandling
af infrarenal abdominal aortaaneurisme. Undersøgelseshypoteser undersøgte,
hvorvidt patienter i standardrisikogrupper oplevede reduceret morbiditet efter
30 dage, samme overlevelse efter 30 dage, samme overlevelse efter 12 måneder
og samme behandlingssucces og forbedrede kliniske hjælpeforanstaltninger
efter 12 måneder sammenlignet med patienter, der blev operativt kontrolleret.
Sikkerheden blev bestemt ved at evaluere, om forsøgspersoner, der modtog
Zenith AAA endovaskulær protese, ville have reduceret morbiditet efter 30 dage,
samme overlevelse efter 30 dage og 12 måneder og samme behandlingssucces
efter 12 måneder, sammenlignet med testpersoner behandlet med åben
kirurgisk behandling. Effektiviteten var baseret på eksklusion af aneurismet,
inklusive fravær af enhver endolækage, fravær af aneurismeforstørrelse (≥ 5 mm)
og fravær af større uønsket hændelse i forbindelse med anordning, evalueret
over ét års opfølgning. Sekundære målsætninger inkluderede vurdering af
kliniske fordele og livskvalitet.
6.2 Undersøgelsens design
Den amerikanske kliniske, ikke- randomiserede multicenterundersøgelse
sammenlignede patienter med standard medicinsk risiko, der modtog en
endovaskulær protese, med åben kirurgisk kontrol. Der var to yderligere
undersøgelsesgrene for behandlingsgrupper med høj medicinsk risiko og
indlæring. Femten centre tilmeldte 200 patienter med standardrisiko,
80 med kirurgisk kontrol, 100 med højrisiko og 52 indlæringspatienter.
Kontrolgruppen inkluderede patienter, hvis vaskulære anatomi muligvis ikke
var egnet til endovaskulær AAA reparation. Opfølgende evalueringer blev
planlagt til før udskrivning, efter 1 måned, 6 måneder, 12 måneder og
24 måneder. Der er vist patientopfølgning og ansvarlighed ved 1 måned,
12 måneder og 24 måneder i Tabel 6.2.1, da disse var de primære tidspunkter
for dataanalyse. Oversigtens billeddiagnostiske data er baseret på resultaterne
fra et uafhængigt centraliseret billedanalyselaboratorium
(centrallaboratorium), der gennemså CT-scanninger og abdominale
røntgenbilleder for at vurdere diameterændringer i aneurismet, migration af
anordning relativ til komponent, integritet af anordning (tråd og protese) og
tilstedeværelsen og typen af endolækager. Kliniske hændelser blev bedømt
af et uafhængigt udvalg for kliniske hændelser, og sikkerheden blev
overvåget af et udvalg for sikkerhedsovervågning af data.
Kirurgiske patienter og Zenith standardrisikopatienter opfyldte identiske
patofysiologiske risikokriterier. De endovaskulære grupper ekskluderede:
periferisk trombe i den proksimale hals, proksimal hals mindre end 15 mm
lang målt fra ydre væg til ydre væg, proksimal halsdiameter mindre end
18 mm eller større end 28 mm, alvorlig proksimal vinkeldannelse i hals,
a. iliaca diameter mindre end 7,5 mm eller større end 20 mm målt fra ydre
væg til ydre væg, ved distale fikseringssted eller a. iliaca distale fikseringssted,
mindre end 10 mm lang.
Patienter blev regnet som havende en højere risiko ved kirurgisk reparation
hvis de var over 80 år, havde baseline kreatinin > 2,0 mg/dl, modtog
iltbehandling i hjemmet, FEV1 < 1 liter, ejektionsfraktion < 25 %, invaliderende
kronisk obstruktiv lungesygdom, klassifikation 3 eller 4 fra New York Heart
Association (NYHA), modvillig abdomen, dialyse, myokardieinfarkt indenfor de
sidste 6 måneder, hypertension, der var vanskelig at behandle medicinsk,
tidligere slagtilfælde med residual deficit, kulturel modvilje mod at modtage
blod eller blodprodukter, tidligere renal bypass kirurgi eller inflammatorisk
aneurisme.
Inden tilmelding af patienter i den afgørende undersøgelse blev centre uden
Zenith AAA endovaskulær protese anmodet om at behandle initiale patienter
under opsyn af en tilsynsførende. Disse indlæringspatienter var en
kombination af standard- og højrisikopatienter og blev fulgt i
overensstemmelse med den samme tidsplan som patienterne i den afgørende
undersøgelse.
52

Tabel 6.2.1 Patientopfølgning og ansvarlighed
1
Behandling Zenith standardrisiko Kirurgisk standardrisiko
Interval
Ingen anordning 1 1
Konvertering til åben reparation 0 2
1 mån. 12 mån. 24 mån. 1 mån. 12 mån. 24 mån.
2
2
2
1
2
2
0 0 Ikke relevant
Ikke relevant Ikke relevant Ikke relevant
Udløbet 1 7 17 2 3 I kke relevant
Tilbagetrukket/mistet til opfølgning 0 0 2 0 4 I kke relevant
Tilgængelige 198 190 178 78 73 I kke relevant
CT-scanning på stedet 191 168 110 69 58 Ikke relevant
CT-scanning på centrallaboratorium 190 165 99 69 59 Ikke relevant
KUB-billeddiagnostik (nyre, urinleder, blære) på stedet 179 153 108 I kke relevant Ik ke relevant Ikke relevant
KUB-billeddiagnostik (nyre, urinleder, blære) på centrallaboratorium 178 149 93 Ikke relevant Ikke relevant Ikke relevant
Stedet evalueret for endolækage 187 163 107 Ikke relevant Ik ke relevant Ikke relevant
Centrallaboratorium evalueret for endolækage 161 148 92 Ikke relevant Ikke relevant Ikke relevant
Stedet evalueret for aneurismeforstørrelse Ikke relevant 149 104 Ikke relevant Ikke relevant Ikke relevant
Centrallaboratorium evalueret for neurismeforstørrelse Ikke relevant 151 94 Ikke relevant Ikke relevant Ikke relevant
1
Dataanalysens prøvestørrelse varierer for hver af tidspunkterne ovenfor og i de følgende tabeller. Denne afvigelse skyldes patienters tilgængelighed ved opfølgning, såvel som
kvantiteten og kvaliteten af billeder fra specifikke tidspunkter, tilgængelige for evalueringen. For eksempel er antallet og kvaliteten af billeder, tilgængelige for evalueringen af
endolækage ved 12 måneder, forskelligt fra antallet og kvaliteten af billeder tilgængelige ved 24 måneder, på grund af variationen af antallet af udførte billedundersøgelser, antallet af
billeder fra det kliniske sted til centrallaboratoriet og/eller antallet af billeder med acceptabel evalueringskvalitet. Samlede (totale) ved tidspunkterne er ikke kumulative, med mindre
det er nævnt.
2
Samlede (totale) ved tidspunkterne er kumulative.
6.3 Demografiske oplysninger om patienter
Tabellerne 6.3.1 og 6.3.2 sammenligner forsøgspersoners karakteristika og aneurismets initiale diameter for Zenith AAA endovaskulær protese og åben
kirurgisk gruppe, respektive.
Tabel 6.3.1 Sammenligning af forsøgspersoners karakteristika
Emne Zenith standardrisiko Kirurgisk standardrisiko P værdi Zenith højrisiko Zenith indlæring
Alder (år) 71 ± 7 69 ± 7 0,03 77 ± 7 74 ± 8
Maskulint køn 94 % (187/200) 89 % (71/80) 0,22 92 % (92/100) 90 % (47/52)
Aktuelle medicinske tilstande
Periferisk vaskulær sygdom
Hypertension 64 % (127/200) 83 % (65/78) 0,001 68 % (67/99) 67 % (35/52)
16 % (31/195) 25 % (19/76) 0,12 24 % (23/96) 9,6 % (5/52)
Nyresvigt 0,0 % (0/197) 0,0 % (0/79) > 0,99 5,2 % (5/97) 1,9 % (1/52)
Kronisk obstruktiv lungesygdom 20 % (39/199) 18 % (14/78) 0,87 34 % (33/98) 22 % (11/51)
Tromboembolisk hændelse 4,5 % (9/199) 7,7 % (6/78) 0,38 7,1 % (7/99) 1,9 % (1/52)
Leversygdom 2,1 % (4/192) 5,1 % (4/79) 0,24 1,0 % (1/99) 1,9 % (1/52)
Diabetes mellitus 12 % (24/199) 15 % (12/79) 0,55 17 % (17/99) 14 % (7/51)
Afhængig af insulin 17 % (4/24) 8,3 % (1/12) 0,65 24 % (4/17) 43 % (3/7)
Tidligere medicinske tilstande
Myokardieinfarkt
39 % (74/192) 29 % (23/80) 0,13 35 % (34/98) 35 % (18/52)
Kongestiv hjerteinsufficiens 5,0 % (10/199) 12 % (9/78) 0,07 16 % (16/100) 10 % (5/50)
Angina 49 % (98/198) 39 % (31/79) 0,14 45 % (44/98) 44 % (23/52)
Arytmi 20 % (40/197) 22 % (17/78) 0,87 28 % (27/98) 24 % (12/51)
Cerebrovaskulær lidelse 9,5 % (19/199) 16 % (13/79) 0,14 20 % (20/99) 9,8 % (5/51)
Systemisk infektion 1,0 % (2/196) 0,0 % (0/78) > 0,99 3,1 % (3/97) 0,0 % (0/49)
Cancer 22 % (43/200) 19 % (15/80) 0,74 31 % (31/99) 29 % (15/51)
Tilfælde af aneurismatisk sygdom i familien 16 % (24/150) 27 % (17/63) 0,09 14 % (11/77) 26 % (10/38)
Tidligere kirurgi på stedet 10 % (20/200) 15 % (12/79) 0,22 10 % (10/99) 14 % (7/51)
Tidligere bestråling på stedet 0,5 % (1/197) 0,0 % (0/79) > 0,99 2,0 % (2/100) 2,0 % (1/51)
Overdrevet forbrug af alkohol 3,6 % (7/193) 10 % (8/77) 0,04 3,1 % (3/96) 4,0 % (2/50)
Brug af tobak
Aldrig røget
Røget tidligere 69 % (133/193) 60 % (48/80) 0,03 69 % (66/96) 57 % (28/49)
Ryger stadig 21 % (40/193) 35 % (28/80) 18 % (17/96) 18 % (9/49)
På grund af medtagelseskriterierne var højrisikopatienterne ældre (P < 0,001), havde mere nyreinsufficiens (P = 0,004), kronisk obstruktiv lungesygdom (P = 0,01), kongestiv
hjerteinsufficiens (P = 0,004) og cerebrovaskulær sygdom (P = 0,02) end standardrisikopatienterne.
10 % (20/193) 5,0 % (4/80)
14 % (13/96) 24 % (12/49)
Tabel 6.3.2 Fordeling af aneurismets diameter
Diameterområde Zenith standardrisiko Kirurgisk standardrisiko Zenith højrisiko Zenith indlæring
< 30 mm
30-39 mm
40-49 mm
50-59 mm
60-69 mm
70-79 mm
80-89 mm
≥ 90 mm
Fordeling af aneurismets diameter blev ikke vurderet i tre højrisiko- og én indlæringspatient.
6.4 Resultater
Data samlet i Tabel 6.4.1 til 6.4.3, blev indsamlet af de kliniske undersøgelsessteder og centrallaboratoriet. 24 måned-data er inkluderet, hvor de var
tilgængelige. Kontrolpatienter blev ikke fulgt efter 12 måneder, og visse data ud over 12 måneder er endnu ikke bedømt. Derfor er visse resultater præsenteret
op til 12 måneder, mens andre resultater er præsenteret op til 24 måneder i dette afsnit. Tabel 6.4.1 beskriver anordningerne implanteret i patienterne i den
kliniske undersøgelse. Tabel 6.4.2 beskriver de primære resultater fra den kliniske undersøgelse. Figurerne 6.4.1 og 6.4.2 er Kaplan-Meier grafer, for total og
AAA-relateret overlevelse op til 24 måneder, respektive. Et uafhængigt udvalg for kliniske hændelser bedømte alle dødsfald for mulig forbindelse til reparation
af aneurisme. Alle tidlige dødsfald (0-30 dage) blev bedømt som AAA-relaterede. Dødsfald efter 30 dage blev bedømt som AAA-relaterede hvis der blev
bekræftet AAA-sygdom eller medvirken af anordning. Tabel 6.4.3 præsenterer Måltal for succes og Figur 6.4.3 er Kaplan-Meier grafen Fravær af morbiditet.
0,0 % (0/199) 0,0 % (0/78) 0,0 % (0/100) 0,0 % (0/52)
0,5 % (1/199) 0,0 % (0/78) 1,0 % (1/100) 0,0 % (0/52)
23 % (45/199) 7,7 % (6/78) 15 % (15/100) 13 % (7/52)
48 % (95/199) 33 % (26/78) 47 % (47/100) 40 % (21/52)
24 % (47/199) 29 % (23/78) 27 % (27/100) 42 % (22/52)
3,0 % (6/199) 21 % (16/78) 5,0 % (5/100) 1,9 % (1/52)
2,5 % (5/199) 6,4 % (5/78) 1,0 % (1/100) 0,0 % (0/52)
0,0 % (0/199) 2,6 % (2/78) 1,0 % (1/100) 0,0 % (0/52)
Tabel 6.4.1 Implanterede anordninger
Emne Zenith standardrisiko Zenith højrisiko Zenith indlæring
Hovedprotese og ben 99,5 % (199/200)* 100 % (100/100)
Hovedproteseforlænger
Ipsilateral iliaca-benforlænger
Kontralateral iliaca-benforlænger
Konverteringsenhed 0,5 % (1/199)** 0,0 % (0/100) 0,0 % (0/52)
Okklusionsenhed 0,0 % (0/199) 0,0 % (0/100) 0,0 % (0/52)
*Én standardrisikopatient modtog ikke en anordning pga. snirklethed og forkalkning af adgangskarret.
**Konverteringsenhed var brugt uden okklusionsenhed.
1
Én anordning var tilpasset.
2
To standardrisiko- og én højrisikopatient modtog hovedproteseforlængere efter proceduren; én standardrisikopatient modtog to hovedproteseforlængere.
3
To standardrisiko- og én højrisikopatient modtog ipsilaterale benforlængere efter proceduren.
4
Fire standardrisiko-, to højrisiko- og én indlæringspatient modtog kontralaterale benforlængere efter proceduren.
5
Tre standardrisiko- og tre højrisikopatienter modtog både ipsilaterale og kontralaterale forlængere under proceduren. Én standardrisikopatient modtog ipsilaterale og kontralaterale
forlængere efter proceduren.
2
3,5
4,5
1,5 % (3/199) 1,0 % (1/100) 5,8 % (3/52)
9,5 % (19/199) 11 % (11/100) 0,0 % (0/52)
11 % (21/199) 11 % (11/100) 13,5 % (7/52)
1
100 % (52/52)
53

Tabel 6.4.2 Primære resultater
Emne Zenith standardrisiko
Alle dødsfald
Alle dødsfald
(0-30 dage)
(31-365 dage)
2
2, 3
0,5 % (1/199) 2,5 % (2/80) 0,20 2,0 % (2/100) 1,9 % (1/52)
3,0 % (6/199) 1,3 % (1/80) 0,68 7,0 % (7/100) 9,6 % (5/52)
1
Kirurgisk standardrisiko P værdi Zenith højrisiko Zenith indlæring
AAA-relaterede 0,0 % (0/199) 1,3 % (1/80) 0,29 3,0 % (3/100) 0,0 % (0/52)
Ikke-AAA-relaterede 3,0 % (6/199) 0,0 % (0/80) 0,19 4,0 % (4/100) 9,6 % (5/52)
Alle dødsfald
(0-365 dage)
2,3,4
3,5 % (7/199) 3,8 % (3/80) > 0,99 9,0 % (9/100) 11,5 % (6/52)
AAA-relaterede 0,5 % (1/199) 3,8 % (3/80) 0,07 5,0 % (5/100) 1,9 % (1/52)
Ikke-AAA-relaterede 3,0 % (6/199) 0,0 % (0/80) 0,19 4,0 % (4/100) 9,6 % (5/52)
Ruptur
(0-30 dage) 0,0 % (0/199) Ik ke relevant Ikke relevant 0,0 % (0/100) 0,0 % (0/52)
(31-365 dage) 0,0 % (0/199) Ikke relevant Ikke relevant 1,0 % (1/100) 0,0 % (0/52)
(0-365 dage) 0,0 % (0/199) Ikke relevant Ikke relevant 1,0 % (1/100) 0,0 % (0/52)
Konvertering
(0-30 dage) 0,0 % (0/199) Ik ke relevant Ikke relevant 0,0 % (0/100) 0,0 % (0/52)
(31-365 dage)
(0-365 dage)
Uønskede hændelser
(0-30 dage)
(31-365 dage)
(0-365 dage)
1
Nævner på 199, da én standardrisikopatient ikke modtog en anordning.
2
Alle dødsfald (0-30 dage) blev regnet som relateret til AAA og proceduren.
3
Af dødsfaldene (31-365 dage) blev fire regnet som AAA-relaterede: 1 kirurgisk (septisk shock fra iskæmisk colitis) og 3 højrisiko (pancreatitis med nyresvigt og sepsis, hæmorragi fra
øvre abdominal aneurisme [ubehandlet AAA] og flere systemsvigt).
4
Af dødsfaldene (0-365 dage) blev ti regnet som AAA-relaterede: 1 standardrisiko (hjertesvigt), 3 kirurgiske (alvorlig hæmorragi, mesenterisk iskæmi og septisk shock fra iskæmisk
colitis), 5 højrisiko (respirationsinsufficiens, hjertesvigt med pulmonær emboli, pancreatitis med nyresvigt og sepsis, hæmorragi fra øvre abdominal aneurisme [ikke AAA-behandlet]
og flere systemsvigt) og 1 indlæring (mistanke om hjertesvigt).
5
Standardrisikopatienter undergik konverteringer pga. en vedvarende, proksimal type I endolækage og en ny suprarenal aortaaneurisme. Tre kirurgiske patienter havde alvorlig
hæmorragi, hvoraf to af patienterne krævede operation igen og én patient døde.
6
Uønskede hændelser inkluderet i morbiditetsindekset.
5
5
6
6
6
1,0 % (2/199) Ikke relevant Ikke relevant 1,0 % (1/100) 0,0 % (0/52)
1,0 % (2/199) Ikke relevant Ikke relevant 1,0 % (1/100) 0,0 % (0/52)
20 % (40/200) 43 % (34/80) < 0,001 32 % (32/100) 27 % (14/52)
8,6 % (17/199) 14 % (11/78) 0,25 21 % (21/98) 14 % (7/51)
25 % (49/200) 51 % (41/80) < 0,001 45 % (45/100) 38 % (20/52)
Patienter med Zenith AAA endovaskulær protese udviste ingen signifikante
forskelle mellem mænd og kvinder angående overlevelse og fravær af
uønskede hændelser (fejllinjerne i Figurerne 6.4.1, 6.4.2 og 6.4.3
repræsenterer konfidensgrænser på 95 %). Figur 6.4.1 præsenterer total
overlevelse op til 24 måneder. Den ledsagende tabel præsenterer KaplanMeier analysen ved 1, 6, 12 og 24 måneder.
Figur 6.4.1 Overlevelse ved 24 måneder
Zenith standardrisiko
(N = 200, 1 dødsfald op til 30 dage
4 dødsfald op til 6 måneder
7 dødsfald op til 12 måneder
17 dødsfald op til 24 måneder
Kirurgisk standardrisiko
(N = 80, 2 dødsfald op til 30 dage
3 dødsfald op til 6 måneder
Procentvis overlevelse
3 dødsfald op til 12 måneder)
Måneder efter procedure
1 måned 6 måneder 12 måneder 24 måneder
% over-
n
levelse n
Zenith
standard-
198* 99,5 194 98,0 190 96,5 97 90,9
risiko
Kirurgisk
standardrisiko
n = Patienter i live og tilgængelige til opfølgning ved intervallets afslutning
P = 0,81
*Én patient døde indenfor 1 måned og én patient modtog ikke en anordning.
78 97,5 73 96,2 67 96,2
% over-
levelse n
% over-
levelse n
relevant
Ikke
% over-
levelse
Ikke
relevant
Figur 6.4.2 præsenterer AAA-relateret overlevelse (bestemt af Clinical Events
Committee) op til 24 måneder. Den ledsagende tabel præsenterer KaplanMeier analysen ved 1, 6, 12 og 24 måneder.
Figur 6.4.2 AAA-relateret overlevelse ved 24 måneder
Zenith standardrisiko
(N = 200, 1 dødsfald op til 30 dage
1 dødsfald op til 6 måneder
1 dødsfald op til 12 måneder
2 dødsfald op til 24 måneder
Kirurgisk standardrisiko
(N = 80, 2 dødsfald op til 30 dage
3 dødsfald op til 6 måneder
relaterede dødsfald
3 dødsfald op til 12 måneder)
Procentvis fravær af aneurisme-
Hændelse: aneurisme-relateret dødsfald
Måneder efter procedure
1 måned 6 måneder 12 måneder 24 måneder
% over-
n
levelse n
Zenith
standard-
198* 99,5 194 99,5 190 99,5 97 99,0
risiko
Kirurgisk
standardrisiko
n = Patienter i live og tilgængelige til opfølgning ved intervallets afslutning
P = 0,04
*Én patient døde indenfor 1 måned og én patient modtog ikke en anordning.
78 97,5 73 96,2 67 96,2
% over-
levelse n
% overlevelse n
relevant
Ikke
% over-
levelse
Ikke
relevant
54

Tabel 6.4.3 Måltal for succes
Emne Zenith standardrisiko Kirurgisk standardrisiko Zenith højrisiko Zenith indlæring
Teknisk succes
Proceduremæssig succes ved
30 dage
Behandlingssucces ved
12 måneder
1
Åben protese efter anlæggelse.
2
Teknisk succes uden større komplikationer, åben protese og ingen type I eller type III endolæk age ved 30 dage.
3
Proceduremæssig succes forlænget til 12 måneder uden aneurismeforstørrelse (> 5 mm).
Figur 6.4.3 præsenterer fravær af morbiditet (hændelser i morbiditetsindekset) op til 12 måneder. Den medfølgende tabel præsenterer Kaplan-Meier analysen ved 1,
6 og 12 måneder.
1
2
3
99,5 % (199/200) 98,8 % (79/80) 100 % (100/100) 100 % (52/52)
95,1 % (155/163) 88 % (60/68) 86 % (70/81) 91 % (30/33)
89 % (122/137) 85 % (52/61) 70 % (44/63) 87 % (26/30)
Figur 6.4.3 Fravær af morbiditet (0-365 dage)
Zenith standardrisiko
(N = 200, 38 patienter med hændelser op til 30 dage
43 patienter med hændelser op til 6 måneder
45 patienter med hændelser op til 12 måneder)
Kirurgisk standardrisiko
(N = 80, 34 patienter med hændelser op til 30 dage
38 patienter med hændelser op til 6 måneder
Procentvis fravær af morbiditet
41 patienter med hændelser op til 12 måneder)
Måneder efter procedure
1 måned 6 måneder 12 måneder
n % n % n %
Zenith standardrisiko 162 81,0 154 78,5 133 77,4
Kirurgisk standardrisiko 45 57,1 39 52,0 33 47,8
n = Patienter i live og uden morbiditet ved intervallets afslutning
P = < 0,001
Tabellerne 6.4.4 til 6.4.7 beskriver resultaterne af forsøgspersoner med Zenith AAA endovaskulær protese, som rapporteret af centrallaboratoriet.
Anordningens præstationsfaktorer, analyseret af centrallaboratoriet, inkluderede anordningens integritet (Tabel 6.4.4), anordningens åbenhed (Tabel 6.4.5),
migration (Tabel 6.4.6) og separation af lem (Tabel 6.4.7).
Tabel 6.4.4 Abdominale fund under røntgen – Anordningens integritet
Emne Zenith standardrisiko Zenith højrisiko Zenith indlæring
Stentfrakturer
1
Før udskrivning 0,0 % (0/172) 0,0 % (0/81) 0,0 % (0/39)
30 dage 0,0 % (0/172) 0,0 % (0/83) 0,0 % (0/43)
6 måneder 0,0 % (0/166) 0,0 % (0/78) 0,0 % (0/35)
12 måneder 0,0 % (0/148) 0,0 % (0/60) 0,0 % (0/28)
24 måneder 0,0 % (0/93) 0,0 % (0/42) 0,0 % (0/19)
Separation af modhage
2
Før udskrivning 0,0 % (0/176) 0,0 % (0/86) 0,0 % (0/39)
30 dage 0,0 % (0/178) 0,0 % (0/86) 0,0 % (0/43)
6 måneder 1,2 % (2/167) 2,5 % (2/80) 0,0 % (0/35)
12 måneder 2,0 % (3/149) 1,7 % (1/60) 0,0 % (0/28)
24 måneder 1,1 % (1/93) 0,0 % (0/42) 0,0 % (0/19)
Ruptur af protesemateriale
Før udskrivning 0,0 % (0/176) 0,0 % (0/86) 0,0 % (0/39)
30 dage 0,0 % (0/178) 0,0 % (0/86) 0,0 % (0/43)
6 måneder 0,0 % (0/167) 0,0 % (0/80) 0,0 % (0/35)
12 måneder 0,0 % (0/149) 0,0 % (0/60) 0,0 % (0/28)
24 måneder 0,0 % (0/93) 0,0 % (0/42) 0,0 % (0/19)
1
Procentdele af stentfraktur er for hovedprotesen. Centrallaboratoriet observerede heller ikke fraktur af højre iliaca-ben, venstre iliaca-ben, okklusionsenhed, konverteringsenhed,
venstre iliaca-forlænger, højre iliaca-forlænger eller hovedproteseforlænger.
2
Patienter med separation af 1 eller 2 modhager (af 10 eller 12 totalt). Ingen uønskede kliniske følgevirkninger.
Tabel 6.4.5 CT fund – Åbenhed af protese
Emne Zenith standardrisiko Zenith højrisiko Zenith indlæring
Åbenhed af protese
30 dage 100 % (185/185) 99 % (85/86) 100 % (47/47)
6 måneder 99 % (183/184) 100 % (74/74) 100 % (39/39)
12 måneder 99 % (153/155) 100 % (62/62) 100 % (30/30)
24 måneder 100 % (96/96) 100 % (33/33) 100 % (25/25)
Tabel 6.4.6 CT fund – Migration af protese (hovedprotese)
Emne Zenith standardrisiko Zenith højrisiko Zenith indlæring
Migration af protese (> 5 mm) ved 12 måneder
med klinisk følgevirkning1 eller intervention 0,0 % (0/162) 0,0 % (0/71) 0,0 % (0/34)
uden klinisk følgevirkning1 eller intervention 2,5 % (4/162) 2,8 % (2/71) 0,0 % (0/34)
Migration af protese (> 10 mm) 0,0 % (0/162) 0,0 % (0/71) 0,0 % (0/34)
1
Migration med klinisk følgevirkning inkluderer endolækage, konvertering, ruptur eller AAA-relateret død.
55

Tabel 6.4.7 Abdominale fund under røntgen – separation af lem
Emne Zenith standardrisiko Zenith højrisiko Zenith indlæring
Separation af lem
Før udskrivning 0,0 % (0/176) 0,0 % (0/86) 0,0 % (0/39)
30 dage 0,0 % (0/178) 0,0 % (0/86) 0,0 % (0/43)
6 måneder 0,0 % (0/167) 0,0 % (0/80) 0,0 % (0/35)
12 måneder 0,0 % (0/149) 0,0 % (0/60) 0,0 % (0/28)
24 måneder 0,0 % (0/93) 0,0 % (0/42) 0,0 % (0/19)
6.5 Styring af endolækage
Under den kliniske undersøgelse blev type I endolækager behandlet under den initiale procedure med installation af ekstra ballon, eller hvis det ikke lykkedes,
ekstra proteser. Type II endolækager blev observeret over en periode på én til seks måneder for at bestemme, om de spontant ville trombosere, eller ved
fraværet af aneurismeforstørrelse blev de behandlet med endovaskulære teknikker efter lægens vurdering. Hvis aneurismet blev større, blev emboli eller
underbinding overvejet, og i visse tilfælde udført. Type III endolækager, forårsaget af protesedefekter, utilstrækkelig forsegling eller frakobling af modulære
komponenter, blev behandlet med yderligere ballon eller protese. Som det blev rapporteret af det angiografiske centrallaboratorie var der ingen type IV
endolækager under den amerikanske kliniske undersøgelse. Protesematerialet, der blev brugt til at fremstille Zenith AAA endovaskulær protese, er af
standardtykkelse, og er det samme materiale, der bruges i åbne kirurgiske procedurer. Tabel 6.5.1 præsenterer tilfældene af endolækager efter evaluerings-
interval, som identificeret af centrallaboratoriet for standardrisiko-, højrisiko- og indlæringspatienter respektive.
Tabel 6.5.1 Endolækager (alle typer, nye og vedvarende)
Emne Zenith standardrisiko Zenith højrisiko Zenith indlæring
Endolækager
Før udskrivning 15 % (23/153) 14 % (11/78) 12 % (3/26)
1
30 dage
1
6 måneder
1
12 måneder
1
Inkluderer både vedvarende endolækager og nye observationer.
Tabellerne 6.5.2 – 6.5.4 præsenterer antallet af første tilfælde af endolækage pr. evalueringsinter val, som identificeret af centrallaboratoriet, inden eller ved 30
dages, 6 måneders og 12 måneders undersøgelser for standardrisiko-, højrisiko- og indlæringspatienter, respektive. Antallet af patienter uden lækage derefter
opgives også.
9,9 % (16/161) 12 % (9/75) 6,3 % (2/32)
8,7 % (15/172) 11 % (8/70) 8,6 % (3/35)
7,4 % (11/148) 8,8 % (5/57) 3,4 % (1/29)
Tabel 6.5.2 Første tilfælde af Endolækage1 for standardrisikopatienter
Emne
Til én-måned undersøgelse
N = 179
% Endolækage
1
Uden lækage
derefter
2
Seks-måned undersøgelse
N = 172
% Endolækage
Uden lækage
1
derefter
2
Tolv-måned undersøgelse
N = 148
% Endolækage
Uden lækage
1
derefter
Endolækager 17 31 17 2,3 4 3 3,4 5 2
Proksimal type I 2,8 5 4 0,0 0 0 0,0 0 0
Distal
Type I 1,7 3 1 0,0 0 0 0,7 1 1
Type II 9,5 17 9 2,3 4 3 1,4 2 1
Type III 1,1 2 2 0,0 0 0 0,7 1 0
Type IV 0,0 0 0 0,0 0 0 0,0 0 0
Flere 1,1 2 1 0,0 0 0 0,0 0 0
Ukendt 1,1 2 0 0,0 0 0 0,7 1 0
1
Identificeret af centrallaboratoriet.
2
Efterfølgende endolækager muligvis anden type end den oprindelige.
3
Kun 2 patienter havde nye endolækager efter 12 måneder. Opfølgning efter 24 måneder ikke tilgængelig.
Tabel 6.5.3 Første tilfælde af endolækage1 for højrisikopatienter
Emne
Til én-måned undersøgelse
N = 88
% Endolækage
Uden lækage
1
derefter
2
Seks-måned undersøgelse
N = 70
% Endolækage
Uden lækage
1
derefter
2
Tolv-måned undersøgelse
N = 57
% Endolækage
Uden lækage
1
Endolækager 18 16 6 2,9 2 0 3,5 2 1
Proksimal type I 2,3 2 1 0,0 0 0 0,0 0 0
Distal
Type I 1,1 1 1 0,0 0 0 0,0 0 0
Type II 9,1 8 3 1,4 1 0 1,8 1 0
Type III 0,0 0 0 1,4 1 0 1,8 1 1
Type IV 0,0 0 0 0,0 0 0 0,0 0 0
Flere 4,5 4 0 0,0 0 0 0,0 0 0
Ukendt 1,1 1 1 0,0 0 0 0,0 0 0
1
Identificeret af centrallaboratoriet.
2
Efterfølgende endolækager muligvis anden type end den oprindelige.
3
Ingen endolækager efter 12 måneder. Opfølgning efter 24 måneder ikke tilgængelig.
3
3
derefter
2
2
Tabel 6.5.4 Første tilfælde af endolækage1 for indlæringspatienter
Emne
Til én-måned undersøgelse
N = 36
% Endolækage
1
Uden lækage
derefter
2
Seks-måned undersøgelse
N = 35
% Endolækage
Uden lækage
1
derefter
2
Tolv-måned undersøgelse
N = 29
% Endolækage
Uden lækage
1
Endolækager 11 4 2 2,9 1 0 0,0 0 0
Proksimal type I 0,0 0 0 0,0 0 0 0,0 0 0
Distal
Type I 0,0 0 0 0,0 0 0 0,0 0 0
Type II 5,6 2 1 2,9 1 0 0,0 0 0
Type III 2,8 1 0 0,0 0 0 0,0 0 0
Type IV 0,0 0 0 0,0 0 0 0,0 0 0
Flere 0,0 0 0 0,0 0 0 0,0 0 0
Ukendt 2,8 1 1 0,0 0 0 0,0 0 0
1
Identificeret af centrallaboratoriet.
2
Efterfølgende endolækager muligvis anden type end den oprindelige.
3
Ingen endolækager efter 12 måneder. Opfølgning efter 24 måneder ikke tilgængelig.
56
3
derefter
2

6.6 Aneurismeændring
Tabellerne 6.6.1 – 6.6.3 præsenterer ændringen i aneurismets diameter i de endovaskulære patienter, som identificeret af centrallaboratoriet. Tabel 6.6.1
præsenterer aneurismets maksimale diameterændring efter interval. Tabellerne 6.6.2 og 6.6.3 præsenterer aneurismets ændring og endolækage ved 12 og
24 måneder, respektive.
Tabel 6.6.1 Ændring i aneurismets maksimale diameter efter interval*
Emne Zenith standardrisiko Zenith højrisiko Zenith indlæring
Fra før udskrivning indenfor 30 dage
Reduceret > 5 mm 1,7 % (3/180) 4,8 % (4/84) 0,0 % (0/40)
Uændret 97 % (174/180) 94 % (79/84) 97,5 % (39/40)
Øget > 5 mm 1,7 % (3/180) 1,2 % (1/84) 2,5 % (1/40)
Ændring fra før udskrivning indenfor 6 måneder
Reduceret > 5 mm 36 % (63/173) 41 % (30/73) 49 % (18/37)
Uændret 62 % (108/173) 59 % (43/73) 51 % (19/37)
Øget > 5 mm 1,2 % (2/173) 0,0 % (0/73) 0,0 % (0/37)
Ændring fra før udskrivning indenfor 12 måneder
Reduceret > 5 mm 68 % (102/151) 63 % (39/62) 67 % (20/30)
Uændret 31 % (47/151) 35 % (22/62) 33 % (10/30)
Øget > 5 mm 1,3 % (2/151) 1,6 % (1/62) 0,0 % (0/30)
Ændring fra før udskrivning indenfor 24 måneder
Reduceret > 5 mm 78 % (74/94) 75 % (27/36) 71 % (17/24)
Uændret 19 % (18/94) 25 % (9/36) 25 % (6/24)
Øget > 5 mm 2,1 % (2/94) 0,0 % (0/36) 4,2 % (1/24)
*Inkluderer kun forsøgspersoner med udlæggelige film og målinger af aneurismets ændring fra 1 til 24 måneder.
Tabel 6.6.2 Ændring af aneurismets størrelse og endolækage ved 12 måneder
Zenith standardrisiko
N = 140
Zenith højrisiko
N = 53
Zenith indlæring
N = 27
Endolækage Endolækage Endolækage
Emne
Ændring af aneurismets størrelse fra før udskrivning til 12 måneder
N n % N n % N n %
Reduceret > 5 mm 96 3 3,0 35 3 9,0 20 0 0,0
Uændret 42 8 19 17 2 12 7 1 14
Øget > 5 mm 2 0 0,0 1 0 0,0 0 0 0,0
Tabel 6.6.3 Ændring af aneurismets størrelse og endolækage ved 24 måneder
Zenith standardrisiko
N = 90
Zenith højrisiko
N = 31
Zenith indlæring
N = 21
Endolækage Endolækage Endolækage
Emne
Ændring af aneurismets størrelse fra før udskrivning til 24 måneder
N n % N n % N n %
Reduceret > 5 mm 71 3 4,0 24 1 4,0 16 0 0,0
Uændret 18 1 6,0 7 3 43 5 0 0,0
Øget > 5 mm 1 1 100 1 0 0,0 0 0 0,0
6.7 AAA-relaterede sekundære interventioner
AAA-relaterede sekundære interventioner indenfor det første år blev udført i 11 % af Zenith standardrisiko-, 13 % Zenith højrisiko- og 5,8 %
indlæringsforsøgspersoner, som vist i Tabel 6.7.1. Mere end 50 % af de sekundære interventioner involverede kateterisering for at behandle en endolækage.
AAA-relaterede sekundære interventioner indenfor det andet år blev udført i 4,2 % af Zenith standardrisiko-, 2,2 % af Zenith højrisiko- og 2,3 % af
indlæringsforsøgspersoner, som vist i Tabel 6.7.2.
Tabel 6.7.1 Sekundære interventioner (til 12 måneder)
Intervention
Konvertering til åben reparation 2 1,0 1 1,0 0 0,0
Zenith standardrisiko
N = 199
Zenith højrisiko
N = 100
Zenith indlæring
N = 52
n % n % n %
Forsøgspersoner med ≥ 1 intervention 21 11 13 13 3 5,8
Behandling af en endolækage
Embolisering 5 2,5 3 3,0 1 2,0
Hjælpekomponent 6 3,0 4 4,0 1 2,0
Stent 1 0,5 0 0,0 0 0,0
Angioplastik 0 0,0 1 1,0 0 0,0
Behandling af en aneurismeforstørrelse
Embolisering 0 0,0 0 0,0 0 0,0
Behandling af en okklusion i lem 1 0,5 3 3,0 1 2,0
Behandling af en stenose i lem 1 0,5 0 0,0 0 0,0
Behandling af en nyrearterie 5 2,5 0 0,0 0 0,0
Behandling af infrainguinal iskæmi 1 0,5 1 1,0 0 0,0
Behandling af flere hændelser 1 0,5 1 1,0 0 0,0
Tabel 6.7.2 Sekundære interventioner (> 12 til 24 måneder)
Intervention
Konvertering til åben reparation 1 0,5 1 1,1 0 0,0
Zenith standardrisiko
N = 190
Zenith højrisiko
N = 90
Zenith indlæring
N = 44
n % n % n %
Forsøgspersoner med ≥ 1 intervention 8 4,2 2 2,2 1 2,3
Behandling af en endolækage
Embolisering 3
1
1,6 0 0,0 1
2
Hjælpekomponent 1 0,5 0 0,0 0 0,0
Stent 0 0,0 1 1,1 0 0,0
Angioplastik 0 0,0 0 0,0 0 0,0
Behandling af en aneurismeforstørrelse
Embolisering 1 0,5 0 0,0 1
2
Behandling af en okklusion i lem 1 0,5 0 0,0 0 0,0
Behandling af en stenose i lem 0 0,0 0 0,0 0 0,0
Behandling af en nyrearterie 0 0,0 1 1,1 0 0,0
Behandling af infrainguinal iskæmi 1 0,5 0 0,0 0 0,0
Behandling af flere hændelser 1 0,5 0 0,0 0 0,0
1
Patient modtog også hjælpekomponent.
2
Patient undergik intervention for at behandle aneurismeforstørrelse og endolækage.
57
2,3
2,3

6.8 Måltal for sekundære resultater
Som beskrevet i Tabel 6.8.1 udviste behandling af AAA med Zenith AAA endovaskulær protese signifikante fordele ved helbredelse og livskvalitet,
sammenlignet med den kirurgiske kontrolgruppe.
Tabel 6.8.1 Sekundære resultater efter behandlingsgruppe
Emne Zenith standardrisiko Kirurgisk standardrisiko P værdi Zenith højrisiko Zenith indlæring
Anæstesitid (min.) 221,6 ± 67,3 304,5 ± 102,7 < 0,001 218,9 ± 69,6 213,9 ± 57,7
Proceduretid (min.) 153,2 ± 56,3 238,7 ± 92,2 < 0,001 153,5 ± 58,6 155,9 ± 43,2
Modtagne blodbanksprodukter 5,0 % (10/200) 84 % (67/80) < 0,001 12 % (12/100) 3,8 % (2/52)
Blodtab (ml) 299 ± 324 1676 ± 1676 < 0,001 356 ± 514 265 ± 226
Dage på intensivafdelingen 0,4 ± 0,9 3,4 ± 4,6 < 0,001 0,5 ± 1,2 0,5 ± 0,9
Dage til udskrivning 2,6 ± 1,7 8,8 ± 5,6 < 0,001 3,0 ± 2,8 2,7 ± 1,5
Dage til orale væsker 0,5 ± 0,8 3,9 ± 2,5 < 0,001 0,5 ± 0,6 0,7 ± 0,5
Dage til normal diæt 1,3 ± 1,2 6,6 ± 4,9 < 0,001 1,3 ± 0,8 1,1 ± 0,7
Dage til normal tarmfunktion 2,6 ± 1,4 4,2 ± 2,1 < 0,001 2,6 ± 1,5 2,0 ± 1,2
Dage til oppegående 1,2 ± 0,7 3,5 ± 3,4 < 0,001 1,2 ± 0,7 1,2 ± 0,6
Intubation i timer 1,9 ± 2,2 11,7 ± 13,6 < 0,001 1,2 ± 1,7 2,6 ± 4,6
Maksimaltemperatur (°F) 101,1 ± 1,3 100,7 ± 1,2 0,06 100,8 ± 1,1 101,0 ± 1,2
7 PATIENTUDVÆLGELSE OG BEHANDLING
Se Afsnit 4, Advarsler og forholdsregler
Individualisering af behandling
Cook anbefaler, at komponenternes diametre til Zenith Flex AAA endovaskulær
protese vælges som beskrevet i Tabellerne 10.5.1 og 10.5.2. Længden af
Zenith Flex AAA endovaskulær protese skal nå fra den laveste nyrearterie til
netop over a. iliaca interna-bifurkaturen (hypogastrisk). Alle de nødvendige
længder og diametre af produktet til at gennemføre proceduren skal være
tilgængelige for lægen, især når de præ-operative planlægningsmål til tilfældet
(behandlings-diametre/længder) ikke er sikre. Denne fremgangsmåde giver
større intraoperativ fleksibilitet til at opnå optimale resultater af proceduren.
Risici og fordele, beskrevet tidligere i
UNDERSØGELSER
Flex AAA endovaskulær protese. Yderligere betragtninger vedr. valg af patienter
omfatter, men er ikke begrænset til:
• Patientens alder og forventet levetid
• Komorbiditeter (f.eks. hjerte-, lunge- eller nyreinsufficiens inden operationen,
morbid obesitas)
• Patienters egnethed til åben kirurgisk reparation
• Patienters anatomiske egnethed for endovaskulær reparation
• Risikoen for aneurismeruptur sammenlignet med risikoen ved behandling med
Zenith Flex AAA endovaskulær protese
• Evne til at tolerere generel-, regional- eller lokalanæstesi
• Størrelse og morfologi af iliofemoral adgangskar (minimal trombe, forkalkning
og/eller snirklethed) skal være kompatibel med vaskulære adgangsteknikker
og tilbehør med den samme diameter som en 14 French til 22 French vaskulær
indføringssheath
• Non-aneurismalt infrarenalt aortasegment (hals) proksimalt for aneurismet:
• med en længde på mindst 15 mm,
• med en diameter, målt ydre væg til ydre væg, på højst 32 mm og mindst
18 mm,
• med en vinkel, der er mindre end 60 grader i forhold til aneurismets
længdeakse, og
• med en vinkel, der er mindre end 45 grader i forhold til den suprarenale
aortaakse
• Et distalt fikseringssted i a. iliaca, der er større end 10 mm i længde og
7,5-20 mm i diameter (målt ydre væg til ydre væg)
• Ingen signifikant okklusiv sygdom i a. femoralis/a. iliaca, som ville hæmme
flowet gennem den endovaskulære protese.
, bør nøje overvejes for hver patient inden brug af Zenith
Afsnit 6, RESUME OVER KLINISKE
Den endelige behandlingsbeslutning tages af lægen og patienten.
8 PATIENTRÅDGIVNINGSINFORMATION
Lægen og patienten (og/eller familiemedlemmer) skal gennemgå risici og fordele
under samtalen om dette endovaskulære produkt og procedure, herunder:
• Risici og forskelle mellem endovaskulær reparation og kirurgisk reparation
• Potentielle fordele ved traditionel åben kirurgi
• Potentielle fordele ved endovaskulær reparation
• Muligheden for, at efterfølgende intervention eller åben kirurgisk reparation af
aneurismet kan være påkrævet efter initial endovaskulær reparation
Ud over risiciene og fordelene ved en endovaskulær reparation skal lægen
vurdere patientens tilsagn og samarbejdsvilje vedrørende postoperativ
opfølgning efter behov for at sikre fortsat sikre og effektive resultater. Herunder
angives yderligere emner, der bør diskuteres med patienten med hensyn til
forventningerne efter en endovaskulær reparation:
• Endovaskulære protesers funktion og sikkerhed på langt sigt er endnu
ikke fastlagt. Alle patienter skal gøres opmærksom på, at endovaskulær
behandling kræver regelmæssig opfølgning resten af deres liv til
bedømmelse af deres helbredstilstand og af deres endovaskulære
protese. Patienter med særlige kliniske fund (f.eks. endolækager, aneurismer,
der forstørres, eller ændringer i den endovaskulære proteses struktur eller
position) bør følges nøjere. Specifikke retningslinjer for opfølgning beskrives i
Afsnit 12, RETNINGSLINJER FOR BILLEDDIAGNOSTIK OG POSTOPERATIV
OPFØLGNING.
• Patienter bør rådgives om vigtigheden af at overholde opfølgningsplanen,
både i det første år og med årlige mellemrum derefter. Patienter bør
informeres om, at regelmæssig og konsekvent opfølgning er en meget vigtig
del af at sikre den fortsatte sikkerhed og effektivitet af den endovaskulære
behandling af AAA’er. Der kræves som minimum årlig gennemlysning og
efterfølgning af rutinemæssige postoperative opfølgningskrav, der bør regnes
som en livslang forpligtelse til patientens sundhed og velvære.
• Patienten bør have besked om, at vellykket reparation af et aneurisme ikke
stopper sygdomsprocessen. Det er stadig muligt at have associeret
degeneration af kar.
• Lægen skal rådgive alle patienter om, at det er vigtigt straks at søge
lægehjælp, hvis han/hun oplever tegn på okklusion i lem,
aneurismeforstørrelse eller ruptur. Tegn på okklusion i protese/lem inkluderer
smerte i hofte(r) eller ben under gang eller i hvile, eller misfarvning eller
kolde ben. Aneurismeruptur kan være asymptomatisk, men opleves
sædvanligvis som: smerte, følelsesløshed, svaghed i ben, smerter i ryg, bryst,
abdomen eller lyske, svimmelhed, besvimelse, hurtigt hjerteslag eller
pludselig svaghed.
• På grund af den billeddiagnostik, der kræves for vellykket placering og
opfølgning af endovaskulære implantater, skal risici for strålingseksponering
for væv under dannelse diskuteres med kvinder, som er gravide eller har
mistanke herom. Mænd, som gennemgår endovaskulær eller åben kirurgisk
reparation, kan opleve impotens.
Læger skal referere patienten til Patientvejledning angående risici under og efter
anordningens implantation. Procedurerelaterede risici inkluderer kardielle,
pulmonale og neurologiske komplikationer samt tarm- og blødningskomplikationer. Anordningsrelaterede risici inkluderede okklusion, endolækage,
aneurismeforstørrelse, fraktur, mulighed for reintervention og åben kirurgisk
konvertering, ruptur og død (se Afsnit 5.1, Observerede uønskede hændelser
og Afsnit 5.2, Mulige uønskede hændelser). Lægen skal udfylde Patientens
ID-kort og give det til patienten, således at han/hun altid kan have det på sig.
Patienter skal referere til kortet, når som helst han/hun besøger andre læger, i
særdeleshed for yderligere diagnostiske procedurer (f.eks. MRI).
9 LEVERING
• Zenith Flex AAA endovaskulær protese leveres steril og præmonteret i peelopen pakninger.
• Produktet er kun beregnet til engangsbrug. Produktet må ikke resteriliseres.
• Se produktet og emballagen efter for at verificere, at de ikke er blevet
beskadiget under transporten. Dette produkt må ikke anvendes, hvis der er
sket beskadigelse, eller hvis steriliseringsbarrieren er beskadiget eller brudt.
Hvis der er indtruffet beskadigelse, må produktet ikke anvendes, det skal
sendes tilbage til COOK.
• Inden brug skal det verificeres, at de korrekte produkter (antal og størrelse) er
blevet leveret til patienten ved at sammenligne produktet med lægens
ordination for den pågældende patient.
• Hovedprotesen er præmonteret på en 18, 20 eller 22 French Flexor®
indføringssheath. Sheathens overflade er behandlet med en hydrofil coating
der, når den er hydreret, forbedrer sporbarheden. Den hydrofile coating
aktiveres ved at aftørre overfladen med et sterilt gazekompres, der er opblødt i
saltvand.
• Produktet må ikke anvendes, efter udløbsdatoen (USE BY) på etiketten er
overskredet.
• Opbevares på et tørt og køligt sted.
10 INFORMATION OM KLINISK ANVENDELSE
10.1 Lægeuddannelse
FORSIGTIG: Der skal altid være et kvalificeret kirurgisk team til rådighed
under implantations- eller reinterventionsprocedurer i tilfælde af, at det
bliver nødvendigt at konvertere til åben kirurgisk reparation.
FORSIGTIG: Zenith Flex AAA endovaskulær protese med H&L-B One-Shot
indføringssystem må kun anvendes af læger og teams, der er uddannet
i vaskulære interventionsteknikker og i brugen af denne anordning. De
anbefalede krav til dygtighed/viden for læger, der bruger Zenith Flex
AAA endovaskulær protese med H&L-B One-Shot indføringssystem,
angives nærmere herunder:
Patientudvælgelse:
• Viden om abdominale aortaaneurismers (AAA) naturlige historie og
komorbiditeter associeret med reparation af AAA.
• Viden om tolkning af røntgenbilleder, produktudvælgelse og
størrelsesbestemmelse.
Et multi-disciplinært team, der har kombineret proceduremæssig
erfaring med:
• Frilægning af a. femoralis, arteriotomi og reparation
• Perkutane adgangs- og lukketeknikker
• Nonselektive og selektive kateterleder- og kateterteknikker
• Tolkning af gennemlysning og angiografi
• Embolisering
• Angioplastik
• Anlæggelse af endovaskulær stent
• Slyngeteknikker
• Hensigtsmæssig brug af røntgenkontraststof
• Teknikker til at minimere røntgeneksponering
• Ekspertise i nødvendige patientopfølgningsmodaliteter
10.2 Inspektion inden brug
Se produktet og emballagen efter for at verificere, at de ikke er blevet
beskadiget under transporten. Dette produkt må ikke anvendes, hvis der er
sket beskadigelse, eller hvis steriliseringsbarrieren er beskadiget eller brudt.
Hvis der er indtruffet beskadigelse, må produktet ikke anvendes, det skal
sendes tilbage til COOK. Inden brug skal det verificeres, at de korrekte
produkter (antal og størrelse) er blevet leveret til patienten ved at
sammenligne produktet med lægens ordination for den pågældende patient.
10.3 Nødvendige materialer
(Ikke inkluderet i det 3-delte modulsystem)
• Zenith hjælpekit til AAA endovaskulær protese
• Fluoroskop med digitale angiografifunktioner (C-arm eller fikseret apparat)
• Kontraststof
• Sprøjte
• Hepariniseret saltvandsopløsning
• Sterile gazeservietter
10.4 Anbefalede materialer
(Ikke inkluderet i det 3-delte modulsystem)
De følgende produkter anbefales til implantation af ethvert produkt i Zenith
produktlinjen. For oplysninger om brugen af disse produkter refereres til de
individuelle produkters Anbefalet brugsanvisning.
• 0,035 tomme (0,89 mm) ekstrastiv kateterleder, 260 cm, f.eks.:
• Cook Lunderquist ekstrastive kateterledere (LES)
• 0,035 tomme (0,89 mm) standard kateterleder, f.eks.:
• Cook 0,035 tomme (0,89 mm) kateterledere
• Cook Nimble™ kateterledere
• Formningsballoner, f.eks.:
• Cook CODA® ballonkateter
• Indføringssæt, f.eks.:
• Cook Check-Flo® indføringssæt
• Cook ekstrastor Check-Flo® indføringssæt
• Cook Flexor® Balkin Up & Over® kontralaterale indførere
• Størrelsesbestemmende katetre, f.eks.:
• Cook Aurous® centimeterstørrelsesbestemmende katetre
• Angiografikatetre med røntgenfast spids, f.eks.:
• Cook angiografikatetre med Beacon® spids
• Cook Royal Flush katetre med Beacon® spids
58

• Punkturkanyler, f.eks.:
• Cook enkeltvægget punkturkanyler
10.5 Retningslinjer for størrelsesbestemmelse af produktdiameter
Diametervalget skal afgøres fra diameteren af karret målt fra ydre væg til ydre
væg, og ikke lumendiameteren. En for lille eller for stor størrelse kan resultere
i ufuldstændig forsegling eller kompromitteret flow.
Tabel 10.5.1 Størrelsesguide for hovedprotesens diameter*
Tilsigtet aortakar
diameter
1
Maksimum diameter langs det proksimale fikseringssted.
2
Rund den målte aortadiameter op til nærmeste mm.
3
Yderligere betragtninger kan få indflydelse på diametervalg.
*Alle dimensioner er nominale.
Hovedprotese-
1,2
(mm)
18-19 22 82/112, 96/126, 111/141,
20-21 24 82/112, 96/126, 111/141,
22 26 82/112, 96/126, 111/141,
23-24 28 82/112, 96/126, 111/141,
25-26 30 82/112, 96/126, 111/141,
27-28 32 82/112, 96/126, 111/141,
29-32 36 95/125, 113/143,
diameter
(mm)
Samlet længde til
kontralateral kant/Samlet
3
længde til ipsilateral kant
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
131/161, 149/179
(mm)
Indførings-
sheath
(French)
18
18
18
20
20
20
22
Tabel 10.5.2 Størrelsesguide for iliaca-benprotesens diameter*
Tilsigtet
iliacakar,
diameter
(mm)
< 8 8 37, 54, 71, 88, 105, 122 14
8-9 10 37, 54, 71, 88, 105, 122 14
10-11 12 37, 54, 71, 88, 105, 122 14
12-13 14 37, 54, 71, 88 14
14-15 16 37, 54, 71, 88 14
16-17 18 37, 54, 71, 88 16
18 20 37, 54, 71, 88 16
19 22 37, 54, 71, 88 16
20 24 37, 54, 71, 88 16
1
Maksimum diameter langs det distale fikseringssted.
2
Rund den målte iliaca-diameter op til nærmeste mm.
3
Yderligere betragtninger kan få indflydelse på diametervalg.
4
Benlængde i alt = arbejdslængde + 22 mm sammenkoblingsstent.
*Alle dimensioner er nominale.
11 VEJLEDNING
Anatomiske krav
• Størrelsen på det iliofemorale adgangskar og mor fologi (minimal trombe,
kalk og/eller snoning) bør være kompatibel med vaskulære
adgangsteknikker og tilbehør. Arteriel kanalteknik kan være nødvendig.
• Proksimale aortahalslængder bør være mindst 15 mm med en diameter
målt fra ydre væg til ydre væg på 18-32 mm.
• Et distalt fikseringssted i iliaca-ar terien, der er mere end 10 mm langt og
7,5-20 mm i diameter (målt fra ydre væg til ydre væg).
Gennemgå denne håndbog Anbefalet brugsanvisning inden brug af Zenith
Flex AAA endovaskulær protese med H&L-B One-Shot indføringssystem.
Følgende instruktioner indeholder en grundlæggende vejledning for
anlæggelse af produktet. Det kan være nødvendigt at variere følgende
procedure. Disse instruktioner er tænkt som en vejledning til lægen og
træder ikke i stedet for lægens skøn.
1,2
Iliaca-
bendiameter
(mm)
3
Arbejdslængde
af iliaca-ben
(mm)
Indførings-
4
sheath
(French)
Generel information om anvendelse
• Der sk al anvendes standard teknikker til placering af arterielle
adgangssheaths, styrekatetre, angiografikatetre og kateterledere ved brug
af Zenith Flex AAA endovaskulær protese med H&L-B One-Shot
indføringssystem. Zenith Flex AAA endovaskulær protese med H&L-B OneShot indføringssystem er kompatibel med kateterledere med en diameter
på 0,035 tomme (0,89 mm).
• Transplantation af en endovaskulær stent er en kirurgisk procedure, og der
kan af forskellige årsager forekomme blodtab, som i sjældne tilfælde kræver
intervention (inklusive transfusion) for at forhindre negative resultater. Det
er vigtigt at overvåge blodtab fra hæmostaseventilen under hele
proceduren, men det er særligt relevant under og efter manipulering af den
grå positioneringsanordning. Hvis der er overdrevent blodtab, når den grå
positioneringsanordning er blevet fjernet, skal det overvejes at anlægge en
ikke-inflateret formningsballon eller en dilatator til indføringssystemet
inden i ventilen, så flowet begrænses.
Afgørende faktorer før implantation
Det skal verificeres, at det korrekte produkt er valgt i planlægningsfasen før
implantationen. Afgørende faktorer inkluderer:
1. Valg af a. femoralis til indføring af hovedprotesesystemet (dvs. de
respektive kontralaterale og ipsilaterale iliaca-arterier skal defineres).
2. Vinkling af aortahals, aneurisme og iliaca-arterier.
3. Aortahalsens kvalitet.
4. Diametrene på infrarenal aortahals og distale iliaca-arterier.
5. Afstand fra nyrearterier til aortabifurkaturen.
6. Længde fra aortabifurkatur til aa. iliacae internae/fikseringssted(er).
7. Aneurisme(r) der går ind i aa. iliacae, kan kræve særlig overvejelse ved
valget af et egnet grænsefladested for protese/arterie.
8. Graden af forkalkning af kar skal overvejes.
Klargøring af patienten
1. Der henvises til hospitalets protokoller vedrørende anæstesi,
antikoagulation og monitorering af livstegn.
2. Patienten lejres på gennemlysningslejet med visualisering fra aortabuen
til femoralisbifurkaturerne.
3. Begge aa. femorales communes blotlægges ved hjælp af standard
kirurgisk teknik.
4. Adækvat proksimal og distal vaskulær kontrol etableres for begge aa.
femorales.
11.1 Bifurkationssystem (Fig. 2)
11.1.1 Forberedelse/skylning af bifurkationshovedprotese
1. Fjern transportstiletten med sort muffe (fra den indre kanyle), kanylens
beskyttelsesrør (fra den indre kanyle) og dilatatorspidsens beskyttelse
(fra dilatatorspidsen). Fjern Peel-Away® sheathen fra den hæmostatiske
ventils bagside. (Fig. 5) Elevér systemets distale spids og gennemskyl det
gennem hanen på den hæmostatiske ventil, indtil væske kommer ud af
sideporten nær spidsen af indføringssheathen. (Fig. 6) Fortsæt med at
injicere alle 20 ml skylleopløsning gennem produktet. Indstil injektionen
og luk stophanen på forbindelsesslangen.
BEMÆRK: Der bruges ofte hepariniseret saltvandsopløsning til at
gennemskylle protesen.
2. Påsæt sprøjten med hepariniseret saltvand til muffen på den indvendige
kanyle. Gennemskyl, indtil væske kommer ud af dilatatorspidsen. (Fig. 7)
BEMÆRK: Når systemet gennemskylles, skal systemets distale ende eleveres
for at lette fjernelse af luft.
3. Gennemvæd sterile gazekompresser i saltvandsopløsning og brug dem
til at aftørre Flexor® indføringssheath for at aktivere den hydrofile coating.
Hydrér både sheath og dilatator rigeligt.
11.1.2 Forberedelse/skylning af kontralateral iliaca-ben
1. Fjern den indre stilet med sort muffe (fra den indre kanyle), kanylens
beskyttelsesrør (fra den indre kanyle) og dilatatorspidsens beskyttelse
(fra dilatatorspidsen). Fjern Peel-Away® sheathen fra den hæmostatiske
ventils bagside. (Fig. 8) Elevér systemets distale spids og gennemskyl det
gennem hanen på den hæmostatiske ventil, indtil væske kommer ud af
sideporten nær spidsen af indføringssheathen. (Fig. 9) Fortsæt med at
injicere alle
og luk stophanen på forbindelsesslangen.
BEMÆRK: Der bruges ofte hepariniseret saltvandsopløsning til at gennemskylle
protesen.
2. Påsæt sprøjten med hepariniseret saltvand til muffen på den distale indre
kanyle. Gennemskyl, indtil væske kommer ud af den distale dilatatorspids.
(Fig. 7)
BEMÆRK: Når systemet gennemskylles, skal systemets distale ende eleveres for
at lette fjernelse af luft.
11.1.3 Forberedelse/skylning af ipsilateral iliaca-ben
Følg vejledningerne i det forrige Afsnit 11.1.2 Forberedelse/skylning af
kontralateral iliaca-ben for at sikre korrekt skylning af ipsilaterale iliaca-
benprotese.
11.1.4 Vaskulær adgang og angiografi
1. Punktér de valgte aa. femorales communes ved hjælp af standard teknik
med en af de to arteriekanyler med ultratynd væg, 18UT eller 19UT. Efter
adgang til karret indføres:
• Kateterledere - standard 0,035 tomme (0,89 mm) diameter, 145 cm lang med
J-spids eller Bentson kateterleder
• Sheaths af hensigtsmæssig størrelse (f.eks. 6,0 eller 8,0 French)
• Gennemskylningskateter (ofte røntgenfaste katetre til
størrelsesbestemmelse - f.eks. katetre til centimeterstørrelsesbestemmelse
eller lige gennemskylningskatetre)
2. Udfør angiografi for at identificere niveau(erne) af nyrearterier,
aortabifurkatur og iliaca-bifurkaturer.
BEMÆRK: Hvis fluoroskopvinkling bruges med en vinklet hals, kan det være
nødvendigt at udføre angiogrammer vha. forskellige projektioner.
11.1.5 Placering af hovedprotese
1. Kontrollér, at fremføringssystemet er blevet gennemskyllet med hepariniseret
saltvand, og at al luft er fjernet fra systemet.
2. Giv systemisk heparin og kontrollér skylleopløsningerne. Gennemskyl efter
hver udskiftning af kateter og/eller kateterleder.
20 ml skylleopløsning gennem anordningen. Indstil injektionen
BEMÆRK: Monitorér patientens koagulationsstatus under hele proceduren.
3. På den ipsilaterale side udskiftes J-lederen med en stiv kateterleder (LES),
0,035 tomme (0,89 mm), 260 cm lang, og før den frem gennem kateteret
og op til thorakale aorta. Fjern gennemskylningskateteret og sheathen.
Oprethold kateterlederens position.
4. Inden indføring placeres det distale fremføringssystem for hovedprotesen
på patientens abdomen under gennemlysning for at afgøre orienteringen
af den kontralaterale kants røntgenfaste markør. Den hæmostatiske
ventils sidearm kan være en ekstern reference for den kontralaterale kants
røntgenfaste markør.
5. Indfør fremføringssystemet for den distale hovedprotese over kateterlederen
ind i a. femoralis med opmærksomhed på sidearmsreferencen.
FORSIGTIG: Oprethold kateterlederens position under indføring af
fremføringssystemet.
FORSIGTIG: Alle komponenter i systemet skal drejes samtidig (fra ydre
sheath til indre kanyle) for at undgå snoninger i den endovaskulære
protese under drejning af fremføringssystemet.
6. Fremfør fremføringssystemet, indtil de fire røntgenfaste guldmarkører (der
er positioneret 2 mm fra det mest proksimale segment af protesematerialet)
(Fig. 10, Illustration 1) er netop inferior for den laveste nyreåbning.
7. Verificér kateterlederens position i den thorakale aorta. Kontrollér, at
protesesystemet er orienteret således, at den kontralaterale kant er
positioneret over og anterior for den kontralaterale iliacas oprindelse. Hvis
den røntgenfaste markør på den kontralaterale kant ikke er korrekt tilpasset,
drejes hele systemet, indtil det er korrekt placeret halvvejs mellem en lateral
og en anterior position på den kontralaterale side.
• En markørformation med et ✓ angiver en anterior position af den korte
(kontralaterale) kant. (Fig. 10, Illustration 4)
• En markørformation med et angiver en posterior position af den korte
(kontralaterale) kant. (Fig. 10, Illustration 5)
• En markørformation med et | angiver en lateral position af den korte
(kontralaterale) kant. (Fig. 10, Illustration 6)
BEMÆRK: Røntgenfast kanyle på hver stent mellem nyrearterierne og den
kontralaterale kant er tilpasset med den kontralaterale røntgenfaste kantmarkør.
8. Gentag angiogrammet for at verificere, at de fire røntgenfaste guldmarkører
er 2 mm eller mere under den mest inferior nyreåbning.
9. Det skal sikres, at Captor hæmostatisk ventil på Flexor® indførings-sheathen
er drejet til åben position. (Fig. 11)
10. Stabilisér den grå positioneringsanordning (fremføringssystemets skaft),
samtidig med at sheathen trækkes tilbage. Anlæg de første to (2) tildækkede
stents ved at trække sheathen tilbage, samtidig med at produktets
lokalisering monitoreres.
11. Uden at bordet flyttes mindskes forstørrelsen for at kunne kontrollere
positionen af den kontralaterale kants røntgenfaste markør og
nyrearterierne. Fortsæt med anlæggelsen, indtil den kontralaterale kant er
fuldstændig anlagt. (Fig. 12) Stop tilbagetrækning af sheath.
BEMÆRK: Kontrollér, at den kontralaterale kant er mindst 5 mm over
aortabifurkaturen og i den ønskede position for kanylering.
11.1.6 Placering af den kontralaterale iliaca-kateterleder
1. Manipuler kateter og kateterleder gennem den åbne ende af den kontralaterale kant og ind i hovedprotesen. Fremfør kateterlederen, indtil den
kurver inden i hovedprotesen. AP og skrå gennemlysninger kan hjælpe med
verifikationen af produktkanyleringen.
2. Efter kanylering fremføres angiografikatetret over kateterlederen og ind i
den endovaskulære hovedprotese. Fjern kateterleder og udfør angiografi
for at bekræfte position. Fremfør kateterleder, indtil den kurver inden i
hovedprotesen. Fjern angiografikatetret. (Fig. 13)
59

11.1.7 Proksimal (top) anlæggelse af hovedprotese
1. Udfør angiografi gennem et angiografikateter for at verificere positionen
af den endovaskulære protese i forhold til nyrearterierne. Om nødvendigt
omplaceres den dækkede del af den endovaskulære protese forsigtigt
i forhold til nyrearterierne (omplacering kan på dette tidspunkt kun
udføres over et lille afstandsområde).
BEMÆRK: Sørg for, at nyrearterierne er åbne ved at bekræfte, at de
proksimale protesemarkører er 2 mm eller mere under den laveste åbne
nyrearterie.
FORSIGTIG: Under fjernelse af proksimal udløser-wire, fremføring af
tophætte og efterfølgende anlæggelse af suprarenal stent skal det
verificeres, at hovedprotesens kateterleder går ud lige distalt på
aortabuen, og at understøtningen af systemet er maksimeret.
2. Fjern sikkerhedslåsen fra den sorte udløsningsmekanisme til udløserwiren. Træk under gennemlysning udløser-wiren tilbage og fjern den ved
at skubbe den sorte udløsningsmekanisme til udløser-wiren af håndtaget,
og fjern den derefter gennem rillen over den indvendige kanyle. (Fig. 14)
Hvis der mærkes modstand eller bemærkes bøjning af systemet, er udløserwiren under spænding. Overdreven kraft kan bevirke, at protesens position
ændres. Stands og vurdér situationen, hvis der bemærkes overdreven
modstand eller bevægelse af indføringssystemet. Hvis det ikke er muligt at
fjerne den sorte udløsningsmekanisme for udløser-wiren fra tophætten, skal
følgende procedure udføres under gennemlysning:
a. Fjern spændingen på udløser-wiren ved at løsne pin visen og trække let
i den indre kanyle for at flytte tophætten ned over den suprarenale stent.
Undgå at trykke Zenith Flex’s hovedprotese sammen.
b. Spænd pin visen igen.
c. Fjern den sorte udløsningsmekanisme for udløser-wiren.
d. Fortsæt med trin 3 i Afsnit 11.1.7, Proksimal (top) anlæggelse af
hovedprotese.
BEMÆRK: Hvis det stadig ikke er muligt at fjerne topstentens
udløsningsmekanisme for udløser-wiren fra tophætten, henvises til
Afsnit 14.1, Fejlfinding ved udløser-wirens frigørelse.
3. Løsn pin visen. (Fig. 15) Kontrollér protesens position ved at stabilisere
indførerens grå placeringsanordning.
4. Anlæg den suprarenale stent ved at føre den indre kanyles tophætte
1 til 2 mm frem ad gangen, samtidig med at hovedprotesens position
kontrolleres, indtil topstenten er fuldstændig anlagt. (Fig. 16 og 17) Før
tophættekanylen frem yderligere 1 til 2 cm og spænd dernæst pin visen
igen for at undgå kontakt med den anlagte suprarenale stent.
BEMÆRK: Hvis det er umuligt at anlægge den suprarenale stent fuldstændigt
ved at fremføre tophættens indvendige kanyle, henvises til Afsnit 14.2,
Fejlfinding ved anlæggelse af den suprarenale stent.
BEMÆRK: Efter den suprarenale stent med modhager er anlagt, anbefales
yderligere forsøg på omplacering ikke.
ADVARSEL: Zenith Flex AAA endovaskulær protese omfatter en
suprarenal stent med fikseringsmodhager. Der skal udvises meget stor
forsigtighed ved manipulering af interventionelle produkter i området
med den suprarenale stent.
5. Fremfør den kontralaterale kateterleder ind i den thorakale aorta.
11.1.8 Placering og anlæggelse af kontralateral iliaca-ben
FORSIGTIG: Verificér inden implantationen, at det forudfastlagte kontralaterale iliaca-ben er valgt til indføring på patientens kontralaterale side.
1. Anbring billedforstærkeren således, at både kontralaterale a. iliaca interna
og kontralaterale a. iliaca communis vises.
2. Før indføringen af fremføringssystemet til det kontralaterale iliaca-ben,
injiceres kontrast gennem den kontralaterale femoralis-sheath for at
lokalisere den kontralaterale a. iliaca interna.
3. Før fremføringssystemet for det kontralaterale iliaca-ben ind i arterien.
Før det langsomt frem, indtil iliaca-benprotesen overlapper mindst én
fuld iliaca-benstent (dvs. proksimale stent af iliaca-benprotesen) inden
i hovedprotesens kontralaterale kant. (Fig. 18) Hvis hovedprotesen har
tendens til at flyttes under denne manøvre, skal den holdes i position ved at
stabilisere den grå positioneringsanordning på den ipsilaterale side.
BEMÆRK: Hvis der opstår vanskeligheder ved fremføringen af fremføringssystemet for iliaca-benet, udskiftes til en mere støttende kateterleder. I snirklede
kar kan anatomien ændres signifikant ved indføringen af de stive kateterledere
og sheathsystemer.
4. Bekræft positionen af den distale ende af iliaca-benprotesen. Omplacér
iliaca-benprotesen, hvis det er nødvendigt, for at sikre både åbenhed i a.
iliaca interna og en minimumsoverlapning på en fuld iliaca-benstent (dvs.
iliaca-benprotesens proksimale stent, maksimumsoverlapning på 1,5 stents)
inden i den endovaskulære hovedprotese.
5. Hold iliaca-benprotesen i position med den grå positioneringsanordning,
samtidig med at sheathen trækkes tilbage, for at anlægge den. (Fig. 19 og
20) Sørg for, at overlapning svarende til en stentlængde bevares.
6. Stop tilbagetrækningen af sheathen, så snart den distale ende af iliacabenprotesen udløses.
7. Under gennemlysning og efter verificering af iliaca-benprotesens position,
løsnes pin vise’n, den indre kanyle trækkes tilbage for at sammenkoble
den konusformede dilatator med den grå positioneringsanordning.
Spænd pin vise’n. Oprethold sheathpositionen, samtidig med at den grå
positioneringsanordning trækkes tilbage med den fastgjorte indre kanyle.
(Fig. 21)
8. Kontrollér igen kateterlederens position.
11.1.9 Distal (bund) anlæggelse af hovedprotese
1. Gå tilbage til den ipsilaterale side.
2. Anlæg hovedprotesens ipsilaterale kant fuldstændigt ved at trække
sheathen tilbage, indtil den mest distale stent er ekspanderet. (Fig. 22 og
23) Stop tilbagetrækning af sheath.
BEMÆRK: Den distale stent fastholdes stadig af udløser-wiren.
3. Fjern sikkerhedslåsen fra den hvide udløsningsmekanisme for udløserwiren. Træk udløser-wiren tilbage, og fjern den ved at føre den hvide
udløsningsmekanisme for udløser-wiren af håndtaget, og fjern den dernæst
via dens rille over produktets indre kanyle. (Fig. 24)
11.1.10 Sammenkobling af tophætten
1. Løsn pin vise’n. (Fig. 25)
2. Fastgør sheath og indre kanyle for at undgå, at disse komponenter
bevæger sig.
3. Før den grå positioneringsanordning over den indre kanyle, indtil den
sammenkobles med tophætten. (Fig. 26, 27 og 28)
BEMÆRK: Hvis der mærkes modstand, drejes den grå positioneringsanordning
en smule, og fremføringen fortsættes forsigtigt.
4. Spænd pin vise’n igen og træk hele tophætten og den grå positioneringsanordning gennem protesen og gennem sheathen ved at trække i den
indre kanyle. (Fig. 29) Lad sheath og kateterleder blive siddende.
BEMÆRK: Oprethold sheathens og kateterlederens position.
5. Luk Captor® hæmostatiske ventil på Flexor® indføringssheathen ved at dreje
den i retning med uret, indtil den standser. (Fig. 30)
11.1.11 Placering og anlæggelse af ipsilateral iliaca-ben
BEMÆRK: Det skal sikres, at Captor hæmostatisk ventil på indføringssheathen
er drejet til åben position. (Fig. 31)
1. Brug hovedprotesens kateterleder og sheathdel til at indføre den ipsilaterale
iliaca-benprotese. Fremfør dilatator og sheathdel ind i hovedprotesens
sheath.
BEMÆRK: I snirklede kar kan aa. iliacae internaes position ændres signifikant
ved indføringen af stive kateterledere og sheathsystemer.
2. Før det langsomt frem, indtil den ipsilaterale iliaca-benprotese overlapper
mindst én fuld iliaca-benstent (dvs. proksimale stent af iliaca-benprotesen)
inden i hovedprotesens ipsilaterale kant. (Fig. 32)
BEMÆRK: Hvis der kræves en overlapning med mere end 3 iliaca-benstents
(mere end 2 iliaca-benstents for benlængder på 37 mm og 54 mm), kan det
være nødvendigt at bruge en benforlænger i den modsatte sides
bifurkationsområde.
3. Bekræft positionen af den distale ende af iliaca-benprotesen. Omplacér om
nødvendigt iliaca-benprotesen for at sikre invendig åbenhed af iliaca.
4. Anlæg ved at stabilisere iliaca-benprotesen med den grå positioneringsanordning, mens iliaca-bensheathen tilbagetrækkes. (Fig. 33 og 34)
Hovedprotesens sheath tilbagetrækkes, om nødvendigt.
5. Under gennemlysning og efter verificering af iliaca-benprotesens position,
løsnes pin vise’n, den indre kanyle trækkes tilbage for at sammenkoble
den konusformede dilatator med den grå positioneringsanordning.
Spænd pin vise’n. Oprethold sheathpositionen, samtidig med at den grå
positioneringsanordning trækkes tilbage med den fastgjorte indre kanyle.
(Fig. 35)
6. Luk Captor hæmostatiske ventil på Flexor® indføringssheath ved at dreje den
i retning med uret, indtil den standser.
7. Kontrollér igen kateterledernes position. Lad sheath og kateterleder blive
siddende.
11.1.12 Indføring af formningsballon
1. Klargør formningsballonen på følgende måde:
• Gennemskyl kateterlederlumenet med hepariniseret saltvand.
• Fjern al luft fra ballonen.
2. Til forberedelse af indføringen af formningsballonen åbnes Captor
hæmostatiske ventil ved at dreje den mod uret.
3. Før formningsballonen frem over kateterlederen og gennem Captor
hæmostatiske ventil på hovedprotesens indføringssystem til nyrearteriernes
niveau. Oprethold korrekt sheathposition.
4. Spænd Captor hæmostatiske ventil med et let tryk rundt om
formningsballonen ved at dreje den med uret.
FORSIGTIG: Inflatér ikke ballonen i iliaca-karret uden for protesen.
5. Ekspandér formningsballonen med fortyndet kontraststof (som anvist af
producenten) i området med den mest proksimale dækkede stent og den
infrarenale hals, idet der startes proksimalt og arbejdes i distal retning.
(Fig. 36)
FORSIGTIG: Kontroller fuld deflation af ballonen inden omplacering.
FORSIGTIG: Captor hæmostatisk ventil skal være åben før
formningsballonen omplaceres.
6. Træk formningsballonen tilbage til den ipsilaterale kants overlapning og
ekspandér.
FORSIGTIG: Captor hæmostatisk ventil skal være åben før
formningsballonen omplaceres.
7. Træk formningsballonen tilbage til det ipsilaterale lems distale fikseringssted
og ekspandér.
FORSIGTIG: Inflater ikke ballonen i iliaca-karret uden for protesen.
8. Deflater og fjern formningsballonen. Over før formningsballonen over
på den kontralaterale kateterleder og ind i indføringssystemet for det
kontralaterale iliaca-ben. Før formningsballonen frem til den kontralaterale
kants overlapning og ekspandér.
FORSIGTIG: Kontroller fuld deflation af ballonen inden omplacering.
9. Træk formningsballonen tilbage til det kontralaterale iliaca-bens/kars distale
fiksering og ekspandér. (Fig. 36)
FORSIGTIG: Inflater ikke ballonen i iliaca-karret uden for protesen.
10. Fjern formningsballonen og udskif t den med et angiografikateter for
at udføre afslutningsangiogrammer.
11. Fjern eller udsk ift alle stive kateterledere for at tillade iliaca-arterierne
at genoptage deres naturlige position.
Slutangiogram
1. Placér angiografikateteret lige netop over nyrearterieniveauet. Udfør
angiografi for at verificere, at nyrearterierne er åbne, og at der ingen
endolækager er til stede. Verificér, at aa. iliacae internae er åbne.
2. Bekræft, at der ingen endolækager eller knæk er til stede, og verificér
positionen af de proksimale røntgenfaste guldmarkører. Fjern sheaths,
kateterledere og katetere.
BEMÆRK: Hvis der observeres endolækager eller andre problemer, skal der
refereres til Anbefalet brugsanvisning for Zenith hjælpekomponenter til AAA
endovaskulær protese.
3. Reparér karrene og luk på sædvanlig kirurgisk vis.
12 RETNINGSLINJER FOR BILLEDDIAGNOSTIK OG POSTOPERATIV
OPFØLGNING
12.1 Generelle
• Endovaskulære protesers funktion og sikkerhed på langt sigt er endnu
ikke fastlagt.
behandling kræver regelmæssig opfølgning resten af deres liv til
bedømmelse af deres helbredstilstand og af deres endovaskulære
protese. Patienter med specifikke kliniske fund (f.eks. endolækager,
aneurismer, der bliver større, eller ændringer i den endovaskulære proteses
struktur eller position) skal følges nøjere.
• Patienter bør rådgives om vigtigheden af at overholde opfølgningsplanen,
både i det første år og med årlige mellemrum derefter. Patienter bør
informeres om, at regelmæssig og konsekvent opfølgning er en meget vigtig
del af at sikre den fortsatte sikkerhed og effektivitet af den endovaskulære
behandling af AAA’er.
• Læger bør evaluere patienter på et individuelt grundlag og ordinere deres
opfølgning i forhold til hver enkelt patients individuelle behov og
omstændigheder. Den anbefalede plan for billediagnostik vises i Tabel 12.1.
Denne plan er fortsat et minimumskrav for patientopfølgning og bør
opretholdes, selv ved fraværet af kliniske symptomer (f.eks. smerte,
følelsesløshed, svaghed). Patienter med specifikke kliniske fund (f.eks.
endolækager, aneurismer, der bliver større, eller ændringer i stentprotesens
struktur eller position) bør følges ved hyppigere intervaller.
• Årlig billedopfølgning bør omfatte abdominalrøntgenbilleder og CT-scanninger
både med og uden kontrast. Hvis nyrekomplikationer eller andre faktorer
Alle patienter skal gøres opmærksom på, at endovaskulær
60

udelukker brugen af billedkontraststoffer, kan abdominalrøntgenbilleder, CT
uden kontrast og duplex ultralyds-scanning bruges.
• Kombinationen af CT-scanning med og uden kontrast giver information om
ændring af aneurismets diameter, endolækage, åbenhed, snirklethed,
fremskridende sygdom, fikseringslængde og andre morfologiske ændringer.
• Abdominale røntgenbilleder giver information om produktets integritet
(separation mellem komponenter, stentbrud og modhageadskillelse).
• Duplex-ultralydsscanning kan give information om ændring i aneurismets
diameter, endolækage, åbenhed, snirklethed og fremskridende sygdom. I dette
tilfælde bør en CT-scanning uden kontrast udføres til brug sammen med
ultralydsscanning. Ultralydsscanning kan være en mindre pålidelig og mindre
følsom diagnostisk metode sammenlignet med CT-scanning.
Tabel 12.1 opfører minimumskravene til opfølgning med billeddiagnostik for
patienter med en Zenith Flex AAA endovaskulær protese. Patienter, hvor mere
intensiv opfølgning er nødvendig, skal have interimevalueringer.
Tabel 12.1 Anbefalet billeddiagnostisk plan for endoprotesepatienter
Angiogram
Før proceduren X
Under proceduren X
Før udskrivning (i løbet af
7 dage)
1 måned X
3 måneder X
6 måneder X
12 måneder (herefter årligt) X
1
Billeddiagnostik skal udføres inden for 6 måneder før proceduren.
2
Duplex-ultralydsscanning kan bruges til de patienter, der har nyresvigt, eller som af
anden årsag er ude af stand til at gennemgå kontrastforstærket CT-scanning. Med
ultralydsscanning anbefales CT-scanning uden kontrast stadigvæk.
3
CT-scanning anbefales enten før udskrivning eller efter 1 måned.
4
Hvis type I eller III endolækage er til stede, anbefales øjeblikkelig intervention og
yderligere opfølgning efter interventionen - se Afsnit 12.6 Yderligere kontrol og behandling.
5
Anbefalet, hvis endolækage rapporteres ved før udskrivning eller 1 måned.
12.2 Anbefalinger for CT-scanning med og uden kontrast
• Filmsæt bør indeholde alle sekventielle billeder ved lavest mulige slice-
tykkelse (≤ 3 mm). Udfør IKKE stor slice-tykkelse (> 3 mm) og/eller
udeladelse af konsekutive CT-billeder/filmsæt, da det forhindrer præcise
anatomiske sammenligninger og produktsammenligninger over tid.
• Alle billeder bør indeholde en målestok for hver film/billede. Billeder bør
arrangeres ikke mindre end 20:1 billeder på 35,5x43,2 cm (14x17 tommer)
ark, hvis der bruges film.
• Der kræves optagelser både med og uden kontrast med matchende eller
tilsvarende lejepositioner.
• Slice-tykkelse og interval for optagelse før kontrast og med kontrast skal
matche.
• Patientens orientering må IKKE ændres og patienten må ikke pilemærkes
om mellem optagelser med og uden kontrast.
Det er vigtigt med basislinje og opfølgningsbilleddiagnostik både uden
kontrast og kontrastforstærket for optimal patientkontrol. Det er vigtigt at
følge acceptable billeddiagnostiske protokoller under CT-scanningen.
Tabel 12.2 viser eksempler på acceptable billeddiagnostikprotokoller.
1
CT
(Kontrast og uden
kontrast)
1
X
2,3,4
X
2,3,4
2,4,5
2,4
2,4
Abdominale
røntgenbilleder
X
X
X
X
Tabel 12.2 Acceptable billeddiagnostiske protokoller
Uden kontrast Kontrast
IV-kontrast Nej Ja
Acceptable maskiner
Injektionsvolumen Ikke relevant 150 ml
Injektionshastighed Ikke relevant > 2,5 ml/s
Injektionsmetode I kke relevant Maskin-
Bolustiming Ikke relevant
Dækning - start Diaphragma 1 cm superior for axis celiaca
Dækning - slut Proksimal femur
Kollimation < 3 mm < 3 mm
Rekonstruktion
Aksial DFOV 32 cm 32 cm
Post-injektions
scanninger
Spiralbilleddiagnostik
i stand til > 40 sekunder
2,5 mm hele vejen -
blød algoritme
Ingen Ingen
Spiralbilleddiagnostik
i stand til > 40 sekunder
Test bolus: SmartPrep, C.A.R.E.
eller tilsvarende
Profunda femoris
begyndelsespunktet
2,5 mm hele vejen -
blød algoritme
12.3 Abdominale røntgenbilleder
Følgende visninger er påkrævet:
• Fire film: rygleje-frontal (AP), lateralt tværs over lejet, 30 grader LPO (venstre
posteriore skråmuskel) og 30 grader RPO (højre posteriore skråmuskel)
visninger centreret på umbilicus.
• Notér afstanden mellem leje og film og brug den samme afstand ved hver
efterfølgende undersøgelse.
Sørg for, at produktet fremstår i hele sin længde på hvert enkelt billedformat.
Hvis der er tvivl om produktets integritet (dvs. knæk, stentbrud,
modhageadskillelse, relativ komponentmigration), anbefales det at
bruge forstørrede visninger. Den ansvarlige læge bør evaluere filmene
til konstatering af produktintegritet (hele produktets længde, inklusive
komponenter) ved hjælp af 2-4X forstørrelse.
12.4 Ultralydsscanning
Ultralydsscanning kan foretages i stedet for CT-scanning med kontrast, når
patientfaktorer udelukker brugen af billedkontraststoffer. Ultralydsscanning kan
foretages sammen med CT-scanning uden kontrast. En komplet aorta duplex
skal videooptages for maksimal aneurismediameter, endolækager,
stentåbenhed og stenose. Følgende information skal være inkluderet på
videoen som anført herunder:
• Tværgående og længdegående scanning bør opnås fra det proksimale aorta
niveau, der viser mesenteriske og nyrearterier til iliaca-bifurkationerne med
henblik på at afgøre, om der er endolækager til stede vha. farveflow og
farveangiografi (hvis tilgængeligt).
• Spektral analysebekræftelse bør foretages for alle mistænkte endolækager.
• Tværgående og længdegående scanning af den maksimale aneurisme skal opnås.
12.5 Sikkerhed og kompatibilitet ved MR-scanning
Ikke-klinisk testning har påvist, at Zenith AAA endovaskulær protese er MR
conditional. Den er ufarlig at scanne på følgende betingelser:
1,5 Tesla systemer:
• Statisk magnetisk feltstyrke på 1,5 Tesla
• Rumlig feltstyrke for gradient på 450 Gauss/cm
• M aksimal helkropsgennemsnitlig specifik absorptionsrate (SAR) på
2,0 W/kg i 15 minutters scanning
I ikke-klinisk testning frembragte Zenith AAA endovaskulær protese en
temperaturstigning på mindre end eller lig med 1,4 °C ved en maksimal
helkropsgennemsnitlig specifik absorptionsrate (SAR) på 2,8 W/kg, bedømt ved
kalorimetri i 15 minutters MR-scanning i en 1,5 Tesla, Siemens Medical
Magnetom, Numaris/4 Software, Version Syngo MR 2002B DHHS MR-scanner.
Den maksimale helkropsgennemsnitlige specifikke absorptionsrate (SAR) var
2,8 W/kg, hvilket svarer til en kalorimetrisk målt værdi på 1,5 W/kg.
3,0 Tesla systemer:
• Statisk magnetisk feltstyrke på 3,0 Tesla
• Rumlig feltstyrke for gradient på 720 Gauss/cm
• M aksimal helkropsgennemsnitlig specifik absorptionsrate (SAR) på
2,0 W/kg i 15 minutters scanning
I ikke-klinisk testning frembragte Zenith AAA endovaskulær protese en
temperaturstigning på mindre end eller lig med 1,9 °C ved en maksimal
helkropsgennemsnitlig specifik absorptionsrate (SAR) på 3,0 W/kg, bedømt ved
kalorimetri i 15 minutters MR-scanning i en 3,0 Tesla Excite, GE Electric
Healthcare, G3.0-052B Software, MR-scanner. Den maksimale
helkropsgennemsnitlige specifikke absorptionsrate (SAR) var 3,0 W/kg, hvilket
svarer til en kalorimetrisk målt værdi på 2,8 W/kg.
Billedartefaktet strækker sig gennem hele den anatomiske region, der
indeholder implantatet, og slører billedet af umiddelbart nærliggende
anatomiske strukturer inden for ca. 20 cm fra implantatet, såvel som hele
implantatet og dets lumen, når der scannes i ikke-klinisk testning ved hjælp af
sekvensen: Hurtigt spinekko i en 3,0 Tesla, Excite, GE Electric Healthcare, med
G3.0-052B software, MR-system med kropsradiofrekvensspiral.
For alle scannere spredes billedartefaktet, når afstanden fra implantatet til
området af interesse øges. MR-scanninger af hoved og hals og
underekstremiteter kan tages uden billedartefakt. Billedartefakt kan findes i
scanninger af abdominalregionen og overekstremiteterne, afhængigt af
afstanden fra implantatet til området af interesse.
Der findes klinisk information om sytten patienter, som fik MR-scanninger efter
implantation af stent-protese. Der har ikke været nogen rapporterede uønskede
hændelser eller implantatproblemer hos nogen af disse patienter som et
resultat af, at de er blevet MR-scannet. Desuden er der blevet implanteret langt
over 50.000 Zenith AAA endovaskulære proteser globalt, hvor der ikke har
været nogen rapporterede uønskede hændelser eller implantatproblemer som
et resultat af MR-scanning.
Cook anbefaler, at patienten registrerer betingelserne for MR-scanning, der er
beskrevet i denne brugsanvisning, hos MedicAlert Foundation. MedicAlert
Foundation kan kontaktes på følgende måder:
Adresse: MedicAlert Foundation International
Telefon: +1 888-633-4298 frikaldsnummer
FAX: +1 209-669-2450
2323 Colorado Avenue
Turlock, CA 95382 USA
+1 209-668-3333 fra uden for USA
Web: w ww.medicalert.org
12.6 Yderligere kontrol og behandling
Yderligere kontrol og mulig behandling anbefales for:
• Aneurismer med type I endolækage
• Aneurismer med type III endolækage
• Aneurismeforstørrelse,
endolækagestatus)
• Migration
• Utilstrækkelig lukkelængde
Overvejelse vedrørende reintervention eller konvertering til åben kirurgi bør
omfatte den ansvarlige læges vurdering af den enkelte patients komorbiditeter,
forventet levetid og patientens personlige valg. Patienter bør rådgives om, at
der er mulighed for efterfølgende reinterventioner, inklusive kateterbaseret og
konvertering til åben kirurgi, efter placering af endoprotesen.
13 OPLYSNINGER OM SPORING AF PATIENTER
Udover disse Brugsanvisninger er Zenith Flex AAA endovaskulær protese med
H&L-B One-Shot indføringssystemet pakket med en Formular til sporing af
anordning, som skal udfyldes af hospitalspersonalet og fremsendes til COOK
med det formål at spore alle patienter, der modtager Zenith Flex AAA
endovaskulær protese (efter amerikansk lovgivning).
≥ 5
mm af maksimal diameter (uanset
14 FEJLSØGNING
BEMÆRK: Teknisk assistance fra en Cook produktspecialist fås ved
henvendelse til den lokale repræsentant for Cook.
14.1 Fejlfinding ved udløser-wirens frigørelse
FORSIGTIG: Følgende trin skal kun udføres, hvis det er umuligt at fjerne
den proksimale udløser-wire som beskrevet i Afsnit 11.1.7.
14.1.1 Proksimal (top) anlæggelse af hovedprotese
1. Hvis den sorte (tophætte) udløsningsmekanismes udløserwire ikke
kan fjernes fra håndtaget, skal udløser-wiren skæres af tæt ved
udløsningsmekanismen (Fig. 37), og udløsningsmekanismen fjernes fra
håndtaget.
2. Stabiliser den grå positioneringsanordning, mens sheathen trækkes
tilbage, for at anlægge den ipsilaterale kant.
3. Fjern sikkerhedslåsen fra den hvide udløsningsmekanisme for udløser-
wiren.
4. Træk udløser-wiren tilbage, fjern den ved at skubbe den hvide
udløsningsmekanisme for udløser-wiren af håndtaget, og fjern den
dernæst via dens rille over produktets indre kanyle.
5. Klem og fastgør den afskårne ende af tophættens udløser-wire ved hjælp
af en låsbar tang. (Fig. 38)
6. Løsn pin vise’n, og idet den indre kanyles og udløser-wirens position
bevares, føres den grå positioneringsanordning og sheathen ind i
protesen, til spidsen af den grå positioneringsanordning er ca. 2 cm fra
guldmarkørerne. (Fig. 39) Den fremførte grå positioneringsanordning
yder ekstra understøtning af den indre kanyle.
BEMÆRK: Hold en let spænding på udløser-wiren for at fjerne eventuelt slæk
i wiren, mens den grå positioneringsanordning og sheathen føres frem.
BEMÆRK: Sørg for, at spidsen af den grå positioneringsanordning ikke føres
ind i tophætten.
7. Lås pin vise’n. Kontroller, at udløser-wiren sidder godt fast på tangen.
8. Stabiliser den grå positioneringsanordning, og før sheathen langsomt
frem, til sheath-spidsen er 2 mm fra guldmarkørerne. (Fig. 40)
BEMÆRK: Undgå at føre selve protesen frem under sheath-fremføringen.
9. Stabiliser sheathen og træk den grå positioneringsanordning tilbage
sammen med den indre kanyle for at trække tophætten over den
suprarenale stent. (Fig. 41)
10. Kontroller guldmarkørernes position inferior for nyrearterierne.
11. Fjern udløser-wiren.
12. Træk sheathen tilbage, indtil den tilspidsede spids på den grå positioner
er synlig.
13. Før en formningsballon gennem hovedprotesens kontralaterale kant, og
placer den lige superior for protesens bifurkation.
14. Pust ballonen op til protesens fulde diameter. (Fig. 42)
15. Løsn pin vise’n.
61

16. Stabiliser den grå positioneringsanordning og ballonkateteret, og før den
indre kanyle frem for at anlægge tophætten.
17. Spænd pin vise’n.
18. Tøm luften af ballonen og skub den kontralaterale kateterleder ind i aorta
thoracica.
BEMÆRK: På grund af risikoen for en for tidlig anlæggelse af den ipsilaterale
benprotese og for tidlig frigørelse af udløser-wiren foreslås det at lade
formningsballonen være i eller lige over den kontralaterale kant for at skabe
protesestabilitet under placeringen af den ipsilaterale kant.
14.1.2 Sammenkobling af tophætten
1. Løsn pin vise’n. (Fig. 25)
2. Fastgør sheath og indre kanyle for at undgå, at disse komponenter
bevæger sig.
3. Før den grå positioneringsanordning over den indre kanyle, indtil den
sammenkobles med tophætten. (Fig. 43, 27, 44)
BEMÆRK: Hvis der mærkes modstand, drejes den grå
positioneringsanordning en smule, og fremføringen fortsættes forsigtigt.
4. Spænd pin vise’n igen og træk hele tophætten og den grå
positioneringsanordning gennem protesen og gennem sheathen ved
at trække i den indre kanyle. (Fig. 45) Lad sheath og kateterleder blive
siddende.
5. Luk Captor® hæmostatisk ventil på Flexor® indføringssheathen ved at
dreje den i retning med uret, indtil den standser. (Fig. 30)
14.1.3 Placering og anlæggelse af ipsilateral iliaca-ben
BEMÆRK: Det skal sikres, at Captor hæmostatisk ventil på indføringssheathen
er drejet til åben position. (Fig. 31)
1. Brug hovedprotesens kateterleder og sheath-del til at indføre den
ipsilaterale benprotese.
BEMÆRK: På grund af ændringer i instruktionerne vedrørende
tophættefrigørelse skal hovedprotesens sheath-enhed trækkes tilbage til et
punkt 1-2 cm inden for den proksimale ipsilaterale kant. Før den ipsilaterale
kants dilatator-sheath ind i hovedprotesens sheath.
BEMÆRK: Formningsballonen kan eventuelt pustes op i hovedprotesens
kontralaterale kant, hvis yderligere stabilisering af protesen er nødvendig.
FORSIGTIG: Pust ikke ballonen op uden for protesen.
BEMÆRK: I snirklede kar kan aa. iliacae internaes position ændres signifikant
ved indføringen af stive kateterledere og sheathsystemer.
2. Før det langsomt frem, indtil den ipsilaterale iliaca-benprotese overlapper
mindst én fuld iliaca-benstent (dvs. proksimale stent af iliaca-benprotesen)
inden i hovedprotesens ipsilaterale kant. (Fig. 46)
BEMÆRK: Hvis det er nødvendigt med en overlapning på mere end tre iliacabenstents (mere end to iliaca-benstents for 37 og 54 mm benlængder), kan
det være nødvendigt at overveje at anvende en benforlænger i den modsatte
sides bifurkationsområde.
3. Bekræft positionen af den distale ende af iliaca-benprotesen. Omplacér
om nødvendigt iliaca-benprotesen for at sikre indvendig åbenhed af
iliaca.
4. For at anlægge iliaca-benprotesen skal den stabiliseres med
den grå positioneringsanordning, mens iliaca-bensheathen og
hovedprotesesheathen trækkes samlet tilbage. (Fig. 33 og 47)
Hovedprotesens sheath tilbagetrækkes om nødvendigt.
5. Under gennemlysning og efter verificering af iliaca-benprotesens position
løsnes pin vise’n, og den indre kanyle trækkes tilbage for at sammenkoble
den konusformede dilatator med den grå positioneringsanordning.
Spænd pin vise’n. Oprethold sheathpositionen, samtidig med at den grå
positioneringsanordning trækkes tilbage med den fastgjorte indre kanyle.
(Fig. 48)
6. Luk Captor hæmostatisk ventil på Flexor indføringssheathen ved at dreje
den i retning med uret, indtil den standser.
7. Kontrollér igen kateterledernes position. Lad sheath og kateterledere blive
siddende.
8. Fjern den tømte ballon fra den kontralaterale side.
14.1.4 Placering og anlæggelse af kontralateral iliaca-ben
FORSIGTIG: Verificer inden implantationen, at det forudfastlagte
kontralaterale iliaca-ben er valgt til indføring på patientens
kontralaterale side.
1. Anbring billedforstærkeren således, at både kontralaterale a. iliaca interna
og kontralaterale a. iliaca communis vises.
2. Før indføringen af fremføringssystemet til det kontralaterale iliaca-ben,
injiceres kontrast gennem den kontralaterale femoralis-sheath for at
lokalisere den kontralaterale a. iliaca interna.
3. Før fremføringssystemet for det kontralaterale iliaca-ben ind i arterien.
Før det langsomt frem, indtil iliaca-benprotesen overlapper mindst én
fuld iliaca-benstent (dvs. proksimale stent af iliaca-benprotesen) inden i
hovedprotesens kontralaterale kant. (Fig. 49)
BEMÆRK: Hvis der opstår vanskeligheder ved fremføringen af
fremføringssystemet for iliaca-benet, udskiftes til en mere støttende
kateterleder. I snirklede kar kan anatomien ændres signifikant ved
indføringen af de stive kateterledere og sheathsystemer.
4. Bekræft positionen af den distale ende af iliaca-benprotesen. Omplacer
iliaca-benprotesen, hvis det er nødvendigt, for at sikre både åbenhed i
a. iliaca interna og en minimumsoverlapning på en fuld iliaca-benstent
(dvs. iliaca-benprotesens proksimale stent, maksimumsoverlapning på
1,5 stenter) inden i den endovaskulære hovedprotese.
5. Hold iliaca-benprotesen i position med den grå positioneringsanordning,
samtidig med at sheathen trækkes tilbage, for at anlægge den. (Fig. 19
og 50) Sørg for, at overlapning svarende til en stentlængde bevares.
6. Stop tilbagetrækningen af sheathen, så snart den distale ende af iliacabenprotesen frigøres.
7. Under gennemlysning og efter kontrol af iliaca-benprotesens position
løsnes pin vise’n, og den indre kanyle trækkes tilbage for at sammenkoble
den konusformede dilatator med den grå positioneringsanordning.
Spænd pin vise’n. Oprethold sheathpositionen, samtidig med at den grå
positioneringsanordning trækkes tilbage med den fastgjorte indre kanyle.
(Fig. 51)
8. Kontrollér igen kateterledernes position.
9. Foretag isætning af formningsballonen og udfør det endelige angiogram
som beskrevet i Afsnit 11.1.12 Indføring af formningsballon.
14.2 Fejlfinding ved anlæggelse af den suprarenale stent
FORSIGTIG: Følgende trin skal kun udføres, hvis det er umuligt at
anlægge tophætten som beskrevet i Afsnit 11.1.7.
14.2.1 Proksimal (top) anlæggelse af hovedprotese
1. Hvis den suprarenale stent ikke kan anlægges fuldstændigt ved at
fremføre tophættens indvendige kanyle, fremføres en formningsballon
gennem hovedprotesens kontralaterale kant og placeres lige netop
superiort for stentprotesens bifurkation.
2. Inflatér ballonen til protesens fulde diameter for at yde understøtning af
den indvendige kanyle.
3. Løsn pin visen. (Fig. 15)
4. Stabilisér den grå positioneringsanordning og ballonkateteret, og før den
indvendige kanyle frem for at anlægge den suprarenale stent.
Hvis den suprarenale stent er fuldstændigt anlagt:
5. Spænd pin visen. Deflatér ballonen og træk den tilbage.
6. Før den kontralaterale kateterleder frem i den thorakale aorta, og gå
tilbage til det relevante trin i brugsanvisningen for at afslutte proceduren.
Hvis den suprarenale stent stadig ikke kan anlægges fuldstændigt:
5. Spænd pin visen og deflatér ballonen. Idet ballonens position bevares,
stabiliseres den grå positioneringsanordning, og sheathen trækkes
tilbage, så den ipsilaterale kant anlægges fuldstændigt.
6. Fjern sikkerhedslåsen fra udløsermekanismen for den ipsilaterale kants
udløser-wire.
7. Træk udløser-wiren tilbage, og fjern den ved at føre udløsermekanismen
for den ipsilaterale kants udløser-wire af håndtaget og fjerne den via dens
spalte over produktets indvendige kanyle.
8. Løsn pin visen (Fig. 25), og idet den indvendige kanyles position
bevares, føres den grå positioneringsanordning og sheathen ind i
protesen, til spidsen af den grå positioneringsanordning er ca. 2 cm
inferiort for de proksimale guldmarkører. (Fig. 52) Den fremførte grå
positioneringsanordning yder ekstra understøtning af den indre kanyle.
BEMÆRK: Undgå at føre protesen frem under fremføringen af den grå
positioneringsanordning og sheathen.
BEMÆRK: Sørg for, at spidsen af den grå positioneringsanordning ikke
føres ind i tophætten.
9. Lås pin visen.
10. Kontroller guldmarkørernes position inferiort for nyrearterierne.
11. Omplacér formningsballonen, så den støder op til bifurkationen.
12. Inflatér ballonen til protesens fulde diameter. (Fig. 42)
13. Løsn pin visen.
14. Stabilisér den grå positioneringsanordning og ballonkateteret, og før den
indvendige kanyle frem for at anlægge den suprarenale stent.
15. Spænd pin visen.
16. Deflatér ballonen og skub den kontralaterale kateterleder ind i aorta
thoracica.
BEMÆRK: På grund af for tidlig anlæggelse af ipsilateralt ben og
frigørelse af udløser-wiren foreslås det at lade formningsballonen være i
eller lige over den kontralaterale kant for at skabe protesestabilitet under
placeringen af den ipsilaterale kant.
14.2.2 Sammenkobling af tophætten
1. Løsn pin visen. (Fig. 25)
2. Fastgør sheath og indre kanyle for at undgå, at disse komponenter
bevæger sig.
3. Før den grå positioneringsanordning over den indre kanyle, indtil den
sammenkobles med tophætten. (Fig. 43, 27 og 44)
BEMÆRK: Hvis der mærkes modstand, drejes den grå
positioneringsanordning en smule, og fremføringen fortsættes forsigtigt.
4. Spænd pin vise’n igen og træk hele tophætten og den grå
positioneringsanordning gennem protesen og gennem sheathen ved
at trække i den indre kanyle. (Fig. 45) Lad sheath og kateterleder blive
siddende.
5. Luk Captor® hæmostaseventilen på Flexor® indføringssheathen ved at
dreje den i retning med uret, indtil den standser. (Fig. 30)
14.2.3 Placering og anlæggelse af ipsilateral iliaca-ben
BEMÆRK: Det skal sikres, at Captor hæmostaseventil på indføringssheathen
er drejet til åben position. (Fig. 31)
1. Anbring billedforstærkeren, så den viser både den ipsilaterale a. iliaca
interna og den ipsilaterale a. iliaca communis.
2. Efter indføring af indføringssystemet til det ipsilaterale iliaca-ben injiceres
kontrast gennem hovedprotesens sheath for at lokalisere den ipsilaterale
a. iliaca interna.
3. Brug hovedprotesens kateterleder og sheathenhed til at indføre den
ipsilaterale benprotese.
BEMÆRK: På grund af ændring i tophættefrigørelse skal hovedprotesens
sheathenhed trækkes tilbage til et punkt 1-2 cm inden for den proksimale
ipsilaterale kant. Før den ipsilaterale kants dilatator-sheath ind i
hovedprotesens sheath.
BEMÆRK: Formningsballonen kan eventuelt inflateres i hovedprotesens
kontralaterale kant, hvis yderligere stabilisering af protesen er nødvendig.
FORSIGTIG: Inflatér ikke ballonen uden for protesen.
BEMÆRK: I snirklede kar kan aa. iliacae internaes position ændres
signifikant ved indføringen af stive kateterledere og sheathsystemer.
4. Før indføringssystemet til det ipsilaterale iliaca-ben langsomt frem, indtil
den ipsilaterale benprotese overlapper mindst én fuld iliaca-benstent
(dvs. proksimale stent af iliaca-benprotesen) inden i hovedprotesens
ipsilaterale kant. (Fig. 46)
BEMÆRK: Hvis det er nødvendigt med en overlapning på mere end
tre iliaca-benstents (mere end to iliaca-benstents for 37 og 54 mm
benlængder), kan det være nødvendigt at overveje at anvende en
benforlænger i den modsatte sides bifurkationsområde.
5. Bekræft positionen af den distale ende af iliaca-benet. Omplacér om
nødvendigt iliaca-benprotesen for at sikre indvendig åbenhed af iliaca.
6. For at anlægge iliaca-benprotesen skal den stabiliseres med
den grå positioneringsanordning, mens iliaca-bensheathen og
hovedprotesesheathen trækkes samlet tilbage. (Fig. 33 og 47)
7. Under gennemlysning og efter verificering af iliaca-benprotesens position
løsnes pin vise’n, og den indre kanyle trækkes tilbage for at sammenkoble
den konusformede dilatator med den grå positioneringsanordning.
Spænd pin visen. Oprethold sheathpositionen, samtidig med at den grå
positioneringsanordning trækkes tilbage med den fastgjorte indre kanyle.
(Fig. 48)
8. Luk Captor hæmostaseventil på Flexor indføringssheathen ved at dreje
den i retning med uret, indtil den standser.
9. Kontrollér igen kateterledernes position. Lad sheath og kateterledere
blive siddende.
10. Fjern den tømte ballon fra den kontralaterale side.
14.2.4 Placering og anlæggelse af kontralateral iliaca-ben
FORSIGTIG: Verificer inden implantationen, at det forudfastlagte
kontralaterale iliaca-ben er valgt til indføring på patientens
kontralaterale side.
1. Anbring billedforstærkeren, så den viser både den kontralaterale a. iliaca
interna og den kontralaterale a. iliaca communis.
2. Før indføringen af indføringssystemet til det kontralaterale iliaca-ben,
injiceres kontrast gennem den kontralaterale femoralis-sheath for at
lokalisere den kontralaterale a. iliaca interna.
3. Indfør indføringssystemet til det kontralaterale iliaca-ben i arterien. Før
det langsomt frem, indtil iliaca-benprotesen overlapper mindst én fuld
iliaca-benstent (dvs. proksimale stent af iliaca-benprotesen) inden i
hovedprotesens kontralaterale kant. (Fig. 49)
BEMÆRK: Hvis der opstår vanskeligheder ved fremføringen af
indføringssystemet for iliaca-benet, udskiftes til en mere støttende
kateterleder. Anatomien i snoede kar kan blive signifikant ændret under
indføring af stive kateterledere og sheathsystemer.
4. Bekræft positionen af den distale ende af iliaca-benet. Omplacér iliacabenprotesen, hvis det er nødvendigt, for at sikre både åbenhed i a. iliaca
interna og en minimumsoverlapning på en fuld iliaca-benstent (dvs.
iliaca-benprotesens proksimale stent, maksimumsoverlapning på
1,5 stents) inden i den endovaskulære hovedprotese.
62

5. Hold iliaca-benprotesen i position med den grå positioneringsanordning,
samtidig med at sheathen trækkes tilbage, for at anlægge den.
(Fig. 19 og 50) Sørg for, at overlapning svarende til en stentlængde
bevares.
6. Stop tilbagetrækningen af sheathen, så snart den distale ende af iliacabenprotesen frigøres.
7. Under gennemlysning og efter kontrol af iliaca-benprotesens position
løsnes pin vise’n, og den indre kanyle trækkes tilbage for at sammenkoble
den konusformede dilatator med den grå positioneringsanordning.
Spænd pin visen. Oprethold sheathpositionen, samtidig med at den grå
positioneringsanordning trækkes tilbage med den fastgjorte indre kanyle.
(Fig. 51)
8. Kontrollér igen kateterledernes position.
9. Udfør indføring af formningsballon og slutangiogram som beskrevet i
afsnittet Indføring af formningsballon i produktets brugsanvisning.
63

DEUTSCH
ENDOVASKULÄRE AAA-PROTHESE ZENITH FLEX® MIT H&L-B
ONE-SHOT™ EINFÜHRSYSTEM
Die Anleitungen sorgfältig und vollständig durchlesen. Nichtbeachtung der
Anleitungen, Warnhinweise und Vorsichtsmaßnahmen kann ernsthafte chirurgische
Konsequenzen haben und zu Verletzungen des Patienten führen.
VORSICHT: Diese Vorrichtung darf nach Bundesgesetzen der USA nur an einen
Arzt oder auf Verordnung eines Arztes verkauft werden.
VORSICHT: Alle im äußeren Beutel enthaltenen Bestandteile (einschließlich
des Einführsystems und der endovaskulären Prothesen) werden steril geliefert
und sind nur für den Einmalgebrauch bestimmt.
Für die Zenith-Produktreihe liegen vier empfohlene Gebrauchsanweisungen
vor. In dieser Gebrauchsanweisung wird das vorgeschlagene Vorgehen für
die endovaskuläre AAA-Prothese Zenith Flex (Hauptteil und iliakale Schenkel)
beschrieben. Informationen zu anderen Zenith-Komponenten sind in den
folgenden Gebrauchsanweisungen enthalten:
• Endovaskuläre AAA-Prothese Zenith (Hauptteil der Zenith endovaskulären AAAProthese und iliakale Schenkel);
• Hilfskomponenten für die endovaskuläre AAA-Prothese Zenith
(Hauptteilverlängerung, iliakale Schenkelverlängerung, Konverter und iliakales
Verschluss-Segment);
• AAA-Hilfsprothese Zenith® Renu™ (Hauptteilverlängerungs- und
Konverterkonfiguration) und
• CODA® Ballonkatheter.
1 BESCHREIBUNG DES INSTRUMENTS
1.1 Hauptteil (Aortenteil) und iliakale Schenkel
Die endovaskuläre AAA-Prothese Zenith Flex ist ein modulares System und
umfasst drei Komponenten: einen gegabelten Hauptteil (Aortenteil) und zwei
iliakale Schenkel. (Abb. 1) Die Prothesenmodule bestehen aus Polyestergewebe
voller Stärke, das mit geflochtenem Polyesterfaden und PolypropylenMonofilamentfaden an selbstexpandierende Cook-Z® Stents aus Edelstahl
angenäht ist. Die Komponenten sind durchgehend mit Stents versehen, um
ausreichende Stabilität zu geben und die zum Öffnen des Prothesenlumens
während der Entfaltung erforderliche Expansionskraft zu liefern. Die Cook-Z Stents
gewährleisten darüber hinaus die erforderliche feste und dichte Verbindung der
Prothese zur Gefäßwand.
Der unbedeckte suprarenale Stent am proximalen Ende der Prothese ist in 3-mmAbständen mit Haken versehen, die der Prothese zusätzlichen Halt geben. Zur
besseren fluoroskopischen Darstellung des Stent-Grafts trägt dieser röntgendichte
Goldmarkierungen an den folgenden Stellen: eine Markierung am lateralen
Aspekt des am weitesten distal liegenden Stents am kontralateralen Ansatz des
gegabelten Abschnitts des Hauptteils und vier weitere in kreisförmiger Anordnung
innerhalb von 2 mm von dem am weitesten superior liegenden Aspekt des
Prothesenmaterials.
1.2 Einführsystem für Hauptteil
Der Hauptteil der endovaskulären AAA-Prothese Zenith Flex wird mit bereits
angebrachtem H&L-B One-Shot Einführsystem geliefert. (Abb. 2) Die integrierten
Funktionen des sequenziellen Entfaltungsvorgangs ermöglichen die kontinuierliche
Steuerung der endovaskulären Prothese während des Entfaltungsvorgangs.
Das H&L-B One-Shot Einführsystem ermöglicht eine genaue Positionierung der
Prothese sowie eine Korrektur der endgültigen Prothesenposition vor Entfaltung
des mit Haken versehenen suprarenalen Stents.
Zum Einführen des Prothesenhauptteils wird ein H&L-B One-Shot Einführsystem
von 18, 20 oder 22 French verwendet. Die endovaskuläre Prothese ist mit einem
doppelten Auslösedrahtmechanismus so lange fest mit dem Einführsystem
verbunden, bis sie vom Arzt freigegeben wird. Alle Systeme sind mit einem
0,035-Inch-Führungsdraht (0,89 mm) kompatibel.
Zur besseren Hämostase kann das Captor®-Hämostaseventil bei der Einführung
bzw. Entfernung von Hilfskomponenten in die bzw. aus der Schleuse gelockert
oder fester angezogen werden. Die Einführsysteme für den Hauptteil weisen
eine knickresistente, hydrophil beschichtete Flexor®-Einführschleuse auf. Beide
Merkmale sollen die Steuerbarkeit in den Aa. iliacae und der Bauchaorta verbessern.
1.3 Einführsystem für iliakale Schenkel
Die iliakalen Schenkel der endovaskulären AAA-Prothese Zenith werden mit bereits
angebrachtem H&L-B One-Shot Einführsystem geliefert. (Abb. 3) Das Einführsystem
ist für hohe Benutzerfreundlichkeit mit minimaler Vorbereitung ausgelegt. Zum
Einbringen der iliakalen Schenkel wird ein H&L-B One-Shot Einführsystem von
14 French oder 16 French verwendet. Alle Systeme sind mit einem 0,035-InchFührungsdraht (0,89 mm) kompatibel.
1.4 Hilfskomponenten für die endovaskuläre AAA-Prothese Zenith
Zusätzliche Hilfskomponenten (Hauptteilverlängerungen, Verlängerungen für die
iliakalen Schenkel, Konverter und iliakale Verschluss-Segmente) sind erhältlich.
(Abb. 4) Nähere Informationen sind in der Gebrauchsanweisung für die jeweilige
Hilfskomponente für die endovaskuläre AAA-Prothese Zenith enthalten.
2 VERWENDUNGSZWECK
Die endovaskuläre AAA-Prothese Zenith Flex mit dem H&L-B One-Shot
Einführsystem ist für die endovaskuläre Behandlung von Patienten mit
abdominalen Aortenaneurysmen oder aortoiliakalen Aneurysmen angezeigt,
die sich nach ihrer Morphologie für eine endovaskuläre Reparatur eignen; dazu
gehören:
• Ausreichender iliakaler/femoraler Zugang, der mit den erforderlichen
Einführsystemen kompatibel ist.
• Nicht-aneurysmatisches infrarenales Aortensegment (Hals) proximal zum
Aneurysma:
• mit einer Länge von mindestens 15 mm,
• mit einem Durchmesser (Außenwand zu Außenwand) von maximal 32 mm
und mindestens 18 mm,
• mit einem Winkel von weniger als 60 Grad relativ zur Längsachse des
Aneurysmas, und
• einem Winkel von weniger als 45 Grad relativ zur Achse der suprarenalen
Aorta.
• Distale Befestigungsstelle an der Iliaka von mindestens 10 mm Länge und
7,5 mm bis 20 mm Durchmesser (Außenwand zu Außenwand gemessen).
3 KONTRAINDIKATIONEN
Es sind keine Gegenanzeigen zu diesen Prothesen bekannt.
4 WARNHINWEISE UND VORSICHTSMASSNAHMEN
4.1 Allgemeines
• Die Anleitungen sorgfältig und vollständig durchlesen. Nichtbeachtung der
Anleitungen, Warnhinweise und Vorsichtsmaßnahmen kann ernsthafte
Konsequenzen haben und zu Verletzungen des Patienten führen.
• Während der Implantation oder einer Reintervention sollte für den Fall, dass eine
Umstellung auf eine offene chirurgische Reparatur erforderlich wird, stets ein
qualifiziertes Chirurgieteam zur Verfügung stehen.
• Die endovaskuläre AAA-Prothese Zenith Flex mit dem H&L-B One-Shot
Einführsystem darf nur von Ärzten und Teams verwendet werden, die in der
Technik von (katheterbasierten und chirurgischen) interventionellen
Gefäßoperationen und der Verwendung dieser Prothese geschult wurden.
Spezifische Schulungsanforderungen werden in Abschnitt 10.1, Ärzteschulung,
beschrieben.
• Für Patienten, bei denen eine Aneurysmavergrößerung, nicht mehr akzeptable
Verkürzung der Befestigungslänge (Überlappung von Gefäß und Komponenten)
und/oder ein Endoleak festgestellt wird, sollte eine weitere endovaskuläre
Intervention oder Umstellung auf eine übliche offene chirurgische Reparatur nach
der anfänglichen endovaskulären Reparatur in Betracht gezogen werden. Eine
Zunahme in der Größe des Aneurysmas und/oder ein persistierendes Endoleak
oder Migration kann zu einer Aneurysmaruptur führen.
• Bei Patienten mit reduziertem Blutfluss durch den Prothesenansatz und/oder mit
Lecks können sekundäre Interventionen und/oder chirurgische Eingriffe
erforderlich werden.
4.2 Auswahl, Behandlung und Nachsorge der Patienten
• Die endovaskuläre AAA-Prothese Zenith Flex ist zur Behandlung eines
Aortenhalses mit einem Durchmesser von mindestens 18 mm und höchstens
32 mm ausgelegt. Die endovaskuläre AAA-Prothese Zenith Flex ist zur
Behandlung eines proximalen Aortenhalses (distal zur untersten Nierenarterie)
mit einer Länge von mindestens 15 mm ausgelegt. Eine distale Befestigungsstelle
an der Iliaka von mindestens 10 mm Länge und 7,5 mm bis 20 mm Durchmesser
(Außenwand zu Außenwand gemessen) ist erforderlich. Diese Maße zur
Größenbestimmung sind für den Erfolg der endovaskulären Reparatur
entscheidend.
• Wichtige anatomische Parameter, die eine erfolgreiche Überbrückung des
Aneurysmas verhindern können, sind: schwere proximale Halsangulation
(>60 Grad für infrarenalen Hals zur Achse des AAA oder >45 Grad für
suprarenalen Hals relativ zum angrenzenden infrarenalen Hals), kurzer proximaler
Aortenhals (<15 mm), invertierte Trichterform (mehr als 10% Zunahme im
Durchmesser über 15 mm Länge des proximalen Aortenhalses),
Zirkumferenzthrombus und/oder -kalzifizierung an den arteriellen
Implantationsstellen, insbesondere am Übergang zwischen proximalem
Aortenhals und der distalen Iliaka. Bei Vorhandensein anatomischer
Beschränkungen ist ggf. ein längerer Hals erforderlich, um eine angemessene
Abdichtung und Befestigung zu erreichen. Ungleichmäßige Verkalkung und/
oder Plaque können die Befestigung und Dichtheit der Befestigungsstellen
beeinträchtigen. Bei einem Hals mit diesen anatomischen Schlüsselfaktoren
besteht ein höheres Risiko der Prothesenmigration oder eines Endoleaks.
• Zum Einführen der Prothese in das Gefäßsystem ist ein angemessener iliakaler
oder femoraler Zugang erforderlich. Der Durchmesser des Zugangsgefäßes
(Innenwand zu Innenwand gemessen) und die Morphologie (minimale
Gewundenheit, Okklusionskrankheit und/oder Verkalkung) sollten mit den
gewählten Gefäßzugangstechniken und Einführsystemen mit dem Profil einer
Gefäßeinführschleuse von 16 French bis 22 French kompatibel sein. Bei Gefäßen
mit signifikanter Verkalkung, Okklusion, Gewundenheit oder vorhandenen
Thromben ist u.U. keine Prothesenimplantation möglich und/oder es besteht ein
erhöhtes Embolierisiko. Damit der Eingriff erfolgreich ist, ist bei manchen
Patienten eine Technik zum Anlegen eines vaskulären Leitungswegs erforderlich.
• Die endovaskuläre AAA-Prothese Zenith Flex mit Einführsystem H&L-B One-Shot
wird nicht für Patienten empfohlen, die keine der für die intraoperative und
postoperative Bildgebung erforderlichen Kontrastmittel vertragen. Alle Patienten
sind engmaschig zu überwachen und regelmäßig hinsichtlich des weiteren
Krankheitsverlaufs und der Integrität der Endoprothese zu kontrollieren.
• Die endovaskuläre AAA-Prothese Zenith Flex mit dem H&L-B One-Shot
Einführsystem wird nicht für Patienten empfohlen, deren Gewicht oder Größe die
für die Bildgebung zulässigen Höchstwerte überschreitet.
• Bei Okklusion eines unentbehrlichen Astes der A. mesenterica inferior oder wenn
nicht mindestens eine A. iliaca interna offen gehalten werden kann, besteht u.U.
ein erhöhtes Risiko einer Becken-/Darmischämie.
• Bei Patienten mit multiplen großen, durchgängigen Lumbalarterien,
Parietalthrombus und einer durchgängigen A. mesenterica inferior kann ein
erhöhtes Risiko eines Endoleaks vom Typ II bestehen. Patienten mit nicht
korrigierbarer Koagulopathie können ebenfalls einem erhöhten Risiko durch
Endoleaks vom Typ II oder Blutungskomplikationen unterliegen.
• Die Sicherheit und Wirksamkeit der endovaskulären AAA-Prothese Zenith Flex
mit dem H&L-B One-Shot Einführsystem wurden in folgenden
Patientenpopulationen nicht untersucht:
• traumatische Aortenverletzung
• Leckagen mit Rupturgefahr oder Aneurysmen nach Ruptur
• mykotische Aneurysmen
• Pseudoaneurysmen infolge einer vorherigen Prothesenimplantation
• Revision einer früher implantierten endovaskulären Prothese
• nicht korrigierbare Koagulopathie
• unentbehrliche A. mesenterica
• genetische Bindegewebserkrankung (z.B. Marfan- oder Ehlers-DanlosSyndrom)
• gleichzeitige thorakoaortale oder thorakoabdominale Aneurysmen
• Patienten mit aktiven systemischen Infektionen
• Schwangerschaft oder Stillzeit
• morbid-adipöse Patienten
• Alter unter 18 Jahre
• Patienten mit proximalem Aortenhals von weniger als 15 mm Länge und mehr
als 60 Grad Angulation relativ zur Längsachse des Aneurysmas
• Die erfolgreiche Patientenauswahl erfordert spezifische Bildgebung und genaue
Messungen; siehe hierzu Abschnitt 4.3 Messtechniken und Bildgebung vor
dem Eingriff.
• Alle Längen und Durchmesser der für die vollständige Durchführung des Eingriffs
erforderlichen Produkte sollten dem Arzt zur Verfügung stehen, insbesondere,
wenn die präoperativen Fallplanungsmessungen (Behandlungsdurchmesser/längen) unsicher sind. Zur Erzielung optimaler Operationsergebnisse erlaubt
dieser Ansatz eine größere intraoperative Flexibilität.
4.3 Messtechniken und Bildgebung vor dem Eingriff
• Wenn die kontrastmittelfreie CT-Untersuchung nicht durchgeführt wird, können
Verkalkungen der Aa. iliacae und der Aorta, die den Zugang oder eine
zuverlässige Fixierung und Abdichtung unmöglich machen, eventuell unerkannt
bleiben.
• Wenn für die präoperative Bildgebungsrekonstruktion Schichtdicken von >3 mm
verwendet werden, kann es zu einer suboptimalen Größenbestimmung oder zur
Nichterkennung von fokalen Stenosen im CT kommen.
• Ausgehend von der klinischen Erfahrung wird als Bildgebungsmodalität zur
genauen Beurteilung der Anatomie des Patienten vor einer Behandlung mit der
endovaskulären AAA-Prothese Zenith Flex dringend eine kontrastmittelverstärkte
Spiral-CT-Angiographie (CTA) mit 3D-Rekonstruktion empfohlen. Ist keine
kontrastmittelverstärkte Spiral-CTA mit 3D-Rekonstruktion verfügbar, sollte der
Patient an eine Einrichtung überwiesen werden, an der dies möglich ist.
• Ärzte empfehlen, den C-Arm des Röntgensystems während der Angiographie so
zu positionieren, dass der Ursprung der Nierenarterien und insbesondere die
unterste durchgängige Nierenarterie vor Entfaltung des proximalen Rands des
Prothesenmaterials (Abdichtungsstent) des Hauptteils gut dargestellt werden.
Darüber hinaus sollte die Angiographie die Gabelungen der Aa. iliacae derart
sichtbar machen, dass die distalen Aa. iliacae communes relativ zum Ursprung
der bilateralen Aa. iliacae internae vor der Entfaltung der iliakalen Schenkel gut
definiert sind.
Durchmesser:
Zur Auswahl der geeigneten Prothesengröße und der geeigneten Prothese sind CTMessungen des Gefäßdurchmessers (von Außenwand zu Außenwand gemessen)
und nicht des Gefäßlumens heranzuziehen. Das kontrastmittelverstärkte Spiral-CT
muss 1 cm oberhalb des Truncus coeliacus beginnen und sich bei einer axialen
Schichtdicke von höchstens 3 mm bis einschließlich zu den Hüftköpfen erstrecken.
Längen:
Zur genauen Bestimmung der infrarenalen proximalen Halslänge sowie
zur Planung der Größe des Hauptteils und der Schenkelkomponenten für
die endovaskuläre AAA-Prothese Zenith Flex sind CT-Längenmessungen
heranzuziehen. Diese Rekonstruktionen sollten in sagittaler und koronarer Ebene
und in 3D durchgeführt werden.
• Über das langfristige Verhalten der endovaskulären Prothese ist derzeit
nichts bekannt. Die Patienten sollten darauf hingewiesen werden, dass die
endovaskuläre Therapie eine lebenslange Nachsorge erforderlich macht,
die regelmäßig zur Überwachung des Gesundheitszustandes und des
Verhaltens der endovaskulären Prothese erforderlich ist. Patienten mit
spezifischen klinischen Befunden (z.B. Endoleaks, Vergrößerung des Aneurysmas
oder Änderungen in der Struktur oder Position der endovaskulären Prothese)
sollten zusätzliche Nachsorgeuntersuchungen erhalten. Spezifische
Nachsorgerichtlinien werden in Abschnitt 12, RICHTLINIEN FÜR DIE
BILDGEBUNG UND POSTOPERATIVE NACHSORGE, besprochen.
• Die endovaskuläre AAA-Prothese Zenith Flex mit Einführsystem H&L-B One-Shot
wird nicht für Patienten empfohlen, die nicht in der Lage sind, sich den
erforderlichen prä- und postoperativen Bildgebungsverfahren und
Implantationsstudien wie in Abschnitt 12, RICHTLINIEN FÜR DIE BILDGEBUNG
UND POSTOPERATIVE NACHSORGE beschrieben zu unterziehen bzw. diese zu
befolgen.
64

• Nach Implantation der endovaskulären Prothese sollte der Patient regelmäßig
hinsichtlich folgender Punkte überwacht werden: Perigraftfluss, Aneurysmawachstum, Änderungen in der Struktur oder Position der endovaskulären
Prothese. Es sind mindestens jährliche bildgebende Untersuchungen
erforderlich, die folgendes umfassen: 1) Röntgenaufnahmen des Abdomens zur
Überwachung der Unversehrtheit des Produkts (Ablösungen von Komponenten,
Stentbruch, Hakenablösung) und 2) CT-Aufnahmen mit und ohne Kontrastmittel
zur Untersuchung von Veränderungen im Aneurysma, Perigraftfluss,
Durchgängigkeit, Gewundenheit und Krankheitsfortschritt. Wenn infolge von
renalen Komplikationen oder anderen Faktoren keine Kontrastmittel verwendet
werden können, kann eine abdominale Röntgenuntersuchung zusammen mit
einem Duplexultraschall die gleichen Informationen liefern.
4.4 Auswahl der Prothese
• Es wird dringend empfohlen, sich bei der Auswahl der geeigneten
Prothesengröße strikt an die Anleitung zur Größenbestimmung in der
Gebrauchsanweisung für die endovaskuläre AAA-Prothese Zenith Flex zu halten
(Tabelle 10.5.1 und 10.5.2). Bei der Anleitung zur Größenbestimmung in der
Gebrauchsanweisung wurde eine angemessene Übergröße der Prothese bereits
berücksichtigt. Die Verwendung von Größen außerhalb dieses Bereichs kann zu
Endoleak, Fraktur, Migration, Einfaltung oder Kompression der Prothese führen.
4.5 Implantationsverfahren
(Siehe Abschnitt 11, GEBRAUCHSANWEISUNG)
• Um die endovaskuläre AAA-Prothese Zenith Flex erfolgreich zu positionieren
und die akkurate Anlagerung an die Aortenwand sicherzustellen, muss der
Eingriff mit einer geeigneten Bildgebungstechnik überwacht werden.
• Das Einführsystem nicht biegen oder knicken. Verbiegungen oder Knicke
können das Einführsystem und die endovaskuläre AAA-Prothese Zenith Flex
beschädigen.
• Um interne Verwindungen in der endovaskulären Prothese zu verhindern, ist
sorgfältig darauf zu achten, dass bei einer Drehung des Einführsystems alle
Komponenten des Systems (von der äußeren Schleuse bis zur inneren Kanüle)
zusammen gedreht werden.
• Tritt beim Vorschieben des Führungsdrahts oder Einführsystems ein fühlbarer
Widerstand auf, muss der Vorgang sofort abgebrochen werden. Anhalten und
der Ursache des Widerstands nachgehen. Das Gefäß, der Katheter oder die
Prothese können verletzt bzw. beschädigt werden. Bei Stenosen oder
intravasalen Thromben oder in kalzifizierten bzw. gewundenen Gefäßen ist
besondere Vorsicht geboten.
• Versehentliche partielle Entfaltung oder Migration der Endoprothese können
eine chirurgische Entfernung erforderlich machen.
• Sofern medizinisch nicht besonders angezeigt, darf die endovaskuläre AAAProthese Zenith Flex nicht an einer Stelle entfaltet werden, an der es zur
Okklusion von Organe oder Extremitäten versorgenden Arterien kommen
würde. Die Endoprothese darf keine wichtigen Nieren- oder Mesenterialarterien
blockieren (mit Ausnahme der A. mesenterica inferior). Dies kann zur
Gefäßokklusion führen. Die Prothese wurde im Rahmen dieser klinischen Studie
nicht an Patienten mit zwei okkludierten Aa. iliacae internae geprüft.
• Nicht versuchen, die Prothese nach partieller oder kompletter Entfaltung in die
Schleuse zurückzuziehen.
• Eine distale Lageveränderung des Stent-Grafts nach partieller Entfaltung des
bedeckten proximalen Stents kann zu einer Beschädigung des Stent-Grafts
und/oder des Gefäßes führen.
• Falsche Positionierung und/oder mangelnde Dichtigkeit der endovaskulären
AAA-Prothese Zenith Flex im Gefäß kann zu einem erhöhten Risiko für
Endoleaks, Migration oder unbeabsichtigte Okklusion der A. renalis oder A.
iliacae internae führen. Um die Gefahr eines Nierenversagens mit seinen
Folgekomplikationen zu minimieren, muss die Durchgängigkeit der Aa. renales
jederzeit gewährleistet sein.
• Unzureichende Befestigung der endovaskulären AAA-Prothese Zenith Flex kann
das Migrationsrisiko des Stent-Grafts erhöhen. Bei fehlerhafter Entfaltung oder
bei Migration der Endoprothese kann eine chirurgische Intervention
erforderlich werden.
• Systemische Antikoagulation sollte während der Implantation entsprechend
den Protokollen der Klinik und der Entscheidung des Arztes angewandt werden.
Wenn Heparin nicht angezeigt ist, sollte ein alternatives Antikoagulanz in
Betracht gezogen werden.
• Zur Aktivierung der hydrophilen Beschichtung an der Außenseite der FlexorEinführschleuse muss diese mit in Kochsalzlösung getränkten Kompressen
abgewischt werden. Um optimale Leistung zu erzielen, muss die Schleuse stets
feucht gehalten werden.
• Um das Kontaminations- und Infektionsrisiko so gering wie möglich zu halten,
sollte die nicht entfaltete Endoprothese bei der Vorbereitung und Einführung
möglichst wenig manipuliert werden.
• Beim Einbringen des Einführsystems die Position des Führungsdrahts
beibehalten.
• Die Einführung und Entfaltung sollte zur Bestätigung der richtigen Funktion des
Platzierungssystems, der richtigen Platzierung der Prothese und des
gewünschten Verfahrensergebnisses unter Durchleuchtung erfolgen.
• Die Verwendung der endovaskulären AAA-Prothese Zenith Flex mit dem H&L-B
One-Shot Einführsystem erfordert die Verabreichung eines intravaskulären
Kontrastmittels. Patienten mit Niereninsuffizienz unterliegen möglicherweise
einem erhöhten Risiko von postoperativem Nierenversagen. Es ist sorgfältig
darauf zu achten, die während des Eingriffs verwendete Kontrastmittelmenge
zu begrenzen und präventive Behandlungsmethoden einzuhalten, um eine
Beeinträchtigung der Nieren zu verringern (z.B. ausreichende
Flüssigkeitszufuhr).
• Beim Zurückziehen der Schleuse und/oder des Führungsdrahts können sich die
Anatomie und die Lage der Prothese verändern. Die Lage der Prothese
fortlaufend kontrollieren und ggf. angiographisch überprüfen.
• Die endovaskuläre AAA-Prothese Zenith Flex umfasst einen suprarenalen Stent
mit Befestigungshaken. Auf Höhe des suprarenalen Stents müssen
interventionelle und angiographische Vorrichtungen äußerst sorgfältig
gehandhabt werden.
• Katheter, Drähte und Schleusen müssen innerhalb eines Aneurysmas sehr
sorgfältig gehandhabt werden. Erhebliche Störungen können zur Ablösung von
Thrombusfragmenten führen, welche eine distale Embolisierung oder eine
Ruptur des Aneurysmas verursachen können.
• Falls ein erneuter Eingriff mit chirurgischen Instrumenten an der Prothese
erforderlich ist, eine Beschädigung der Prothese oder eine Lageveränderung der
Prothese vermeiden.
• Vor Entfalten des suprarenalen Stents sicherstellen, dass der Führungsdraht
gerade noch distal des Aortenbogens endet.
• Vor der Implantation sicherstellen, dass der gewählte kontralaterale iliakale
Schenkel auch zum Einführen auf der kontralateralen Seite des Patienten
vorgesehen ist.
4.6 Verwendung des Modellierungsballons
• Den Ballon nicht außerhalb der Prothese im Gefäß insufflieren, da dies
Gefäßverletzungen verursachen kann. Den Ballon gemäß der zugehörigen
Dokumentation verwenden.
• Beim Insufflieren des Ballons in der Prothese vorsichtig vorgehen, wenn
Verkalkungen vorliegen, da eine übermäßige Insufflation Gefäßverletzungen
verursachen kann.
• Vor einer Umpositionierung ist sicherzustellen, dass der Ballon vollkommen
entleert ist.
• Zur zusätzlichen Hämostasekontrolle kann das Captor Hämostaseventil beim
Einführen und späteren Entfernen eines Modellierungsballons entsprechend
gelockert oder festgezogen werden.
4.7 Sicherheit und Kompatibilität mit MRT
Nicht klinische Tests haben ergeben, dass die endovaskuläre AAA-Prothese Zenith
bedingt MRT-kompatibel ist. Unter folgenden Bedingungen kann eine MRT ohne
Bedenken durchgeführt werden:
Systeme mit 1,5 Tesla:
• Statisches Magnetfeld mit 1,5 Tesla
• Feld mit einem räumlichen Gradienten von 450 Gauss/cm
• Maximale ganzkörpergemittelte spezifische Absorptionsrate (SAR) von
2,0 W/kg während einer Scan-Dauer von 15 Minuten
In nicht klinischen Tests verursachte die endovaskuläre AAA-Prothese Zenith
während eines 15-minütigen MRT-Scans in einem Magnetom-Scanner von
Siemens Medical Magnetom mit 1,5 Tesla, Numaris/4 Software, Version Syngo
MR 2002B DHHS, bei einer maximalen ganzkörpergemittelten spezifischen
Absorptionsrate (SAR) von 2,8 W/kg einen kalorimetrisch gemessenen
Temperaturanstieg von höchstens 1,4 °C. Die maximale ganzkörpergemittelte
spezifische Absorptionsrate (SAR) betrug 2,8 W/kg, was einem kalorimetrischen
Messwert von 1,5 W/kg entspricht.
Systeme mit 3,0 Tesla:
• Statisches Magnetfeld von 3,0 Tesla
• Feld mit einem räumlichen Gradienten von 720 Gauss/cm
• Maximale ganzkörpergemittelte spezifische Absorptionsrate (SAR) von
2,0 W/kg während einer Scan-Dauer von 15 Minuten
In nicht klinischen Tests verursachte die endovaskuläre AAA-Prothese Zenith
während eines 15-minütigen MRT-Scans in einem Excite-Scanner von GE
Electric Healthcare mit 3,0 Tesla, G3.0-052B Software, bei einer maximalen
ganzkörpergemittelten spezifischen Absorptionsrate (SAR) von 3,0 W/kg einen
kalorimetrisch gemessenen Temperaturanstieg von höchstens 1,9 °C. Die
maximale ganzkörpergemittelte spezifische Absorptionsrate (SAR) betrug
3,0 W/kg, was einem kalorimetrischen Messwert von 2,8 W/kg entspricht.
Bei Scans in nicht klinischen Tests mit folgender Sequenz verläuft das Bildartefakt
durch die gesamte anatomische Region, in der sich die Prothese befindet, und
verdeckt innerhalb von etwa 20 cm der Prothese die Sicht auf unmittelbar
angrenzende anatomische Strukturen sowie die gesamte Prothese und ihr Lumen:
Fast-Spin-Echo-Impulssequenz in einem Excite-Scanner von GE Electric Healthcare
mit 3,0 Tesla, G3.0-052B Software mit Körper-Hochfrequenzspule.
Bei allen Scannern verschwindet das Bildartefakt mit zunehmendem Abstand
zwischen Prothese und relevantem Bereich. MRT-Aufnahmen von Kopf und
Hals und unteren Gliedmaßen können ohne Bildartefakt erhalten werden. Je
nach Abstand der Prothese vom relevanten Bereich können in Aufnahmen der
Abdominalregion und der oberen Gliedmaßen Bildartefakte vorhanden sein.
Es liegen klinische Angaben über siebzehn Patienten vor, bei denen nach
Implantation eines Stent-Grafts MRT-Untersuchungen durchgeführt wurden.
Bei keinem dieser Patienten wurde im Nachfeld der MRT-Untersuchung über
unerwünschte Ereignisse oder Probleme mit der Prothese berichtet. Außerdem
sind weltweit weit über 50.000 endovaskuläre AAA-Prothesen Zenith implantiert
worden, ohne dass als Ergebnis von MRT-Untersuchungen unerwünschte
Ereignisse oder Probleme mit der Prothese gemeldet worden sind.
Cook empfiehlt, dass der Patient die in dieser Gebrauchsanweisung angegebenen
MR-Bedingungen bei der MedicAlert Foundation registriert. Die MedicAlert
Foundation kann folgenderweise kontaktiert werden:
Anschrift: MedicAler t Foundation International
Telefon: +1 888-633-4298 Gebührenfrei (Inland)
Fax: +1 209-669-2450
2323 Colorado Avenue
Turlock, CA 95382 USA
+1 209-668-3333 Von außerhalb der USA
Internet: w ww.medicalert.org
5 UNERWÜNSCHTE EREIGNISSE
5.1 Beobachtete unerwünschte Ereignisse
Eine in den USA in 15 Zentren mit 352 endovaskulären Patienten (200 Standardrisiko-, 100 Hochrisiko- und 52 Studienpatienten sowie 80 Kontrollpatienten)
durchgeführte prospektive multizentrische Studie zu einer früheren Ausführung
der Prothese (endovaskuläre AAA-Prothese Zenith) stellt die Basis für die
beobachtete und in Tabelle 5.1.1 beschriebene Häufigkeit unerwünschter
Ereignisse dar. Patienten, die physiologisch den Belastungen durch eine offene
oder endovaskuläre Reparatur widerstehen konnten und von der Anatomie her
für eine Therapie mit der endovaskulären AAA-Prothese Zenith mit dem H&L-B
One-Shot Einführsystem in Frage kamen, wurden in den Standardrisikoarm der
Studie aufgenommen. Von der Anatomie her zwar geeignete Patienten, die
jedoch hinsichtlich der offenen Reparatur ein höheres Morbiditäts- oder
Mortalitätsrisiko aufwiesen, wurden in den Hochrisikoarm der Studie
aufgenommen. Anfänglich im Rahmen der Studie behandelte Patienten wurden
in den Studienarm der Studie aufgenommen. Die Kontrollgruppe umfasste
Patienten, die von der Anatomie ihrer Gefäßsysteme her nicht uneingeschränkt
für eine endovaskuläre Reparatur in Frage kamen.
Tabelle 5.1.1 Todesfälle und Rupturen in der klinischen Studie
Todesfälle und Rupturen Zenith Standardrisiko
Alle Todesfälle
2
(0–30 Tage)
(31–365 Tage)
(0–365 Tage)
Ruptur
1
Nenner ist 199, da ein Standardrisikopatient keine Prothese erhielt.
2
Alle Todesfälle (0–30 Tage) wurden als mit dem AAA und dem Verfahren in Verbindung stehend betrachtet.
3
Von den Todesfällen (31–365 Tage) wurden vier als mit dem AAA in Verbindung stehend betrachtet: 1 chirurgische Komplikation (septischer Schock durch ischämische Kolitis) und
3 Hochrisikofälle (Pankreatitis mit Nierenversagen und Sepsis, Blutung von Aneurysma im oberen Abdomen [unbehandeltes AAA] und multiples Systemversagen).
4
Von den Todesfällen (0–365 Tage) wurden zehn als mit dem AAA in Verbindung stehend betrachtet: 1 Standardrisikofall (Herzversagen), 3 chirurgische Komplikationen (massive
Blutung, mesenterische Ischämie und septischer Schock durch ischämische Kolitis), 5 Hochrisikofälle (akute respiratorische Insuffizienz, Herzversagen mit Lungenembolie, Pankreatitis
mit Nierenversagen und Sepsis, Blutung von Aneurysma im oberen Abdomen [unbehandeltes AAA] und multiples Systemversagen), sowie 1 Studienfall (Verdacht auf Herzversagen).
3
mit AAA in Verbindung stehend
nicht im Zusammenhang mit
AAA stehend
2,3,4
mit AAA in Verbindung stehend
nicht im Zusammenhang mit
AAA stehend
(0–30 Tage) 0,0% (0/199) n. zutr. n. zutr. 0,0% (0/100) 0,0% (0/52)
(31–365 Tage) 0,0% (0/198) n. zutr. n. zutr. 1,0% (1/98) 0,0% (0/51)
(0–365 Tage) 0,0% (0/199) n. zutr. n. zutr. 1,0% (1/100) 0,0% (0/52)
0,5% (1/199) 2,5% (2/80) 0,20 2,0% (2/100) 1,9% (1/52)
3,0% (6/198) 1,3% (1/78) 0,68 7,1% (7/98) 9,8% (5/51)
0,0% (0/198) 1,3% (1/78) 0,29 3,1% (3/98) 0,0% (0/51)
3,0% (6/198) 0,0% (0/78) 0,19 4,1% (4/98) 9,8% (5/51)
3,5% (7/199) 3,8% (3/80) > 0,99 9,0% (9/100) 11,5% (6/52)
0,5% (1/199) 3,8% (3/80) 0,07 5,0% (5/100) 1,9% (1/52)
3,0% (6/199) 0,0% (0/80) 0,19 4,0% (4/100) 9,6% (5/52)
1
Oene Chirurgie Standardrisiko P-Wert Zenith hohes Risiko Zenith Studienarm
65

Tabelle 5.1.2 Unerwünschte Ereignisse1 in der klinischen Studie
Zenith Standardrisiko Oene Chirurgie Standardrisiko P- Wert Zenith hohes Risiko Zenith Studienarm
Abwesenheit von Morbidität
(0–30 Tage)
3
4, 9
6
7
8,11
(31–365 Tage)
3
4, 10
6
7
8
(0–365 Tage)
3
4, 9,10
6
7
8,11
2
5
2
5
2
5
Herz/Kreislauf
Lunge
Nieren
Verdauungssystem
Wunde
Neurologie
Gefäße
Abwesenheit von Morbidität
Herz/Kreislauf
Lunge
Nieren
Verdauungssystem
Wunde
Neurologie
Gefäße
Abwesenheit von Morbidität
Herz/Kreislauf
Lunge
Nieren
Verdauungssystem
Wunde
Neurologie
Gefäße
1
Vom Morbiditätsindex.
2
Herz/Kreislauf beinhaltete: Myokardinfarkte mit und ohne Q-Welle, dekompensierte Herzinsuffizienz, eine Neubehandlung erforderlich machende Arrhythmien, eine Intervention
erforderlich machende Herzischämie, inotrope Unterstützung, therapierefraktäre Hypertonie.
3
Lunge beinhaltete: Reintubation oder Beatmung > 24 Stunden, Antibiotika erfordernde Pneumonie, Sauerstoffunterstützung bei Entlassung.
4
Nieren beinhaltete: Dialyse bei Patienten mit normaler Nierenfunktion präoperativ, Kreatininspiegel > 30% über Basislinie bei mindestens zwei Nachsorgetests.
5
Verdauungssystem beinhaltete: Darmverschluss, Darmischämie, aortoenterische Fistel, paralytischer Ileus > 4 Tage.
6
Wunde beinhaltete: Antibiotika erfordernde Infektion, Hernie, Lymphfistel, Dehiszenz, Debridement erfordernde Nekrose.
7
Neurologie beinhaltete: Schlaganfall, TIA, Rückenmarkischämie/-paralyse.
8
Gefäße beinhaltete: Gliederthrombose, zu Gewebeverlust führende oder eine Intervention erfordernde distale Embolie, postoperative Transfusion (infolge von Pseudoaneurysma,
Gefäßverletzung, Aneurysmaleck oder anderen verfahrensbezogenen Faktoren), Pseudoaneurysma, Gefäßverletzung (wie unbeabsichtigte Okklusion, Dissektion oder andere
verfahrensbezogene Ursachen), Aneurysmaleck oder Ruptur, Aneurysmawachstum um mehr als 0,5 cm relativ zur kleinsten aller vorherigen Messungen.
9
Zwei weitere Hochrisikopatienten (einmal mit Okklusion einer akzessorischen Nierenarterie, ein weiterer mit chronischer Niereninsuffizienz) wurden „anderen“ Ereignissen zugeordnet.
10
Ein weiterer Studienpatient mit Niereninsuffizienz und ein weiterer chirurgischer Patient mit Niereninsuffizienz wurden „anderen“ Ereignissen zugeordnet.
11
Ein weiterer Studienpatient mit intraoperativ auftretender aortaler Plaqueruptur, die zur Okklusion der A. renalis führte, wurde „anderen“ Ereignissen zugeordnet.
5.2 Mögliche unerwünschte Ereignisse
Mögliche und/oder eine Intervention erfordernde unerwünschte Ereignisse
umfassen unter anderem:
• Amputation
• Aneurysmaruptur mit Todesfolge
• Aortaverletzung einschl. Perforation, Dissektion, Blutung, Ruptur mit
Todesfolge
• Arterielle oder venöse Thrombose und/oder Pseudoaneurysma
• Arteriovenöse Fistel
• Blutung, Hämatom oder Koagulopathie
• Claudicatio (z.B. Gesäß, untere Gliedmaßen)
• Embolie (Mikro und Makro) mit transienter oder permanenter Ischämie oder
Infarzierung
• Endoleak
• Endoprothese: falsche Komponentenplatzierung, unvollständige
Komponentenentfaltung, Migration von Komponenten, Nahtbruch,
Okklusion, Infekt, Stentfraktur, Verschleiß des Prothesenmaterials, Dilatation,
Erosion, Punktion, Perigraftfluss, Hakenablösung und Korrosion
• Fieber und lokale Entzündung
• Gefäßspasmen oder Gefäßtrauma (z.B. iliofemorale Gefäßdissektion, Blutung,
Ruptur, Tod)
• Gefäßverletzung
• Impotenz
• Infektion des Aneurysmas, der Prothese oder der Zugangsstelle einschl.
Abszessbildung, transientem Fieber und Schmerzen
• Kardiologische Komplikationen mit den entsprechenden Folgeproblemen
(z.B. Arrhythmie, Myokardinfarkt, dekompensierte Herzinsuffizienz,
Hypotonie, Hypertonie)
• Komplikationen an der Gefäßzugangsstelle einschl. Infektion, Schmerzen,
Hämatom, Pseudoaneurysma, arteriovenöse Fistel
• Komplikationen mit den entsprechenden Folgeproblemen (z.B. Aspiration)
durch die Anästhesie
• Leberversagen
• Lymphatische Komplikationen mit den entsprechenden Folgeproblemen (z.B.
Lymphfistel)
• Neurologische lokale oder systemische Komplikationen mit den
entsprechenden Folgeproblemen (z.B. Schlaganfall, transitorische
ischämische Attacke, Paraplegie, Paraparese, Paralyse)
• Ödem
• Okklusion der Prothese oder eines nativen Gefäßes
• Pulmonale/respiratorische Komplikationen mit den entsprechenden
Folgeproblemen (z.B. Pneumonie, akute respiratorische Insuffizienz, längere
Intubation)
• Renale Komplikationen mit den entsprechenden Folgeproblemen (z.B.
Arterienokklusion, Kontrastmitteltoxizität, Insuffizienz, Versagen)
• Tod
• Umstellung auf offene chirurgische Reparatur
• Urogenitale Komplikationen mit den entsprechenden Folgeproblemen (z.B.
Ischämie, Erosion, Fistel, Inkontinenz, Hämaturie, Infektion)
• Verdauungssystemkomplikationen (z.B. Ileus, transiente Ischämie, Infarkt,
Nekrose)
• Vergrößerung des Aneurysmas
• Wundkomplikationen mit den entsprechenden Folgeproblemen (z.B.
Dehiszenz, Infektion)
Bericht zu prothesenbezogenen unerwünschten Ereignissen
Alle unerwünschten Ereignisse (klinische Vorfälle), bei denen die endovaskuläre
AAA-Prothese Zenith Flex eine Rolle spielt, sind umgehend COOK zu melden.
Meldung eines klinischen Vorfalls: Kunden in den USA wenden sich bitte an das
Customer Relations Department unter der Nummer 1-800-457-4500 (rund um die
Uhr) oder 1-812-339-2235. Kunden in anderen Ländern als den USA wenden sich
bitte an ihren Händler.
80% (160/200) 58% (46/80) < 0,001 68% (68/100) 73% (38/52)
3,0% (6/200) 11% (9/80) 0,02 14% (14/100) 1,9% (1/52)
1,0% (2/200) 15% (12/80) < 0,001 2,0% (2/100) 0,0% (0/52)
2,5% (5/200) 10% (8/80) 0,01 6,0% (6/100) 5,8% (3/52)
1,0% (2/200) 3,8% (3/80) 0,14 1,0% (1/100) 1,9% (1/52)
4,5% (9/200) 7,5% (6/80) 0,38 2,0% (2/100) 3,8% (2/52)
0,0% (0/200) 2,5% (2/80) 0,08 0,0% (0/100) 0,0% (0/52)
11% (21/200) 31% (25/80) < 0,001 20% (20/100) 19% (10/52)
91% (181/198) 86% (67/78) 0,25 79% (77/98) 86% (44/51)
2,5% (5/198) 3,8% (3/78) 0,69 5,1% (5/98) 2,0% (1/51)
0,5% (1/198) 1,3% (1/78) 0,49 4,1% (4/98) 0,0% (0/51)
0,5% (1/198) 0,0% (0/78) > 0,99 3,1% (3/98) 0,0% (0/51)
0,5% (1/198) 1,3% (1/78) 0,49 0,0% (0/98) 0,0% (0/51)
2,0% (4/198) 5,1% (4/78) 0,23 3,1% (3/98) 2,0% (1/51)
1,0% (2/198) 0,0% (0/78) > 0,99 1,0% (1/98) 3,9% (2/51)
3,0% (6/198) 3,8% (3/78) 0,72 8,2% (8/98) 5,9% (3/51)
76% (151/200) 49% (39/80) < 0,001 55% (55/100) 62% (32/52)
5,0% (10/200) 14% (11/80) 0,02 19% (19/100) 3,8% (2/52)
1,5% (3/200) 16% (13/80) < 0,001 6,0% (6/100) 0,0% (0/52)
2,5% (5/200) 10% (8/80) 0,01 9,0% (9/100) 5,8% (3/52)
1,5% (3/200) 3,8% (3/80) 0,36 1,0% (1/100) 1,9% (1/52)
5,5% (11/200) 13% (10/80) 0,08 5,0% (5/100) 5,8% (3/52)
1,0% (2/200) 2,5% (2/80) 0,32 1,0% (1/100) 3,8% (2/52)
12% (24/200) 33% (26/80) < 0,001 25% (25/100) 23% (12/52)
6 ZUSAMMENFASSUNG DER KLINISCHEN STUDIEN
6.1 Ziele
Das primäre Ziel der klinischen Studie bestand in der Bewertung der Sicherheit
und Wirksamkeit einer früheren Ausführung der Prothese (endovaskuläre AAAProthese Zenith) als Alternative zur offenen chirurgischen Reparatur bei der
Primärbehandlung des infrarenalen abdominalen Aortenaneurysmas. Als
Studienhypothesen wurde untersucht, ob Standardrisikopatienten im Vergleich
mit den chirurgischen Kontrollpatienten eine geringere 30-Tage-M orbidität, nicht
schlechtere 30-Tage -Überlebensraten, nicht schlechtere 12-MonatsÜberlebensraten, nicht schlechteren 12-Monats-Behandlungserfolg sowie ein
besseres klinisches Gesamtbild zeigten. Zur Ermittlung der Sicherheit wurde
untersucht, ob die mit der endovaskulären AAA-Prothese Zenith behandelten
Patienten im Vergleich mit den offen chirurgisch behandelten Patienten eine
geringere 30-Tage-Morbidität, nicht schlechtere 30-Tage- und nicht schlechtere
12-Monats-Überlebensraten sowie nicht schlechteren 12-MonatsBehandlungserfolg zeigten. Die Wirksamkeitsbeurteilung beruhte auf dem
Ausschluss des Aneurysmas einschließlich Abwesenheit eines jeglichen
Endoleaks, Abwesenheit von Aneurysmawachstum (≥ 5 mm) und Abwesenheit
von wichtigen unerwünschten Ereignissen bezüglich der Prothese über einen
Nachbeobachtungszeitraum von einem Jahr. Sekundäre Ziele beinhalteten eine
Bewertung des klinischen Nutzens und der Maßstäbe zur Lebensqualität.
6.2 Studiendesign
Bei der in den USA durchgeführten klinischen Studie handelte es sich um eine
nicht-randomisierte multizentrische Studie zum Vergleich von
Standardrisikopatienten, die eine endovaskuläre Prothese erhielten, mit einer
offen chirurgisch behandelten Kontrollgruppe. Zwei weitere Studienarme
umfassten Hochrisikopatienten und Studienpatienten. In insgesamt
15 Zentren wurden 200 Standardrisikopatienten, 80 chirurgische
Kontrollpatienten, 100 Hochrisikopatienten und 52 Studienpatienten in die Studie
aufgenommen. Die Kontrollgruppe umfasste Patienten, die von der Anatomie
ihrer Gefäßsysteme her nicht uneingeschränkt für eine endovaskuläre Reparatur
in Frage kamen. Nachsorgeuntersuchungen wurden für folgende Zeitpunkte
angesetzt: vor der Entlassung, nach 1 Monat, 6 Monaten, 12 Monaten und
24 Monaten. Tabelle 6.2.1 zeigt Daten zu Patientennachsorge und
Verantwortlichkeit nach 1 Monat, 12 Monaten und 24 Monaten (den primären
Zeitpunkten für die Datenanalyse). In dieser Zusammenfassung enthaltene
Bildgebungsdaten beruhen auf Befunden aus einem unabhängigen zentralen
Bildanalyselabor (Zentrallabor), in dem CT-Bilder und abdominale Röntgenbilder
im Hinblick auf Veränderungen des Aneurysmadurchmessers, die relative
Migration von Prothese und Komponenten, die Unversehrtheit des Implantats
(Draht und Prothese) und die Anwesenheit und Art von Endoleaks analysiert
wurden. Klinische Vorfälle wurden in einer dafür zuständigen unabhängigen
Kommission besprochen und bewertet; die Sicherheit wurde von einer
Kommission für Daten und Sicherheit überwacht.
Die Patienten für offene Chirurgie und die Standardrisikopatienten für die ZenithProthese erfüllten die gleichen pathophysiologischen Risikokriterien. Für die
endovaskuläre Gruppe ausgeschlossen wurden Patienten mit peripherem
Thrombus im proximalen Hals, mit einem proximalen Hals von weniger als 15 mm
Länge, mit einem proximalen Halsdurchmesser von weniger als 18 mm oder
mehr als 28 mm (Außenwand zu Außenwand), mit starker Angulation des
proximalen Halses, mit einem Iliakadurchmesser von weniger als 7,5 mm oder
mehr als 20 mm (Außenwand zu Außenwand) an der distalen Befestigungsstelle
oder einer Befestigungsstelle an der Iliaka von weniger als 10 mm Länge.
Als Hochrisikopatienten für chirurgische Reparatur wurden Probanden mit
folgenden Merkmalen betrachtet: Alter über 80 Jahre, Kreatiningrundspiegel
> 2,0 mg/dl, Heimsauerstofftherapie, FEV1 < 1 Liter, Ejektionsfraktion < 25%, stark
behindernde COPD, NYHA-Klassifikation 3 oder 4, Verwachsungsbauch, Dialyse,
MI innerhalb der letzten 6 Monate, therapierefraktäre Hypertonie, früherer
Schlaganfall mit Restdefizit, kulturelle Einwände gegen Transfusion von Blut oder
Blutprodukten, frühere renale Bypass-Operation oder entzündliches Aneurysma.
Bevor ein Zentrum ohne Erfahrungen mit der endovaskulären AAA-Prothese
Zenith Patienten in die Hauptstudie aufnehmen konnte, mussten zuerst Patienten
unter Aufsicht eines erfahrenen Arztes behandelt werden. Bei diesen
Studienpatienten handelte es sich um Standardrisiko- und Hochrisikopatienten,
die den gleichen Nachsorgemaßnahmen wie die als echte Studienteilnehmer
operierten Patienten unterlagen.
66

Tabelle 6.2.1 Patientennachsorge und Verantwortlichkeiten
1
Behandlung Zenith Standardrisiko Oene Chirurgie Standardrisiko
Intervall
Keine Prothese 1 1
Umstellung auf offene Operation 0 2
1 Mo. 12 Mo. 24 Mo. 1 Mo. 12 Mo. 24 Mo.
2
2
2
1
2
2
0 0 n. zutr.
n. zutr. n. zutr. n. zutr.
Verstorben 1 7 17 2 3 n. zutr.
Aus Studie ausgeschieden/keine Nachsorge 0 0 2 0 4 n. zutr.
Verfügbar 198 190 178 78 73 n. zutr.
CT-Aufnahmen im Studienzentrum 191 168 110 69 58 n. zutr.
CT-Aufnahmen im Zentrallabor 190 165 99 69 59 n. zutr.
Aufnahmen der ableitenden Harnwege im Studienzentrum 179 153 108 n. zutr. n. zutr. n. zutr.
Aufnahmen der ableitenden Harnwege im Zentrallabor 178 149 93 n. zutr. n. zutr. n. zutr.
Vom Studienzentrum auf Endoleaks bewertet 187 163 107 n. zutr. n. zutr. n. zutr.
Vom Zentrallabor auf Endoleaks bewertet 161 148 92 n. zutr. n. zutr. n. zutr.
Vom Studienzentrum auf Aneurysmawachstum bewertet n. zutr. 149 104 n. zutr. n. zutr. n. zutr.
Vom Zentrallabor auf Aneurysmawachstum bewertet n. zutr. 151 94 n. zutr. n. zutr. n. zutr.
1
Der Probenumfang für die Datenanalyse ist für die o.g. Zeitpunkte und in den folgenden Tabellen verschieden. Diese Unterschiede beruhen auf der Verfügbarkeit der Patienten für die
Nachsorgeuntersuchungen sowie der Anzahl und der Qualität der für jeden Zeitpunkt für die Auswertung verfügbaren Bilder. Beispiel: Die Anzahl und Qualität der für die Bewertung
auf Endoleaks nach 12 Monaten verfügbaren Bilder sind nicht gleich der Anzahl und Qualität nach 24 Monaten, da sich die einzelnen Zeitpunkte hinsichtlich der Anzahl durchgeführter
bildgebender Untersuchungen, der Anzahl der von der Klinik dem Zentrallabor zur Verfügung gestellten Bilder und/oder der Anzahl Bilder mit einer für die Bewertung ausreichenden
Qualität unterscheiden. Sofern nicht anderweitig angegeben, ist der Gesamtwert für einen Zeitpunkt nicht kumulativ.
2
Der Gesamtwert für einen Zeitpunkt ist kumulativ.
6.3 Patientendemographische Daten
In den Tabellen 6.3.1 und 6.3.2 werden die Merkmale der Patienten und die anfänglichen Aneurysmadurchmesser in den mit offener Chirurgie oder der
endovaskulären AAA-Prothese Zenith behandelten Populationen einander gegenübergestellt.
Tabelle 6.3.1 Vergleich der Patientenmerkmale
Faktor Zenith Standardrisiko Oene Chirurgie Standardrisiko P-Wer t Zenith hohes Risiko Zenith Studienarm
Alter (Jahre) 71 ± 7 69 ± 7 0,03 77 ± 7 74 ± 8
Männl. Geschlecht 94% (187/200) 89% (71/80) 0,22 92% (92/100) 90% (47/52)
Aktuelle Gesundheitsprobleme
Periphere Verschlusskrankheit
Hypertonie 64% (127/200) 83% (65/78) 0,001 68% (67/99) 67% (35/52)
Nierenversagen 0,0% (0/197) 0,0% (0/79) > 0,99 5,2% (5/97) 1,9% (1/52)
16% (31/195) 25%
(19/76)
0,12 24% (23/96) 9,6% (5/52)
COPD 20% (39/199) 18% (14/78) 0,87 34% (33/98) 22% (11/51)
Thromboembolie 4,5% (9/199) 7,7% (6/78) 0,38 7,1% (7/99) 1,9% (1/52)
Lebererkrankung 2,1% (4/192) 5,1% (4/79) 0,24 1,0% (1/99) 1,9% (1/52)
Diabetes mellitus 12% (24/199) 15% (12/79) 0,55 17% (17/99) 14% (7/51)
Insulinabhängig 17% (4/24) 8,3% (1/12) 0,65 24% (4/17) 43% (3/7)
Frühere Gesundheitsprobleme
MI
39% (74/192) 29%
(23/80)
0,13 35% (34/98) 35% (18/52)
Dekompensierte Herzinsuffizienz 5,0% (10/199) 12% (9/78) 0,07 16% (16/100) 10% (5/50)
Angina 49% (98/198) 39% (31/79) 0,14 45% (44/98) 44% (23/52)
Arrhythmie 20% (40/197) 22% (17/78) 0,87 28% (27/98) 24% (12/51)
Zerebrovaskuläre Verschlusskrankheit 9,5% (19/199) 16% (13/79) 0,14 20% (20/99) 9,8% (5/51)
Systemische Infektion 1,0% (2/196) 0,0% (0/78) > 0,99 3,1% (3/97) 0,0% (0/49)
Krebs 22% (43/200) 19% (15/80) 0,74 31% (31/99) 29% (15/51)
Familiäre Aneurysmakrankheit 16% (24/150) 27% (17/63) 0,09 14% (11/77) 26% (10/38)
Frühere Operation an der Eingriffsstelle 10% (20/200) 15% (12/79) 0,22 10% (10/99) 14% (7/51)
Frühere Strahlentherapie an der Eingriffsstelle 0,5% (1/197) 0,0% (0/79) > 0,99 2,0% (2/100) 2,0% (1/51)
Alkoholmissbrauch 3,6% (7/193) 10% (8/77) 0,04 3,1% (3/96) 4,0% (2/50)
Nikotingebrauch
Noch nie geraucht
Rauchen aufgegeben 69% (133/193) 60% (48/80) 0,03 69% (66/96) 57% (28/49)
Aktiver Raucher 21% (40/193) 35% (28/80) 18% (17/96) 18% (9/49)
Auf Grund der Einschlusskriterien waren die Hochrisikopatienten älter (P < 0,001) und wiesen mehr Fälle von Nierenversagen (P = 0,004), von COPD (P = 0,01), von dekompensierter
Herzinsuffizienz (P = 0,004) und von zerebrovaskulärer Verschlusskrankheit (P = 0,02) als die Standardrisikopatienten auf.
10% (20/193) 5,0%
(4/80)
14% (13/96) 24% (12/49)
Tabelle 6.3.2 Ver teilung der Aneurysmadurchmesser
Durchmesserbereich Zenith Standardrisiko Oene Chirurgie Standardrisiko Zenith hohes Risiko Zenith Studienarm
< 30 mm
30–39 mm
40–49 mm
50–59 mm
60–69 mm
70–79 mm
80–89 mm
≥ 90 mm
Drei Hochrisikopatienten und ein Studienpatient wurden hinsichtlich der Verteilung der Aneurysmadurchmesser nicht evaluiert.
6.4 Ergebnisse
Die in den Tabellen 6.4.1 bis 6.4.3 zusammengestellten Daten wurden in den teilnehmenden Kliniken und im Zentrallabor gesammelt. Soweit verfügbar,
werden 24-Monatsdaten zur Verfügung gestellt. Die Kontrollpatienten wurden nicht über 12 Monate hinaus verfolgt, und einige Daten nach dem
12-Monatstermin wurden noch nicht beurteilt. Daher werden in diesem Abschnitt in einigen Fällen nur Daten bis zu 12 Monaten und in anderen Fällen bis zu
24 Monaten angegeben. In Tabelle 6.4.1 werden die im Rahmen der klinischen Studie implantierten Prothesen beschrieben. Tabelle 6.4.2 enthält Angaben
zu den primären Ergebnissen der Studie. Abbildung 6.4.1 und 6.4.2 stellen die Kaplan-Meier-Diagramme zu den 24-Monats-Überlebensraten hinsichtlich aller
Ursachen und hinsichtlich AAA-bezogener Ursachen dar. Eine unabhängige Kommission für klinische Vorfälle beurteilte alle Todesfälle auf mögliche
Zusammenhänge mit der Aneurysmareparatur. Alle früh eingetretenen Todesfälle (0–30 Tage) wurden als mit dem AAA in Verbindung stehend eingestuft.
Später als 30 Tage eingetretene Todesfälle wurden als mit dem AAA in Verbindung stehend eingestuft, wenn eine Beziehung zum AAA oder zur Prothese
hergestellt werden konnte. Tabelle 6.4.3 zeigt die Erfolgsmaßstäbe und Abbildung 6.4.3 zeigt das Kaplan-Meier-Diagramm zur Abwesenheit von Morbidität.
0,0% (0/199) 0,0% (0/78) 0,0% (0/100) 0,0% (0/52)
0,5% (1/199) 0,0% (0/78) 1,0% (1/100) 0,0% (0/52)
23% (45/199) 7,7% (6/78) 15% (15/100) 13% (7/52)
48% (95/199) 33% (26/78) 47% (47/100) 40% (21/52)
24% (47/199) 29% (23/78) 27% (27/100) 42% (22/52)
3,0% (6/199) 21% (16/78) 5,0% (5/100) 1,9% (1/52)
2,5% (5/199) 6,4% (5/78) 1,0% (1/100) 0,0% (0/52)
0,0% (0/199) 2,6% (2/78) 1,0% (1/100) 0,0% (0/52)
67

Tabelle 6.4.1 Implantierte Geräte
Faktor Zenith Standardrisiko Zenith hohes Risiko Zenith Studienarm
Hauptteil und Schenkel 99,5% (199/200)* 100% (100/100)
Verlängerung des Hauptteils
Verlängerung des ipsilateralen iliakalen Schenkels
Verlängerung des kontralateralen iliakalen Schenkels
Konverter 0,5% (1/199)** 0,0% (0/100) 0,0% (0/52)
Okkluder 0,0% (0/199) 0,0% (0/100) 0,0% (0/52)
*Ein Standardrisikopatient erhielt auf Grund der Gewundenheit und Verkalkung des Zugangsgefäßes keine Prothese.
**Der Konverter wurde ohne Okkluder verwendet.
1
Eine Prothese war Sonderanfertigung.
2
Zwei Standardrisikopatienten und ein Hochrisikopatient erhielten postoperativ Hauptteilverlängerungen; ein Standardrisikopatient erhielt zwei Hauptteilverlängerungen.
3
Zwei Standardrisikopatienten und ein Hochrisikopatient erhielten postoperativ ipsilaterale Schenkelverlängerungen.
4
Vier Standardrisikopatienten, zwei Hochrisikopatienten und ein Studienpatient erhielten postoperativ kontralaterale Schenkelverlängerungen.
5
Drei Standardrisiko- und drei Hochrisikopatienten erhielten intraoperativ sowohl ipsilaterale als auch kontralaterale Schenkelverlängerungen; ein Standardrisikopatient erhielt
postoperativ sowohl ipsilaterale als auch kontralaterale Schenkelverlängerungen.
2
3,5
4,5
1,5% (3/199) 1,0% (1/100) 5,8% (3/52)
9,5% (19/199) 11% (11/100) 0,0% (0/52)
11% (21/199) 11% (11/100) 13,5% (7/52)
1
100% (52/52)
Tabelle 6.4.2 Primärergebnisse
Faktor Zenith Standardrisiko
Alle Todesfälle
Alle Todesfälle
Alle Todesfälle
2
(0–30 Tage)
(31–365 Tage)
mit AAA in Verbindung stehend 0,0% (0/199) 1,3% (1/80) 0,29 3,0% (3/100) 0,0% (0/52)
nicht im Zusammenhang mit AAA
stehend
(0–365 Tage)
2, 3
2,3,4
0,5% (1/199) 2,5% (2/80) 0,20 2,0% (2/100) 1,9% (1/52)
3,0% (6/199) 1,3% (1/80) 0,68 7,0% (7/100) 9,6% (5/52)
3,0% (6/199) 0,0% (0/80) 0,19 4,0% (4/100) 9,6% (5/52)
3,5% (7/199) 3,8% (3/80) > 0,99 9,0% (9/100) 11,5% (6/52)
1
Oene Chirurgie Standardrisiko P-Wer t Zenith hohes Risiko Zenith Studienarm
mit AAA in Verbindung stehend 0,5% (1/199) 3,8% (3/80) 0,07 5,0% (5/100) 1,9% (1/52)
nicht im Zusammenhang mit AAA
stehend
3,0% (6/199) 0,0% (0/80) 0,19 4,0% (4/100) 9,6% (5/52)
Ruptur
(0–30 Tage) 0,0% (0/199) n. zutr. n. zutr. 0,0% (0/100) 0,0% (0/52)
(31–365 Tage) 0,0% (0/199) n. zutr. n. zutr. 1,0% (1/100) 0,0% (0/52)
(0–365 Tage) 0,0% (0/199) n. zutr. n. zutr. 1,0% (1/100) 0,0% (0/52)
Umstellung
(0–30 Tage) 0,0% (0/199) n. zutr. n. zutr. 0,0% (0/100) 0,0% (0/52)
(31–365 Tage)
(0–365 Tage)
Unerwünschte Ereignisse
(0–30 Tage)
(31–365 Tage)
(0–365 Tage)
1
Nenner ist 199, da ein Standardrisikopatient keine Prothese erhielt.
2
Alle Todesfälle (0–30 Tage) wurden als mit dem AAA und dem Verfahren in Verbindung stehend betrachtet.
3
Von den Todesfällen (31–365 Tage) wurden vier als mit dem AAA in Verbindung stehend betrachtet: 1 chirurgische Komplikation (septischer Schock durch ischämische Kolitis)
und 3 Hochrisikofälle (Pankreatitis mit Nierenversagen und Sepsis, Blutung von Aneurysma im oberen Abdomen [unbehandeltes AAA] und multiples Systemversagen).
4
Von den Todesfällen (0–365 Tage) wurden zehn als mit dem AAA in Verbindung stehend betrachtet: 1 Standardrisikofall (Herzversagen), 3 chirurgische Komplikationen (massive
Blutung, mesenterische Ischämie und septischer Schock durch ischämische Kolitis), 5 Hochrisikofälle (akute respiratorische Insuffizienz, Herzversagen mit Lungenembolie,
Pankreatitis mit Nierenversagen und Sepsis, Blutung von Aneurysma im oberen Abdomen [unbehandeltes AAA] und multiples Systemversagen), sowie 1 Studienfall (Verdacht auf
Herzversagen).
5
Standardrisikopatienten wurden auf Grund eines persistierenden proximalen Endoleaks vom Typ I und eines neuen suprarenalen Aortenaneurysmas umgestellt. Drei offenchirurgische Patienten hatten massive Blutungen; in zwei Fällen war eine Nachoperation erforderlich und ein Patient verstarb.
6
Unerwünschte Ereignisse im Morbiditätsindex enthalten.
5
5
6
6
6
1,0% (2/199) n. zutr. n. zutr. 1,0% (1/100) 0,0% (0/52)
1,0% (2/199) n. zutr. n. zutr. 1,0% (1/100) 0,0% (0/52)
20% (40/200) 43% (34/80) < 0,001 32% (32/100) 27% (14/52)
8,6% (17/199) 14% (11/78) 0,25 21% (21/98) 14% (7/51)
25% (49/200) 51% (41/80) < 0,001 45% (45/100) 38% (20/52)
Es gab keine signifikanten geschlechtsbezogenen Unterschiede zwischen
männlichen und weiblichen Patienten mit endovaskulärer AAA-Prothese Zenith
im Hinblick auf Überlebensrate und Abwesenheit von wichtigen unerwünschten
Ereignissen. (Fehlerbalken in den Abbildungen 6.4.1, 6.4.2 und 6.4.3 stellen
die 95%-Vertrauensgrenzen dar.) Abbildung 6.4.1 zeigt die Überlebensrate
nach 24 Monaten mit Bezug auf alle Ursachen. Die Begleittabelle beschreibt die
Kaplan-Meier-Analyse nach 1, 6, 12 und 24 Monaten.
Abbildung 6.4.1 Überlebensrate nach 24 Monaten
Zenith Standardrisiko
(N = 200, 1 Todesfall bis zum 30. Tag
4 Todesfälle bis zum 6. Monat
7 Todesfälle bis zum 12. Monat
17 Todesfälle bis zum 24. Monat)
Offene Chirurgie Standardrisiko
(N = 80, 2 Todesfälle bis zum 30. Tag
3 Todesfälle bis zum 6. Monat
Überlebende in Prozent
3 Todesfälle bis zum 12. Monat)
Monate nach dem Eingri
1 Monat 6 Monate 12 Monate 24 Monate
n % Überleben n % Überleben n % Überleben n % Überleben
Zenith
Standard-
198* 99,5 194 98,0 190 96,5 97 90,9
risiko
Offene
Chirurgie
Standardrisiko
n = überlebende und für die Nachsorge am Ende des Intervalls verfügbare Patienten
P = 0,81
*Ein Patient vor Ablauf eines Monats verstorben, ein weiterer Patient erhielt keine
Prothese.
78 97,5 73 96,2 67 96,2
zutr.
n.
n.
zutr.
Abbildung 6.4.2 zeigt die mit dem AAA in Verbindung stehende 24-MonatsÜberlebensrate (vom Ausschuss für klinische Vorfälle ermittelt). Die
Begleittabelle beschreibt die Kaplan-Meier-Analyse nach 1, 6, 12 und 24
Monaten.
Abbildung 6.4.2 AAA-bezogene 24-Monats-Überlebensrate
Zenith Standardrisiko
(N = 200, 1 Todesfall bis zum 30. Tag
1 Todesfall bis zum 6. Monat
1 Todesfall bis zum 12. Monat
2 Todesfälle bis zum 24. Monat)
Offene Chirurgie Standardrisiko
(N = 80, 2 Todesfälle bis zum 30. Tag
3 Todesfälle bis zum 6. Monat
Todesfällen, in Prozent
3 Todesfälle bis zum 12. Monat)
Frei von aneurysmenbedingten
Ereignis: aneurysmabedingter Exitus
Monate nach dem Eingri
1 Monat 6 Monate 12 Monate 24 Monate
n % Überleben n % Überleben n % Überleben n % Überleben
Zenith
Standard-
198* 99,5 194 99,5 190 99,5 97 99,0
risiko
Offene
Chirurgie
Standardrisiko
n = überlebende und für die Nachsorge am Ende des Intervalls verfügbare Patienten
P= 0,04
*Ein Patient vor Ablauf eines Monats verstorben, ein weiterer Patient erhielt keine
Prothese.
78 97,5 73 96,2 67 96,2
zutr.
n.
n.
zutr.
68

Tabelle 6.4.3 Erfolgsmaßstäbe
Faktor Zenith Standardrisiko Oene Chirurgie Standardrisiko Zenith hohes Risiko Zenith Studienarm
Technischer Erfolg
Verfahrenserfolg nach
30 Tagen
Behandlungserfolg nach
12 Monaten
1
Durchgängige Prothese nach Entfaltung.
2
Technischer Erfolg ohne große Komplikationen, durchgängige Prothese und keine Endoleaks von Typ I oder III nach 30 Tagen.
3
Verfahrenserfolg auf 12 Monate ausgedehnt, ohne Aneurysmawachstum (> 5 mm).
Abbildung 6.4.3 zeigt die Abwesenheit von Morbidität (Vorfälle im Morbiditätsindex) bis 12 Monate. Die Begleittabelle beschreibt die Kaplan-Meier-Analyse nach 1,
6 und 12 Monaten.
1
2
3
99,5% (199/200) 98,8% (79/80) 100% (100/100) 100% (52/52)
95,1% (155/163) 88% (60/68) 86% (70/81) 91% (30/33)
89% (122/137) 85% (52/61) 70% (44/63) 87% (26/30)
Abbildung 6.4.3 Abwesenheit von Morbidität (0–365 Tage)
Zenith Standardrisiko
(N = 200, 38 Patienten mit Ereignissen bis zum 30. Tag
43 Patienten mit Ereignissen bis zum 6. Monat
45 Patienten mit Ereignissen bis zum 12. Monat)
Offene Chirurgie Standardrisiko
(N = 80, 34 Patienten mit Ereignissen bis zum 30. Tag
Frei von Morbidität, in Prozent
38 Patienten mit Ereignissen bis zum 6. Monat
41 Patienten mit Ereignissen bis zum 12. Monat)
Monate nach dem Eingriff
1 Monat 6 Monate 12 Monate
n % n % n %
Zenith Standardrisiko 162 81,0 154 78,5 133 77,4
Offene Chirurgie Standardrisiko 45 57,1 39 52,0 33 47,8
n = überlebende und am Ende des Intervalls morbiditätsfreie Patienten
P = <0,001
Die Tabellen 6.4.4 bis 6.4.7 enthalten Angaben zu den Ergebnissen von Patienten mit der endovaskulären AAA-Prothese Zenith entsprechend den
Informationen vom Zentrallabor. Das Zentrallabor analysierte folgende Geräteleistungsfaktoren: Unversehrtheit der Prothese (Tabelle 6.4.4), Durchgängigkeit
der Prothese (Tabelle 6.4.5), Migration (Tabelle 6.4.6) und Ablösung des Ansatzes (Tabelle 6.4.7).
Tabelle 6.4.4 Abdominale Röntgenbefunde – Unversehrtheit der Prothese
Faktor Zenith Standardrisiko Zenith hohes Risiko Zenith Studienarm
Stentfrakturen
1
Vor der Entlassung 0,0% (0/172) 0,0% (0/81) 0,0% (0/39)
30 Tage 0,0% (0/172) 0,0% (0/83) 0,0% (0/43)
6 Monate 0,0% (0/166) 0,0% (0/78) 0,0% (0/35)
12 Monate 0,0% (0/148) 0,0% (0/60) 0,0% (0/28)
24 Monate 0,0% (0/93) 0,0% (0/42) 0,0% (0/19)
Hakenablösung
2
Vor der Entlassung 0,0% (0/176) 0,0% (0/86) 0,0% (0/39)
30 Tage 0,0% (0/178) 0,0% (0/86) 0,0% (0/43)
6 Monate 1,2% (2/167) 2,5% (2/80) 0,0% (0/35)
12 Monate 2,0% (3/149) 1,7% (1/60) 0,0% (0/28)
24 Monate 1,1% (1/93) 0,0% (0/42) 0,0% (0/19)
Ruptur des Prothesenmaterials
Vor der Entlassung 0,0% (0/176) 0,0% (0/86) 0,0% (0/39)
30 Tage 0,0% (0/178) 0,0% (0/86) 0,0% (0/43)
6 Monate 0,0% (0/167) 0,0% (0/80) 0,0% (0/35)
12 Monate 0,0% (0/149) 0,0% (0/60) 0,0% (0/28)
24 Monate 0,0% (0/93) 0,0% (0/42) 0,0% (0/19)
1
Prozentangaben zu Stentfrakturen beziehen sich auf den Hauptteil. Das Zentrallabor stellte weiterhin keine Frakturen am rechten iliakalen Schenkel, linken iliakalen Schenkel,
Okkluder, Konverter, der linken iliakalen Verlängerung, rechten iliakalen Verlängerung oder der Hauptteilverlängerung fest.
2
Patienten mit Ablösung von 1 oder 2 Haken (von insgesamt 10 oder 12); keine schädigenden Folgen.
Tabelle 6.4.5 CT-Befunde – Durchgängigkeit der Prothese
Faktor Zenith Standardrisiko Zenith hohes Risiko Zenith Studienarm
Durchgängigkeit der Prothese
30 Tage 100% (185/185) 99% (85/86) 100% (47/47)
6 Monate 99% (183/184) 100% (74/74) 100% (39/39)
12 Monate 99% (153/155) 100% (62/62) 100% (30/30)
24 Monate 100% (96/96) 100% (33/33) 100% (25/25)
Tabelle 6.4.6 CT-Befunde – Migration der Prothese (Hauptteil)
Faktor Zenith Standardrisiko Zenith hohes Risiko Zenith Studienarm
Prothesenmigration (> 5 mm) nach 12 Monaten
mit klinischen Folgen1 oder Intervention 0,0% (0/162) 0,0% (0/71) 0,0% (0/34)
ohne klinische Folgen1 oder Intervention 2,5% (4/162) 2,8% (2/71) 0,0% (0/34)
Prothesenmigration (> 10 mm) 0,0% (0/162) 0,0% (0/71) 0,0% (0/34)
1
Migration mit klinischen Folgen würde Endoleaks, Umstellung, Ruptur oder mit AAA in Verbindung stehende Todesfälle umfassen.
69

Tabelle 6.4.7 Abdominale Röntgenbefunde – Ansatzablösung
Faktor Zenith Standardrisiko Zenith hohes Risiko Zenith Studienarm
Ansatzablösung
Vor der Entlassung 0,0% (0/176) 0,0% (0/86) 0,0% (0/39)
30 Tage 0,0% (0/178) 0,0% (0/86) 0,0% (0/43)
6 Monate 0,0% (0/167) 0,0% (0/80) 0,0% (0/35)
12 Monate 0,0% (0/149) 0,0% (0/60) 0,0% (0/28)
24 Monate 0,0% (0/93) 0,0% (0/42) 0,0% (0/19)
6.5 Endoleak-Management
Während der klinischen Studie wurden Endoleaks vom Typ I beim ursprünglichen Eingriff mit Hilfe eines Ballons behandelt; bei ausbleibendem Erfolg wurden
zusätzliche Prothesen verwendet. Endoleaks vom Typ II wurden über einen Zeitraum von 1 bis 6 Monaten verfolgt, um festzustellen, ob sich eine spontane
Thrombose ergab; wenn sich das Aneurysma nicht vergrößerte wurden sie nach Maßgabe des verantwortlichen Arztes mit endovaskulären Techniken
behandelt. Wenn das Aneurysma Wachstum zeigte, wurde zur Behandlung eine Embolisation oder Ligation in Betracht gezogen und in einigen Fällen auch
durchgeführt. Durch Prothesendefekte verursachte Endoleaks des Typs III, mangelnde Dichtigkeit oder Ablösung der modularen Komponenten wurden durch
Anwendung eines Ballons oder mit Prothesen behandelt. Nach Angaben des angiographischen Zentrallabors traten in der klinischen US-Studie keine
Endoleaks vom Typ IV auf. Das zur Herstellung der endovaskulären AAA-Prothese Zenith verwendete Material besitzt Standardstärke und ist mit dem bei der
offenen Chirurgie verwendeten Material identisch. Tabelle 6.5.1 zeigt die Inzidenz von Endoleaks nach Bewertungsintervall, wie vom Zentrallabor bei den
Standardrisiko-, Hochrisiko- und Studienpatienten festgestellt.
Tabelle 6.5.1 Endoleaks (alle Typen, neu und persistierend)
Faktor Zenith Standardrisiko Zenith hohes Risiko Zenith Studienarm
Endoleaks
Vor der Entlassung 15% (23/153) 14% (11/78) 12% (3/26)
1
30 Tage
1
6 Monate
1
12 Monate
1
Beinhaltet neu aufgetretene und persistierende Endoleaks.
Die Tabellen 6.5.2 bis 6.5.4 zeigen die Inzidenz des erstmaligen Auftretens eines Endoleaks nach Bewertungsintervall, wie vom Zentrallabor bei oder vor der
30-Tage-Untersuchung, 6-Monatsuntersuchung und 12-Monatsuntersuchung für die Standardrisiko-, Hochrisiko- und Studienpatienten festgestellt. Die Anzahl der
Patienten ohne Lecks ist ebenfalls angegeben.
9,9% (16/161) 12% (9/75) 6,3% (2/32)
8,7% (15/172) 11% (8/70) 8,6% (3/35)
7,4% (11/148) 8,8% (5/57) 3,4% (1/29)
Tabelle 6.5.2 Erstes Auftreten eines Endoleaks1 bei Standardrisikopatienten
Faktor
Bis 1-Monatsuntersuchung
N = 179
% Endoleak
1
Anschließend
2
leckfrei
6-Monatsuntersuchung
N = 172
% Endoleak
Anschließend
1
leckfrei
12-Monatsuntersuchung
N = 148
2
% Endoleak
Anschließend
1
Endoleaks 17 31 17 2,3 4 3 3,4 5 2
Proximal Typ I 2,8 5 4 0,0 0 0 0,0 0 0
Distal
Typ I 1,7 3 1 0,0 0 0 0,7 1 1
Typ II 9,5 17 9 2,3 4 3 1,4 2 1
Typ III 1,1 2 2 0,0 0 0 0,7 1 0
Typ IV 0,0 0 0 0,0 0 0 0,0 0 0
Mehrfach 1,1 2 1 0,0 0 0 0,0 0 0
Unbekannt 1,1 2 0 0,0 0 0 0,7 1 0
1
Identifikation durch Zentrallabor.
2
Spätere Endoleaks können von anderem Typ als das erste Endoleak gewesen sein.
3
Nur 2 Patienten zeigten neue Endoleaks nach 12 Monaten; keine Nachsorgedaten für 24 Monate.
Tabelle 6.5.3 Erstes Auftreten eines Endoleaks1 bei Hochrisikopatienten
Faktor
Bis 1-Monatsuntersuchung
N = 88
% Endoleak
Anschließend
1
leckfrei
2
6-Monatsuntersuchung
N = 70
% Endoleak
Anschließend
1
leckfrei
2
12-Monatsuntersuchung
N = 57
% Endoleak
Anschließend
1
Endoleaks 18 16 6 2,9 2 0 3,5 2 1
Proximal Typ I 2,3 2 1 0,0 0 0 0,0 0 0
Distal
Typ I 1,1 1 1 0,0 0 0 0,0 0 0
Typ II 9,1 8 3 1,4 1 0 1,8 1 0
Typ III 0,0 0 0 1,4 1 0 1,8 1 1
Typ IV 0,0 0 0 0,0 0 0 0,0 0 0
Mehrfach 4,5 4 0 0,0 0 0 0,0 0 0
Unbekannt 1,1 1 1 0,0 0 0 0,0 0 0
1
Identifikation durch Zentrallabor.
2
Spätere Endoleaks können von anderem Typ als das erste Endoleak gewesen sein.
3
Keine Endoleaks nach 12 Monaten; keine Nachsorgedaten für 24 Monate.
3
leckfrei
3
leckfrei
2
2
Tabelle 6.5.4 Erstes Auftreten eines Endoleaks1 bei Studienpatienten
Faktor
Bis 1-Monatsuntersuchung
N = 36
% Endoleak
1
Anschließend
2
leckfrei
6-Monatsuntersuchung
N = 35
% Endoleak
Anschließend
1
leckfrei
2
12-Monatsuntersuchung
N = 29
% Endoleak
Anschließend
1
Endoleaks 11 4 2 2,9 1 0 0,0 0 0
Proximal Typ I 0,0 0 0 0,0 0 0 0,0 0 0
Distal
Typ I 0,0 0 0 0,0 0 0 0,0 0 0
Typ II 5,6 2 1 2,9 1 0 0,0 0 0
Typ III 2,8 1 0 0,0 0 0 0,0 0 0
Typ IV 0,0 0 0 0,0 0 0 0,0 0 0
Mehrfach 0,0 0 0 0,0 0 0 0,0 0 0
Unbekannt 2,8 1 1 0,0 0 0 0,0 0 0
1
Identifikation durch Zentrallabor.
2
Spätere Endoleaks können von anderem Typ als das erste Endoleak gewesen sein.
3
Keine Endoleaks nach 12 Monaten; keine Nachsorgedaten für 24 Monate.
70
3
leckfrei
2

6.6 Aneurysmaänderungen
Die Tabellen 6.6.1 bis 6.6.3 zeigen die Änderungen des Aneurysmadurchmessers bei den endovaskulären Patienten, wie vom Zentrallabor festgestellt.
Tabelle 6.6.1 zeigt die maximale Änderung des Aneurysmadurchmessers nach Intervall. Die Tabellen 6.6.2 und 6.6.3 zeigen die Aneurysmaänderungen und
Endoleaks nach 12 bzw. 24 Monaten.
Tabelle 6.6.1 Änderungen des maximalen Aneurysmadurchmessers nach Intervall*
Faktor Zenith Standardrisiko Zenith hohes Risiko Zenith Studienarm
Von vor Entlassung 30 Tage
Verkleinerung > 5 mm 1,7% (3/180) 4,8% (4/84) 0,0% (0/40)
Unverändert 97% (174/180) 94% (79/84) 97,5% (39/40)
Vergrößerung > 5 mm 1,7% (3/180) 1,2% (1/84) 2,5% (1/40)
Änderung von vor Entlassung 6 Monate
Verkleinerung > 5 mm 36% (63/173) 41% (30/73) 49% (18/37)
Unverändert 62% (108/173) 59% (43/73) 51% (19/37)
Vergrößerung > 5 mm 1,2% (2/173) 0,0% (0/73) 0,0% (0/37)
Änderung von vor Entlassung 12 Monate
Verkleinerung > 5 mm 68% (102/151) 63% (39/62) 67% (20/30)
Unverändert 31% (47/151) 35% (22/62) 33% (10/30)
Vergrößerung > 5 mm 1,3% (2/151) 1,6% (1/62) 0,0% (0/30)
Änderung von vor Entlassung 24 Monate
Verkleinerung > 5 mm 78% (74/94) 75% (27/36) 71% (17/24)
Unverändert 19% (18/94) 25% (9/36) 25% (6/24)
Vergrößerung > 5 mm 2,1% (2/94) 0,0% (0/36) 4,2% (1/24)
*Umfasst nur Patienten mit auswertbaren Filmen und Messungen der Aneurysmaänderung von 1 Monat bis 24 Monate.
Tabelle 6.6.2 Änderung der Aneurysmagröße und Endoleak nach 12 Monaten
Zenith Standardrisiko
N = 140
Zenith hohes Risiko
N = 53
Zenith Studienarm
N = 27
Endoleak Endoleak Endoleak
Faktor
Änderung der Aneurysmagröße von vor Entlassung bis 12 Monate
N n % N n % N n %
Verkleinerung > 5 mm 96 3 3,0 35 3 9,0 20 0 0,0
Unverändert 42 8 19 17 2 12 7 1 14
Vergrößerung > 5 mm 2 0 0,0 1 0 0,0 0 0 0,0
Tabelle 6.6.3 Änderung der Aneurysmagröße und Endoleak nach 24 Monaten
Zenith Standardrisiko
N = 90
Zenith hohes Risiko
N = 31
Zenith Studienarm
N = 21
Endoleak Endoleak Endoleak
Faktor
Änderung der Aneurysmagröße von vor Entlassung bis 24 Monate
N n % N n % N n %
Verkleinerung > 5 mm 71 3 4,0 24 1 4,0 16 0 0,0
Unverändert 18 1 6,0 7 3 43 5 0 0,0
Vergrößerung > 5 mm 1 1 100 1 0 0,0 0 0 0,0
6.7 AAA-bezogene Sekundärinterventionen
Mit dem AAA in Verbindung stehende sekundäre Interventionen innerhalb des ersten Jahres waren bei 11% der Zenith-Standardrisikopatienten, 13% der
Zenith-Hochrisikopatienten und 5,8% der Zenith-Studienpatienten erforderlich; siehe Tabelle 6.7.1. Über 50% der sekundären Interventionen beinhalteten
eine Katheterisierung zur Behandlung eines Endoleaks. Mit dem AAA in Verbindung stehende sekundäre Interventionen innerhalb des zweiten Jahres waren
bei 4,2% der Zenith-Standardrisikopatienten, 2,2% der Zenith-Hochrisikopatienten und 2,3% der Zenith-Studienpatienten erforderlich; siehe Tabelle 6.7.2.
Tabelle 6.7.1 Sekundäre Interventionen (bis 12 Monate)
Intervention
Umstellung auf offene Operation 2 1,0 1 1,0 0 0,0
Zenith Standardrisiko
N = 199
Zenith hohes Risiko
N = 100
Zenith Studienarm
N = 52
n % n % n %
Patienten mit ≥ 1 Intervention 21 11 13 13 3 5,8
Behandlung eines Endoleaks
Embolisation 5 2,5 3 3,0 1 2,0
Hilfskomponente 6 3,0 4 4,0 1 2,0
Stent 1 0,5 0 0,0 0 0,0
Angioplastie 0 0,0 1 1,0 0 0,0
Behandlung eines wachsenden Aneurysmas
Embolisation 0 0,0 0 0,0 0 0,0
Behandlung eines Schenkelverschlusses 1 0,5 3 3,0 1 2,0
Behandlung einer Ansatzstenose 1 0,5 0 0,0 0 0,0
Behandlung einer Nierenarterie 5 2,5 0 0,0 0 0,0
Behandlung einer infrainguinalen Ischämie 1 0,5 1 1,0 0 0,0
Behandlung von mehreren Vorkommnissen 1 0,5 1 1,0 0 0,0
Tabelle 6.7.2 Sekundäre Interventionen (> 12 bis 24 Monate)
Intervention
Umstellung auf offene Operation 1 0,5 1 1,1 0 0,0
Zenith Standardrisiko
N = 190
Zenith hohes Risiko
N = 90
Zenith Studienarm
N = 44
n % n % n %
Patienten mit ≥ 1 Intervention 8 4,2 2 2,2 1 2,3
Behandlung eines Endoleaks
Embolisation 3
1
1,6 0 0,0 1
2
Hilfskomponente 1 0,5 0 0,0 0 0,0
Stent 0 0,0 1 1,1 0 0,0
Angioplastie 0 0,0 0 0,0 0 0,0
Behandlung eines wachsenden Aneurysmas
Embolisation 1 0,5 0 0,0 1
2
Behandlung eines Schenkelverschlusses 1 0,5 0 0,0 0 0,0
Behandlung einer Ansatzstenose 0 0,0 0 0,0 0 0,0
Behandlung einer Nierenarterie 0 0,0 1 1,1 0 0,0
Behandlung einer infrainguinalen Ischämie 1 0,5 0 0,0 0 0,0
Behandlung von mehreren Vorkommnissen 1 0,5 0 0,0 0 0,0
1
Patient erhielt ebenfalls eine Hilfskomponente.
2
Intervention zur Behandlung eines wachsenden Aneurysmas und eines Endoleaks.
71
2,3
2,3

6.8 Maßstäbe für sekundäre Ergebnisse
Wie in Tabelle 6.8.1 beschrieben, zeigte die Behandlung eines AAA mit der endovaskulären AAA-Prothese Zenith im Vergleich mit der chirurgischen
Kontrollgruppe signifikante Vorteile hinsichtlich Rekonvaleszenz und Lebensqualität.
Tabelle 6.8.1 Sekundäre Ergebnisse nach Behandlungsgruppe
Faktor Zenith Standardrisiko Oene Chirurgie Standardrisiko P-Wert Zenith hohes Risiko Zenith Studienarm
Anästhesiezeit (Min.) 221,6 ± 67,3 304,5 ± 102,7 < 0,001 218,9 ± 69,6 213,9 ± 57,7
Verfahrenszeit (Min.) 153,2 ± 56,3 238,7 ± 92,2 < 0,001 153,5 ± 58,6 155,9 ± 43,2
Verabreichte Blutbankprodukte 5,0% (10/200) 84% (67/80) < 0,001 12% (12/100) 3,8% (2/52)
Blutverlust (ml) 299 ± 324 1676 ± 1676 < 0,001 356 ± 514 265 ± 226
Tage auf der Intensivstation 0,4 ± 0,9 3,4 ± 4,6 < 0,001 0,5 ± 1,2 0,5 ± 0,9
Tage bis Entlassung 2,6 ± 1,7 8,8 ± 5,6 < 0,001 3,0 ± 2,8 2,7 ± 1,5
Tage bis orale Flüssigkeiten 0,5 ± 0,8 3,9 ± 2,5 < 0,001 0,5 ± 0,6 0,7 ± 0,5
Tage bis Normalkost 1,3 ± 1,2 6,6 ± 4,9 < 0,001 1,3 ± 0,8 1,1 ± 0,7
Tage bis zur normalen Darmfunktion 2,6 ± 1,4 4,2 ± 2,1 < 0,001 2,6 ± 1,5 2,0 ± 1,2
Tage bis Gehfähigkeit 1,2 ± 0,7 3,5 ± 3,4 < 0,001 1,2 ± 0,7 1,2 ± 0,6
Stunden der Intubation 1,9 ± 2,2 11,7 ± 13,6 < 0,001 1,2 ± 1,7 2,6 ± 4,6
Maximale Temperatur (°F) 101,1 ± 1,3 100,7 ± 1,2 0,06 100,8 ± 1,1 101,0 ± 1,2
7 AUSWAHL UND BEHANDLUNG DER PATIENTEN
Siehe Abschnitt 4, Warnhinweise und Vorsichtsmaßnahmen
Individuelle Gestaltung der Behandlung
Cook empfiehlt, die Durchmesser der endovaskulären AAA-Prothese Zenith
Flex so auszuwählen, wie in den Tabellen 10.5.1 und 10.5.2 beschrieben. Die
endovaskuläre AAA-Prothese Zenith Flex sollte sich von der untersten
Nierenarterie bis knapp über die Gabelung der inneren Beckenarterie
(Hypogastrika) erstrecken. Alle Längen und Durchmesser der für die
vollständige Durchführung des Eingriffs erforderlichen Produkte sollten dem
Arzt zur Verfügung stehen, insbesondere wenn die präoperativen
Fallplanungsmessungen (Behandlungsdurchmesser/-längen) unsicher sind.
Dieser Ansatz ermöglicht eine größere intraoperative Flexibilität zur Erzielung
optimaler Operationsergebnisse. Die weiter oben in Abschnitt 6,
ZUSAMMENFASSUNG DER KLINISCHEN STUDIEN, beschriebenen Risiken
und Nutzen sind vor Verwendung der endovaskulären AAA-Prothese Zenith
Flex für jeden einzelnen Patienten sorgfältig abzuwägen. Weitere bei der
Patientenauswahl zu beachtende Faktoren sind unter anderem:
• Alter und Lebenserwartung des Patienten
• Komorbiditäten (z.B. Herz-, Lungen- oder Niereninsuffizienz vor dem Eingriff,
krankhafte Adipositas)
• Eignung des Patienten für eine offene chirurgische Reparatur
• Anatomische Eignung des Patienten für eine endovaskuläre Reparatur
• Das Risiko einer Aneurysmaruptur im Vergleich zum Risiko der Behandlung
mit der endovaskulären AAA-Prothese Zenith Flex
• Verträglichkeit einer Voll-, Regional- oder Lokalanästhesie durch den
Patienten
• Größe und Morphologie des iliofemoralen Zugangsgefäßes (minimaler
Thrombus, Kalziumablagerungen und/oder Gewundenheit) sollten mit den
Gefäßzugangsmethoden und Geräten kompatibel sein, die dem
Einführprofil einer Gefäßeinführschleuse von 14–22 French entsprechen.
• Nicht-aneurysmatisches infrarenales Aortensegment (Hals) proximal zum
Aneurysma:
• mit einer Länge von mindestens 15 mm,
• mit einem Durchmesser (Außenwand zu Außenwand) von maximal
32 mm und mindestens 18 mm,
• mit einem Winkel von weniger als 60 Grad relativ zur Längsachse des
Aneurysmas und
• mit einem Winkel von weniger als 45 Grad relativ zur Achse der
suprarenalen Aorta
• Distale Befestigungsstelle an der Iliaka von mindestens 10 mm Länge und
7,5 mm bis 20 mm Durchmesser (Außenwand zu Außenwand)
• Keine signifikanten Verschlusskrankheiten der Aa. femorales oder Aa. iliacae,
die den Blutstrom durch die endovaskuläre Prothese einschränken würden
Die endgültige Behandlungsentscheidung liegt im Ermessen von Arzt und
Patient.
8 INFORMATIONEN ZUR AUFKLÄRUNG DES PATIENTEN
Arzt und Patient (und/oder dessen Angehörige) sollten bei der Erwägung
dieser endovaskulären Prothese die Risiken und Nutzen eines Eingriffs
besprechen. Diese sind u.a.:
• Risiken der endovaskulären Reparatur und Unterschiede zwischen
endovaskulärer und chirurgischer Reparatur
• Potenzielle Vorteile einer herkömmlichen offenen chirurgischen Reparatur
• Potenzielle Vorteile einer endovaskulären Reparatur
• Die Möglichkeit, dass nach der ursprünglichen endovaskulären Reparatur
eine weitere interventionelle oder offene chirurgische Reparatur des
Aneurysmas erforderlich werden kann.
Zusätzlich zu den Risiken und Nutzen einer endovaskulären Reparatur sollte
der Arzt beurteilen, inwieweit der Patient willens und in der Lage ist, der
postoperativen Nachsorge Folge zu leisten, da diese für ein sicheres und
effektives Ergebnis notwendig ist. Im Folgenden sind weitere Themen
aufgeführt, die mit dem Patienten hinsichtlich der Erwartungen für die Zeit
nach der endovaskulären Reparatur zu besprechen sind:
• Über das langfristige Verhalten der endovaskulären Prothese ist derzeit
nichts bekannt. Die Patienten sind darauf hinzuweisen, dass die
endovaskuläre Therapie eine lebenslange regelmäßige Nachsorge zur
Überwachung ihres Gesundheitszustands und des Verhaltens der
endovaskulären Prothese erforderlich macht. Patienten mit spezifischen
klinischen Befunden (z.B. Endoleaks, Vergrößerung des Aneurysmas oder
Änderungen in der Struktur oder Position der endovaskulären Prothese)
sollten zusätzliche Nachsorgeuntersuchungen erhalten. Spezifische
Nachsorgerichtlinien werden in Abschnitt 12, RICHTLINIEN FÜR DIE
BILDGEBUNG UND DIE POSTOPERATIVE NACHSORGE, besprochen.
• Der Patient ist darüber aufzuklären, dass die Einhaltung der
Nachsorgetermine sowohl während des ersten Jahrs nach der Operation als
auch in jährlichen Abständen darüber hinaus wichtig ist. Der Patient ist
darüber zu informieren, dass die regelmäßige und konsequente Nachsorge
unabdingbar für die dauernde Sicherheit und Wirksamkeit der
endovaskulären Behandlung eines AAA ist. Die Minimalnachsorge umfasst
jährliche Untersuchungen mit bildgebenden Verfahren und die Einhaltung
der routinemäßigen OP-Nachsorgevorschriften. Die Nachsorge ist als
lebenslange Verpflichtung zu Gesundheit und Wohlbefinden des Patienten
anzusehen.
• Der Patient ist darauf hinzuweisen, dass eine erfolgreiche Aneurysmareparatur den Krankheitsprozess nicht aufhält. Es ist weiterhin möglich, dass
eine damit zusammenhängende Gefäßdegeneration stattfindet.
• Der Arzt muss den Patienten darüber aufklären, dass bei Anzeichen eines
Ansatzverschlusses, einer Vergrößerung oder Ruptur des Aneurysmas sofort
ärztliche Hilfe gesucht werden muss. Anzeichen eines
Prothesenansatzverschlusses sind u.a. Schmerzen in Becken oder Bein(en)
beim Laufen oder in Ruhelage, oder eine Verfärbung oder Kälte des Beins.
Aneurysmenrupturen können asymptomatisch sein, gehen aber
üblicherweise mit Schmerzen, Taubheitsgefühl; Schwächegefühl in den
Beinen; Schmerzen in Rücken, Brustkorb, Abdomen oder Leiste;
Schwindelgefühl; Ohnmacht; Herzrasen oder plötzlicher Schwäche einher.
• Aufgrund der für eine erfolgreiche Platzierung und Nachkontrolle
endovaskulärer Prothesen erforderlichen Bildgebungsverfahren sollten
Schwangere oder Frauen, die bei sich eine Schwangerschaft vermuten, auf
die Risiken der Strahlenexposition für sich entwickelndes Gewebe
hingewiesen werden. Bei Männern, die sich einer endovaskulären oder
offen-chirurgischen Reparatur unterziehen, kann es zu Impotenz kommen.
Der Arzt sollte den Patienten auf die Patienteninformation verweisen, in der
die während oder nach der Implantation der Prothese bestehenden Risiken
beschrieben werden. Mit dem Eingriff in Zusammenhang stehende Risiken
sind u.a. Herz-, Lungen-, Nerven-, Darm- und Blutungskomplikationen. Mit
der Prothese in Verbindung stehende Risiken sind u.a. Okklusion, Endoleak,
Vergrößerung des Aneurysmas, Bruch, mögliche Reintervention und
Umstellung auf offene Chirurgie, Ruptur und Exitus (siehe Abschnitt 5.1,
Beobachtete unerwünschte Ereignisse, und Abschnitt 5.2, Mögliche
unerwünschte Ereignisse). Der Arzt sollte die Patientenkarte ausfüllen und
dem Patienten aushändigen, damit dieser sie immer bei sich tragen kann. Der
Patient sollte bei jedem Arztbesuch die Karte vorzeigen, insbesondere dann,
wenn weitere diagnostische Verfahren durchgeführt werden (z.B. MRT).
9 LIEFERFORM
• Die endovaskuläre AAA-Prothese Zenith Flex wird steril und bereits geladen
in einer Aufreißverpackung geliefert.
• Das Produkt ist für den einmaligen Gebrauch vorgesehen. Produkt nicht
erneut sterilisieren.
• Produkt und Packung auf Schäden überprüfen, die während des Transports
aufgetreten sein können. Produkt nicht verwenden, wenn es beschädigt ist
oder die Sterilbarriere geschädigt oder durchbrochen wurde. Ein
beschädigtes Produkt nicht verwenden und an COOK zurücksenden.
• Vor dem Gebrauch sicherstellen, dass die richtigen Produkte (Anzahl und
Größe) für den Patienten geliefert wurden. Dazu das Produkt mit der
Verordnung des Arztes für den jeweiligen Patienten vergleichen.
• Der Hauptteil der Prothese ist bereits in eine Flexor® Einführschleuse von 18,
20 oder 22 French geladen. Die Oberfläche der Schleuse ist außen hydrophil
beschichtet, was nach Befeuchten die Gleitfähigkeit verbessert. Zur
Aktivierung dieser hydrophilen Beschichtung muss die Außenseite mit einer
in Kochsalzlösung getränkten Kompresse abgewischt werden.
• Nicht nach dem Ablauf des auf dem Etikett angegebenen Verfallsdatum
(„USE BY“) verwenden.
• Kühl und trocken aufbewahren.
10 INFORMATIONEN ZUM KLINISCHEN EINSATZ
10.1 Ärzteschulung
VORSICHT: Während der Implantation oder einer Reintervention sollte
für den Fall, dass eine Umstellung auf eine offene chirurgische Reparatur
erforderlich wird, stets ein qualifiziertes Chirurgieteam zur Verfügung
stehen.
VORSICHT: Die endovaskuläre AAA-Prothese Zenith Flex mit dem H&L-B
One-Shot Einführsystem darf nur von Ärzten und Teams verwendet
werden, die in der Technik von interventionellen Gefäßoperationen und
der Verwendung dieser Prothese geschult wurden. Die empfohlenen
Anforderungen bezüglich der Kenntnisse des Arztes, der die
endovaskuläre AAA-Prothese Zenith Flex mit dem H&L-B One-Shot
Einführsystem verwendet, sind im Folgenden aufgeführt.
Patientenauswahl:
• Kenntnis des natürlichen Verlaufs abdominaler Aortenaneurysmen (AAA)
und mit der AAA-Reparatur assoziierter Komorbiditäten.
• Die für die Interpretation von Röntgenbildern, für Produktauswahl und
-größenbestimmung erforderlichen Kenntnisse.
Ein multidisziplinäres Team mit Erfahrung mit den folgenden Verfahren:
• Chirurgischer Zugang zur A. femoralis, Arteriotomie und Reparatur
• Perkutane Zugangs- und Verschlusstechniken
• Nicht-selektive und selektive Führungsdraht- und Kathetertechniken
• Interpretation fluoroskopischer und angiographischer Aufnahmen
• Embolisation
• Angioplastie
• Platzierung endovaskulärer Stents
• Schlingentechniken
• Angemessener Einsatz von Röntgenkontrastmitteln
• Methoden zur Minimierung der Strahlenbelastung
• Kompetenz in den erforderlichen Nachsorgemodalitäten
10.2 Überprüfung vor dem Gebrauch
Produkt und Packung auf Schäden überprüfen, die während des Transports
aufgetreten sein können. Produkt nicht verwenden, wenn es beschädigt ist
oder die Sterilbarriere geschädigt oder durchbrochen wurde. Ein beschädigtes
Produkt nicht verwenden und an COOK zurücksenden. Vor dem Gebrauch
sicherstellen, dass die richtigen Produkte (Anzahl und Größe) für den Patienten
geliefert wurden. Dazu das Produkt mit der Verordnung des Arztes für den
jeweiligen Patienten vergleichen.
10.3 Erforderliche Materialien
(Im 3-teiligen modularen System nicht enthalten)
• Hilfsmittel-Kit für die endovaskuläre AAA-Prothese Zenith
• Für die digitale Angiographie geeignetes Fluoroskop (mit C-Bogen oder
feststehendes Gerät)
• Kontrastmittel
• Spritze
• Heparinisierte Kochsalzlösung
• Sterile Kompressen
10.4 Empfohlene Materialien
(Im 3-teiligen modularen System nicht enthalten)
Die folgenden Produkte werden für die Implantation aller Komponenten der
Zenith-Produktpalette empfohlen. Informationen zur Verwendung dieser
72

Produkte sind der empfohlenen Gebrauchsanweisung für das jeweilige
Produkt zu entnehmen.
• Extrasteifer Führungsdraht der Stärke 0,035 Inch (0,89 mm), 260 cm, z.B.:
• Extrasteife Cook Lunderquist (LES) Führungsdrähte
• Standard-Führungsdraht der Stärke 0,035 Inch (0,89 mm), z.B.:
• Führungsdrähte der Stärke 0,035 Inch (0,89 mm) von Cook
• Cook Nimble™ Führungsdrähte
• Modellierungsballons, z.B.:
• Cook CODA® Ballonkatheter
• Einführsets, z.B.:
• Cook Check-Flo® Einführsets
• Extra große Cook Check-Flo® Einführsets
• Kontralaterale Einführinstrumente Flexor® Balkin Up & Over® von Cook
• Messkatheter, z.B.:
• Cook Aurous® Messkatheter (in cm)
• Angiographiekatheter mit röntgendichter Spitze, z.B.:
• Cook Angiographiekatheter mit Beacon® Spitze
• Cook Royal Flush-Katheter mit Beacon® Spitze
• Einführnadeln, z.B.:
• Einwandige Punktionskanülen von Cook
10.5 Richtlinien zur Bestimmung des Prothesendurchmessers
Die Wahl des Durchmessers sollte sich am Gefäßdurchmesser von
Außenwand zu Außenwand, nicht am Durchmesser des Lumens orientieren.
Bei der Wahl einer zu geringen oder zu großen Größe kann es zu
unvollständiger Abdichtung oder einer Beeinträchtigung der Durchblutung
kommen.
Tabelle 10.5.1 Anleitung zur Bestimmung des Durchmessers
Durchmesser der
zu behandelnden
1,2
Aorta
(mm)
18–19 22 82/112, 96/126, 111/141,
20–21 24 82/112, 96/126, 111/141,
22 26 82/112, 96/126, 111/141,
23–24 28 82/112, 96/126, 111/141,
25–26 30 82/112, 96/126, 111/141,
27–28 32 82/112, 96/126, 111/141,
29–32 36 95/125, 113/143,
1
Maximaldurchmesser entlang der proximalen Befestigungsstelle.
2
Den gemessenen Aortendurchmesser auf den nächsten mm-Wert runden.
3
Die Wahl des Durchmessers kann durch weitere Überlegungen beeinflusst werden.
*Bei allen Abmessungen handelt es sich um Nennwerte.
Tabelle 10.5.2 Anleitung zur Bestimmung des Durchmessers
Durchmesser
der zu
behandelnden
1,2
Iliaka
(mm)
< 8 8 37, 54, 71, 88, 105, 122 14
8–9 10 37, 54, 71, 88, 105, 122 14
10–11 12 37, 54, 71, 88, 105, 122 14
12–13 14 37, 54, 71, 88 14
14–15 16 37, 54, 71, 88 14
16–17 18 37, 54, 71, 88 16
18 20 37, 54, 71, 88 16
19 22 37, 54, 71, 88 16
20 24 37, 54, 71, 88 16
1
Maximaldurchmesser entlang der distalen Befestigungsstelle.
2
Den gemessenen Iliakadurchmesser auf den nächsten mm-Wert runden.
3
Die Wahl des Durchmessers kann durch weitere Überlegungen beeinflusst werden.
4
Gesamtschenkellänge = Arbeitslänge + 22 mm für den Ansatzstent.
*Bei allen Abmessungen handelt es sich um Nennwerte.
11 GEBRAUCHSANWEISUNG
Anatomische Voraussetzungen
• Die Größe der iliofemoralen Zugangsgefäße und ihre Morphologie
(minimale Thrombusbildung, Verk alkung und/oder Gewundenheit) sollten
mit den Gefäßzugangstechniken und Instrumenten kompatibel sein.
Gegebenenfalls ist eine Technik zum Anlegen eines arteriellen
Leitungswegs erforderlich.
• Der proximale Aortenhals sollte eine Länge von mindestens 15 mm und
einen Durchmesser (Außenwand zu Außenwand) von 18–32 mm aufweisen
• Distale Befestigungsstelle an der Iliak a von mindestens 10 mm Länge und
7,5 mm bis 20 mm Durchmesser (Außenwand zu Außenwand gemessen).
Vor Gebrauch der endovaskulären AAA-Prothese Zenith Flex mit dem H&L-B
One-Shot Einführsystem diese Broschüre mit den Empfehlungen zum
Gebrauch durchlesen. Die folgenden Anweisungen stellen grundlegende
Richtlinien für die Platzierung der Prothese dar. Abweichungen von den
nachfolgend beschriebenen Verfahren können erforderlich sein. Diese
Anweisungen sollen den Arzt bei seinen Entscheidungen unterstützen, nicht
dessen Fachkompetenz ersetzen.
des Prothesenhauptteils*
Durchmesser
Hauptteils
Gesamtlänge bis zum kontralater-
des
alen Ansatz/Gesamtlänge bis zum
3
(mm)
ipsilateralen Ansatz
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
131/161, 149/179
(mm)
des iliakalen Prothesenschenkels*
Durchmesser
des iliakalen
Schenkels
(mm)
3
Arbeitslänge des
iliakalen Schenkels
(mm)
Einführschleuse
(French)
18
18
18
20
20
20
22
Einführ-
4
schleuse
(French)
Allgemeine Informationen zum Gebrauch
• Bei der Ar beit mit der endovaskulären AAA-Prothese Zenith Flex und dem
H&L-B One-Shot-Einführsystem sind die Standardtechniken zur Platzierung
von arteriellen Zugangsschleusen, Führungskathetern, Angiographiekathetern und Führungsdrähten zu verwenden. Die AAA-Prothese Zenith
Flex mit dem H&L-B One-Shot Einführsystem ist mit Führungsdrähten der
Stärke 0,035 Inch (0,89 mm) kompatibel.
• Bei der endovaskulären Stent-Graft-Platzierung handelt es sich um einen
chirurgischen Eingriff, bei dem es aufgrund verschiedener Ursachen zu
einem Blutverlust kommen kann. Zur Vermeidung unerwünschter Folgen
bedarf dieser in seltenen Fällen einer Intervention (einschl. Transfusion). Es
ist wichtig, den Blutverlust durch das Hämostaseventil während des
gesamten Eingriffs zu überwachen, besonders während und nach der
Manipulation des grauen Positionierers. Ist der Blutverlust nach Entfernung
des grauen Positionierers übermäßig, sollte zur Eingrenzung des Flusses die
Platzierung eines nicht inflatierten Modellierungsballons oder eines
Einführsystemdilatators innerhalb des Ventils in Betracht gezogen werden.
Die Präimplantationsphase bestimmende Faktoren
Anhand der Präimplantationsplanung sicherstellen, dass die richtige Prothese
ausgewählt wurde. Die Richtgrößen sind:
1. Die zur Einführung des Hauptteils gewählte A. femoralis (also die
Festlegung, welche Iliaka die kontra- und welche die ipsilaterale Arterie ist).
2. Die Winkel zwischen Aortenhals, Aneurysma und Aa. iliacae.
3. Die Qualität des Aortenhalses.
4. Die Durchmesser des infrarenalen Aortenhalses und der distalen Aa. iliacae.
5. Abstand zwischen Aa. renales und der Aortengabelung.
6. Abstand von Aortengabelung zu Aa. iliacae internae/Befestigungsstelle(n).
7. Aneurysmen, die sich bis in die Aa. iliacae erstrecken, machen u.U.
besondere Erwägungen bei der Wahl einer geeigneten Verbindungsstelle
zwischen Prothese und Arterie erforderlich.
8. Der Grad der Gefäßverkalkung ist zu berücksichtigen.
Vorbereitung des Patienten
1. Die Vorschriften der jeweiligen Einrichtung zu Anästhesie,
Antikoagulanzien und Vitalzeichenüberwachung beachten.
2. Den Patienten so auf dem Aufnahmetisch positionieren, dass eine
fluoroskopische Darstellung vom Aortenbogen bis zu den femoralen
Gabelungen möglich ist.
3. Unter Verwendung der normalen chirurgischen Verfahren beide
Oberschenkelschlagadern freilegen.
4. Für angemessene proximale und distale Kontrolle in beiden
Oberschenkelschlagadern sorgen.
11.1 Gegabeltes System (Abb. 2)
11.1.1 Vorbereitung/Spülen des gegabelten Hauptteils
1. Den Transportmandrin mit dem schwarzen Ansatz (aus der inneren
Kanüle), den Kanülenschutzschlauch (von der inneren Kanüle) und den
Dilatatorspitzenschutz (von der Dilatatorspitze) entfernen. Die Peel-Away®Schleuse von der Rückseite des Hämostaseventils abnehmen. (Abb. 5)
Die distale Spitze des Systems anheben und durch den Absperrhahn am
Hämostaseventil spülen, bis Flüssigkeit aus dem Seitenloch in der Nähe
der Spitze der Einführschleuse austritt. (Abb. 6) Fortfahren, bis 20 ml
Spüllösung vollständig durch die Prothese gespült wurden. Die Spülung
beenden und den Absperrhahn am Verbindungsschlauch schließen.
HINWEIS: Zum Spülen der Prothese wird oft heparinisierte physiologische
Kochsalzlösung verwendet.
2. Eine Spritze mit heparinisierter physiologischer Kochsalzlösung am
Ansatz der inneren Kanüle anschließen. Spülen, bis Flüssigkeit an der
Dilatatorspitze austritt. (Abb. 7)
HINWEIS: Beim Spülen des Systems das distale Ende des Systems hoch
halten, damit die Luft besser entweichen kann.
3. Sterile Kompressen in Kochsalzlösung tränken und die Flexor®
Einführschleuse damit abwischen, um die hydrophile Beschichtung zu
aktivieren. Sowohl Schleuse als auch Dilatator reichlich hydratisieren.
11.1.2 Vorbereitung/Spülen des kontralateralen iliakalen Schenkels
1. Den Innenmandrin mit dem schwarzen Ansatz (aus der inneren
Kanüle), den Kanülenschutzschlauch (von der inneren Kanüle) und den
Dilatatorspitzenschutz (von der Dilatatorspitze) entfernen. Die Peel-Away®Schleuse von der Rückseite des Hämostaseventils abnehmen. (Abb. 8)
Die distale Spitze des Systems anheben und durch den Absperrhahn am
Hämostaseventil spülen, bis Flüssigkeit aus dem Seitenloch in der Nähe
der Spitze der Einführschleuse austritt. (Abb. 9) Fortfahren, bis 20 ml
Spüllösung vollständig durch die Prothese gespült wurden. Die Spülung
beenden und den Absperrhahn am Verbindungsschlauch schließen.
HINWEIS: Zum Spülen der Prothese wird oft heparinisierte physiologische
Kochsalzlösung verwendet.
2. Eine Spritze mit heparinisierter physiologischer Kochsalzlösung am Ansatz
der distalen inneren Kanüle anschließen. Spülen, bis Flüssigkeit an der
distalen Dilatatorspitze austritt. (Abb. 7)
HINWEIS: Beim Spülen des Systems das distale Ende des Systems hoch
halten, damit die Luft besser entweichen kann.
11.1.3 Vorbereitung/Spülen des ipsilateralen iliakalen Schenkels
Den Anweisungen im vorhergehenden Abschnitt 11.1.2, Vorbereiten/Spülen
des kontralateralen iliakalen Schenkels, befolgen, um die ordnungsgemäße
Spülung der ipsilateralen iliakalen Schenkelprothese zu gewährleisten.
11.1.4 Gefäßzugang und Angiographie
1. Die ausgewählten gemeinsamen Oberschenkelschlagadern unter
Verwendung der normalen Methoden mit einer ultradünnen
Arterienkanüle der Größe 18UT oder 19UT punktieren. Nach dem
Gefäßzugang folgende Vorrichtungen einführen:
• Führungsdrähte – Standarddurchmesser 0,035 Inch (0,89 mm), Länge
145 cm, J-Spitzen- oder Bentson-Führungsdraht
• Schleusen der geeigneten Größe (z.B. 6,0 oder 8,0 French)
• Spülkatheter (oft röntgendichte Messkatheter, z.B. ZentimeterMesskatheter oder gerader Spülkatheter)
2. Angiographie durchführen, um die Höhe(n) der Aa. renales,
Aortengabelung und Iliakagabelungen zu bestimmen.
HINWEIS: Wenn bei einem abgewinkelten Hals das Fluoroskop anguliert
verwendet wird, müssen u.U. Angiogramme in verschiedenen Projektionen
durchgeführt werden.
11.1.5 Positionieren des Hauptteils
1. Sicherstellen, dass das Einführsystem mit heparinisierter physiologischer
Kochsalzlösung gespült und sämtliche Luft daraus entfernt wurde.
2. Systemisch Heparin geben und die Spüllösungen überprüfen. Nach jedem
Katheter- und/oder Führungsdrahtwechsel spülen.
HINWEIS: Während des gesamten Eingriffs den Gerinnungsstatus des
Patienten überwachen.
3. Auf der ipsilateralen Seite den J-Draht durch einen steifen Führungsdraht
(LES) mit 0,035 Inch (0,89 mm) Durchmesser und 260 cm Länge ersetzen
und diesen durch den Katheter und bis in die Brustschlagader (Aorta
thoracica) vorschieben. Den Spülkatheter und die Schleuse entfernen. Die
Führungsdrahtposition beibehalten.
4. Vor dem Einbringen das Einführsystem des Hauptteils unter
Durchleuchtungskontrolle auf den Bauch des Patienten legen, um die
Ausrichtung der röntgendichten Markierung auf der kontralateralen
Verlängerung zu bestimmen. Der Seitenarm des Hämostaseventils
kann als externer Bezugspunkt für die röntgendichte Markierung des
kontralateralen Ansatzes verwendet werden.
5. Das Hauptteileinführsystem über den Draht in die Oberschenkel-schlagader
einführen und dabei auf den Seitenarm als Bezugspunkt achten.
VORSICHT: Beim Einbringen des Einführsystems die Position des
Führungsdrahts beibehalten.
VORSICHT: Um interne Ver windungen in der endovaskulären Prothese
zu verhindern, ist sorgfältig darauf zu achten, dass bei einer Drehung
des Einführsystems alle Komponenten des Systems (von der äußeren
Schleuse bis zur inneren Kanüle) zusammen gedreht werden.
6. Das Einführsystem so weit vorschieben, bis die vier röntgendichten
Goldmarkierungen (in 2 mm Abstand vom proximalsten Segment der
Prothese) sich gerade inferior zu der am weitesten inferior gelegenen
Nierenöffnung befinden (Abb. 10, Bild 1).
73

7. Die Position des Führungsdrahtes in der Brustschlagader überprüfen.
Sicherstellen, dass das Prothesensystem so ausgerichtet ist, dass sich
der kontralaterale Ansatz oberhalb und anterior zum Ursprung der
kontralateralen Iliaka befindet. Wenn die röntgendichte Markierung
des kontralateralen Ansatzes nicht korrekt ausgerichtet ist, das gesamte
System drehen, bis sie auf halbem Wege zwischen einer lateralen und
einer anterioren Position auf der kontralateralen Seite zu liegen kommt.
• Wenn die Markierungen ein ✓ bilden, zeigen sie eine anteriore Position
des kurzen (kontralateralen) Ansatzes an. (Abb. 10, Bild 4)
• Wenn die Markierungen ein bilden, zeigen sie eine posteriore Position
des kurzen (kontralateralen) Ansatzes an. (Abb. 10, Bild 5)
• Wenn die Markierungen ein | bilden, zeigen sie eine laterale Position des
kurzen (kontralateralen) Ansatzes an. (Abb. 10, Bild 6)
HINWEIS: Die röntgendichten Kanülen auf jedem Stent zwischen den
Nierenarterien und dem kontralateralen Ansatz liegen auf einer Linie mit der
röntgendichten Markierung des kontralateralen Ansatzes.
8. Ein weiteres Angiogramm aufnehmen, um sicherzustellen, dass die vier
röntgendichten Goldmarkierungen sich mindestens 2 mm unterhalb der
untersten Nierenöffnung befinden.
9. Sicherstellen, dass das Captor-Hämostaseventil an der Flexor®Einführschleuse geöffnet ist. (Abb. 11)
10. Den grauen Positionierer (Schaft des Einführsystems) während
des Zurückziehens der Schleuse stabilisieren. Die ersten beiden
abgedeckten Stents entfalten. Dazu die Schleuse unter Beobachtung der
Vorrichtungsposition zurückziehen.
11. Ohne den Tisch zu bewegen die Vergrößerung verringern und die
Position der röntgendichten Markierung des kontralateralen Ansatzes
und der Nierenarterien überprüfen. Mit dem Entfalten fortfahren, bis der
kontralaterale Ansatz sich vollständig entfaltet hat. (Abb. 12) Die Schleuse
nicht weiter zurückziehen.
HINWEIS: Sicherstellen, dass sich der kontralaterale Ansatz mindestens 5 mm
oberhalb der Aortengabelung und an der für die Kanülierung gewünschten
Position befindet.
11.1.6 Positionieren des kontralateralen iliakalen Führungsdrahts
1. Den Katheter und Führungsdraht durch das offene Ende des
kontralateralen Ansatzes in den Hauptteil der Prothese manövrieren. Den
Führungsdraht vorschieben, bis er sich im Hauptteil der Prothese krümmt.
Fluoroskopische AP- und Schrägansichten können bei der Überprüfung
der Kanülierung der Vorrichtung hilfreich sein.
2. Nach der Kanülierung den Angiographiekatheter über den Draht in das
Hauptteil der endovaskulären Prothese vorschieben. Den Draht entfernen
und die richtige Position angiographisch bestätigen. Den Führungsdraht
vorschieben, bis er sich im Hauptteil der Prothese krümmt. Den
Angiographiekatheter entfernen. (Abb. 13)
11.1.7 Entfalten des proximalen (oberen) Hauptkörpers
1. Mittels eines Angiographiekatheters die Position der endovaskulären
Prothese relativ zu den Nierenarterien angiographisch überprüfen. Falls
nötig, den gecoverten Teil der endovaskulären Prothese relativ zu den
Nierenarterien vorsichtig neu positionieren. (In diesem Stadium kann die
Neupositionierung nur über sehr geringe Distanzen erfolgen.)
HINWEIS: Die Durchgängigkeit der Nierenarterien sicherstellen. Dazu
überprüfen, ob sich die proximalen Prothesenmarkierungen mindestens
2 mm unterhalb der tiefstliegenden durchgängigen Nierenarterie befinden.
VORSICHT: Bei der Entfernung des proximalen Auslösedrahtes, dem
Vorschub der oberen Kappe und der anschließenden suprarenalen
Stententfaltung ist die Position des Führungsdrahtes für den
Hauptkörper dahingehend zu überprüfen, dass er gerade noch distal des
Aortenbogens endet und das System maximal gestützt wird.
2. Die Sicherheitssperre des schwarzen Auslösedrahtmechanismus
entfernen. Unter fluoroskopischer Beobachtung den
Auslösedraht zurückziehen und entfernen. Dazu den schwarzen
Auslösedrahtmechanismus vom Griff herunterschieben und dann durch
dessen Schlitz über die innere Kanüle entfernen. (Abb. 14)
Ist ein Widerstand zu spüren oder wölbt sich das System, steht der
Auslösedraht unter Spannung. Durch übermäßigen Kraftaufwand kann die
Position der Prothese verändert werden. Wird ein starker Widerstand oder
eine Bewegung des Platzierungssystems festgestellt, muss das Verfahren
unterbrochen und die Situation beurteilt werden. Wenn sich der schwarze
Auslösedrahtmechanismus nicht von der oberen Kappe abnehmen lässt,
unter fluoroskopischer Beobachtung die folgenden Schritte ausführen:
a. Den Zug am Auslösedraht lockern, indem die Klemmschraube gelöst
und leicht an der inneren Kanüle gezogen wird, bis die obere Kappe
nach unten über den suprarenalen Stent gleitet. Hauptkörper der Zenith
Flex-Prothese nicht zusammendrücken.
b. Die Klemmschraube wieder anziehen.
c. Den schwarzen Auslösedrahtmechanismus abnehmen.
d. Mit Schritt 3 in Abschnitt 11.1.7, Entfalten des proximalen (oberen)
Hauptkörpers, fortfahren.
HINWEIS: Wenn sich der Auslösedrahtmechanismus für den oberen Stent
immer noch nicht von der oberen Kappe abnehmen lässt, siehe Abschnitt
14.1, Fehlerbehebung am Auslösedrahtmechanismus.
3. Die Klemmschraube lösen. (Abb. 15) Durch Stabilisieren des grauen
Positionierers des Einführinstruments die Position der Prothese steuern.
4. Den suprarenalen Stent entfalten. Dazu die innere Kanüle der oberen
Kappe in Schritten von 1 bis 2 mm vorschieben, bis der obere Stent
vollständig entfaltet ist; die Position des Hauptkörpers laufend
kontrollieren. (Abb. 16 und 17) Die Kanüle der oberen Kappe weitere 1
bis 2 cm vorschieben und dann die Klemmschraube wieder anziehen, um
jede Berührung mit dem entfalteten suprarenalen Stent zu vermeiden.
HINWEIS: Wenn sich der suprarenale Stent durch Vorschub der inneren
Kanüle der oberen Kappe nicht vollständig entfalten lässt, siehe Abschnitt
14.2, Fehlerbehebung bei der suprarenalen Stententfaltung.
HINWEIS: Wenn der mit Haken versehene suprarenale Stent einmal
eingesetzt wurde, sollte die Prothese nicht mehr neu positioniert werden.
WARNHINWEIS: Die Zenith Flex endovaskuläre AAA-Prothese umfasst
einen suprarenalen Stent mit Befestigungshaken. Auf Höhe des
suprarenalen Stents müssen interventionelle Instrumente äußerst
sorgfältig gehandhabt werden.
5. Den kontralateralen Führungsdraht in die thorakale Aorta vorschieben.
11.1.8 Positionieren und Entfalten des kontralateralen iliakalen Schenkels
VORSICHT: Vor der Implantation sicherstellen, dass der gewählte
kontralaterale iliakale Schenkel auch zum Einführen auf der
kontralateralen Seite des Patienten vorgesehen ist.
1. Den Bildverstärker so positionieren, dass sowohl die kontralaterale A.
iliaca interna als auch die kontralaterale A. iliaca communis zu sehen sind.
2. Vor dem Einbringen des Einführsystems für den kontralateralen iliakalen
Schenkel Kontrastmittel durch die kontralaterale femorale Schleuse
injizieren, um so die kontralaterale A. iliaca interna zu lokalisieren.
3. Das Einführsystem für den kontralateralen iliakalen Schenkel in die Arterie
einbringen. Langsam vorschieben, bis der iliakale Schenkel der Prothese
mindestens einen ganzen Stent des iliakalen Schenkels (d.h. einen proximalen
Stent des iliakalen Schenkels der Prothese) im kontralateralen Ansatz des
Hauptteils überlappt. (Abb. 18) Wenn der Hauptteil der Prothese während
dieses Manövers dazu neigt, sich zu bewegen, ist er durch Stabilisieren des
grauen Positionierers auf der ipsilateralen Seite in Position zu halten.
HINWEIS: Wenn es beim Vorschieben des Einführsystems für den iliakalen
Schenkel zu Schwierigkeiten kommt, auf einen steiferen Führungsdraht
ausweichen. In stark gewundenen Gefäßen kann es durch die Einführung der
steifen Drähte und Schleusensysteme zu erheblichen Änderungen in der
Anatomie kommen.
4. Die Position des distalen Endes des iliakalen Schenkels der Prothese
bestätigen. Falls nötig, den iliakalen Schenkel der Prothese neu
positionieren, um sowohl die Durchgängigkeit der A. iliaca interna als
auch eine Überlappung von mindestens einem vollen Stent des iliakalen
Schenkels (d.h. eines proximalen Stents des iliakalen Prothesenschenkels,
Maximalüberlappung von 1,5 Stents) innerhalb des Hauptteils der
endovaskulären Prothese sicherzustellen.
5. Zum Entfalten den iliakalen Prothesenschenkel mit dem grauen
Positionierer in Position halten und gleichzeitig die Schleuse
zurückziehen. (Abb. 19 und 20) Sicherstellen, dass eine Überlappung von
einem Stent erhalten bleibt.
6. Sobald das distale Ende des iliakalen Prothesenschenkels freigegeben
wird, mit dem Zurückziehen der Schleuse aufhören.
7. Unter fluoroskopischer Beobachtung und nach Überprüfung der Position
des iliakalen Prothesenschenkels die Klemmschraube lockern und die
innere Kanüle so zurückziehen, dass der sich verjüngende Dilatator am
grauen Positionierer andockt. Die Klemmschraube wieder anziehen. Beim
Zurückziehen des grauen Positionierers mit gesicherter innerer Kanüle die
Schleusenposition beibehalten. (Abb. 21)
8. Die Position des Führungsdrahtes erneut überprüfen.
11.1.9 Entfalten des distalen (untersten) Stents des Hauptteils
1. Zurück auf die ipsilaterale Seite wechseln.
2. Den ipsilateralen Ansatz des Hauptteils vollständig entfalten. Dazu die
Schleuse zurückziehen, bis sich der am weitesten distal gelegene Stent
entfaltet hat. (Abb. 22 und 23) Die Schleuse nicht weiter zurückziehen.
HINWEIS: Der distale Stent wird noch durch den Auslösedraht gesichert.
3. Die Sicherheitssperre des weißen Auslösedrahtmechanismus entfernen.
Den Auslösedraht zurückziehen und entfernen. Dazu den weißen
Auslösedrahtmechanismus vom Griff herunter schieben und dann über
den Schlitz über die innere Kanüle der Prothese entfernen. (Abb. 24)
11.1.10 Andocken der oberen Kappe
1. Die Klemmschraube lösen. (Abb. 25)
2. Die Schleuse und innere Kanüle so sichern, dass sie sich nicht bewegen
können.
3. Den grauen Positionierer über die innere Kanüle vorschieben, bis er an
der oberen Kappe andockt. (Abb. 26, 27 und 28)
HINWEIS: Wenn ein Widerstand auftritt, den grauen Positionierer leicht
drehen und weiter vorsichtig vorschieben.
4. Die Klemmschraube erneut anziehen und an der inneren Kanüle ziehen,
um die gesamte obere Kappe und den grauen Positionierer durch die
Prothese und durch die Schleuse zurückzuziehen. (Abb. 29) Die Schleuse
und den Führungsdraht an ihrer jeweiligen Position belassen.
HINWEIS: Die Positionen von Schleuse und Führungsdraht beibehalten.
5. Das Captor®-Hämostaseventil an der Flexor®-Einführschleuse bis zum
Anschlag im Uhrzeigersinn drehen, um es zu schließen. (Abb. 30)
11.1.11 Positionieren und Entfalten des ipsilateralen iliakalen Schenkels
HINWEIS: Sicherstellen, dass das Captor-Hämostaseventil an der
Einführschleuse geöffnet ist. (Abb. 31)
1. Die Führungsdraht- und Schleusenvorrichtung des Prothesenhauptteils zur
Einführung des ipsilateralen iliakalen Prothesenschenkels verwenden. Die
Einheit aus Dilatator und Schleuse in die Schleuse des Hauptteils vorschieben.
HINWEIS: Bei stark gewundenen Gefäßen kann sich die Position der Iliaka
interna durch das Einführen der starren Draht- und Schleusensysteme
deutlich verschieben.
2. Langsam vorschieben, bis der ipsilaterale iliakale Prothesenschenkel
mindestens einen ganzen Stent des iliakalen Schenkels (d.h., einen
proximalen Stent des iliakalen Prothesenschenkels) innerhalb des
ipsilateralen Ansatzes des Hauptteils überlappt. (Abb. 32)
HINWEIS: Wenn eine Überlappung von mehr als 3 iliakalen Stentschenkeln
erforderlich ist (mehr als 2 iliakale Stentschenkel bei Schenkellängen von
37 mm und 54 mm), muss u.U. der Einsatz einer Schenkelverlängerung im
Gabelungsbereich der gegenüberliegenden Seite in Betracht gezogen werden.
3. Die Position des distalen Endes des iliakalen Schenkels der Prothese
bestätigen. Falls erforderlich, den iliakalen Prothesenschenkel
so um-positionieren, dass die Durchgängigkeit der Iliaka interna
gewährleistet ist.
4. Den iliakalen Prothesenschenkel zum Entfalten mit dem grauen
Positionierer stabilisieren und gleichzeitig die Schleuse des iliakalen
Schenkels zurückziehen. (Abb. 33 und 34) Falls nötig, die Schleuse des
Hauptteils zurückziehen.
5. Unter fluoroskopischer Beobachtung und nach Überprüfung der Position
des iliakalen Prothesenschenkels die Klemmschraube lockern und die
innere Kanüle so zurückziehen, dass der sich verjüngende Dilatator am
grauen Positionierer andockt. Die Klemmschraube wieder anziehen. Beim
Zurückziehen des grauen Positionierers mit gesicherter innerer Kanüle die
Schleusenposition beibehalten. (Abb. 35)
6. Das Captor-Hämostaseventil an der Flexor®-Einführschleuse bis zum
Anschlag im Uhrzeigersinn drehen, um es zu schließen.
7. Die Position der Führungsdrähte erneut überprüfen. Schleuse und
Führungsdrähte in ihren Positionen belassen.
11.1.12 Einführen des Modellierungsballons
1. Den Modellierungsballon wie folgt vorbereiten:
• Das Drahtlumen mit heparinisierter physiologischer Kochsalzlösung
spülen.
• Luft vollständig aus dem Ballon entfernen.
2. Zur Vorbereitung der Einführung des Modellierungsballons das CaptorHämostaseventil durch Drehen gegen den Uhrzeigersinn öffnen.
3. Den Modellierungsballon über den Führungsdraht und durch das
Captor-Hämostaseventil des Hauptteileinführsystems bis auf Höhe der
Nierenarterien vorschieben. Die richtige Schleusenposition beibehalten.
4. Das Captor-Hämostaseventil durch Drehen im Uhrzeigersinn mit sanftem
Druck um den Modellierungsballon festziehen.
VORSICHT: Den Ballon nicht außerhalb der Prothese im Gefäß
insufflieren.
5. Den Modellierungsballon mit entsprechend den Herstellerangaben
verdünntem Kontrastmittel im Bereich des am weitesten proximal
gelegenen bedeckten Stents und des infrarenalen Halses aufweiten.
Dabei von proximal nach distal vorgehen. (Abb. 36)
VORSICHT: Vor einer Umpositionierung ist sicherzustellen, dass der
Ballon vollkommen entleert ist.
VORSICHT: Vor der Neupositionierung des Modellierungsballons muss
das Captor-Hämostaseventil geöffnet sein.
74

6. Den Modellierungsballon zur Überlappung des ipsilateralen Ansatzes
zurückziehen und aufweiten.
VORSICHT: Vor der Neupositionierung des Modellierungsballons muss
das Captor-Hämostaseventil geöffnet sein.
7. Den Modellierungsballon zur ipsilateralen distalen Befestigungsstelle
zurückziehen und aufweiten.
VORSICHT: Den Ballon nicht außerhalb der Prothese im Gefäß
insufflieren.
8. Den Modellierungsballon entleeren und entfernen. Den Modellierungsballon auf den kontralateralen Führungsdraht setzen und in das
Einführsystem für den kontralateralen iliakalen Schenkel einführen. Den
Modellierungsballon bis zur Überlappung des kontralateralen Ansatzes
vorschieben und aufweiten.
VORSICHT: Vor einer Umpositionierung ist sicherzustellen, dass der
Ballon vollkommen entleert ist.
9. Den Modellierungsballon zur distalen Befestigungsstelle des kontralateralen
iliakalen Schenkels/Gefäßes zurückziehen und aufweiten. (Abb. 36)
VORSICHT: Den Ballon nicht außerhalb der Prothese im Gefäß
insufflieren.
10. Den Modellierungsballon entfernen und durch einen
Angiographiekatheter für die Durchführung der abschließenden
Angiographie ersetzen.
11. Alle steifen Führungsdrähte entfernen oder ersetzen, so dass die
Aa. iliacae wieder ihre natürliche Lage einnehmen können.
Abschließendes Angiogramm
1. Den Angiographiekatheter knapp oberhalb der Höhe der Aa. renales
positionieren. Eine Angiographie durchführen, um sicherzustellen, dass
die Aa. renales durchgängig sind und keine Endoleaks vorliegen. Die
Durchgängigkeit der Aa. iliacae internae überprüfen.
2. Sicherstellen, dass keine Endoleaks oder Knicke vorliegen und dass sich
die proximalen röntgendichten Goldmarkierungen an den richtigen
Stellen befinden. Schleusen, Drähte und Katheter entfernen.
HINWEIS: Wenn Endolecks oder andere Probleme beobachtet werden, die
empfohlene Gebrauchsanweisung für die Hilfskomponenten der
endovaskulären AAA-Prothese Zenith konsultieren.
3. Gefäße verschließen und mit normalen chirurgischen Verfahren den
Wundverschluss durchführen.
12 RICHTLINIEN FÜR DIE BILDGEBUNG UND POSTOPERATIVE
NACHSORGE
12.1 Allgemeines
• Über das langfristige Verhalten der endovaskulären Prothese ist derzeit
nichts bekannt. Die Patienten sind darauf hinzuzweisen, dass die
endovaskuläre Therapie eine lebenslange regelmäßige Nachsorge zur
Überwachung ihres Gesundheitszustands und des Verhaltens der
endovaskulären Prothese erforderlich macht. Patienten mit spezifischen
klinischen Befunden (z.B. Endoleaks, Vergrößerung des Aneurysmas oder
Änderungen der Struktur oder Position der endovaskulären Prothese) sollten
zusätzliche Nachsorgeuntersuchungen erhalten.
• Der Patient ist darüber aufzuklären, dass die Einhaltung der
Nachsorgetermine sowohl während des ersten Jahrs nach der Operation als
auch in jährlichen Abständen darüber hinaus wichtig ist. Der Patient ist
darüber zu informieren, dass die regelmäßige und konsequente Nachsorge
unabdingbar für die dauernde Sicherheit und Wirksamkeit der
endovaskulären Behandlung eines AAA ist.
• Jeder Patient ist vom Arzt individuell zu bewerten, und die
Nachsorgeverordnung muss sich an den Bedürfnissen und Umständen des
einzelnen Patienten orientieren. Der empfohlene Zeitplan für bildgebende
Untersuchungen ist in Tabelle 12.1 aufgeführt. Bei diesem Zeitplan handelt
es sich um die Minimalanforderungen an die Patientennachsorge, und er ist
auch einzuhalten, wenn klinische Symptome (z.B. Schmerzen,
Taubheitsgefühl, Schwäche) fehlen. Patienten mit spezifischen klinischen
Befunden (z.B. Endoleaks, Vergrößerung des Aneurysmas oder Änderungen
in der Struktur oder Position der endovaskulären Prothese) sollten
zusätzliche Nachsorgeuntersuchungen erhalten.
• Die jährlichen bildgebenden Nachsorgeuntersuchungen sollten
Röntgenaufnahmen des Abdomens sowie CT-Untersuchungen mit und
ohne Kontrastmittel umfassen. Wenn durch Nierenkomplikationen oder
andere Faktoren der Gebrauch von Kontrastmitteln ausgeschlossen ist,
können Röntgenaufnahmen des Abdomens, CT-Untersuchungen ohne
Kontrastmittel und Duplexultraschalluntersuchungen eingesetzt werden.
• Die Kombination von CT-Untersuchungen mit und ohne Kontrastmittel
liefert Informationen zu Veränderungen im Aneurysmadurchmesser,
Endoleaks, Durchgängigkeit, Gewundenheit, Krankheitsverlauf,
Befestigungslänge und anderen morphologischen Veränderungen.
• Die Röntgenaufnahmen des Abdomens liefern Informationen zur Integrität
der Prothese (Trennung zwischen den Komponenten, Stent-Frakturen,
Ablösung von Haken).
• Duplexultraschalluntersuchungen können Informationen zu Änderungen
des Aneurysmadurchmessers, Endoleaks, Durchgängigkeit, Gewundenheit
und Krankheitsverlauf liefern. In diesem Fall sollte eine CT-Untersuchung
ohne Kontrastmittel in Verbindung mit dem Ultraschall durchgeführt
werden. Im Vergleich zur CT ist der Ultraschall u.U. die weniger zuverlässige
und weniger empfindliche diagnostische Methode.
In Tabelle 12.1 sind die mindestens durchzuführenden bildgebenden
Nachuntersuchungen für Patienten mit der endovaskulären AAA-Prothese
Zenith Flex aufgeführt. Bei Patienten, die einer zusätzlichen Nachsorge
bedürfen, sollten auch zwischen diesen Terminen Untersuchungen stattfinden.
Tabelle 12.1 Empfohlener Bildgebungsplan für Patienten
Vor dem Eingriff X
Während des Eingriffs X
Vor der Entlassung
(innerhalb von 7 Tagen)
1 Monat X
3 Monate X
6 Monate X
12 Monate
(anschließend jährlich)
1
Die Aufnahmen sollten innerhalb von 6 Monaten vor dem Eingriff gemacht werden.
2
Bei Patienten mit Nierenversagen oder Patienten, bei denen aus anderen
Gründen keine CT-Untersuchung mit Kontrastmittel möglich ist, kann auch eine
Duplexultraschalluntersuchung durchgeführt werden. In Kombination mit dem Ultraschall
wird eine kontrastmittelfreie CT-Untersuchung empfohlen.
3
Vor der Entlassung oder nach einem Monat wird eine CT-Untersuchung empfohlen.
4
Bei Endoleaks vom Typ I oder III wird eine zügige Intervention und eine verstärkte
Nachsorge nach der Intervention empfohlen; siehe Abschnitt 12.6, Zusätzliche
Überwachung und Behandlung.
5
Empfohlen, wenn vor der Entlassung oder beim 1-Monatstermin ein Endoleak beobachtet
wird.
mit Endoprothese
Angiographie
1
CT
(mit und ohne
Kontrastmittel)
1
X
2,3,4
X
2,3,4
2,4,5
2,4
2,4
X
Röntgenaufnahmen des
Abdomens
X
X
X
X
12.2 Empfehlungen für CT mit und ohne Kontrastmittel
• Die Filmsätze sollten alle sequenziellen Bilder mit der geringstmöglichen
Schichtdicke enthalten (≤ 3 mm). Aufnahmen NICHT mit hohen
Schichtdicken (> 3 mm) durchführen und/oder aufeinander folgende
CT-Bilder/Filmsätze auslassen, da sonst kein genauer zeitabhängiger
Vergleich von Anatomie und Prothese möglich ist.
• Auf allen Aufnahmen sollte der jeweilige Maßstab angegeben sein. Bei
Verwendung von Filmen sollten die Bilder nicht kleiner sein als bei
Anordnung von 20:1 Aufnahmen auf Filmblättern von 35,5 x 43,2 cm
(14 Inch x 17 Inch).
• Es sind Aufnahmen sowohl mit als auch ohne Kontrastmittel erforderlich,
wobei die Tischpositionen einander jeweils entsprechen oder ergänzen
müssen.
• Die Schichtdicken und Intervalle der Aufnahmeserien mit und ohne
Kontrastmittel müssen einander entsprechen.
• Zwischen den Aufnahmen ohne und mit Kontrastmittel die Ausrichtung des
Patienten oder die Messpunkte am Patienten NICHT ändern.
Für die optimale Patientenüberwachung sind Aufnahmen des
Ausgangszustandes und zu Nachsorgeterminen mit und ohne Kontrastmittel
wichtig. Bei der CT-Untersuchung müssen akzeptable Protokolle eingehalten
werden. Tabelle 12.2 enthält Beispiele für akzeptable Bildgebungsprotokolle.
Tabelle 12.2 Akzeptable Bildgebungsprotokolle
Ohne Kontrastmittel Mit Kontrastmittel
IV-Kontrastmittel Nein Ja
Geeignete Geräte
Injektionsvolumen n. zutr. 150 ml
Injektionsrate n. zutr. > 2,5 ml/s
Injektionsmodus n. zutr. Leistung
Bolus-Timing n. zutr.
Abgedeckter Bereich -
Beginn
Abgedeckter Bereich
- Ende
Kollimation < 3 mm < 3 mm
Rekonstruktion
Axiales DFOV 32 cm 32 cm
Aufnahmen nach
Injektion
Spiral-CT mit
Fähigkeit zu mehr als
40 Sekunden
Zwerchfell
Proximaler Femur
Durchweg 2,5 mm –
weicher Algorithmus
Keine Keine
Spiral-CT mit
Fähigkeit zu mehr als
40 Sekunden
Testbolus: SmartPrep, C.A.R.E
oder entsprechend
1 cm oberhalb des Truncus
coeliacus
Ursprung der Profunda
femoris
Durchweg 2,5 mm –
weicher Algorithmus
12.3 Röntgenaufnahmen des Abdomens
Die folgenden Ansichten sind erforderlich:
• Vier Filme: supiniert-frontal (AP), seitlich-nativ, 30 Grad LPO und 30 Grad
RPO, jeweils auf den Nabel zentriert.
• Den Abstand zwischen Tisch und Film festhalten und bei jeder weiteren
Untersuchung verwenden.
Sicherstellen, dass es Längsaufnahmen der gesamten Prothese in jedem
Bildformat gibt.
Bestehen Bedenken hinsichtlich der Unversehrtheit der Prothese
(z.B. Knicke, Stentbrüche, Ablösung von Haken, relative Migration
von Komponenten) wird die Verwendung von vergrößerten Ansichten
empfohlen. Der behandelnde Arzt sollte Filme auf die Unversehrtheit
der Prothese (entlang der gesamten Prothese einschließlich ihrer
Komponenten) mit einer 2-4fach vergrößernden Sehhilfe auswerten.
12.4 Ultraschall
Wenn Patientenfaktoren den Einsatz von Kontrastmitteln ausschließen, kann
anstelle der CT-Untersuchung mit Kontrastmittel eine Ultraschallaufnahme
gemacht werden. Ultraschalluntersuchungen können mit CT-Untersuchungen
ohne Kontrastmittel kombiniert werden. Zur Bestimmung des maximalen
Aneurysmadurchmessers sowie von Endoleaks, Stentdurchgängigkeit und
Stenosen ist ein vollständiger Aortenduplex aufzuzeichnen. Die
Videoaufzeichnung sollte die folgenden Informationen umfassen:
• Transversale und longitudinale Aufnahmen aus Höhe der proximalen Aorta,
die die A. mesenterica und Aa. renalis bis zu den Iliakagabelungen zeigen,
um eventuell vorhandene Endoleaks aufzuspüren. Dazu nach Möglichkeit
Farbdoppler- und Power-Doppler-Angiographie verwenden.
• Jeder Verdacht auf ein Endoleak ist durch Spektralanalyse zu bestätigen.
• Transversale und longitudinale Aufnahmen des maximalen
Aneurysmadurchmessers sind anzufertigen.
12.5 Sicherheit und Kompatibilität mit MRT
Nicht klinische Tests haben ergeben, dass die endovaskuläre AAA-Prothese
Zenith bedingt MRT-kompatibel ist. Unter folgenden Bedingungen kann eine
MRT ohne Bedenken durchgeführt werden:
Systeme mit 1,5 Tesla:
• Statisches Magnetfeld mit 1,5 Tesla
• Feld mit einem räumlichen Gradienten von 450 Gauss/cm
• M aximale ganzkörpergemittelte spezifische Absorptionsrate (SAR) von
2,0 W/kg während einer Scan-Dauer von 15 Minuten
In nicht klinischen Tests verursachte die endovaskuläre AAA-Prothese Zenith
während eines 15-minütigen MRT-Scans in einem Magnetom-Scanner von
Siemens Medical Magnetom mit 1,5 Tesla, Numaris/4 Software, Version Syngo
MR 2002B DHHS, bei einer maximalen ganzkörpergemittelten spezifischen
Absorptionsrate (SAR) von 2,8 W/kg einen kalorimetrisch gemessenen
Temperaturanstieg von höchstens 1,4 °C. Die maximale ganzkörpergemittelte
spezifische Absorptionsrate (SAR) betrug 2,8 W/kg, was einem
kalorimetrischen Messwert von 1,5 W/kg entspricht.
Systeme mit 3,0 Tesla:
• Statisches Magnetfeld von 3,0 Tesla
• Feld mit einem räumlichen Gradienten von 720 Gauss/cm
• M aximale ganzkörpergemittelte spezifische Absorptionsrate (SAR) von
2,0 W/kg während einer Scan-Dauer von 15 Minuten
In nicht klinischen Tests verursachte die endovaskuläre AAA-Prothese Zenith
während eines 15-minütigen MRT-Scans in einem Excite-Scanner von GE
Electric Healthcare mit 3,0 Tesla, G3.0-052B Software, bei einer maximalen
ganzkörpergemittelten spezifischen Absorptionsrate (SAR) von 3.0 W/kg
einen kalorimetrisch gemessenen Temperaturanstieg von höchstens 1,9 °C.
Die maximale ganzkörpergemittelte spezifische Absorptionsrate (SAR) betrug
3,0 W/kg, was einem kalorimetrischen Messwert von 2,8 W/kg entspricht.
Bei Scans in nicht klinischen Tests mit folgender Sequenz verläuft das
Bildartefakt durch die gesamte anatomische Region, in der sich die Prothese
befindet, und verdeckt innerhalb von etwa 20 cm der Prothese die Sicht auf
unmittelbar angrenzende anatomische Strukturen sowie die gesamte
Prothese und ihr Lumen: Fast-Spin-Echo-Impulssequenz in einem ExciteScanner von GE Electric Healthcare mit 3,0 Tesla, G3.0-052B Software mit
Körper-Hochfrequenzspule.
75

Bei allen Scannern verschwindet das Bildartefakt mit zunehmendem Abstand
zwischen Prothese und relevantem Bereich. MRT-Aufnahmen von Kopf und
Hals und unteren Gliedmaßen können ohne Bildartefakt erhalten werden. Je
nach Abstand der Prothese vom relevanten Bereich können in Aufnahmen
der Abdominalregion und der oberen Gliedmaßen Bildartefakte vorhanden
sein.
Es liegen klinische Angaben über siebzehn Patienten vor, bei denen nach
Implantation eines Stent-Grafts MRT-Untersuchungen durchgeführt wurden.
Bei keinem dieser Patienten wurde im Nachfeld der MRT-Untersuchung über
unerwünschte Ereignisse oder Probleme mit der Prothese berichtet.
Außerdem sind weltweit weit über 50.000 endovaskuläre AAA-Prothesen
Zenith implantiert worden, ohne dass als Ergebnis von MRT-Untersuchungen
unerwünschte Ereignisse oder Probleme mit der Prothese gemeldet worden
sind.
Cook empfiehlt, dass der Patient die in dieser Gebrauchsanweisung
angegebenen MR-Bedingungen bei der MedicAlert Foundation registriert.
Die MedicAlert Foundation kann folgenderweise kontaktiert werden:
Anschrift: MedicAlert Foundation International
Telefon: +1 888-633-4298 Gebührenfrei (Inland)
Fax: +1 209-669-2450
2323 Colorado Avenue
Turlock, CA 95382 USA
+1 209-668-3333 Von außerhalb der USA
Internet: w ww.medicalert.org
12.6 Zusätzliche Überwachung und Behandlung
Zusätzliche Überwachungstermine und möglicherweise auch Behandlungen
werden in folgenden Fällen empfohlen:
• Aneurysmen mit Endoleak vom Typ I
• Aneurysmen mit Endoleak vom Typ III
• Vergrößerung des Aneurysmas ≥ 5 mm des maximalen Durchmessers
(unabhängig vom Endoleakstatus)
• Migration
• Unzureichende Länge der Abdichtung
Die Erwägung einer Reintervention oder Umstellung auf offen-chirurgische
Reparatur sollte die Beurteilung des behandelnden Arztes hinsichtlich
Komorbiditäten, Lebenserwartung und persönliche Wünsche des Patienten
berücksichtigen. Die Patienten sind über die Möglichkeit späterer
Reinterventionen einschließlich katheterbasierter Eingriffe und Umstellung
auf offene Chirurgie nach der Implantation einer endovaskulären Prothese
aufzuklären.
13 INFORMATIONEN ZUR PATIENTENVERFOLGUNG
Zusätzlich zu dieser Gebrauchsanweisung liegt der endovaskulären AAAProthese Zenith Flex mit dem H&L-B One-Shot-Einführsystem ein Formular zur
Produktnachverfolgung bei, das vom Krankenhauspersonal ausgefüllt und an
COOK eingesandt werden muss, damit überwacht werden kann, welche
Patienten die endovaskuläre AAA-Prothese Zenith Flex erhalten (Vorschrift
nach US-Bundesgesetz).
14 FEHLERBEHEBUNG
HINWEIS: Technische Unterstützung durch einen Cook-Produktspezialisten
kann über den zuständigen Cook- Außendienstmitarbeiter angefordert
werden.
14.1 Fehlerbehebung am Auslösedrahtmechanismus
VORSICHT: Die folgenden Schritte nur dann ausführen, wenn der
proximale Auslösedraht sich nicht wie in Abschnitt 11.1.7 beschrieben
abnehmen lässt.
14.1.1 Entfalten des proximalen (oberen) Hauptkörpers
1. Wenn sich der schwarze Auslösedrahtmechanismus (obere Kappe) nicht
vom Griff abnehmen lässt, den Draht neben dem Auslösedrahtmechanismus
kappen (Abb. 37) und den Auslösedrahtmechanismus vom Griff abnehmen.
2. Zur vollen Entfaltung des ipsilateralen Ansatzes den grauen Positionierer
stabilisieren und gleichzeitig die Schleuse zurückziehen.
3. Die Sicherheitssperre des weißen Auslösedrahtmechanismus entfernen.
4. Den Auslösedraht zurückziehen und entfernen. Dazu den weißen
Auslösedrahtmechanismus vom Griff herunterschieben und dann über den
Schlitz über die innere Kanüle der Prothese entfernen.
5. Das abgeschnittene Ende des Auslösedrahts der oberen Kappe mit einer
feststellbaren Pinzette einklemmen und sichern. (Abb. 38)
6. Die Klemmschraube lösen und – ohne die innere Kanüle und den
Auslösedraht zu bewegen – den grauen Positionierer und die Schleuse in
die Prothese vorschieben, bis die Spitze des grauen Positionierers etwa
2 cm von den Goldmarkierungen entfernt ist. (Abb. 39) Der vorgeschobene
graue Positionierer gibt der inneren Kanüle zusätzlichen Halt.
HINWEIS: Beim Vorschieben des grauen Positionierers und der Schleuse
leichten Zug auf den Auslösedraht ausüben, damit er straff bleibt.
HINWEIS: Darauf achten, dass die Spitze des grauen Positionierers nicht in die
obere Kappe vorgeschoben wird.
7. Die Klemmschraube festziehen. Bestätigen, dass der Auslösedraht mit der
Pinzette gesichert ist.
8. Den grauen Positionierer stabilisieren und die Schleuse langsam
vorschieben, bis die Spitze der Schleuse etwa 2 mm von den Goldmarkierungen entfernt ist. (Abb. 40)
HINWEIS: Beim Vorschieben der Schleuse darauf achten, nicht die Prothese
selbst vorzuschieben.
9. Die Schleuse stabilisieren und den grauen Positionierer zusammen mit der
inneren Kanüle zurückziehen, um die obere Kappe über den suprarenalen
Stent zu ziehen. (Abb. 41)
10. Die Position der Goldmarkierungen unterhalb der Aa. renales bestätigen.
11. Den Auslösedraht entfernen.
12. Die Schleuse zurückziehen, bis die konische Spitze des grauen
Positionierers freiliegt.
13. Einen Modellierungsballon durch den kontralateralen Ansatz des
Hauptteils vorschieben und gerade oberhalb der Gabelung der Prothese
positionieren.
14. Den Ballon auf den vollen Durchmesser der Prothese insufflieren. (Abb. 42)
15. Die Klemmschraube lösen.
16. Den grauen Positionierer und den Ballonkatheter stabilisieren und die
innere Kanüle vorschieben, um die obere Kappe zu entfalten.
17. Die Klemmschraube anziehen.
18. Den Ballon deflatieren und den kontralateralen Führungsdraht in die
Aorta thoracica vorschieben.
HINWEIS: Weil eine frühzeitige Entfaltung des ipsilateralen Schenkels und
Freigabe des Auslösedrahts möglich ist, wird vorgeschlagen, den
Modellierungsballon im kontralateralen Ansatz bzw. gerade oberhalb davon
zu belassen, damit die Prothese während der Platzierung des ipsilateralen
Ansatzes stabilisiert werden kann.
14.1.2 Andocken der oberen Kappe
1. Die Klemmschraube lösen. (Abb. 25)
2. Die Schleuse und innere Kanüle so sichern, dass sie sich nicht bewegen
können.
3. Den grauen Positionierer über die innere Kanüle vorschieben, bis er an der
oberen Kappe andockt. (Abb. 43, 27, 44)
HINWEIS: Wenn ein Widerstand auftritt, den grauen Positionierer leicht
drehen und weiter vorsichtig vorschieben.
4. Die Klemmschraube erneut anziehen und an der inneren Kanüle ziehen,
um die gesamte obere Kappe und den grauen Positionierer durch die
Prothese und durch die Schleuse zurückzuziehen. (Abb. 45) Die Schleuse
und den Führungsdraht an ihrer jeweiligen Position belassen.
5. Das Captor® Hämostaseventil an der Flexor® Einführschleuse schließen.
Dazu bis zum Anschlag im Uhrzeigersinn drehen. (Abb. 30)
14.1.3 Positionieren und Entfalten des ipsilateralen iliakalen Schenkels
HINWEIS: Sicherstellen, dass das Captor-Hämostaseventil an der
Einführschleuse geöffnet ist. (Abb. 31)
1. Die Führungsdraht- und Schleusenvorrichtung des Prothesenhauptteils
zur Einführung des ipsilateralen Prothesenschenkels verwenden.
HINWEIS: Aufgrund einer Modifikation der Freigabe der oberen Kappe muss
die komplette Schleuse des Hauptteils bis an einen Punkt etwa 1 bis 2 cm
innerhalb des proximalen ipsilateralen Ansatzes zurückgezogen werden. Die
Dilatator-/Schleuseneinheit des ipsilateralen Ansatzes in die Schleuse des
Hauptteils vorschieben.
HINWEIS: Falls eine zusätzliche Stabilisierung der Prothese erforderlich ist,
kann der Modellierungsballon im kontralateralen Ansatz des
Prothesenhauptteils insuffliert werden.
VORSICHT: Den Ballon nicht außerhalb der Prothese insufflieren.
HINWEIS: Bei stark gewundenen Gefäßen kann sich die Position der Iliaka
interna durch das Einführen der starren Draht- und Schleusensysteme
deutlich verschieben.
2. Langsam vorschieben, bis der ipsilaterale iliakale Prothesenschenkel
mindestens einen ganzen Stent des iliakalen Schenkels (d.h. einen
proximalen Stent des iliakalen Prothesenschenkels) innerhalb des
ipsilateralen Ansatzes des Hauptteils überlappt. (Abb. 46)
HINWEIS: Ist eine Überlappung von mehr als drei Stents des iliakalen
Schenkels (mehr als zwei Stents des iliakalen Schenkels für eine
Schenkellänge von 37 mm und 54 mm) erforderlich, muss eventuell eine
Schenkelverlängerung in den Gabelungsbereich der gegenüberliegenden
Seite in Betracht gezogen werden.
3. Die Position des distalen Endes des iliakalen Schenkels der Prothese
bestätigen. Falls erforderlich, den iliakalen Prothesenschenkel so
umpositionieren, dass die Durchgängigkeit der A. iliaca interna
gewährleistet ist.
4. Zum Entfalten den iliakalen Prothesenschenkel mit dem grauen
Positionierer stabilisieren und gleichzeitig die Schleuse des iliakalen
Schenkels und die Schleuse des Hauptteils zurückziehen. (Abb. 33 und 47)
Falls nötig, die Schleuse des Hauptteils zurückziehen.
5. Unter fluoroskopischer Beobachtung und nach Überprüfung der Position
des iliakalen Prothesenschenkels die Klemmschraube lockern und die
innere Kanüle so zurückziehen, dass der sich verjüngende Dilatator am
grauen Positionierer andockt. Die Klemmschraube wieder anziehen. Beim
Zurückziehen des grauen Positionierers mit gesicherter innerer Kanüle die
Schleusenposition beibehalten. (Abb. 48)
6. Das Captor Hämostaseventil an der Flexor Einführschleuse schließen. Dazu
bis zum Anschlag im Uhrzeigersinn drehen.
7. Die Position der Führungsdrähte erneut überprüfen. Schleuse und
Führungsdrähte in ihren Positionen belassen.
8. Den deflatierten Ballon aus der kontralateralen Seite entfernen.
14.1.4 Positionieren und Entfalten des kontralateralen iliakalen
Schenkels
VORSICHT: Vor der Implantation sicherstellen, dass der gewählte
kontralaterale iliakale Schenkel auch zum Einführen auf der
kontralateralen Seite des Patienten vorgesehen ist.
1. Den Bildverstärker so positionieren, dass sowohl die kontralaterale A. iliaca
interna als auch die kontralaterale A. iliaca communis zu sehen sind.
2. Vor dem Einbringen des Einführsystems für den kontralateralen iliakalen
Schenkel Kontrastmittel durch die kontralaterale femorale Schleuse
injizieren, um so die kontralaterale A. iliaca interna zu lokalisieren.
3. Das Einführsystem für den kontralateralen iliakalen Schenkel in die Arterie
einbringen. Langsam vorschieben, bis der iliakale Schenkel der Prothese
mindestens einen ganzen Stent des iliakalen Schenkels (d.h. einen
proximalen Stent des iliakalen Schenkels der Prothese) im kontralateralen
Ansatz des Hauptteils überlappt. (Abb. 49)
HINWEIS: Wenn es beim Vorschieben des Einführsystems für den iliakalen
Schenkel zu Schwierigkeiten kommt, auf einen steiferen Führungsdraht
ausweichen. In stark gewundenen Gefäßen kann es durch die Einführung der
steifen Drähte und Schleusensysteme zu erheblichen Änderungen in der
Anatomie kommen.
4. Die Position des distalen Endes des iliakalen Schenkels der Prothese
bestätigen. Falls nötig, den iliakalen Schenkel der Prothese neu
positionieren, um sowohl die Durchgängigkeit der A. iliaca interna als
auch eine Überlappung von mindestens einem vollen Stent des iliakalen
Schenkels (d.h. eines proximalen Stents des iliakalen Prothesenschenkels,
Maximalüberlappung von 1,5 Stents) innerhalb des Hauptteils der
endovaskulären Prothese sicherzustellen.
5. Zum Entfalten den iliakalen Prothesenschenkel mit dem grauen
Positionierer in Position halten und gleichzeitig die Schleuse
zurückziehen. (Abb. 19 und 50) Sicherstellen, dass eine Überlappung von
einer Stentlänge erhalten bleibt.
6. Sobald das distale Ende des iliakalen Prothesenschenkels freigegeben
wird, mit dem Zurückziehen der Schleuse aufhören.
7. Unter fluoroskopischer Beobachtung und nach Überprüfung der Position
des iliakalen Prothesenschenkels die Klemmschraube lockern und die
innere Kanüle so zurückziehen, dass der sich verjüngende Dilatator am
grauen Positionierer andockt. Die Klemmschraube wieder anziehen. Beim
Zurückziehen des grauen Positionierers mit gesicherter innerer Kanüle die
Schleusenposition beibehalten. (Abb. 51)
8. Die Position der Führungsdrähte erneut überprüfen.
9. Die Einführung des Modellierungsballons und das abschließende
Angiogramm wie in Abschnitt 11.1.12, Einführen des
Modellierungsballons, beschrieben durchführen.
14.2 Fehlerbehebung bei der suprarenalen Stententfaltung
VORSICHT: Die folgenden Schritte sollten nur dann durchgeführt
werden, wenn sich die Entfaltung mithilfe der oberen Kappe nicht wie in
Abschnitt 11.1.7 beschrieben durchführen lässt.
14.2.1 Entfalten des proximalen (oberen) Hauptkörpers
1. Lässt sich der suprarenale Stent durch Vorschub der inneren Kanüle der
oberen Kappe nicht vollständig entfalten, einen Modellierungsballon
durch den kontralateralen Ansatz des Hauptkörpers vorschieben und
gerade noch superior zur Stent-Graft-Gabelung positionieren.
2. Den Ballon zur zusätzlichen Unterstützung der inneren Kanüle auf den
vollen Durchmesser der Prothese inflatieren.
3. Die Klemmschraube lösen. (Abb. 15)
4. Den grauen Positionierer und den Ballonkatheter stabilisieren, und die
innere Kanüle zur Entfaltung des suprarenalen Stents vorschieben.
76

Bei voller Entfaltung des suprarenalen Stents:
5. Die Klemmschraube festziehen; den Ballon deflatieren und zurückziehen.
6. Den kontralateralen Führungsdraht in die Aorta thoracica vorschieben
und zum Abschluss des Verfahrens zum entsprechenden Schritt in der
Gebrauchsanweisung zurückkehren.
Ist die vollständige Entfaltung des suprarenalen Stents noch immer nicht
möglich:
5. Die Klemmschraube festziehen und den Ballon deflatieren. Den grauen
Positionierer unter Beibehaltung der Ballonposition stabilisieren und
die Schleuse zur vollständigen Entfaltung des ipsilateralen Ansatzes
zurückziehen.
6. Die Sicherheitssperre vom Auslösedrahtmechanismus für den
ipsilateralen Ansatz abnehmen.
7. Den Auslösedraht zurückziehen und entfernen. Dazu den
Auslösedrahtmechanismus für den ipsilateralen Ansatz vom Griff
herunter schieben und dann durch dessen Schlitz über die innere Kanüle
entfernen.
8. Die Klemmschraube lösen (Abb. 25). Den grauen Positionierer und die
Schleuse unter Beibehaltung der Position der inneren Kanüle in die
Prothese vorschieben, bis die Spitze des grauen Positionierers etwa
2 cm unterhalb der proximalen Goldmarkierungen liegt. (Abb. 52) Der
vorgeschobene graue Positionierer gibt der inneren Kanüle zusätzlichen
Halt.
HINWEIS: Es muss sorgfältig darauf geachtet werden, dass die Prothese
während des Vorschubes des grauen Positionierers und der Schleuse
nicht vorgeschoben wird.
HINWEIS: Es ist sicherzustellen, dass die Spitze des grauen Positionierers
nicht in die obere Kappe vorgeschoben wird.
9. Die Klemmschraube festziehen.
10. Die Position der Goldmarkierungen unterhalb der Aa. renales bestätigen.
11. Den Modellierungsballon so umpositionieren, dass er an der Gabelung
anliegt.
12. Den Ballon auf den vollen Durchmesser der Prothese inflatieren.
(Abb. 42)
13. Die Klemmschraube lösen.
14. Den grauen Positionierer und den Ballonkatheter stabilisieren, und die
innere Kanüle zur Entfaltung des suprarenalen Stents vorschieben.
15. Die Klemmschraube anziehen.
16. Den Ballon deflatieren und den kontralateralen Führungsdraht in die
Aorta thoracica vorschieben.
HINWEIS: Aufgrund frühzeitiger Entfaltung des ipsilateralen Schenkels
und Auslösedraht-Freigabe wird empfohlen, den Modellierungsballon
zur Stabilisierung der Prothese während der Platzierung des ipsilateralen
Schenkels in oder knapp oberhalb des kontralateralen Ansatzes zu
belassen.
14.2.2 Andocken der oberen Kappe
1. Die Klemmschraube lösen. (Abb. 25)
2. Die Schleuse und innere Kanüle so sichern, dass sie sich nicht bewegen
können.
3. Den grauen Positionierer über die innere Kanüle vorschieben, bis er an
der oberen Kappe andockt. (Abb. 43, 27 und 44)
HINWEIS: Wenn ein Widerstand auftritt, den grauen Positionierer leicht
drehen und weiter vorsichtig vorschieben.
4. Die Klemmschraube erneut anziehen und an der inneren Kanüle ziehen,
um die gesamte obere Kappe und den grauen Positionierer durch die
Prothese und durch die Schleuse zurückzuziehen. (Abb. 45) Die Schleuse
und den Führungsdraht an ihrer jeweiligen Position belassen.
5. Das Captor®-Hämostaseventil an der Flexor®-Einführschleuse bis zum
Anschlag im Uhrzeigersinn drehen, um es zu schließen. (Abb. 30)
14.2.3 Positionieren und Entfalten des ipsilateralen iliakalen Schenkels
HINWEIS: Sicherstellen, dass das Captor-Hämostaseventil an der
Einführschleuse geöffnet ist. (Abb. 31)
1. Den Bildverstärker so positionieren, dass er sowohl die ipsilaterale A. iliaca
interna als auch die ipsilaterale A. iliaca communis zeigt.
2. Nach der Einführung des Platzierungssystems für den ipsilateralen
iliakalen Schenkel, Kontrastmittel zur Lokalisierung der ipsilateralen A.
iliaca interna durch die Schleuse des Hauptkörpers injizieren.
3. Die Einheit aus Führungsdraht und Schleuse des Prothesenhauptkörpers
zur Einführung des ipsilateralen Prothesenschenkels verwenden.
HINWEIS: Aufgrund einer Modifikation der Freigabe der oberen Kappe
muss die Schleuseneinheit des Hauptkörpers bis an einen Punkt etwa
1 bis 2 cm innerhalb des proximalen ipsilateralen Ansatzes
zurückgezogen werden. Die Dilatator-/Schleuseneinheit des ipsilateralen
Ansatzes in die Schleuse des Hauptkörpers vorschieben.
HINWEIS: Falls eine zusätzliche Stabilisierung der Prothese erforderlich
ist, kann der Modellierungsballon im kontralateralen Ansatz des
Prothesenhauptkörpers inflatiert werden.
VORSICHT: Den Ballon nicht außerhalb der Prothese inflatieren.
HINWEIS: Bei stark gewundenen Gefäßen kann sich die Position
der Aa. iliacae internae durch das Einführen der starren Draht- und
Schleusensysteme deutlich verschieben.
4. Das Platzierungssystem für den ipsilateralen iliakalen Schenkel der
Prothese langsam vorschieben, bis der ipsilaterale iliakale Schenkel
der Prothese mindestens einen ganzen Stent des iliakalen Schenkels
(d.h. einen proximalen Stent des iliakalen Schenkels der Prothese) im
ipsilateralen Ansatz des Hauptkörpers überlappt. (Abb. 46)
HINWEIS: Ist eine Überlappung von mehr als drei Stents des iliakalen
Schenkels (mehr als zwei Stents des iliakalen Schenkels für eine
Länge von 37 mm und 54 mm) erforderlich, muss eventuell eine
Schenkelverlängerung in den Gabelungsbereich der gegenüberliegenden
Seite in Betracht gezogen werden.
5. Die Position des distalen Endes des iliakalen Prothesenschenkels
bestätigen. Falls erforderlich, den iliakalen Schenkel der Prothese
so umpositionieren, dass die Durchgängigkeit der A. iliaca interna
gewährleistet ist.
6. Zum Entfalten den iliakalen Schenkel der Prothese mit dem grauen
Positionierer stabilisieren und gleichzeitig die Schleuse des iliakalen
Schenkels und die Schleuse des Hauptkörpers zurückziehen.
(Abb. 33 und 47)
7. Unter fluoroskopischer Beobachtung und nach Überprüfung der Position
des iliakalen Schenkels der Prothese die Klemmschraube lockern und die
innere Kanüle so zurückziehen, dass der sich verjüngende Dilatator am
grauen Positionierer andockt. Die Klemmschraube wieder anziehen. Beim
Zurückziehen des grauen Positionierers mit gesicherter innerer Kanüle die
Schleusenposition beibehalten. (Abb. 48)
8. Das Captor-Hämostaseventil an der Flexor-Einführschleuse bis zum
Anschlag im Uhrzeigersinn drehen, um es zu schließen.
9. Die Position der Führungsdrähte erneut überprüfen. Schleuse und
Führungsdrähte in ihren Positionen belassen.
10. Den deflatierten Ballon von der kontralateralen Seite aus entfernen.
14.2.4 Positionieren und Entfalten des kontralateralen iliakalen
Schenkels
VORSICHT: Vor der Implantation sicherstellen, dass der vorgesehene
kontralaterale iliakale Schenkel zum Einführen auf der kontralateralen
Seite des Patienten ausgewählt wird.
1. Den Bildverstärker so positionieren, dass sowohl die kontralaterale A.
iliaca interna als auch die kontralaterale A. iliaca communis zu sehen sind.
2. Vor dem Einbringen des Platzierungssystems für den kontralateralen
iliakalen Schenkel Kontrastmittel durch die kontralaterale femorale
Schleuse injizieren, um so die kontralaterale A. iliaca interna zu
lokalisieren.
3. Das Platzierungssystem für den kontralateralen iliakalen Schenkel in
die Arterie einbringen. Langsam vorschieben, bis der iliakale Schenkel
der Prothese mindestens einen ganzen Stent des iliakalen Schenkels
(d.h. einen proximalen Stent des iliakalen Schenkels der Prothese) im
kontralateralen Ansatz des Hauptkörpers überlappt. (Abb. 49)
HINWEIS: Wenn es beim Vorschieben des Platzierungssystems für
den iliakalen Schenkel zu Schwierigkeiten kommt, auf einen steiferen
Führungsdraht ausweichen. In stark gewundenen Gefäßen kann es durch
die Einführung der steifen Drähte und Schleusensysteme zu erheblichen
Änderungen in der Anatomie kommen.
4. Die Position des distalen Endes des iliakalen Prothesenschenkels
bestätigen. Falls nötig, den iliakalen Schenkel der Prothese neu
positionieren, um sowohl die Durchgängigkeit der A. iliaca interna als
auch eine Überlappung von mindestens einem vollen Stent des iliakalen
Schenkels (d.h. eines proximalen Stents des iliakalen Prothesenschenkels,
Maximalüberlappung von 1,5 Stents) innerhalb des Hauptkörpers der
endovaskulären Prothese sicherzustellen.
5. Zum Entfalten den iliakalen Schenkel der Prothese mit dem grauen
Positionierer in Position halten und gleichzeitig die Schleuse
zurückziehen. (Abb. 19 und 50) Sicherstellen, dass eine Überlappung von
einer Stentlänge erhalten bleibt.
6. Sobald das distale Ende des iliakalen Schenkels der Prothese freigegeben
wird, die Schleuse nicht weiter zurückziehen.
7. Unter fluoroskopischer Beobachtung und nach Überprüfung der Position
des iliakalen Schenkels der Prothese die Klemmschraube lockern und die
innere Kanüle so zurückziehen, dass der sich verjüngende Dilatator am
grauen Positionierer andockt. Die Klemmschraube wieder anziehen. Beim
Zurückziehen des grauen Positionierers mit gesicherter innerer Kanüle die
Schleusenposition beibehalten. (Abb. 51)
8. Die Position der Führungsdrähte erneut überprüfen.
9. Wie im Abschnitt Einführen des Modellierungsballons der
Gebrauchsanweisung zum Produkt einen Modellierungsballon einführen
und ein Abschlussangiogramm aufnehmen.
77

ΕΛΛΗΝΙΚΑ
ΕΝΔΑΓΓΕΙΑΚΟ ΜΟΣΧΕΥΜΑ AAA ZENITH FLEX® ΜΕ ΤΟ ΣΥΣΤΗΜΑ
ΕΙΣΑΓΩΓΗΣ H&L-B ONE-SHOT™
Διαβάστε όλες τις οδηγίες προσεκτικά. Εάν δεν ακολουθήσετε σωστά τις οδηγίες,
τις προειδοποιήσεις και τις προφυλάξεις, ενδέχεται να προκληθούν σοβαρές
χειρουργικές συνέπειες ή τραυματισμός του ασθενούς.
ΠΡΟΣΟΧΗ: Η ομοσπονδιακή νομοθεσία (των Η.Π.Α.) περιορίζει την πώληση
της συσκευής αυτής μόνον από ιατρό ή κατόπιν εντολής ιατρού.
ΠΡΟΣΟΧΗ: Όλα τα περιεχόμενα της εξωτερικής θήκης
(συμπεριλαμβανομένων του συστήματος εισαγωγής και των ενδαγγειακών
μοσχευμάτων) παρέχονται στείρα, για μία μόνο χρήση.
Για τη σειρά προϊόντων Zenith υπάρχουν τέσσερις ισχύουσες προτεινόμενες
οδηγίες χρήσης. Αυτές οι οδηγίες χρήσης περιγράφουν τις προτεινόμενες οδηγίες
χρήσης για το ενδαγγειακό μόσχευμα AAA Zenith Flex (κύριο σώμα και λαγόνια
σκέλη). Για πληροφορίες σχετικά με άλλα εξαρτήματα Zenith, παρακαλούμε
ανατρέξτε στις ακόλουθες προτεινόμενες οδηγίες χρήσης:
• Ενδαγγειακό μόσχευμα AAA Zenith (κύριο σώμα και λαγόνια σκέλη
ενδαγγειακού μοσχεύματος AAA Zenith),
• Βοηθητικά εξαρτήματα ενδαγγειακού μοσχεύματος AAA Zenith (προέκταση
κύριου σώματος, προέκταση λαγόνιου σκέλους, μετατροπέας και λαγόνιο
βύσμα),
• Βοηθητικό μόσχευμα AAA Zenith® Renu™ (διαμορφώσεις προέκτασης κύριου
σώματος και μετατροπέα) και
• Καθετήρας με μπαλόνι CODA®.
1 ΠΕΡΙΓΡΑΦΗ ΤΗΣ ΣΥΣΚΕΥΗΣ
1.1 Εξαρτήματα αορτικού κύριου σώματος και λαγόνιου σκέλους
Το ενδαγγειακό μόσχευμα AAA Zenith Flex είναι ένα αρθρωτό σύστημα που
αποτελείται από τρία εξαρτήματα, ένα διχαλωτό αορτικό κύριο σώμα και δύο
λαγόνια σκέλη. (Εικ. 1) Τα τμήματα του μοσχεύματος κατασκευάζονται από
υφαντό πολυεστερικό ύφασμα πλήρους πάχους, ραμμένο σε αυτοεκτεινόμενες
ενδοπροσθέσεις Cook-Z® από ανοξείδωτο χάλυβα με ράμμα από πλεκτό
πολυεστέρα και μονόκλωνο πολυπροπυλένιο. Τα τμήματα είναι πλήρη
ενδοπροσθέσεων για την παροχή σταθερότητας και της δύναμης επέκτασης που
είναι απαραίτητες για τη διάνοιξη του αυλού του μοσχεύματος κατά τη διάρκεια
της έκπτυξης. Επιπλέον, οι ενδοπροσθέσεις Cook-Z παρέχουν την απαραίτητη
προσάρτηση και στεγανοποίηση του μοσχεύματος στο αγγειακό τοίχωμα.
Η μη καλυμμένη υπερνεφρική ενδοπρόσθεση στο εγγύς άκρο του μοσχεύματος
φέρει ακίδες, οι οποίες είναι τοποθετημένες σε βήματα των 3 mm για επιπλέον
καθήλωση της συσκευής. Για τη διευκόλυνση της ακτινοσκοπικής απεικόνισης του
μοσχεύματος ενδοπρόσθεσης, υπάρχουν τοποθετημένοι χρυσοί ακτινοσκιεροί
δείκτες ως εξής: ένας στην πλευρική επιφάνεια της πλέον περιφερικής
ενδοπρόσθεσης στο ετερόπλευρο μέλος του διχασμένου τμήματος του κυρίου
σώματος και τέσσερις σε περιφερικό προσανατολισμό εντός 2 mm από την
ανώτερη πλευρά του υλικού του μοσχεύματος.
1.2 Σύστημα τοποθέτησης κύριου σώματος
Το κύριο σώμα του ενδαγγειακού μοσχεύματος AAA Zenith Flex
αποστέλλεται προτοποθετημένο στο σύστημα εισαγωγής H&L-B One-Shot.
(Εικ. 2) Περιλαμβάνει μια μέθοδο διαδοχικής έκπτυξης με ενσωματωμένα
χαρακτηριστικά για την παροχή συνεχούς ελέγχου του ενδαγγειακού
μοσχεύματος καθόλη τη διάρκεια της διαδικασίας έκπτυξης. Το σύσ τημα
εισαγωγής H&L-B One-Shot καθιστά δυνατή την ακριβή τοποθέτηση και επιτρέπει
την επαναπροσαρμογή της τελικής θέσης του μοσχεύματος πριν από την έκπτυξη
της ακιδωτής υπερνεφρικής ενδοπρόσθεσης.
Το σύστημα τοποθέτησης μοσχεύματος κύριου σώματος χρησιμοποιεί
ένα σύστημα εισαγωγής H&L-B One-Shot 18, 20 ή 22 French. Μηχανισμοί
απελευθέρωσης διπλού σύρματος ενεργοποίησης ασφαλίζουν το ενδαγγειακό
μόσχευμα στο σύστημα τοποθέτησης έως ότου απελευθερωθεί από τον ιατρό.
Όλα τα συστήματα είναι συμβατά με συρμάτινο οδηγό 0,035 ιντσών (0,89 mm).
Για πρόσθετη αιμόσταση, μπορείτε να ξεσφίξετε ή να σφίξετε την αιμοστατική
βαλβίδα Captor® για την εισαγωγή ή/και την αφαίρεση βοηθητικών συσκευών
εντός και εκτός του θηκαριού. Τα συστήματα τοποθέτησης κύριου σώματος
διαθέτουν ένα θηκάρι εισαγωγέα Flexor®, το οποίο ανθίσταται στη στρέβλωση και
φέρει υδρόφιλη επικάλυψη. Αμφότερα τα χαρακτηριστικά προορίζονται για την
ενίσχυση της στρεπτικότητας στις λαγόνιες αρτηρίες και στην κοιλιακή αορτή.
1.3 Σύστημα τοποθέτησης λαγόνιου σκέλους
Τα λαγόνια σκέλη του ενδαγγειακού μοσχεύματος AAA Zenith αποστέλλονται
προτοποθετημένα στο σύστημα εισαγωγής H&L-B One-Shot. (Εικ. 3) Το σύστημα
τοποθέτησης έχει σχεδιαστεί για ευκολία χρήσης με ελάχιστη προετοιμασία.
Το σύστημα τοποθέτησης λαγόνιου σκέλους χρησιμοποιεί σύστημα εισαγωγής
H&L-B One-Shot 14 ή 16 French. Όλα τα συστήματα είναι συμβατά με συρμάτινο
οδηγό 0,035 ιντσών (0,89 mm).
1.4 Βοηθητικά εξαρτήματα ενδαγγειακού μοσχεύματος AAA Zenith
Διατίθενται επιπλέον βοηθητικά ενδαγγειακά εξαρτήματα (προεκτάσεις
κύριου σώματος, προεκτάσεις λαγόνιου σκέλους, μετατροπείς και λαγόνια
βύσματα). (Εικ. 4) Ανατρέξτε στις οδηγίες χρήσης των βοηθητικών εξαρτημάτων
ενδαγγειακού μοσχεύματος AAA Zenith για περισσότερες πληροφορίες.
2 ΧΡΗΣΗ ΓΙΑ ΤΗΝ ΟΠΟΙΑ ΠΡΟΟΡΙΖΕΤΑΙ
Το ενδαγγειακό μόσχευμα AAA Zenith Flex με το σύστημα εισαγωγής H&L-B
One-Shot ενδείκνυται για την ενδαγγειακή θεραπεία ασθενών με ανευρύσματα
κοιλιακής αορτής ή αορτολαγόνια ανευρύσματα που έχουν μορφολογία
κατάλληλη για ενδαγγειακή αποκατάσταση, συμπεριλαμβανομένων των
ακολούθων:
• Επαρκή λαγόνια/μηριαία προσπέλαση, συμβατή με τα απαιτούμενα συστήματα
εισαγωγής,
• Μη ανευρυσματικό, υπονεφρικό αορτικό τμήμα (αυχένας) εγγύς προς το
ανεύρυσμα:
• με περιφέρεια τουλάχιστον 15 mm,
• με διάμετρο που μετράται από εξωτερικό τοίχωμα σε εξωτερικό τοίχωμα το
πολύ 32 mm και τουλάχιστον 18 mm,
• με γωνία μικρότερη από 60 μοίρες σε σχέση με τον μακρύ άξονα του
ανευρύσματος και
• με γωνία μικρότερη από 45 μοίρες σε σχέση με τον άξονα της επινεφρικής
αορτής.
• Περιφερική θέση καθήλωσης λαγόνιας αρτηρίας μεγαλύτερη από 10 mm σε
μήκος και 7,5-20 mm σε διάμετρο (μετρημένη από εξωτερικό τοίχωμα σε
εξωτερικό τοίχωμα).
3 ΑΝΤΕΝΔΕΙΞΕΙΣ
Δεν υπάρχουν γνωστές αντενδείξεις για τις συσκευές αυτές.
4 ΠΡΟΕΙΔΟΠΟΙΗΣΕΙΣ ΚΑΙ ΠΡΟΦΥΛΑΞΕΙΣ
4.1 Γενικά
• Διαβάστε όλες τις οδηγίες προσεκτικά. Εάν δεν ακολουθήσετε σωστά τις
οδηγίες, τις προειδοποιήσεις και τις προφυλάξεις, ενδέχεται να προκληθούν
σοβαρές συνέπειες ή τραυματισμός του ασθενούς.
• Να έχετε πάντοτε διαθέσιμη μια έμπειρη χειρουργική ομάδα κατά τη διάρκεια
των διαδικασιών εμφύτευσης ή επανεπέμβασης, σε περίπτωση που καταστεί
απαραίτητη η μετατροπή σε ανοικτή χειρουργική αποκατάσταση.
• Το ενδαγγειακό μόσχευμα AAA Zenith Flex με το σύστημα εισαγωγής H&L-B
One-Shot πρέπει να χρησιμοποιείται μόνον από ιατρούς και ομάδες που έχουν
εκπαιδευτεί σε αγγειακές επεμβατικές τεχνικές (με καθετήρα και χειρουργικές)
και στη χρήση της συσκευής αυτής. Οι ειδικές προσδοκίες από την εκπαίδευση
περιγράφονται στην ενότητα 10.1, Εκπαίδευση ιατρού.
• Πρόσθετες ενδαγγειακές επεμβάσεις ή μετατροπή σε τυπική ανοικτή
χειρουργική αποκατάσταση μετά από αρχική ενδαγγειακή αποκατάσταση θα
πρέπει να εξετάζεται για ασθενείς που παρουσιάζουν διευρυνόμενο ανεύρυσμα,
μη αποδεκτή μείωση του μήκους καθήλωσης (αλληλεπικάλυψη αγγείου και
εξαρτήματος) ή/και ενδοδιαφυγή. Μια αύξηση στο μέγεθος του ανευρύσματος
ή/και επίμονη ενδοδιαφυγή ή μετατόπιση ενδέχεται να οδηγήσει σε ρήξη
ανευρύσματος.
• Ασθενείς που παρουσιάζουν μειωμένη ροή αίματος μέσω του μέλους του
μοσχεύματος ή/και διαφυγές ενδέχεται να χρειαστεί να υποβληθούν σε
δευτερεύουσες επεμβάσεις ή χειρουργικές διαδικασίες.
4.2 Επιλογή, θεραπεία και παρακολούθηση ασθενούς
• Το ε νδαγγειακό μόσχευμα AAA Zenith Flex έχει σχεδιαστεί για τη θεραπεία
αυχένων αορτής με διάμετρο όχι μικρότερη από 18 mm και όχι μεγαλύτερη
από 32 mm. Το ενδαγγειακό μόσχευμα AAA Zenith Flex έχει σχεδιαστεί για τη
θεραπεία εγγύς αυχένων αορτής (περιφερικά της κατώτατης νεφρικής
αρτηρίας) μήκους τουλάχιστον 15 mm. Απαιτείται περιφερική θέση καθήλωσης
λαγόνιας αρτηρίας μεγαλύτερη από 10 mm σε μήκος και 7,5-20 mm σε
διάμετρο (μετρημένη από εξωτερικό τοίχωμα σε εξωτερικό τοίχωμα). Αυτές οι
μετρήσεις προσδιορισμού μεγέθους είναι κρίσιμες για τη σωστή εκτέλεση της
ενδαγγειακής αποκατάστασης.
• Τα βασ ικά ανατομικά στοιχεία που ενδέχεται να επηρεάσουν την επιτυχή
εξαίρεση του ανευρύσματος περιλαμβάνουν σοβαρή γωνίωση του εγγύς
αυχένα (>60 μοίρες για τον υπονεφρικό αυχένα έως τον άξονα του AAA ή >45
μοίρες για τον επινεφρικό αυχένα σε σχέση με τον άμεσο υπονεφρικό αυχένα),
βραχύ εγγύς αυχένα αορτής (<15 mm), σχήμα ανεστραμμένης χοάνης (αύξηση
της διαμέτρου μεγαλύτερη από 10% σε μήκος 15 mm του εγγύς αυχένα
αορτής) και περιφερειακό θρόμβο ή/και αποτιτάνωση στις θέσεις αρτηριακής
εμφύτευσης, ειδικά στην επιφάνεια επαφής του εγγύς αυχένα αορτής και της
περιφερικής λαγόνιας αρτηρίας. Παρουσία ανατομικών περιορισμών, μπορεί να
απαιτείται αυχένας μεγαλύτερου μήκους για την επίτευξη επαρκούς
στεγανοποίησης και καθήλωσης. Η ακανόνιστη αποτιτάνωση ή/και πλάκα
ενδέχεται να διακυβεύσει την προσάρτηση και τη στεγανοποίηση των θέσεων
καθήλωσης. Οι αυχένες που εμφανίζουν αυτά τα βασικά ανατομικά στοιχεία
ενδέχεται να προάγουν περισσότερο τη μετατόπιση ή την ενδοδιαφυγή του
μοσχεύματος.
• Απαιτείται επαρκής πρόσβαση στη λαγόνια ή μηριαία αρτηρία για την εισαγωγή
της συσκευής στο αγγειακό σύστημα. Η διάμετρος (μετρημένη από εσωτερικό
τοίχωμα σε εσωτερικό τοίχωμα) και η μορφολογία (ελάχιστη ελίκωση,
αποφρακτική νόσος ή/και αποτιτάνωση) του αγγείου προσπέλασης πρέπει να
είναι συμβατές με τις τεχνικές αγγειακής προσπέλασης και τα συστήματα
τοποθέτησης ενός θηκαριού αγγειακού εισαγωγέα 16 έως 22 French. Αγγεία
που φέρουν σημαντική αποτιτάνωση, απόφραξη, ελίκωση ή επένδυση με
θρόμβο ενδέχεται να αποκλείσουν την τοποθέτηση του ενδαγγειακού
μοσχεύματος ή/και να αυξήσουν τον κίνδυνο εμβολής. Ενδέχεται να απαιτείται
η χρήση τεχνικής αγγειακού αγωγού για την επίτευξη πρόσβασης σε
ορισμένους ασθενείς.
• Το ε νδαγγειακό μόσχευμα AAA Zenith Flex με το σύστημα εισαγωγής H&L-B
One-Shot δε συνιστάται σε ασθενείς, οι οποίοι παρουσιάζουν δυσανεξία στους
σκιαγραφικούς παράγοντες που είναι απαραίτητοι για διεγχειρητική και
μετεγχειρητική απεικόνιση παρακολούθησης. Όλοι οι ασθενείς θα πρέπει να
παρακολουθούνται στενά και να ελέγχονται περιοδικά για τυχόν αλλαγή της
κατάστασης της νόσου τους και για την ακεραιότητα της ενδοπρόσθεσης.
• Το ε νδαγγειακό μόσχευμα AAA Zenith Flex με το σύστημα εισαγωγής H&L-B
One-Shot δε συνιστάται σε ασθενείς που υπερβαίνουν τα όρια βάρους ή/και
μεγέθους, τα οποία διακυβεύουν ή αποτρέπουν τις απαραίτητες απαιτήσεις
απεικόνισης.
• Αδυναμία διατήρησης της βατότητας τουλάχιστον μίας έσω λαγόνιας αρτηρίας
ή απόφραξη μιας απαραίτητης κάτω μεσεντέριας αρτηρίας ενδέχεται να
αυξήσει τον κίνδυνο πυελικής/εντερικής ισχαιμίας.
• Πολλαπλές μεγάλες, βατές οσφυικές αρτηρίες, τοιχωματικός θρόμβος και βατή
κάτω μεσεντέρια αρτηρία ενδέχεται να προδιαθέτουν έναν ασθενή σε
ενδοδιαφυγές τύπου II. Ασθενείς με μη αποκαταστάσιμη διαταραχή της πήξης
ενδέχεται επίσης να διατρέχουν αυξημένο κίνδυνο ενδοδιαφυγής τύπου II ή
επιπλοκών αιμορραγίας.
• Η ασφάλεια και η αποτελεσματικότητα του ενδαγγειακού μοσχεύματος AAA
Zenith Flex με το σύστημα εισαγωγής H&L-B One-Shot δεν έχει αξιολογηθεί
στους ακόλουθους πληθυσμούς ασθενών:
• τραυματική αορτική κάκωση
• ανευρύσματα με διαρροή, επικείμενη ρήξη ή ραγέντα
• μυκητιασικά ανευρύσματα
• ψευδοανευρύσματα που προκύπτουν από προηγούμενη τοποθέτηση
μοσχεύματος
• αναθεώρηση προηγουμένως τοποθετημένων ενδαγγειακών μοσχευμάτων
• μη διορθώσιμη διαταραχή της πηκτικότητας του αίματος
• απαραίτητη μεσεντέρια αρτηρία
• γενετική νόσος συνδετικού ιστού (π.χ. σύνδρομα Marfan ή Ehlers-Danlos)
• συνοδά ανευρύσματα θωρακικής αορτής ή θωρακοκοιλιακά ανευρύσματα
• ασθενείς με ενεργές συστηματικές λοιμώξεις
• έγκυοι ή θηλάζουσες γυναίκες
• ασθενείς με νοσηρή παχυσαρκία
• ηλικίας κάτω των 18 ετών
• ασθενείς με εγγύς αορτικό αυχένα μήκους μικρότερου από 15 mm ή
γωνίωσης μεγαλύτερης από 60 μοίρες σε σχέση με το μακρύ άξονα του
ανευρύσματος.
• Η επιτυχής επιλογή των ασθενών απαιτεί συγκεκριμένες απεικονιστικές
εξετάσεις και ακριβείς μετρήσεις. Δείτε την ενότητα 4.3 Τεχνικές μέτρησης
και απεικόνιση πριν από τη διαδικασία.
• Όλα τα μήκη και οι διάμετροι των συσκευών που είναι απαραίτητα για την
ολοκλήρωση της διαδικασίας πρέπει να είναι διαθέσιμα στον ιατρό, ειδικά όταν
δεν είναι βέβαιες οι μετρήσεις προεγχειρητικού σχεδιασμού περίπτωσης
(διάμετροι/μήκη θεραπείας). Η προσέγγιση αυτή επιτρέπει μεγαλύτερη
διεγχειρητική ευελιξία για την επίτευξη βέλτιστων εκβάσεων διαδικασίας.
4.3 Τεχνικές μέτρησ ης και απεικόνιση πριν από τη διαδικασία
• Η έλλειψη απεικόνισης αξονικής τομογραφίας χωρίς σκιαγραφικό μέσο
ενδέχεται να οδηγήσει σε αποτυχία εκτίμησης της λαγόνιας ή της αορτικής
αποτιτάνωσης, η οποία ενδέχεται να αποκλείσει την προσπέλαση ή την
αξιόπιστη καθήλωση και στεγανοποίηση της συσκευής.
• Πάχος ανακατασκευής κατά την απεικόνιση πριν από τη διαδικασία >3 mm
ενδέχεται να έχει ως αποτέλεσμα υποβέλτιστο προσδιορισμό μεγέθους της
συσκευής ή αποτυχία εκτίμησης των εστιακών στενώσεων από την αξονική
τομογραφία.
• Η κλινική εμπειρία υποδεικνύει ότι η σπειροειδής υπολογιστική αγγειογραφία
τομογραφίας (CTA) με σκιαγραφική ενίσχυση και τρισδιάστατη ανακατασκευή
αποτελεί την ιδιαίτερα συνιστώμενη μέθοδο απεικόνισης για την ακριβή
εκτίμηση της ανατομίας των ασθενών πριν από τη θεραπεία με το ενδαγγειακό
μόσχευμα AAA Zenith Flex. Εάν δεν είναι διαθέσιμη η σπειροειδής CTA με
σκιαγραφική ενίσχυση και τρισδιάστατη ανακατασκευή, ο ασθενής θα πρέπει
να παραπεμφθεί σε ίδρυμα που διαθέτει αυτές τις δυνατότητες.
• Οι κλινικοί ιατροί συνιστούν την τοποθέτηση του βραχίονα C του
ακτινογραφικού μηχανήματος κατά τη διάρκεια της επεμβατικής αγγειογραφίας
με τέτοιο τρόπο ώστε οι εκφύσεις των νεφρικών αρτηριών, και ειδικά της
κατώτατης βατής νεφρικής αρτηρίας, να αναδεικνύονται καλά πριν από την
έκπτυξη του εγγύς άκρου του υλικού του μοσχεύματος (ενδοπρόσθεση
στεγανοποίησης) του κυρίου σώματος. Επιπλέον, η αγγειογραφία θα πρέπει να
αναδεικνύει τους διχασμούς των λαγονίων αρτηριών με τέτοιο τρόπο ώστε τα
περιφερικά τμήματα των κοινών λαγόνιων αρτηριών να ορίζονται σαφώς σε
σχέση με την έκφυση των έσω λαγόνιων αρτηριών άμφω, πριν από την έκπτυξη
των εξαρτημάτων λαγόνιων σκελών.
Διάμετροι:
Με χρήση CT, θα πρέπει να προσδιορίζονται οι μετρήσεις των διαμέτρων από
τη διάμετρο του αγγείου από εξωτερικό τοίχωμα σε εξωτερικό τοίχωμα (όχι με
μέτρηση της διαμέτρου του αυλού) ώστε να διευκολυνθεί ο προσδιορισμός του
78

σωστού μεγέθους της συσκευής και η σωστή επιλογή συσκευής. Η σπειροειδής
CT με σκιαγραφική ενίσχυση πρέπει να ξεκινήσει σε απόσταση 1 cm άνω από
τον κοιλιακό άξονα και να συνεχίσει διαμέσου των μηριαίων κεφαλών σε πάχος
αξονικής τομής 3 mm ή χαμηλότερο.
Μήκη:
Με χρήση CT, θα πρέπει να προσδιορίζονται οι μετρήσεις των μηκών για την
ακριβή εκτίμηση του μήκους του υπονεφρικού εγγύς αυχένα, καθώς επίσης και ο
προγραμματισμός των μεγεθών του κυρίου σώματος και των εξαρτημάτων του
σκέλους για το ενδαγγειακό μόσχευμα AAA Zenith Flex. Αυτές οι ανακατασκευές
θα πρέπει να διενεργούνται σε οβελιαίο, στεφανιαίο και τρισδιάστατο επίπεδο.
• Η μακροχρόνια απόδοση και ασφάλεια των ενδαγγειακών μοσχευμάτων
δεν έχει ακόμα επιβεβαιωθεί. Όλοι οι ασθενείς πρέπει να ενημερώνονται
ότι η ενδαγγειακή θεραπεία απαιτεί μακροχρόνια, τακτική
παρακολούθηση για την εκτίμηση της υγείας τους και της απόδοσης του
ενδαγγειακού μοσχεύματός τους. Ασθενείς με ειδικά κλινικά ευρήματα (π.χ.
ενδοδιαφυγές, διευρυνόμενο ανεύρυσμα ή μεταβολές της δομής ή της θέσης
του ενδαγγειακού μοσχεύματος) πρέπει να τελούν υπό αυξημένη
παρακολούθηση. Ειδικές κατευθυντήριες οδηγίες παρακολούθησης
περιγράφονται στην ενότητα 12, ΚΑΤΕΥΘΥΝΤΗΡΙΕΣ ΟΔΗΓΙΕΣ ΑΠΕΙΚΟΝΙΣΗΣ
ΚΑΙ ΜΕΤΕΓΧΕΙΡΗΤΙΚΗ ΠΑΡΑΚΟΛΟΥΘΗΣΗ.
• Το ενδαγγειακό μόσχευμα AAA Zenith Flex με το σύστημα εισαγωγής H&L-B
One-Shot δεν συνιστάται σε ασθενείς οι οποίοι δεν είναι σε θέση να
υποβληθούν ή οι οποίοι δεν θα συμμορφωθούν με τις απαραίτητες
προεγχειρητικές και μετεγχειρητικές μελέτες απεικόνισης και εμφύτευσης,
όπως περιγράφονται στην ενότητα 12, ΚΑΤΕΥΘΥΝΤΗΡΙΕΣ ΟΔΗΓΙΕΣ
ΑΠΕΙΚΟΝΙΣΗΣ ΚΑΙ ΜΕΤΕΓΧΕΙΡΗΤΙΚΗ ΠΑΡΑΚΟΛΟΥΘΗΣΗ.
• Μετά την τοποθέτηση του ενδαγγειακού μοσχεύματος, οι ασθενείς πρέπει να
παρακολουθούνται τακτικά για ροή περί του μοσχεύματος, ανάπτυξη
ανευρύσματος ή μεταβολές της δομής ή της θέσης του ενδαγγειακού
μοσχεύματος. Ως ελάχιστο, απαιτείται ετήσια απεικόνιση, η οποία περιλαμβάνει:
1) κοιλιακές ακτινογραφίες για την εξέταση της ακεραιότητας της συσκευής
(διαχωρισμός μεταξύ εξαρτημάτων, θραύση της ενδοπρόσθεσης ή
διαχωρισμός των ακίδων) και 2) αξονική τομογραφία με και χωρίς σκιαγραφικό
μέσο για την εξέταση τυχόν μεταβολών του ανευρύσματος, της ροής περί του
μοσχεύματος, της βατότητας, της ελίκωσης των αγγείων και της προοδευτικής
νόσου. Σε περίπτωση που αποκλείεται η χρήση σκιαγραφικού μέσου
απεικόνισης λόγω νεφρικών επιπλοκών ή άλλων παραγόντων, παρόμοιες
πληροφορίες μπορούν να ληφθούν με κοιλιακές ακτινογραφίες και υπέρηχο
duplex.
4.4 Επιλογή συσκευής
• Συνιστάται με έμφαση η αυστηρή τήρηση του οδηγού προσδιορισμού
μεγέθους των οδηγιών χρήσης του ενδαγγειακού μοσχεύματος AAA Zenith Flex
κατά την επιλογή του κατάλληλου μεγέθους συσκευής (Πίνακες 10.5.1 και
10.5.2). Η δέουσα επιλογή μεγαλύτερου μεγέθους συσκευής έχει ενσωματωθεί
στον οδηγό προσδιορισμού μεγέθους των οδηγιών χρήσης. Η επιλογή
μεγέθους εκτός αυτού του εύρους μπορεί να προκαλέσει ενδοδιαφυγή,
θραύση, μετατόπιση, πτύχωση προς τα έσω ή συμπίεση της συσκευής.
4.5 Διαδικασία εμφύτευσης
(Ανατρέξτε στην ενότητα 11, ΟΔΗΓΙΕΣ ΧΡΗΣΗΣ)
• Απαιτείται κατάλληλη διεγχειρητική απεικόνιση για την επιτυχή τοποθέτηση
του ενδαγγειακού μοσχεύματος AAA Zenith Flex και τη διασφάλιση της
κατάλληλης εναπόθεσης επάνω στο αορτικό τοίχωμα.
• Μην κάμπτετε και μη στρεβλώνετε το σύστημα τοποθέτησης. Με την ενέργεια
αυτή ενδέχεται να προκληθεί ζημιά στο σύστημα τοποθέτησης και στο
ενδαγγειακό μόσχευμα AAA Zenith Flex.
• Για να αποφύγετε τυχόν συστροφή του ενδαγγειακού μοσχεύματος, κατά τη
διάρκεια οποιασδήποτε περιστροφής του συστήματος τοποθέτησης,
προσέχετε έτσι ώστε να περιστρέφετε όλα τα εξαρτήματα του συστήματος ως
ενιαία μονάδα (από το εξωτερικό θηκάρι έως την εσωτερική κάνουλα).
• Εάν αισθανθείτε αντίσταση κατά τη διάρκεια της προώθησης του συρμάτινου
οδηγού ή του συστήματος τοποθέτησης, μη συνεχίζετε την προώθηση
οποιουδήποτε τμήματος του συστήματος τοποθέτησης. Σταματήστε και
εκτιμήστε την αιτία της αντίστασης. Υπάρχει κίνδυνος να προκληθεί βλάβη στο
αγγείο, τον καθετήρα ή το μόσχευμα. Προσέχετε ιδιαίτερα σε περιοχές
στένωσης, ενδαγγειακής θρόμβωσης ή σε αποτιτανωμένα ή ελικοειδή αγγεία.
• Η εσφαλμένη μερική έκπτυξη ή η μετατόπιση της ενδοπρόσθεσης ενδέχεται να
απαιτήσει χειρουργική επέμβαση.
• Εκτός εάν ενδείκνυται για ιατρικούς λόγους, μην εκπτύσσετε το ενδαγγειακό
μόσχευμα AAA Zenith Flex σε θέση που θα προκαλέσει απόφραξη αρτηριών
που είναι απαραίτητες για την παροχή ροής αίματος σε όργανα ή άκρα. Μην
καλύπτετε σημαντικές νεφρικές ή μεσεντέριες αρτηρίες (εξαίρεση αποτελεί η
κάτω μεσεντέρια αρτηρία) με την ενδοπρόσθεση. Ενδέχεται να επισυμβεί
απόφραξη του αγγείου. Κατά τη διάρκεια της κλινικής μελέτης, η συσκευή αυτή
δε μελετήθηκε σε ασθενείς με δύο αποφραγμένες έσω λαγόνιες αρτηρίες.
• Μην επιχειρήσετε να τοποθετήσετε πάλι το μόσχευμα στο θηκάρι μετά από
μερική ή πλήρη έκπτυξή του.
• Η επανατοποθέτηση του μοσχεύματος ενδοπρόσθεσης περιφερικά μετά από
την μερική έκπτυξη της επικαλυμμένης εγγύς ενδοπρόσθεσης μπορεί να
προκαλέσει ζημιά στο μόσχευμα της ενδοπρόσθεσης ή/και τραυματισμό του
αγγείου.
• Η μη ακριβής τοποθέτηση ή/και η ατελής στεγανοποίηση του ενδαγγειακού
μοσχεύματος AAA Zenith Flex εντός του αγγείου ενδέχεται να έχει ως
αποτέλεσμα αυξημένο κίνδυνο ενδοδιαφυγής, μετανάστευσης ή ακούσιας
απόφραξης των νεφρικών ή των έσω λαγόνιων αρτηριών. Πρέπει να διατηρείται
η βατότητα της νεφρικής αρτηρίας για την πρόληψη/μείωση του κινδύνου
νεφρικής βλάβης και επακόλουθων επιπλοκών.
• Η ανεπαρκής καθήλωση του ενδαγγειακού μοσχεύματος AAA Zenith Flex
ενδέχεται να έχει ως αποτέλεσμα αυξημένο κίνδυνο μετανάστευσης του
μοσχεύματος της ενδοπρόσθεσης. Η εσφαλμένη έκπτυξη ή η μετανάστευση
της ενδοπρόσθεσης ενδέχεται να απαιτήσει χειρουργική επέμβαση.
• Κατά τη διαδικασία εμφύτευσης πρέπει να χρησιμοποιείται συστηματική
αντιπηκτική αγωγή με βάση το προτιμώμενο πρωτόκολλο του νοσοκομείου και
του ιατρού. Εάν αντενδείκνυται η χρήση ηπαρίνης, πρέπει να εξετάζεται το
ενδεχόμενο εναλλακτικού αντιπηκτικού.
• Για να ενεργοποιήσετε την υδρόφιλη επικάλυψη στην εξωτερική επιφάνεια του
θηκαριού εισαγωγέα Flexor, πρέπει να σκουπίσετε την επιφάνεια με στείρα
επιθέματα γάζας εμποτισμένα σε φυσιολογικό ορό. Διατηρείτε πάντοτε το
θηκάρι ενυδατωμένο για βέλτιστη απόδοση.
• Κατά τη διάρκεια της προετοιμασίας και της εισαγωγής, ελαχιστοποιήστε το
χειρισμό της περιοριζόμενης ενδοπρόσθεσης, έτσι ώστε να μειωθεί ο κίνδυνος
μόλυνσης και λοίμωξης της ενδοπρόσθεσης.
• Διατηρείτε τη θέση του συρμάτινου οδηγού κατά τη διάρκεια της εισαγωγής
του συστήματος τοποθέτησης.
• Θα πρέπει να χρησιμοποιείται ακτινοσκόπηση κατά την εισαγωγή και την
απελευθέρωση για την επιβεβαίωση της σωστής λειτουργίας των εξαρτημάτων
του συστήματος τοποθέτησης, της σωστής τοποθέτησης του μοσχεύματος και
της επιθυμητής έκβασης της επέμβασης.
• Η χρήση του ενδαγγειακού μοσχεύματος AAA Zenith Flex με το σύστημα
εισαγωγής H&L-B One-Shot απαιτεί τη χορήγηση ενδαγγειακού σκιαγραφικού
μέσου. Ασθενείς με προϋπάρχουσα νεφρική ανεπάρκεια ενδέχεται να
διατρέχουν αυξημένο κίνδυνο νεφρικής βλάβης μετεγχειρητικά. Θα πρέπει να
προσέχετε ώστε να περιορίσετε την ποσότητα του σκιαγραφικού μέσου που
χρησιμοποιείτε κατά τη διάρκεια της επέμβασης και να εφαρμόζετε
προληπτικές μεθόδους θεραπείας για να μειώσετε τη νεφρική δυσλειτουργία
(π.χ. επαρκή ενυδάτωση).
• Καθώς αποσύρεται το θηκάρι ή/και ο συρμάτινος οδηγός, μπορεί να αλλάξει η
ανατομία και η θέση του μοσχεύματος. Να παρακολουθείτε συνεχώς τη θέση
του μοσχεύματος και να διενεργείτε αγγειογραφία για να ελέγχετε τη θέση,
όποτε είναι απαραίτητο.
• Το ε νδαγγειακό μόσχευμα AAA Zenith Flex φέρει ενσωματωμένη μια
επινεφρική ενδοπρόσθεση με ακίδες καθήλωσης. Πρέπει να δίνετε ιδιαίτερη
προσοχή κατά το χειρισμό επεμβατικών και αγγειογραφικών συσκευών στην
περιοχή της υπερνεφρικής ενδοπρόσθεσης.
• Προσέχετε ιδιαίτερα κατά τη διάρκεια του χειρισμού καθετήρων, συρμάτων και
θηκαριών εντός ενός ανευρύσματος. Οι σημαντικές διαταραχές μπορεί να
αποκολλήσουν τεμάχια του θρόμβου, τα οποία μπορεί να προκαλέσουν
περιφερική εμβολή ή ρήξη του ανευρύσματος.
• Προσέχετε να μην προκαλέσετε ζημιά στο μόσχευμα και να μη διαταράξετε τη
θέση του μοσχεύματος μετά την τοποθέτηση, σε περίπτωση που καταστεί
απαραίτητη πρόσθετη χρήση χειρουργικών εργαλείων (δευτερογενής
παρέμβαση) στην περιοχή του μοσχεύματος.
• Πριν από την έκπτυξη της υπερνεφρικής ενδοπρόσθεσης, επαληθεύστε ότι η
θέση του συρμάτινου οδηγού προσπέλασης προεκτείνεται μόλις περιφερικά
προς το αορτικό τόξο.
• Επαληθεύστε ότι επιλέγεται το προκαθορισμένο ετερόπλευρο λαγόνιο σκέλος
για εισαγωγή στην ετερόπλευρη πλευρά του ασθενούς πριν από την
εμφύτευση.
4.6 Χρήση μπαλονιού διαμόρφωσης
• Μη φουσκώνετε το μπαλόνι σε αγγείο εκτός του μοσχεύματος, καθώς με αυτόν
τον τρόπο ενδέχεται να προκαλέσετε βλάβη στο αγγείο. Χρησιμοποιήστε το
μπαλόνι σύμφωνα με την επισήμανσή του.
• Προσέξτε κατά το φούσκωμα του μπαλονιού εντός του μοσχεύματος παρουσία
αποτιτάνωσης, καθώς το υπερβολικό φούσκωμα ενδέχεται να προκαλέσει
ζημιά στο αγγείο.
• Επιβεβαιώστε το πλήρες ξεφούσκωμα του μπαλονιού πριν από την
επανατοποθέτηση.
• Για πρόσθετη αιμόσταση, μπορείτε να ξεσφίξετε ή να σφίξετε την αιμοστατική
βαλβίδα Captor, έτσι ώστε να καταστεί δυνατή η εισαγωγή και η επακόλουθη
απόσυρση ενός μπαλονιού διαμόρφωσης.
4.7 Ασφάλεια και συμβατότητα με μαγνητική τομογραφία (MRI)
Μη κλινικές δοκιμές έχουν καταδείξει ότι το ενδαγγειακό μόσχευμα AAA Zenith
είναι ασφαλές για μαγνητική τομογραφία υπό προϋποθέσεις. Μπορεί να σαρωθεί
με ασφάλεια υπό τις ακόλουθες προϋποθέσεις:
Συστήματα 1,5 Tesla
• Στατικό μαγνητικό πεδίο 1,5 Tesla
• Πεδίο χωρικής βαθμίδωσης 450 Gauss/cm
• Μέγιστος μεσοτιμημένος ρυθμός ειδικής ολοσωματικής απορρόφησης (SAR)
2,0 W/kg για 15 λεπτά σάρωσης
Σε μη κλινικές δοκιμές, το ενδαγγειακό μόσχευμα AAA Zenith προκάλεσε αύξηση
θερμοκρασίας χαμηλότερη από ή ίση με 1,4 °C σε μέγιστο μεσοτιμημένο ρυθμό
ειδικής ολοσωματικής απορρόφησης (SAR) 2,8 W/kg, όπως υπολογίστηκε βάσει
θερμιδομετρίας για 15 λεπτά σάρωσης μαγνητικής τομογραφίας σε μαγνητικό
τομογράφο 1,5 Tesla, Siemens Medical Magnetom, με λογισμικό Numaris/4,
έκδοσης Syngo MR 2002B DHHS. Ο μέγιστος μεσοτιμημένος ρυθμός ειδικής
ολοσωματικής απορρόφησης (SAR) ήταν 2,8 W/kg, που αντιστοιχεί σε τιμή
μετρούμενη με θερμιδομετρία ίση με 1,5 W/kg.
Συστήματα 3,0 Tesla:
• Στατικό μαγνητικό πεδίο 3,0 Tesla
• Πεδίο χωρικής βαθμίδωσης 720 Gauss/cm
• Μέγιστος μεσοτιμημένος ρυθμός ειδικής ολοσωματικής απορρόφησης (SAR)
2,0 W/kg για 15 λεπτά σάρωσης
Σε μη κλινικές δοκιμές, το ενδαγγειακό μόσχευμα AAA Zenith προκάλεσε αύξηση
θερμοκρασίας χαμηλότερη από ή ίση με 1,9 °C σε μέγιστο μεσοτιμημένο ρυθμό
ειδικής ολοσωματικής απορρόφησης (SAR) 3,0 W/kg, όπως υπολογίστηκε βάσει
θερμιδομετρίας για 15 λεπτά σάρωσης μαγνητικής τομογραφίας σε μαγνητικό
τομογράφο 3,0 Tesla Excite, GE Electric Healthcare, με λογισμικό G3.0-052B. Ο
μέγιστος μεσοτιμημένος ρυθμός ειδικής ολοσωματικής απορρόφησης (SAR) ήταν
3,0 W/kg, που αντιστοιχεί σε τιμή μετρούμενη με θερμιδομετρία ίση με 2,8 W/kg.
Το τέχνημα εικόνας εκτείνεται σε όλη την ανατομική περιοχή που περιέχει τη
συσκευή, ασαφοποιώντας τις παρακείμενες ανατομικές δομές σε ακτίνα περίπου
20 cm περιμετρικά της συσκευής, καθώς επίσης και ολόκληρη τη συσκευή και
τον αυλό της, κατά τη διενέργεια σάρωσης σε μη κλινικές δοκιμές με χρήση της
ακολουθίας: ταχείας στροφορμικής ηχούς, σε σύστημα μαγνητικής τομογραφίας
(MR) 3,0 Tesla, Excite, GE Electric Healthcare, με λογισμικό G3.0-052B, με πηνίο
ραδιοσυχνοτήτων σώματος.
Για όλους τους τομογράφους, το τέχνημα εικόνας εξασθενεί όσο μεγαλώνει
η απόσταση της συσκευής από την περιοχή ενδιαφέροντος. Μαγνητικές
τομογραφίες της κεφαλής, του αυχένα και των κάτω άκρων είναι δυνατόν
να ληφθούν χωρίς τέχνημα εικόνας. Τέχνημα εικόνας μπορεί να υπάρχει σε
τομογραφίες της κοιλιακής χώρας και των άνω άκρων, ανάλογα με την απόσταση
της συσκευής από την περιοχή ενδιαφέροντος.
Διατίθενται κλινικές πληροφορίες για δεκαεπτά ασθενείς οι οποίοι υποβλήθηκαν
σε μαγνητική τομογραφία μετά από την εμφύτευση του μοσχεύματος
ενδοπρόσθεσης. Δεν έχει αναφερθεί κανένα ανεπιθύμητο συμβάν ή πρόβλημα
με τη συσκευή σε οποιονδήποτε από αυτούς τους ασθενείς λόγω της διενέργειας
μαγνητικής τομογραφίας. Επιπλέον, έχουν εμφυτευθεί πολύ περισσότερα από
50.000 ενδαγγειακά μοσχεύματα AAA Zenith παγκοσμίως, στα οποία δεν έχει
αναφερθεί κανένα ανεπιθύμητο συμβάν ή πρόβλημα με τη συσκευή λόγω
διενέργειας μαγνητικής τομογραφίας.
Η Cook συνιστά την καταχώρηση εκ μέρους του ασθενούς των όρων μαγνητικής
τομογραφίας που δίνονται σε αυτές τις οδηγίες χρήσης στο MedicAlert
Foundation. Τα σ τοιχεία επικοινωνίας με το MedicAlert Foundation είναι τα εξής:
Ταχυδρομική διεύθυνση: MedicAlert Foundation International
Τηλέφωνο: +1 888-633-4298 (γραμμή χωρίς χρέωση)
Φαξ: +1 209-669-2450
Ιστοσελίδα: www.medicalert.org
2323 Colorado Avenue
Turlock, CA 95382 Η.Π.Α.
+1 209-668-3333 για κλήσεις εκτός των Η.Π.Α.
5 ΑΝΕΠΙΘΥΜΗΤΕΣ ΕΝΕΡΓΕΙΕΣ
5.1 Παρατηρούμενες ανεπιθύμητες ενέργειες
Μια πολυκεντρική, προοπτική μελέτη των Η.Π.Α. μιας προηγούμενης έκδοσης της
συσκευής (Ενδαγγειακό μόσχευμα AAA Zenith) που διεξήχθη σε 15 κέντρα, τα
οποία συμπεριέλαβαν 352 ενδαγγειακούς ασθενείς (200 τυπικού κινδύνου,
100 υψηλού κινδύνου και 52 εισαγωγικής περιόδου) και 80 ασθενείς ελέγχου,
παρέχει τη βάση των ποσοστών παρατηρούμενων ανεπιθύμητων ενεργειών στον
πίνακα 5.1.1. Οι ασθενείς εγγράφηκαν στο σκέλος τυπικού κινδύνου εάν ήταν
ικανοί από φυσιολογική άποψη να αντέξουν μια ανοικτή ή ενδαγγειακή
αποκατάσταση και είχαν ανατομία κατάλληλη για θεραπεία με το ενδαγγειακό
μόσχευμα AAA Zenith με το σύστημα εισαγωγής H&L-B One-Shot. Ασθενείς με
κατάλληλη ανατομία, αλλά σε υψηλότερο κίνδυνο νοσηρότητας ή θνησιμότητας με
ανοικτή αποκατάσταση εγγράφηκαν στο σκέλος υψηλού κινδύνου. Οι αρχικοί
ασθενείς που υποβλήθηκαν σε θεραπεία στη μελέτη εγγράφηκαν στο σκέλος
εισαγωγικής περιόδου. Η ομάδα ελέγχου συμπεριέλαβε ασθενείς των οποίων η
αγγειακή ανατομία ενδέχεται να μην ήταν κατάλληλη για ενδαγγειακή
αποκατάσταση AAA.
79

Πίνακας 5.1.1 Θάνατος και ρήξη από την κλινική μελέτη
Θάνατος και ρήξη
Θάνατος κάθε αιτίας
(0-30 ημέρες)
(31-365 ημέρες)
(0-365 ημέρες)
Ρήξη
1
Ο παρανομαστής είναι 199 επειδή ένας ασθενής τυπικού κινδύνου δεν έλαβε μια συσκευή.
2
Όλοι οι θάνατοι (0-30 ημέρες) θεωρήθηκαν ότι σχετίζονταν με το AAA και τη διαδικασία.
3
Από τους θανάτους (31-365 ημέρες), τέσσερις θεωρήθηκαν ότι σχετίζονταν με το AAA: 1 χειρουργικού κινδύνου (σηπτικό σοκ από ισχαιμική κολίτιδα) και 3 υψηλού κινδύνου
(παγκρεατίτιδα με νεφρική βλάβη και σηψαιμία, αιμορραγία από άνω κοιλιακό ανεύρυσμα (μη θεραπευμένο AAA) και ανεπάρκεια πολλαπλών συστημάτων).
4
Από τους θανάτους (0-365 ημέρες), δέκα θεωρήθηκαν ότι σχετίζονταν με το AAA: 1 τυπικού κινδύνου (καρδιακή ανεπάρκεια), 3 χειρουργικού κινδύνου (μαζική αιμορραγία, μεσεντέρια
ισχαιμία και σηπτικό σοκ από ισχαιμική κολίτιδα), 5 υψηλού κινδύνου (αναπνευστική ανεπάρκεια, καρδιακή ανεπάρκεια με πνευμονική εμβολή, παγκρεατίτιδα με νεφρική ανεπάρκεια
και σηψαιμία, αιμορραγία από άνω κοιλιακό ανεύρυσμα (μη θεραπευμένο AAA) και ανεπάρκεια πολλαπλών συστημάτων) και 1 εισαγωγικής περιόδου (πιθανολογούμενη καρδιακή
ανεπάρκεια).
2
Σχετικός με το AAA 0,0% (0/198) 1,3% (1/78) 0,29 3,1% (3/98) 0,0% (0/51)
Μη σχετικός με το AAA 3,0% (6/198) 0,0% (0/78) 0,19 4,1% (4/98) 9,8% (5/51)
Σχετικός με το AAA 0,5% (1/199) 3,8% (3/80) 0,07 5,0% (5/100) 1,9% (1/52)
Μη σχετικός με το AAA 3,0% (6/199) 0,0% (0/80) 0,19 4,0% (4/100) 9,6% (5/52)
(0-30 ημέρες) 0,0% (0/199) μη διαθ. μη διαθ. 0,0% (0/100) 0,0% (0/52)
(31-365 ημέρες) 0,0% (0/198) μη διαθ. μη διαθ. 1,0% (1/98) 0,0% (0/51)
(0-365 ημέρες) 0,0% (0/199) μη διαθ. μη διαθ. 1,0% (1/100) 0,0% (0/52)
3
2,3,4
Τυπικός κίνδυνος από το
μόσχευμα Zenith
0,5% (1/199) 2,5% (2/80) 0,20 2,0% (2/100) 1,9% (1/52)
3,0% (6/198) 1,3% (1/78) 0,68 7,1% (7/98) 9,8% (5/51)
3,5% (7/199) 3,8% (3/80) >0,99 9,0% (9/100) 11,5% (6/52)
1
Χειρουργικός τυπικός
κίνδυνος Τιμή P
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με το
μόσχευμα Zenith
Τυπικός κίνδυνος από το
Περίοδος ελεύθερη νοσηρότητας
(0-30 ημέρες)
Καρδιαγγειακές
Πνευμονικές
Νεφρικές
Εντερικές
Τραυματικές
Νευρολογικές
Αγγειακές
Περίοδος ελεύθερη νοσηρότητας
(31-365 ημέρες)
Καρδιαγγειακές
Πνευμονικές
Νεφρικές
Εντερικές
Τραυματικές
Νευρολογικές
Αγγειακές
Περίοδος ελεύθερη νοσηρότητας
(0-365 ημέρες)
Καρδιαγγειακές
Πνευμονικές
Νεφρικές
Εντερικές
Τραυματικές
Νευρολογικές
Αγγειακές
1
Από το δείκτη νοσηρότητας.
2
Οι καρδιαγγειακές περιελάμβαναν: εμφράγματα μυοκαρδίου κύματος Q και μη κύματος Q, συμφορητική καρδιακή ανεπάρκεια, αρρυθμίες που χρήζουν νέας φαρμακευτικής αγωγής ή
θεραπείας, καρδιακή ισχαιμία που χρήζει επέμβασης, υποστήριξης με ινότροπα φάρμακα, ιατρικώς δυσίατη υπέρταση.
3
Οι πνευμονικές περιελάμβαναν: επαναδιασωλήνωση ή αερισμό >24 ώρες, πνευμονία που χρήζει αντιβιοτικών, συμπληρωματικό οξυγόνο κατά το εξιτήριο.
4
Οι νεφρικές περιελάμβαναν: αιμοκάθαρση σε ασθενείς με φυσιολογική προεγχειρητική νεφρική λειτουργία, αύξηση τιμής κρεατινίνης >30% από τη γραμμή βάσης σε δύο ή
περισσότερες εξετάσεις παρακολούθησης.
5
Οι εντερικές περιελάμβαναν: απόφραξη εντέρου, εντερική ισχαιμία, αορτοεντερικά συρίγγια, παραλυτικό ειλεό >4 ημέρες.
6
Οι τραυματικές περιελάμβαναν: λοίμωξη που χρήζει αντιβιοτικής θεραπείας, κήλη, συρίγγια λέμφου, διάνοιξη τραύματος, νέκρωση που χρήζει χειρουργικού καθαρισμού.
7
Οι νευρολογικές περιελάμβαναν: αγγειακό εγκεφαλικό επεισόδιο, παροδικό ισχαιμικό επεισόδιο, ισχαιμία/παράλυση νωτιαίου μυελού.
8
Οι αγγειακές περιελάμβαναν: θρόμβωση άκρου, περιφερική εμβολή με αποτέλεσμα απώλεια ιστού ή χρήζουσα επέμβασης, μετάγγιση μετά τη διαδικασία (που προκύπτει από
ψευδοανεύρυσμα, αγγειακή βλάβη, διαρροή ανευρύσματος ή άλλες αιτίες που σχετίζονται με τη διαδικασία), ψευδοανεύρυσμα, αγγειακή βλάβη (όπως ακούσια απόφραξη, διαχωρισμό
ή άλλες αιτίες που σχετίζονται με τη διαδικασία), διαρροή ή ρήξη ανευρύσματος, αύξηση στο μέγεθος του ανευρύσματος κατά περισσότερο από 0,5 cm σε σχέση με τη μικρότερη από
οποιαδήποτε προηγούμενη μέτρηση.
9
Οι ερευνητές ανέφεραν ότι ένας επιπλέον ασθενής υψηλού κινδύνου είχε απόφραξη επικουρικής νεφρικής αρτηρίας και ένας επιπλέον ασθενής υψηλού κινδύνου είχε χρόνια νεφρική
ανεπάρκεια ως “άλλες” ανε πιθύμητες ενέργειες.
10
Οι ερευνητές ανέφεραν ότι ένας επιπλέον ασθενής εισαγωγικής περιόδου είχε νεφρική ανεπάρκεια και ένας επιπλέον χειρουργικός ασθενής είχε νεφρική ανεπάρκεια ως “άλλες”
ανεπιθύμητες ενέργειες.
11
Οι ερευνητές ανέφεραν ότι ένας επιπλέον ασθενής εισαγωγικής περιόδου παρουσίασε διεγχειρητική ρήξη αορτικής πλάκας, με αποτέλεσμα απόφραξη νεφρικής αρτηρίας ως “άλλη”
ανεπιθύμητη ενέργεια.
4, 9
5
4, 10
5
4, 9,10
5
2
3
6
7
8,11
2
3
6
7
8
2
3
6
7
8,11
μόσχευμα Zenith
80% (160/200) 58% (46/80) <0,001 68% (68/100) 73% (38/52)
3,0% (6/200) 11% (9/80) 0,02 14% (14/100) 1,9% (1/52)
1,0% (2/200) 15% (12/80) <0,001 2,0% (2/100) 0,0% (0/52)
2,5% (5/200) 10% (8/80) 0,01 6,0% (6/100) 5,8% (3/52)
1,0% (2/200) 3,8% (3/80) 0,14 1,0% (1/100) 1,9% (1/52)
4,5% (9/200) 7,5% (6/80) 0,38 2,0% (2/100) 3,8% (2/52)
0,0% (0/200) 2,5% (2/80) 0,08 0,0% (0/100) 0,0% (0/52)
11% (21/200) 31% (25/80) <0,001 20% (20/100) 19% (10/52)
91% (181/198) 86% (67/78) 0,25 79% (77/98) 86% (44/51)
2,5% (5/198) 3,8% (3/78) 0,69 5,1% (5/98) 2,0% (1/51)
0,5% (1/198) 1,3% (1/78) 0,49 4,1% (4/98) 0,0% (0/51)
0,5% (1/198) 0,0% (0/78) >0,99 3,1% (3/98) 0,0% (0/51)
0,5% (1/198) 1,3% (1/78) 0,49 0,0% (0/98) 0,0% (0/51)
2,0% (4/198) 5,1% (4/78) 0,23 3,1% (3/98) 2,0% (1/51)
1,0% (2/198) 0,0% (0/78) >0,99 1,0% (1/98) 3,9% (2/51)
3,0% (6/198) 3,8% (3/78) 0,72 8,2% (8/98) 5,9% (3/51)
76% (151/200) 49% (39/80) <0,001 55% (55/100) 62% (32/52)
5,0% (10/200) 14% (11/80) 0,02 19% (19/100) 3,8% (2/52)
1,5% (3/200) 16% (13/80) <0,001 6,0% (6/100) 0,0% (0/52)
2,5% (5/200) 10% (8/80) 0,01 9,0% (9/100) 5,8% (3/52)
1,5% (3/200) 3,8% (3/80) 0,36 1,0% (1/100) 1,9% (1/52)
5,5% (11/200) 13% (10/80) 0,08 5,0% (5/100) 5,8% (3/52)
1,0% (2/200) 2,5% (2/80) 0,32 1,0% (1/100) 3,8% (2/52)
12% (24/200) 33% (26/80) <0,001 25% (25/100) 23% (12/52)
5.2 Δυνητικές ανεπιθύμητες ενέργειες
Οι ανεπιθύμητες ενέργειες που ενδέχεται να συμβούν ή/και να απαιτήσουν
επέμβαση, περιλαμβάνουν μεταξύ άλλων:
• Αγγειοσπασμός ή αγγειακό τραύμα (π.χ. διαχωρισμός λαγονομηριαίου
αγγείου, αιμορραγία, ρήξη, θάνατος)
• Αιμορραγία, αιμάτωμα ή διαταραχή της πηκτικότητας του αίματος
• Ακρωτηριασμός
• Ανικανότητα
• Απόφραξη μοσχεύματος ή αυτόχθονου αγγείου
• Αρτηριακή ή φλεβική θρόμβωση ή/και ψευδοανεύρυσμα
• Αρτηριοφλεβώδης επικοινωνία
• Βλάβη αγγείου
• Βλάβη αορτής, συμπεριλαμβανομένης της διάτρησης, του διαχωρισμού, της
αιμορραγίας, της ρήξης και θάνατος
• Διεύρυνση ανευρύσματος
• Εμβολή (μικροεμβολή και μακροεμβολή) με παροδική ή μόνιμη ισχαιμία ή
έμφρακτο
• Ενδοδιαφυγή
• Ενδοπρόσθεση: λανθασμένη τοποθέτηση εξαρτήματος, ατελής έκπτυξη
εξαρτήματος, μετατόπιση εξαρτήματος, κοπή ράμματος, απόφραξη, μόλυνση,
θραύση της ενδοπρόσθεσης, φθορά του υλικού του μοσχεύματος, διαστολή,
διάβρωση, τρώση, περιμοσχευματική ροή, διαχωρισμός και διάβρωση ακίδων
• Εντερικές επιπλοκές (π.χ. ειλεός, παροδική ισχαιμία, έμφρακτο, νέκρωση)
• Επιπλοκές αναισθησίας και επακόλουθα συνοδά προβλήματα (π.χ.
εισρόφηση)
• Επιπλοκές λεμφικού συστήματος και επακόλουθα συνοδά προβλήματα (π.χ.
συρίγγιο λέμφου)
• Επιπλοκές στη θέση αγγειακής προσπέλασης, συμπεριλαμβανομένης της
λοίμωξης, του πόνου, του αιματώματος, του ψευδοανευρύσματος, της
αρτηριοφλεβώδους επικοινωνίας
• Επιπλοκές τραύματος και επακόλουθα συνοδά προβλήματα (π.χ. διάνοιξη,
λοίμωξη)
• Ηπατική ανεπάρκεια
• Θάνατος
• Καρδιακές επιπλοκές και επακόλουθα συνοδά προβλήματα (π.χ. αρρυθμία,
έμφραγμα του μυοκαρδίου, συμφορητική καρδιακή ανεπάρκεια, υπόταση,
υπέρταση)
• Λοίμωξη του ανευρύσματος, της συσκευής ή της θέσης προσπέλασης,
συμπεριλαμβανομένου του σχηματισμού αποστήματος, του παροδικού
πυρετού και του πόνου
• Νευρολογικές τοπικές ή συστηματικές επιπλοκές και επακόλουθα συνοδά
προβλήματα (π.χ. αγγειακό εγκεφαλικό επεισόδιο, παροδικό ισχαιμικό
επεισόδιο, παραπληγία, παραπάρεση, παράλυση)
• Νεφρικές επιπλοκές και επακόλουθα συνοδά προβλήματα (π.χ. απόφραξη
αρτηρίας, τοξικότητα σκιαγραφικού μέσου, ανεπάρκεια, βλάβη)
• Οίδημα
Χειρουργικός τυπικός
κίνδυνος
• Ουρογεννητικές επιπλοκές και επακόλουθα συνοδά προβλήματα (π.χ.
• Πνευμονικές/αναπνευστικές επιπλοκές και επακόλουθα συνοδά προβλήματα
• Πυρετός και εντοπισμένη φλεγμονή
• Ρήξη ανευρύσματος και θάνατος
• Χειρουργική μετατροπή σε ανοικτή αποκατάσταση
• Χωλότητα (π.χ. σε γλουτό, κάτω άκρο)
Αναφορά ανεπιθύμητων ενεργειών που σχετίζονται με τη συσκευή
Οποιαδήποτε ανεπιθύμητη ενέργεια (κλινικό συμβάν) που αφορά το ενδαγγειακό
μόσχευμα AAA Zenith Flex πρέπει να αναφέρεται στην COOK αμέσως. Για
πελάτες στις Η.Π.Α., για την αναφορά ενός συμβάντος, επικοινωνήστε με το
τμήμα σχέσεων πελατών στο τηλέφωνο 1-800-457-4500 (24 ώρες το 24ωρο) ή
στο τηλέφωνο 1-812-339-2235. Για πελάτες εκτός των Η.Π.Α., παρακαλούμε
επικοινωνήστε με το διανομέα σας.
6 ΠΕΡΙΛΗΨΗ ΤΩΝ ΚΛΙΝΙΚΩΝ ΜΕΛΕΤΩΝ
6.1 Στόχοι
Ο πρωτεύων στόχος της κλινικής μελέτης ήταν η αξιολόγηση της ασφάλειας
και της αποτελεσματικότητας μιας προηγούμενης έκδοσης της συσκευής
(ενδαγγειακό μόσχευμα AAA Zenith) ως εναλλακτική λύση σε ανοικτή
χειρουργική αποκατάσταση στην πρωτεύουσα θεραπεία υπονεφρικών
ανευρυσμάτων κοιλιακής αορτής. Υποθέσεις μελετών εξέτασαν εάν οι ασθενείς
που διέτρεχαν τυπικό κίνδυνο είχαν μειωμένη νοσηρότητα στις 30 ημέρες,
ισοδύναμα ποσοστά επιβίωσης στις 30 ημέρες, ισοδύναμα ποσοστά επιβίωσης
στους 12 μήνες, ισοδύναμη επιτυχία θεραπείας στους 12 μήνες και βελτιωμένες
μετρήσεις εκτίμησης της κλινικής χρησιμότητας σε σύγκριση με χειρουργικούς
ασθενείς ομάδας ελέγχου. Η ασφάλεια προσδιορίστηκε με αξιολόγηση εάν οι
ασθενείς με ενδαγγειακό μόσχευμα AAA Zenith είχαν μειωμένη νοσηρότητα
30 ημερών, ισοδύναμη επιβίωση 30 ημερών και 12 μηνών και ισοδύναμη
επιτυχία θεραπείας 12 μηνών σε σύγκριση με τους ασθενείς που υποβλήθηκαν
σε ανοικτή χειρουργική θεραπεία. Η αποτελεσματικότητα βασίστηκε στην
εξαίρεση του ανευρύσματος, συμπεριλαμβανομένης της απουσίας τυχόν
ενδοδιαφυγής, της απουσίας της διεύρυνσης ανευρύσματος (≥5 mm) και της
απουσίας μειζόνων ανεπιθύμητων ενεργειών της συσκευής που αξιολογήθηκαν
κατά τη διάρκεια ενός έτους παρακολούθησης. Οι δευτερεύοντες στόχοι
περιελάμβαναν μια εκτίμηση των μέτρων κλινικού οφέλους και ποιότητας ζωής.
6.2 Σχεδιασμός της μελέτης
Η κλινική μελέτη των Η.Π.Α. ήταν πολυκεντρική, μη τυχαιοποιημένη μελέτη
σύγκρισης ασθενών τυπικού ιατρικού κινδύνου, οι οποίοι έλαβαν ενδαγγειακό
μόσχευμα με ανοικτό χειρουργικό έλεγχο. Υπήρχαν δύο επιπλέον σκέλη της
μελέτης για τις ομάδες υψηλού ιατρικού κινδύνου και θεραπείας εισαγωγικής
περιόδου. Σε δεκαπέντε κέντρα εγγράφηκαν 200 ασθενείς τυπικού κινδύνου,
80 ασθενείς χειρουργικού ελέγχου, 100 ασθενείς υψηλού κινδύνου και
52 ασθενείς εισαγωγικής περιόδου. Η ομάδα ελέγχου συμπεριέλαβε ασθενείς των
οποίων η αγγειακή ανατομία ενδέχεται να μην ήταν κατάλληλη για ενδαγγειακή
αποκατάσταση AAA. Αξιολογήσεις παρακολούθησης προγραμματίστηκαν για
πριν το εξιτήριο, 1 μήνα, 6 μήνες, 12 μήνες και 24 μήνες. Η παρακολούθηση και η
υπευθυνότητα των ασθενών στον 1 μήνα, 12 μήνες και 24 μήνες παρουσιάζονται
80
Τιμή P
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με το
μόσχευμα Zenith
ισχαιμία, διάβρωση, συρίγγιο, ακράτεια, αιματουρία, λοίμωξη)
(π.χ. πνευμονία, αναπνευστική βλάβη, παρατεταμένη διασωλήνωση)
Πίνακας 5.1.2 Ανεπιθύμητες ενέργειες1 στην κλινική μελέτη

στον πίνακα 6.2.1 καθώς αυτές ήταν τα χρονικά σημεία ανάλυσης των
πρωτευόντων δεδομένων. Τα δεδομένα απεικόνισης που παρέχονται σε αυτή την
περίληψη βασίζονται σε ευρήματα από ένα ανεξάρτητο κεντρικοποιημένο
εργαστήριο ανάλυσης εικόνων (κεντρικό εργαστήριο), το οποίο ανασκόπησε
αξονικές τομογραφίες και κοιλιακές ακτινογραφίες για την εκτίμηση των
μεταβολών διαμέτρου ανευρύσματος, της μετανάστευσης της συσκευής και των
σχετικών εξαρτημάτων, της ακεραιότητας της συσκευής (σύρμα και μόσχευμα),
καθώς και της παρουσίας και του τύπου ενδοδιαφυγών. Τα κλινικά συμβάντα
κρίθηκαν τελικά από μια ανεξάρτητη επιτροπή κλινικών συμβάντων και η
ασφάλεια παρακολουθήθηκε από μια επιτροπή παρακολούθησης ασφάλειας
δεδομένων.
Οι χειρουργικοί ασθενείς και οι ασθενείς τυπικού κινδύνου από το μόσχευμα
Zenith κάλυψαν πανόμοια κριτήρια παθοφυσιολογικού κινδύνου. Οι ενδαγγειακές
ομάδες απέκλεισαν περιφερειακό θρόμβο στον εγγύς αυχένα, εγγύς αυχένα
μήκους μικρότερου από 15 mm, διάμετρο εγγύς αυχένα από εξωτερικό τοίχωμα
προς εξωτερικό τοίχωμα μικρότερη από 18 mm ή μεγαλύτερη από 28 mm,
σοβαρή γωνίωση εγγύς αυχένα, διάμετρο λαγόνιας αρτηρίας από εξωτερικό
τοίχωμα προς εξωτερικό τοίχωμα μικρότερη από 7,5 mm ή μεγαλύτερη από
20 mm στην περιφερική θέση καθήλωσης ή περιφερική θέση καθήλωσης στη
λαγόνια αρτηρία μήκους μικρότερου από 10 mm.
Οι ασθενείς θεωρήθηκαν σε υψηλότερο κίνδυνο για χειρουργική αποκατάσταση
εάν είχαν ηλικία άνω των 80 ετών, τιμή κρεατινίνης γραμμής βάσης >2,0 mg/dl,
υποβάλλονταν σε κατ’ οίκον θεραπεία με οξυγόνο, είχαν FEV1 <1 λίτρο, κλάσμα
εξώθησης <25%, χρόνια αποφρακτική πνευμονοπάθεια (COPD) που προκαλεί
αναπηρία, Κατάταξη Καρδιολογικής Ένωσης Νέας Υόρκης 3 ή 4, εχθρική κοιλία,
αιμοκάθαρση, έμφραγμα του μυοκαρδίου εντός των τελευταίων 6 μηνών,
ιατρικώς δυσίατη υπέρταση, προηγούμενο αγγειακό εγκεφαλικό επεισόδιο με
υπολειμματικό έλλειμμα, πολιτισμική αντίρρηση στη λήψη αίματος ή προϊόντων
αίματος, προηγούμενη χειρουργική επέμβαση παράκαμψης νεφρικής αρτηρίας ή
φλεγμονώδες ανεύρυσμα.
Πριν από την εγγραφή ασθενών στην κεντρική μελέτη, κέντρα χωρίς εμπειρία με
το ενδαγγειακό μόσχευμα AAA Zenith ήταν απαραίτητο να υποβάλλουν σε
θεραπεία αρχικούς ασθενείς υπό την επίβλεψη επόπτη. Αυτοί οι ασθενείς
εισαγωγικής περιόδου ήταν ένας συνδυασμός ασθενών τυπικού και υψηλού
κινδύνου και παρακολουθήθηκαν σύμφωνα με το ίδιο πρόγραμμα, όπως οι
ασθενείς στην κεντρική μελέτη.
Πίνακας 6.2.1 Παρακολούθηση και υπευθυνότητα ασθενών
1
Θεραπεία Τυπικός κίνδυνος από το μόσχευμα Zenith Χειρουργικός τυπικός κίνδυνος
Διάστημα
Χωρίς συσκευή 1 1
Μετατροπή σε ανοικτή αποκατάσταση 0 2
Κατέληξε 1 7 17 2 3 μη διαθ.
Αποσύρθηκε/χάθηκε κατά την παρακολούθηση 0 0 2 0 4 μη διαθ.
Διαθέσιμος 198 190 178 78 73 μη διαθ.
Αξονική τομογραφία στο κέντρο 191 168 110 69 58 μη διαθ.
Αξονική τομογραφία στο κεντρικό εργαστήριο 190 165 99 69 59 μη διαθ.
Απεικόνιση νεφρών, ουρητήρων, ουροδόχου κύστης (ΝΟΚ) στο κέντρο 179 153 108 μη διαθ. μη διαθ. μη διαθ.
Απεικόνιση νεφρών, ουρητήρων, ουροδόχου κύστης (ΝΟΚ) στο κεντρικό
εργαστήριο
Αξιολόγηση στο κέντρο για ενδοδιαφυγή 187 163 107 μη διαθ. μη διαθ. μη διαθ.
Αξιολόγηση στο κεντρικό εργαστήριο για ενδοδιαφυγή 161 148 92 μη διαθ. μη διαθ. μη διαθ.
Αξιολόγηση στο κέντρο για διεύρυνση ανευρύσματος μη διαθ. 149 104 μη διαθ. μη διαθ. μη διαθ.
Αξιολόγηση στο κεντρικό εργαστήριο για διεύρυνση ανευρύσματος μη διαθ. 151 94 μη διαθ. μη διαθ. μη διαθ.
1
Το μέγεθος δείγματος ανάλυσης δεδομένων ποικίλει για καθένα από τα παραπάνω χρονικά σημεία και στους ακόλουθους πίνακες. Η μεταβλητότητα αυτή οφείλεται στη διαθεσιμότητα
του ασθενούς για παρακολούθηση, καθώς και στην ποσότητα και την ποιότητα των εικόνων που είναι διαθέσιμες από ειδικά χρονικά σημεία για αξιολόγηση. Για παράδειγμα, ο αριθμός
και η ποιότητα των εικόνων που είναι διαθέσιμες για αξιολόγηση της ενδοδιαφυγής στους 12 μήνες είναι διαφορετικός σε σχέση με τον αριθμό και την ποιότητα των εικόνων που
είναι διαθέσιμες στους 24 μήνες λόγω της διακύμανσης του αριθμού των εξετάσεων εικόνων που εκτελούνται, του αριθμού των εικόνων που παρέχονται από το κέντρο διεξαγωγής
της κλινικής μελέτης στο κεντρικό εργαστήριο ή/και του αριθμού των εικόνων με αποδεκτή ποιότητα αξιολόγησης. Τα σύνολα στα χρονικά σημεία δεν είναι αθροιστικά, εκτός εάν
σημειώνεται διαφορετικά.
2
Τα σύνολα στα χρονικά σημεία είναι αθροιστικά.
1 μήνας 12 μήνες 24 μήνες 1 μήνας 12 μήνες 24 μήνες
178 149 93 μη διαθ. μη διαθ. μη διαθ.
2
2
2
1
2
2
0 0 μη διαθ.
μη διαθ. μη διαθ. μη διαθ.
6.3 Δημογραφικά στοιχεία των ασθενών
Στους πίνακες 6.3.1 και 6.3.2 συγκρίνονται τα χαρακτηριστικά των ασθενών και η αρχική διάμετρος του ανευρύσματος του ενδαγγειακού μοσχεύματος AAA Zenith
και του πληθυσμού ανοικτής χειρουργικής επέμβασης, αντίστοιχα.
Πίνακας 6.3.1 Σύγκριση των χαρακτηριστικών των ασθενών
Στοιχείο
Ηλικία (έτη) 71 ± 7 69 ± 7 0,03 77 ± 7 74 ± 8
Φύλο - άρρεν 94% (187/200) 89% (71/80) 0,22 92% (92/100) 90% (47/52)
Τρέχουσες ιατρικές συνθήκες
Περιφερική αγγειακή νόσος
Υπέρταση 64% (127/200) 83% (65/78) 0,001 68% (67/99) 67% (35/52)
Νεφρική ανεπάρκεια 0,0% (0/197) 0,0% (0/79) >0,99 5,2% (5/97) 1,9% (1/52)
Χρόνια αποφρακτική πνευμονοπάθεια
(COPD)
Θρομβοεμβολικό επεισόδιο 4,5% (9/199) 7,7% (6/78) 0,38 7,1% (7/99) 1,9% (1/52)
Ηπατική νόσος 2,1% (4/192) 5,1% (4/79) 0,24 1,0% (1/99) 1,9% (1/52)
Σακχαρώδης διαβήτης 12% (24/199) 15% (12/79) 0,55 17% (17/99) 14% (7/51)
Ινσουλινοεξαρτώμενος 17% (4/24) 8,3% (1/12) 0,65 24% (4/17) 43% (3/7)
Προηγούμενες ιατρικές καταστάσεις
Έμφραγμα του μυοκαρδίου
Συμφορητική καρδιακή ανεπάρκεια 5,0% (10/199) 12% (9/78) 0,07 16% (16/100) 10% (5/50)
Στηθάγχη 49% (98/198) 39% (31/79) 0,14 45% (44/98) 44% (23/52)
Αρρυθμία 20% (40/197) 22% (17/78) 0,87 28% (27/98) 24% (12/51)
Εγκεφαλοαγγειακή νόσος 9,5% (19/199) 16% (13/79) 0,14 20% (20/99) 9,8% (5/51)
Συστηματική λοίμωξη 1,0% (2/196) 0,0% (0/78) >0,99 3,1% (3/97) 0,0% (0/49)
Καρκίνος 22% (43/200) 19% (15/80) 0,74 31% (31/99) 29% (15/51)
Οικογενειακό ιστορικό ανευρυσματικής νόσου 16% (24/150) 27% (17/63) 0,09 14% (11/77) 26% (10/38)
Προηγούμενη χειρουργική επέμβαση στην
ίδια θέση
Προηγούμενη ακτινοβολία στην ίδια θέση 0,5% (1/197) 0,0% (0/79) >0,99 2,0% (2/100) 2,0% (1/51)
Υπερβολική χρήση αλκοόλ 3,6% (7/193) 10% (8/77) 0,04 3,1% (3/96) 4,0% (2/50)
Χρήση καπνού
Δεν κάπνισε ποτέ
Τέως καπνιστής 69% (133/193) 60% (48/80) 0,03 69% (66/96) 57% (28/49)
Καπνίζει ακόμα 21% (40/193) 35% (28/80) 18% (17/96) 18% (9/49)
Λόγω των κριτηρίων ένταξης, οι ασθενείς υψηλού κινδύνου ήταν μεγαλύτερης ηλικίας (P<0,001), είχαν μεγαλύτερο ποσοστό νεφρικής βλάβης (P=0,004), χρόνιας αποφρακτικής
πνευμονοπάθειας (P=0,01), συμφορητικής καρδιακής ανεπάρκειας (P=0,004) και εγκεφαλοαγγειακής νόσου (P=0,02) σε σχέση με ασθενείς τυπικού κινδύνου.
Τυπικός κίνδυνος από το
μόσχευμα Zenith
16%
20% (39/199) 18% (14/78) 0,87 34% (33/98) 22% (11/51)
39%
10% (20/200) 15% (12/79) 0,22 10% (10/99) 14% (7/51)
10%
(31/195) 25% (19/76) 0,12 24% (23/96) 9,6% (5/52)
(74/192) 29% (23/80) 0,13 35% (34/98) 35% (18/52)
(20/193) 5,0% (4/80)
Χειρουργικός τυπικός
κίνδυνος Τιμή P
Υψηλός κίνδυνος από το
μόσχευμα Zenith
14% (13/96) 24% (12/49)
Εισαγωγική περίοδος με
το μόσχευμα Zenith
Εύρος διαμέτρου
<30 mm
30-39 mm
40-49 mm
50-59 mm
60-69 mm
70-79 mm
80-89 mm
≥90 mm
Η κατανομή διαμέτρων ανευρύσματος δεν εκτιμήθηκε σε τρεις ασθενείς υψηλού κινδύνου και σε έναν ασθενή εισαγωγικής περιόδου.
6.4 Αποτελέσματα
Τα δεδομένα που συγκεντρώθηκαν στους πίνακες 6.4.1 έως 6.4.3 συλλέχθηκαν από τα κέντρα διεξαγωγής των κλινικών μελετών και το κεντρικό εργαστήριο. Όπου
είναι διαθέσιμα, παρέχονται δεδομένα 24 μηνών. Οι ασθενείς ελέγχου δεν παρακολουθήθηκαν πέρα από τους 12 μήνες και ορισμένα δεδομένα δεν έχουν ακόμα
κριθεί τελικά πέρα από τους 12 μήνες. Επομένως, μερικά αποτελέσματα παρουσιάζονται έως τους 12 μήνες, ενώ άλλα αποτελέσματα παρουσιάζονται έως τους 24
μήνες στην ενότητα αυτή. Ο πίνακας 6.4.1 περιγράφει τις συσκευές που εμφυτεύτηκαν σε ασθενείς της κλινικής μελέτης. Ο πίνακας 6.4.2 περιγράφει τα πρωτεύοντα
αποτελέσματα από την κλινική μελέτη. Οι εικόνες 6.4.1 και 6.4.2 είναι τα γραφήματα Kaplan-Meier όλων των αιτιών και της επιβίωσης που σχετίζεται με AAA έως
τους 24 μήνες, αντίστοιχα. Μια ανεξάρτητη επιτροπή κλινικών συμβάντων έκρινε τελικά όλους τους θανάτους για πιθανή σχέση με την αποκατάσταση του
ανευρύσματος. Όλοι οι πρώιμοι θάνατοι (0-30 ημέρες) θεωρήθηκαν ότι σχετίζονταν με AAA. Οι θάνατοι μετά από 30 ημέρες θεωρήθηκαν ότι σχετίζονταν με AAA, εάν
επιβεβαιώθηκε νόσος AAA ή συμμετοχή της συσκευής. Ο πίνακας 6.4.3 παρουσιάζει τα μέτρα της επιτυχίας και η εικόνα 6.4.3 είναι το γράφημα Kaplan-Meier της
ελεύθερης νοσηρότητας περιόδου.
Τυπικός κίνδυνος από το
μόσχευμα Zenith Χειρουργικός τυπικός κίνδυνος
0,0% (0/199) 0,0% (0/78) 0,0% (0/100) 0,0% (0/52)
0,5% (1/199) 0,0% (0/78) 1,0% (1/100) 0,0% (0/52)
23% (45/199) 7,7% (6/78) 15% (15/100) 13% (7/52)
48% (95/199) 33% (26/78) 47% (47/100) 40% (21/52)
24% (47/199) 29% (23/78) 27% (27/100) 42% (22/52)
3,0% (6/199) 21% (16/78) 5,0% (5/100) 1,9% (1/52)
2,5% (5/199) 6,4% (5/78) 1,0% (1/100) 0,0% (0/52)
0,0% (0/199) 2,6% (2/78) 1,0% (1/100) 0,0% (0/52)
81
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με το
μόσχευμα Zenith
Πίνακας 6.3.2 Κατανομή διαμέτρων ανευρύσματος

Πίνακας 6.4.1 Συσκευές που εμφυτεύτηκαν
Στοιχείο
Κύριο σώμα και σκέλη 99,5% (199/200)* 100% (100/100)
Προέκταση κύριου σώματος
Προέκταση σύστοιχου λαγόνιου σκέλους
Προέκταση ετερόπλευρου λαγόνιου σκέλους
Μετατροπέας 0,5% (1/199)** 0,0% (0/100) 0,0% (0/52)
Εργαλείο απόφραξης 0,0% (0/199) 0,0% (0/100) 0,0% (0/52)
*Ένας ασθενής τυπικού κινδύνου δεν έλαβε μια συσκευή λόγω ελίκωσης και αποτιτάνωσης του αγγείου προσπέλασης.
**Ο μετατροπέας χρησιμοποιήθηκε χωρίς εργαλείο απόφραξης.
1
Μία συσκευή ήταν ειδικά προσαρμοσμένη.
2
Δύο ασθενείς τυπικού κινδύνου και ένας ασθενής υψηλού κινδύνου έλαβαν προεκτάσεις κύριου σώματος μετά τη διαδικασία, ένας ασθενής τυπικού κινδύνου έλαβε δύο προεκτάσεις
κύριου σώματος.
3
Δύο ασθενείς τυπικού κινδύνου και ένας ασθενής υψηλού κινδύνου έλαβαν προεκτάσεις σύστοιχου σκέλους μετά τη διαδικασία.
4
Τέσσερις ασθενείς τυπικού κινδύνου, δύο ασθενείς υψηλού κινδύνου και ένας ασθενής εισαγωγικής περιόδου έλαβαν προεκτάσεις ετερόπλευρου σκέλους μετά τη διαδικασία.
5
Τρεις ασθενείς τυπικού κινδύνου και τρεις ασθενείς υψηλού κινδύνου έλαβαν τόσο σύστοιχη όσο και ετερόπλευρη προέκταση κατά τη διάρκεια της διαδικασίας, ένας ασθενής τυπικού
κινδύνου έλαβε τόσο σύστοιχη όσο και ετερόπλευρη προέκταση μετά τη διαδικασία.
2
3,5
4,5
Τυπικός κίνδυνος από το
μόσχευμα Zenith
1,5% (3/199) 1,0% (1/100) 5,8% (3/52)
9,5% (19/199) 11% (11/100) 0,0% (0/52)
11% (21/199) 11% (11/100) 13,5% (7/52)
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με το
1
μόσχευμα Zenith
100% (52/52)
Πίνακας 6.4.2 Πρωτεύοντα αποτελέσματα
Στοιχείο
Θάνατος κάθε αιτίας
(0-30 ημέρες)
2
Θάνατος κάθε αιτίας
(31-365 ημέρες)
Τυπικός κίνδυνος από το
μόσχευμα Zenith
0,5% (1/199) 2,5% (2/80) 0,20 2,0% (2/100) 1,9% (1/52)
2, 3
3,0% (6/199) 1,3% (1/80) 0,68 7,0% (7/100) 9,6% (5/52)
1
Χειρουργικός τυπικός
κίνδυνος Τιμή P
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με το
μόσχευμα Zenith
Σχετικός με το AAA 0,0% (0/199) 1,3% (1/80) 0,29 3,0% (3/100) 0,0% (0/52)
Μη σχετικός με το AAA 3,0% (6/199) 0,0% (0/80) 0,19 4,0% (4/100) 9,6% (5/52)
Θάνατος κάθε αιτίας
(0-365 ημέρες)
2,3,4
3,5% (7/199) 3,8% (3/80) >0,99 9,0% (9/100) 11,5% (6/52)
Σχετικός με το AAA 0,5% (1/199) 3,8% (3/80) 0,07 5,0% (5/100) 1,9% (1/52)
Μη σχετικός με το AAA 3,0% (6/199) 0,0% (0/80) 0,19 4,0% (4/100) 9,6% (5/52)
Ρήξη
(0-30 ημέρες) 0,0% (0/199) μη διαθ. μη διαθ. 0,0% (0/100) 0,0% (0/52)
(31-365 ημέρες) 0,0% (0/199) μη διαθ. μη διαθ. 1,0% (1/100) 0,0% (0/52)
(0-365 ημέρες) 0,0% (0/199) μη διαθ. μη διαθ. 1,0% (1/100) 0,0% (0/52)
Μετατροπή
(0-30 ημέρες) 0,0% (0/199) μη διαθ. μη διαθ. 0,0% (0/100) 0,0% (0/52)
(31-365 ημέρες)
(0-365 ημέρες)
Ανεπιθύμητες ενέργειες
(0-30 ημέρες)
(31-365 ημέρες)
(0-365 ημέρες)
1
Ο παρανομαστής είναι 199 επειδή ένας ασθενής τυπικού κινδύνου δεν έλαβε μια συσκευή.
2
Όλοι οι θάνατοι (0-30 ημέρες) θεωρήθηκαν ότι σχετίζονταν με το AAA και τη διαδικασία.
3
Από τους θανάτους (31-365 ημέρες), τέσσερις θεωρήθηκαν ότι σχετίζονταν με το AAA: 1 χειρουργικού κινδύνου (σηπτικό σοκ από ισχαιμική κολίτιδα) και 3 υψηλού κινδύνου
(παγκρεατίτιδα με νεφρική βλάβη και σηψαιμία, αιμορραγία από άνω κοιλιακό ανεύρυσμα (μη θεραπευμένο AAA) και ανεπάρκεια πολλαπλών συστημάτων).
4
Από τους θανάτους (0-365 ημέρες), δέκα θεωρήθηκαν ότι σχετίζονταν με το AAA: 1 τυπικού κινδύνου (καρδιακή ανεπάρκεια), 3 χειρουργικού κινδύνου (μαζική αιμορραγία,
μεσεντέρια ισχαιμία και σηπτικό σοκ από ισχαιμική κολίτιδα), 5 υψηλού κινδύνου (αναπνευστική ανεπάρκεια, καρδιακή ανεπάρκεια με πνευμονική εμβολή, παγκρεατίτιδα με
νεφρική ανεπάρκεια και σηψαιμία, αιμορραγία από άνω κοιλιακό ανεύρυσμα (μη θεραπευμένο AAA) και ανεπάρκεια πολλαπλών συστημάτων) και 1 εισαγωγικής περιόδου
(πιθανολογούμενη καρδιακή ανεπάρκεια).
5
Οι ασθενείς τυπικού κινδύνου υποβλήθηκαν σε μετατροπές λόγω επίμονης, εγγύς ενδοδιαφυγής τύπου I και νέου επινεφρικού αορτικού ανευρύσματος. Τρεις χειρουργικοί ασθενείς
είχαν μαζική αιμορραγία, από τους οποίους οι 2 χρειάστηκαν επανεπέμβαση και ένας κατέληξε.
6
Οι ανεπιθύμητες ενέργειες συμπεριελήφθησαν στο δείκτη νοσηρότητας.
5
5
6
6
6
1,0% (2/199) μη διαθ. μη διαθ. 1,0% (1/100) 0,0% (0/52)
1,0% (2/199) μη διαθ. μη διαθ. 1,0% (1/100) 0,0% (0/52)
20% (40/200) 43% (34/80) <0,001 32% (32/100) 27% (14/52)
8,6% (17/199) 14% (11/78) 0,25 21% (21/98) 14% (7/51)
25% (49/200) 51% (41/80) <0,001 45% (45/100) 38% (20/52)
Οι ασθενείς με ενδαγγειακό μόσχευμα AAA Zenith δεν παρουσίασαν καμία
σημαντική διαφορά μεταξύ ανδρών και γυναικών για επιβίωση και περίοδο
ελεύθερη από μείζονες ανεπιθύμητες ενέργειες. (Οι ράβδοι σφάλματος στις
εικόνες 6.4.1, 6.4.2 και 6.4.3 αντιπροσωπεύουν όρια εμπιστοσύνης 95%.) Η
εικόνα 6.4.1 παρουσιάζει την επιβίωση κάθε αιτίας έως 24 μήνες. Ο
συνοδευτικός πίνακας παρουσιάζει την ανάλυση Kaplan-Meier στους 1, 6, 12 και
24 μήνες.
Εικόνα 6.4.1 Επιβίωση στους 24 μήνες
Τυπικός κίνδυνος από το μόσχευμα Zenith
(N=200, 1 θάνατος έως 30 ημέρες
4 θάνατοι έως 6 μήνες
7 θάνατοι έως 12 μήνες
17 θάνατοι έως 24 μήνες)
Χειρουργικός τυπικός κίνδυνος
(N=80, 2 θάνατοι έως 30 ημέρες
3 θάνατοι έως 6 μήνες
Εκατοστιαία επιβίωση
3 θάνατοι έως 12 μήνες)
Μήνες μετά τη διαδικασία
1 μήνας 6 μήνες 12 μήνες 24 μήνες
n % επιβίωσης n % επιβίωσης n % επιβίωσης n % επιβίωσης
Τυπικός
κίνδυνος από
το μόσχευμα
Zenith
Χειρουργικός
τυπικός
κίνδυνος
n= Ασθενείς εν ζωή και διαθέσιμοι για παρακολούθηση στο τέλος του διαστήματος
P = 0,81
*Ένας ασθενής κατέληξε πριν από 1 μήνα και ένας ασθενής δεν έλαβε συσκευή.
198* 99,5 194 98,0 190 96,5 97 90,9
78 97,5 73 96,2 67 96,2
μη
διαθ.
διαθ.
μη
Η εικόνα 6.4.2 παρουσιάζει την επιβίωση που σχετίζεται με AAA (προσδιορίζεται
από την Επιτροπή κλινικών συμβάντων) έως τους 24 μήνες.
Ο συνοδευτικός πίνακας παρουσιάζει την ανάλυση Kaplan-Meier στους
1, 6, 12 και 24 μήνες.
Εικόνα 6.4.2 Επιβίωση που σχετίζεται με AAA στους 24 μήνες
Τυπικός κίνδυνος από το μόσχευμα Zenith
(N=200, 1 θάνατος έως 30 ημέρες
1 θάνατος έως 6 μήνες
1 θάνατος έως 12 μήνες
2 θάνατοι έως 24 μήνες)
Χειρουργικός τυπικός κίνδυνος
(N=80, 2 θάνατοι έως 30 ημέρες
3 θάνατοι έως 6 μήνες
3 θάνατοι έως 12 μήνες)
σχετίζεται με ανεύρυσμα
Εκατοστιαία περίοδος ελεύθερη θανάτου που
Συμβάν: Θάνατος που σχετίζεται με ανεύρυσμα
Μήνες μετά τη διαδικασία
1 μήνας 6 μήνες 12 μήνες 24 μήνες
n% επιβίωσης n% επιβίωσης n% επιβίωσης n% επιβίωσης
Τυπικός
κίνδυνος από
το μόσχευμα
Zenith
Χειρουργικός
τυπικός
κίνδυνος
n= Ασθενείς εν ζωή και διαθέσιμοι για παρακολούθηση στο τέλος του διαστήματος
P = 0,04
*Ένας ασθενής κατέληξε πριν από 1 μήνα και ένας ασθενής δεν έλαβε συσκευή.
198* 99,5 194 99,5 190 99,5 97 99,0
78 97,5 73 96,2 67 96,2
μη
διαθ.
διαθ.
μη
82

Πίνακας 6.4.3 Μετρήσεις επιτυχίας
Στοιχείο
Τεχνική επιτυχία
Επιτυχία της διαδικασίας στις
30 ημέρες
Επιτυχία της θεραπείας στους
12 μήνες
1
Βατό μόσχευμα μετά την έκπτυξη.
2
Τεχνική επιτυχία χωρίς μείζονες επιπλοκές, βατό μόσχευμα και χωρίς ενδοδιαφυγές τύπου I ή τύπου III στις 30 ημέρες.
3
Η επιτυχία της διαδικασίας προεκτάθηκε στους 12 μήνες χωρίς διεύρυνση του ανευρύσματος (>5 mm).
Η εικόνα 6.4.3 παρουσιάζει την περίοδο ελεύθερη νοσηρότητας (συμβάντα στο δείκτη νοσηρότητας) έως τους 12 μήνες. Ο συνοδευτικός πίνακας παρουσιάζει την ανάλυση
Kaplan-Meier στους 1, 6 και 12 μήνες.
1
2
3
Τυπικός κίνδυνος από το
μόσχευμα Zenith Χειρουργικός τυπικός κίνδυνος
99,5% (199/200) 98,8% (79/80) 100% (100/100) 100% (52/52)
95,1% (155/163) 88% (60/68) 86% (70/81) 91% (30/33)
89% (122/137) 85% (52/61) 70% (44/63) 87% (26/30)
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με το
μόσχευμα Zenith
Εικόνα 6.4.3 Περίοδος ελεύθερη νοσηρότητας (0-365 ημέρες)
Τυπικός κίνδυνος από το μόσχευμα Zenith
(N=200, 38 ασ θενείς με συμβάντα έως 30 ημέρες
43 ασθενείς με συμβάντα έως 6 μήνες
45 ασθενείς με συμβάντα έως 12 μήνες)
Χειρουργικός τυπικός κίνδυνος
(N=80, 34 ασθενείς με συμβάντα έως 30 ημέρες
38 ασθενείς με συμβάντα έως 6 μήνες
41 ασθενείς με συμβάντα έως 12 μήνες)
Εκατοστιαία περίοδος ελεύθερη νοσηρότητας
Μήνες μετά τη διαδικασία
1 μήνας 6 μήνες 12 μήνες
n % n % n %
Τυπικός κίνδυνος από το μόσχευμα Zenith 162 81,0 154 78,5 133 77,4
Χειρουργικός τυπικός κίνδυνος 45 57,1 39 52,0 33 47,8
n= Ασθενείς εν ζωή και περίοδος ελεύθερη νοσηρότητας στο τέλος του διαστήματος
P = <0,001
Οι πίνακες 6.4.4 έως 6.4.7 περιγράφουν αποτελέσματα των ασθενών με ενδαγγειακό μόσχευμα AAA Zenith, όπως αναφέρονται από το κεντρικό εργαστήριο. Οι
παράγοντες απόδοσης της συσκευής που αναλύθηκαν από το κεντρικό εργαστήριο περιλαμβάνουν την ακεραιότητα της συσκευής (πίνακας 6.4.4), τη βατότητα της
συσκευής (πίνακας 6.4.5), τη μετανάστευση (πίνακας 6.4.6) και το διαχωρισμό μέλους (πίνακας 6.4.7).
Πίνακας 6.4.4 Κοιλιακά ακτινογραφικά ευρήματα – Ακεραιότητα της συσκευής
Στοιχείο
Θραύσεις της ενδοπρόσθεσης
Τυπικός κίνδυνος από το
1
μόσχευμα Zenith
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με το
μόσχευμα Zenith
Προ του εξιτηρίου 0,0% (0/172) 0,0% (0/81) 0,0% (0/39)
30 ημέρες 0,0% (0/172) 0,0% (0/83) 0,0% (0/43)
6 μήνες 0,0% (0/166) 0,0% (0/78) 0,0% (0/35)
12 μήνες 0,0% (0/148) 0,0% (0/60) 0,0% (0/28)
24 μήνες 0,0% (0/93) 0,0% (0/42) 0,0% (0/19)
Διαχωρισμός ακίδας
2
Προ του εξιτηρίου 0,0% (0/176) 0,0% (0/86) 0,0% (0/39)
30 ημέρες 0,0% (0/178) 0,0% (0/86) 0,0% (0/43)
6 μήνες 1,2% (2/167) 2,5% (2/80) 0,0% (0/35)
12 μήνες 2,0% (3/149) 1,7% (1/60) 0,0% (0/28)
24 μήνες 1,1% (1/93) 0,0% (0/42) 0,0% (0/19)
Ρήξη υλικού μοσχεύματος
Προ του εξιτηρίου 0,0% (0/176) 0,0% (0/86) 0,0% (0/39)
30 ημέρες 0,0% (0/178) 0,0% (0/86) 0,0% (0/43)
6 μήνες 0,0% (0/167) 0,0% (0/80) 0,0% (0/35)
12 μήνες 0,0% (0/149) 0,0% (0/60) 0,0% (0/28)
24 μήνες 0,0% (0/93) 0,0% (0/42) 0,0% (0/19)
1
Τα ποσοστά θραύσης της ενδοπρόσθεσης είναι για το κύριο σώμα. Δεν υπήρχαν επίσης θραύσεις δεξιού λαγόνιου σκέλους, αριστερού λαγόνιου σκέλους, εργαλείου απόφραξης,
μετατροπέα, προέκτασης αριστερού λαγόνιου σκέλους, προέκτασης δεξιού λαγόνιου σκέλους ή προέκτασης κύριου σώματος που παρατηρήθηκαν από το κεντρικό εργαστήριο.
2
Ασθενείς με διαχωρισμό 1 ή 2 ακίδων (από 10 ή 12 συνολικά), χωρίς ανεπιθύμητα κλινικά επακόλουθα.
Πίνακας 6.4.5 Ευρήματα αξονικής τομογραφίας – Βατότητα μοσχεύματος
Στοιχείο
Τυπικός κίνδυνος από το
μόσχευμα Zenith
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με το
μόσχευμα Zenith
Βατότητα μοσχεύματος
30 ημέρες 100% (185/185) 99% (85/86) 100% (47/47)
6 μήνες 99% (183/184) 100% (74/74) 100% (39/39)
12 μήνες 99% (153/155) 100% (62/62) 100% (30/30)
24 μήνες 100% (96/96) 100% (33/33) 100% (25/25)
Πίνακας 6.4.6 Ευρήματα αξονικής τομογραφίας – Μετανάστευση μοσχεύματος (κύριο σώμα)
Στοιχείο
Τυπικός κίνδυνος από το
μόσχευμα Zenith
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με το
μόσχευμα Zenith
Μετανάστευση μοσχεύματος (>5 mm) στους 12 μήνες
με κλινικά επακόλουθα1 ή επέμβαση 0,0% (0/162) 0,0% (0/71) 0,0% (0/34)
χωρίς κλινικά επακόλουθα1 ή επέμβαση 2,5% (4/162) 2,8% (2/71) 0,0% (0/34)
Μετανάστευση μοσχεύματος (>10 mm) 0,0% (0/162) 0,0% (0/71) 0,0% (0/34)
1
Η μετανάστευση με κλινικά επακόλουθα θα περιελάμβανε ενδοδιαφυγή, μετατροπή, ρήξη ή θάνατο που σχετίζεται με AAA.
83

Πίνακας 6.4.7 Κοιλιακά ακτινογραφικά ευρήματα – Διαχωρισμός μέλους
Στοιχείο
Τυπικός κίνδυνος από το
μόσχευμα Zenith
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με το
μόσχευμα Zenith
Διαχωρισμός μέλους
Προ του εξιτηρίου 0,0% (0/176) 0,0% (0/86) 0,0% (0/39)
30 ημέρες 0,0% (0/178) 0,0% (0/86) 0,0% (0/43)
6 μήνες 0,0% (0/167) 0,0% (0/80) 0,0% (0/35)
12 μήνες 0,0% (0/149) 0,0% (0/60) 0,0% (0/28)
24 μήνες 0,0% (0/93) 0,0% (0/42) 0,0% (0/19)
6.5 Διαχείριση ενδοδιαφυγής
Κατά τη διάρκεια της κλινικής μελέτης, οι ενδοδιαφυγές τύπου I αντιμετωπίστηκαν κατά τη διάρκεια της αρχικής διαδικασίας με τη χρήση επιπλέον εφαρμογής
μπαλονιού ή, εάν δεν ήταν επιτυχής, με επιπλέον προσθέσεις. Ενδοδιαφυγές τύπου II παρατηρήθηκαν για χρονική περίοδο ενός έως έξι μηνών, έτσι ώστε να
προσδιοριστεί εάν θα παρουσίαζαν αυτόματη θρόμβωση ή απουσία διευρυνόμενων ανευρυσμάτων, υποβλήθηκαν σε θεραπεία με ενδαγγειακές τεχνικές κατά την
κρίση του θεράποντος ιατρού. Εάν το ανεύρυσμα διευρύνθηκε, εξετάστηκε το ενδεχόμενο θεραπείας με εμβολισμό ή απολίνωση και, σε μερικές περιπτώσεις, αυτή
εκτελέστηκε. Οι ενδοδιαφυγές τύπου III που προκλήθηκαν από ελαττώματα του μοσχεύματος, ανεπαρκή στεγανοποίηση ή αποσύνδεση των αρθρωτών εξαρτημάτων
αντιμετωπίστηκαν με επιπλέον εφαρμογή μπαλονιού ή με επιπλέον προσθέσεις. Όπως αναφέρθηκε από το αγγειογραφικό κεντρικό εργαστήριο, δεν υπήρχαν
ενδοδιαφυγές τύπου IV κατά τη διάρκεια της κλινικής μελέτης των Η.Π.Α. Το υλικό μοσχεύματος που χρησιμοποιήθηκε για την κατασκευή του ενδαγγειακού
μοσχεύματος AAA Zenith είναι τυπικού πάχους και είναι το ίδιο υλικό που χρησιμοποιήθηκε σε ανοικτές χειρουργικές διαδικασίες. Ο πίνακας 6.5.1 παρουσιάζει την
επίπτωση των ενδοδιαφυγών κατά διάστημα αξιολόγησης, όπως αναγνωρίζεται από το κεντρικό εργαστήριο για τους ασθενείς τυπικού κινδύνου, υψηλού κινδύνου
και εισαγωγικής περιόδου, αντίστοιχα.
Πίνακας 6.5.1 Ενδοδιαφυγές (Όλοι οι τύποι, νέες και επίμονες)
Στοιχείο
Τυπικός κίνδυνος από το
μόσχευμα Zenith
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με το
μόσχευμα Zenith
Ενδοδιαφυγές
Προ του εξιτηρίου 15% (23/153) 14% (11/78) 12% (3/26)
1
30 ημέρες
1
6 μήνες
1
12 μήνες
1
Περιλαμβάνει τόσο επίμονες ενδοδιαφυγές όσο και νέες παρατηρήσεις.
Οι πίνακες 6.5.2 – 6.5.4 παρουσιάζουν την επίπτωση της πρώτης εμφάνισης μιας ενδοδιαφυγής σύμφωνα με το διάστημα αξιολόγησης, όπως αναγνωρίζεται από το
κεντρικό εργαστήριο στις εξετάσεις που πραγματοποιούνται στο διάστημα 30 ημερών, 6 μηνών και 12 μηνών ή πριν από αυτό, για τους ασθενείς τυπικού κινδύνου, υψηλού
κινδύνου και εισαγωγικής περιόδου, αντίστοιχα. Παρέχεται επίσης ο αριθμός ασθενών που δεν παρουσιάζουν διαρροή ακολούθως.
9,9% (16/161) 12% (9/75) 6,3% (2/32)
8,7% (15/172) 11% (8/70) 8,6% (3/35)
7,4% (11/148) 8,8% (5/57) 3,4% (1/29)
Πίνακας 6.5.2 Πρώτη εμφάνιση ενδοδιαφυγής1 για ασθενείς τυπικού κινδύνου
Στοιχείο
Έως εξέταση ενός μήνα
N=179
% Ενδοδιαφυγή
Χωρίς
διαρροή
1
ακολούθως
2
Εξέταση έξι μηνών
N=172
% Ενδοδιαφυγή
διαρροή
1
ακολούθως
Χωρίς
2
Εξέταση δώδεκα μηνών
N=148
% Ενδοδιαφυγή
1
ακολούθως
Ενδοδιαφυγές 17 31 17 2,3 4 3 3,4 5 2
Εγγύς τύπου I 2,8 5 4 0,0 0 0 0,0 0 0
Περιφερική
Τύπου I 1,7 3 1 0,0 0 0 0,7 1 1
Τύπου II 9,5 17 9 2,3 4 3 1,4 2 1
Τύπου III 1,1 2 2 0,0 0 0 0,7 1 0
Τύπου IV 0,0 0 0 0,0 0 0 0,0 0 0
Πολλαπλή 1,1 2 1 0,0 0 0 0,0 0 0
Άγνωστη 1,1 2 0 0,0 0 0 0,7 1 0
1
Αναγνωρίστηκε από το κεντρικό εργαστήριο.
2
Οι επακόλουθες ενδοδιαφυγές ενδέχεται να ήταν διαφορετικού τύπου από την αρχική.
3
Μόνον 2 ασθενείς είχαν νέες ενδοδιαφυγές μετά από 12 μήνες. Δεν υπάρχουν διαθέσιμα στοιχεία παρακολούθησης μετά από 24 μήνες.
Πίνακας 6.5.3 Πρώτη εμφάνιση ενδοδιαφυγής1 για ασθενείς υψηλού κινδύνου
Στοιχείο
Έως εξέταση ενός μήνα
N=88
% Ενδοδιαφυγή
Χωρίς διαρροή
1
ακολούθως
2
Εξέταση έξι μηνών
N=70
% Ενδοδιαφυγή
Χωρίς διαρροή
1
ακολούθως
2
% Ενδοδιαφυγή
Εξέταση δώδεκα μηνών
N=57
1
Χωρίς διαρροή
ακολούθως
Ενδοδιαφυγές 18 16 6 2,9 2 0 3,5 2 1
Εγγύς τύπου I 2,3 2 1 0,0 0 0 0,0 0 0
Περιφερική
Τύπου I 1,1 1 1 0,0 0 0 0,0 0 0
Τύπου II 9,1 8 3 1,4 1 0 1,8 1 0
Τύπου III 0,0 0 0 1,4 1 0 1,8 1 1
Τύπου IV 0,0 0 0 0,0 0 0 0,0 0 0
Πολλαπλή 4,5 4 0 0,0 0 0 0,0 0 0
Άγνωστη 1,1 1 1 0,0 0 0 0,0 0 0
1
Αναγνωρίστηκε από το κεντρικό εργαστήριο.
2
Οι επακόλουθες ενδοδιαφυγές ενδέχεται να ήταν διαφορετικού τύπου από την αρχική.
3
Χωρίς ενδοδιαφυγές μετά από 12 μήνες. Δεν υπάρχουν διαθέσιμα στοιχεία παρακολούθησης μετά από 24 μήνες.
3
Χωρίς
διαρροή
3
2
2
Πίνακας 6.5.4 Πρώτη εμφάνιση ενδοδιαφυγής1 για ασθενείς εισαγωγικής περιόδου
Στοιχείο
Έως εξέταση ενός μήνα
N=36
% Ενδοδιαφυγή
Χωρίς διαρροή
1
ακολούθως2% Ενδοδιαφυγή
Εξέταση έξι μηνών
N=35
1
Χωρίς διαρροή
ακολούθως
2
% Ενδοδιαφυγή
Εξέταση δώδεκα μηνών
N=29
Χωρίς διαρροή
1
ακολούθως
Ενδοδιαφυγές 11 4 2 2,9 1 0 0,0 0 0
Εγγύς τύπου I 0,0 0 0 0,0 0 0 0,0 0 0
Περιφερική
Τύπου I 0,0 0 0 0,0 0 0 0,0 0 0
Τύπου II 5,6 2 1 2,9 1 0 0,0 0 0
Τύπου III 2,8 1 0 0,0 0 0 0,0 0 0
Τύπου IV 0,0 0 0 0,0 0 0 0,0 0 0
Πολλαπλή 0,0 0 0 0,0 0 0 0,0 0 0
Άγνωστη 2,8 1 1 0,0 0 0 0,0 0 0
1
Αναγνωρίστηκε από το κεντρικό εργαστήριο.
2
Οι επακόλουθες ενδοδιαφυγές ενδέχεται να ήταν διαφορετικού τύπου από την αρχική.
3
Χωρίς ενδοδιαφυγές μετά από 12 μήνες. Δεν υπάρχουν διαθέσιμα στοιχεία παρακολούθησης μετά από 24 μήνες.
84
3
2

6.6 Μεταβολή ανευρύσματος
Οι πίνακες 6.6.1 – 6.6.3 παρουσιάζουν τη μεταβολή στη διάμετρο του ανευρύσματος για τους ενδαγγειακούς ασθενείς, όπως αναγνωρίζεται από το κεντρικό
εργαστήριο. Ο πίνακας 6.6.1 παρουσιάζει τη μέγιστη μεταβολή στη διάμετρο του ανευρύσματος κατά διάστημα. Οι πίνακες 6.6.2 και 6.6.3 παρουσιάζουν τη
μεταβολή του ανευρύσματος και την ενδοδιαφυγή στους 12 και 24 μήνες, αντίστοιχα.
Πίνακας 6.6.1 Μεταβολή στη μέγιστη διάμετρο ανευρύσματος κατά διάστημα*
Στοιχείο
Τυπικός κίνδυνος από το
μόσχευμα Zenith
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με το
μόσχευμα Zenith
Από προ του εξιτηρίου 30 ημέρες
Μείωση >5 mm 1,7% (3/180) 4,8% (4/84) 0,0% (0/40)
Αμετάβλητη 97% (174/180) 94% (79/84) 97,5% (39/40)
Αύξηση >5 mm 1,7% (3/180) 1,2% (1/84) 2,5% (1/40)
Μεταβολή από προ του εξιτηρίου 6 μήνες
Μείωση >5 mm 36% (63/173) 41% (30/73) 49% (18/37)
Αμετάβλητη 62% (108/173) 59% (43/73) 51% (19/37)
Αύξηση >5 mm 1,2% (2/173) 0,0% (0/73) 0,0% (0/37)
Μεταβολή από προ του εξιτηρίου 12 μήνες
Μείωση >5 mm 68% (102/151) 63% (39/62) 67% (20/30)
Αμετάβλητη 31% (47/151) 35% (22/62) 33% (10/30)
Αύξηση >5 mm 1,3% (2/151) 1,6% (1/62) 0,0% (0/30)
Μεταβολή από προ του εξιτηρίου 24 μήνες
Μείωση >5 mm 78% (74/94) 75% (27/36) 71% (17/24)
Αμετάβλητη 19% (18/94) 25% (9/36) 25% (6/24)
Αύξηση >5 mm 2,1% (2/94) 0,0% (0/36) 4,2% (1/24)
*Περιλαμβάνει μόνον ασθενείς με ερμηνεύσιμα φιλμ και μετρήσεις μεταβολής του ανευρύσματος από 1 έως 24 μήνες.
Πίνακας 6.6.2 Μεταβολή στο μέγεθος του ανευρύσματος και ενδοδιαφυγή στους 12 μήνες
Τυπικός κίνδυνος από το
μόσχευμα Zenith
N=140
Υψηλός κίνδυνος από το
μόσχευμα Zenith
N=53
Εισαγωγική περίοδος με το
μόσχευμα Zenith
N=27
Ενδοδιαφυγή Ενδοδιαφυγή Ενδοδιαφυγή
Στοιχείο
Μεταβολή στο μέγεθος του ανευρύσματος από προ του εξιτηρίου έως τους 12 μήνες
N n % N n % N n %
Μείωση >5 mm 96 3 3,0 35 3 9,0 20 0 0,0
Αμετάβλητη 42 8 19 17 2 12 7 1 14
Αύξηση >5 mm 2 0 0,0 1 0 0,0 0 0 0,0
Πίνακας 6.6.3 Μεταβολή στο μέγεθος του ανευρύσματος και ενδοδιαφυγή στους 24 μήνες
Τυπικός κίνδυνος από το
μόσχευμα Zenith
N=90
Υψηλός κίνδυνος από το
μόσχευμα Zenith
N=31
Εισαγωγική περίοδος με το
μόσχευμα Zenith
N=21
Ενδοδιαφυγή Ενδοδιαφυγή Ενδοδιαφυγή
Στοιχείο
Μεταβολή στο μέγεθος ανευρύσματος από προ του εξιτηρίου έως τους 24 μήνες
N n % N n % N n %
Μείωση >5 mm 71 3 4,0 24 1 4,0 16 0 0,0
Αμετάβλητη 18 1 6,0 7 3 43 5 0 0,0
Αύξηση >5 mm 1 1 100 1 0 0,0 0 0 0,0
6.7 Δευτερεύουσες επεμβάσεις που σχετίζονται με AAA
Δευτερεύουσες επεμβάσεις που σχετίζονται με AAA εντός του πρώτου έτους εκτελέστηκαν στο 11% των ασθενών τυπικού κινδύνου με μόσχευμα Zenith,
στο 13% των ασθενών υψηλού κινδύνου με μόσχευμα Zenith και στο 5,8% των ασθενών εισαγωγικής περιόδου με μόσχευμα Zenith όπως φαίνεται στον πίνακα
6.7.1. Περισσότερες από το 50% των δευτερευουσών επεμβάσεων ενείχαν καθετηριασμό για τη θεραπεία ενδοδιαφυγής. Δευτερεύουσες επεμβάσεις που σχετίζονται
με AAA εντός του δεύτερου έτους εκτελέστηκαν στο 4,2% των ασθενών τυπικού κινδύνου με μόσχευμα Zenith, στο 2,2% των ασθενών υψηλού κινδύνου με
μόσχευμα Zenith και στο 2,3% των ασθενών εισαγωγικής περιόδου με μόσχευμα Zenith όπως φαίνεται στον πίνακα 6.7.2.
Πίνακας 6.7.1 Δευτερεύουσες επεμβάσεις (έως 12 μήνες)
Επέμβαση
Μετατροπή σε ανοικτή αποκατάσταση 2 1,0 1 1,0 0 0,0
Τυπικός κίνδυνος από το
μόσχευμα Zenith
N=199
Υψηλός κίνδυνος από το
μόσχευμα Zenith
N=100
Εισαγωγική περίοδος με το
μόσχευμα Zenith
N=52
n % n % n %
Ασθενείς με ≥1 επέμβαση 21 11 13 13 3 5,8
Θεραπεία ενδοδιαφυγοής
Εμβολισμός 5 2,5 3 3,0 1 2,0
Βοηθητικό εξάρτημα 6 3,0 4 4,0 1 2,0
Ενδοπρόσθεση 1 0,5 0 0,0 0 0,0
Αγγειοπλαστική 0 0,0 1 1,0 0 0,0
Θεραπεία αύξησης ανευρύσματος
Εμβολισμός 0 0,0 0 0,0 0 0,0
Θεραπεία απόφραξης μέλους 1 0,5 3 3,0 1 2,0
Θεραπεία στένωσης μέλους 1 0,5 0 0,0 0 0,0
Θεραπεία νεφρικής αρτηρίας 5 2,5 0 0,0 0 0,0
Θεραπεία υποβουβωνικής ισχαιμίας 1 0,5 1 1,0 0 0,0
Θεραπεία πολλαπλών συμβάντων 1 0,5 1 1,0 0 0,0
Πίνακας 6.7.2 Δευτερεύουσες επεμβάσεις (>12 έως 24 μήνες)
Επέμβαση
Μετατροπή σε ανοικτή αποκατάσταση 1 0,5 1 1,1 0 0,0
Τυπικός κίνδυνος από το
μόσχευμα Zenith
N=190
Υψηλός κίνδυνος από το
μόσχευμα Zenith
N=90
Εισαγωγική περίοδος με το
μόσχευμα Zenith
N=44
n % n % n %
Ασθενείς με ≥1 επέμβαση 8 4,2 2 2,2 1 2,3
Θεραπεία ενδοδιαφυγοής
Εμβολισμός 3
1
1,6 0 0,0 1
2
2,3
Βοηθητικό εξάρτημα 1 0,5 0 0,0 0 0,0
Ενδοπρόσθεση 0 0,0 1 1,1 0 0,0
Αγγειοπλαστική 0 0,0 0 0,0 0 0,0
Θεραπεία αύξησης ανευρύσματος
Εμβολισμός 1 0,5 0 0,0 1
2
2,3
Θεραπεία απόφραξης μέλους 1 0,5 0 0,0 0 0,0
Θεραπεία στένωσης μέλους 0 0,0 0 0,0 0 0,0
Θεραπεία νεφρικής αρτηρίας 0 0,0 1 1,1 0 0,0
Θεραπεία υποβουβωνικής ισχαιμίας 1 0,5 0 0,0 0 0,0
Θεραπεία πολλαπλών συμβάντων 1 0,5 0 0,0 0 0,0
1
Ο ασθενής έλαβε επίσης βοηθητικό εξάρτημα.
2
Ο ασθενής υποβλήθηκε σε επέμβαση για τη θεραπεία αύξησης ανευρύσματος και ενδοδιαφυγής.
85

6.8 Μετρήσεις δευτερεύουσας έκβασης
Όπως περιγράφεται στον πίνακα 6.8.1, η θεραπεία του AAA με το ενδαγγειακό μόσχευμα AAA Zenith σε σύγκριση με την ομάδα χειρουργικού ελέγχου κατέδειξε
σημαντικά οφέλη στις μετρήσεις ανάνηψης και ποιότητας ζωής.
Πίνακας 6.8.1 Δευτερεύουσες εκβάσεις κατά ομάδα θεραπείας
Στοιχείο
Χρόνος αναισθησίας (min) 221,6 ± 67,3 304,5 ± 102,7 <0,001 218,9 ± 69,6 213,9 ± 57,7
Χρόνος διαδικασίας (min) 153,2 ± 56,3 238,7 ± 92,2 <0,001 153,5 ± 58,6 155,9 ± 43,2
Προϊόντα τράπεζας αίματος που ελήφθησαν 5,0% (10/200) 84% (67/80) <0,001 12% (12/100) 3,8% (2/52)
Απώλεια αίματος (ml) 299 ± 324 1676 ± 1676 <0,001 356 ± 514 265 ± 226
Ημέρες στη ΜΕΘ 0,4 ± 0,9 3,4 ± 4,6 <0,001 0,5 ± 1,2 0,5 ± 0,9
Ημέρες έως το εξιτήριο 2,6 ± 1,7 8,8 ± 5,6 <0,001 3,0 ± 2,8 2,7 ± 1,5
Ημέρες έως τη λήψη υγρών από του στόματος 0,5 ± 0,8 3,9 ± 2,5 <0,001 0,5 ± 0,6 0,7 ± 0,5
Ημέρες έως τη φυσιολογική δίαιτα 1,3 ± 1,2 6,6 ± 4,9 <0,001 1,3 ± 0,8 1,1 ± 0,7
Ημέρες έως τη φυσιολογική λειτουργία του εντέρου 2,6 ± 1,4 4,2 ± 2,1 <0,001 2,6 ± 1,5 2,0 ± 1,2
Ημέρες έως την κινητοποίηση 1,2 ± 0,7 3,5 ± 3,4 <0,001 1,2 ± 0,7 1,2 ± 0,6
Ώρες διασωλήνωσης 1,9 ± 2,2 11,7 ± 13,6 <0,001 1,2 ± 1,7 2,6 ± 4,6
Μέγιστη θερμοκρασία (°F) 101,1 ± 1,3 100,7 ± 1,2 0,06 100,8 ± 1,1 101,0 ± 1,2
Τυπικός κίνδυνος από το
μόσχευμα Zenith
7 ΕΠΙΛΟΓΗ ΚΑΙ ΘΕΡΑΠΕΙΑ ΑΣΘΕΝΩΝ
Δείτε την ενότητα 4, Προειδοποιήσεις και προφυλάξεις
Εξατομίκευση της θεραπείας
Η Cook συνιστά την επιλογή των διαμέτρων των εξαρτημάτων του ενδαγγειακού
μοσχεύματος AAA Zenith Flex όπως περιγράφεται στους πίνακες 10.5.1 και
10.5.2. Το μήκος του ενδαγγειακού μοσχεύματος AAA Zenith Flex πρέπει να
εκτείνεται από την κατώτατη νεφρική αρτηρία, έως ακριβώς πάνω από το
διχασμό της έσω λαγόνιας (υπογάστριας) αρτηρίας. Όλα τα μήκη και οι διάμετροι
των συσκευών που είναι απαραίτητα για την ολοκλήρωση της διαδικασίας πρέπει
να είναι διαθέσιμα στον ιατρό, ειδικά όταν δεν είναι βέβαιες οι μετρήσεις
προεγχειρητικού σχεδιασμού περίπτωσης (διάμετροι/μήκη θεραπείας). Η
προσέγγιση αυτή επιτρέπει μεγαλύτερη διεγχειρητική ευελιξία για την επίτευξη
βέλτιστων εκβάσεων διαδικασίας. Οι κίνδυνοι και τα οφέλη που περιγράφηκαν
προηγουμένως στην ενότητα 6, ΠΕΡΊΛΗΨΗ ΤΩΝ ΚΛΙΝΙΚΏΝ ΜΕΛΕΤΏΝ πρέπει
να εξετάζονται προσεκτικά για κάθε ασθενή, πριν από τη χρήση του
ενδαγγειακού μοσχεύματος AAA Zenith Flex. Επιπλέον ζητήματα για την επιλογή
του ασθενούς περιλαμβάνουν μεταξύ άλλων:
• Ηλικία και προσδόκιμο ζωής του ασθενούς
• Συνυπάρχουσες νόσοι (π.χ. καρδιακή, πνευμονική ή νεφρική ανεπάρκεια πριν
από τη χειρουργική επέμβαση, νοσηρή παχυσαρκία)
• Καταλληλότητα του ασθενούς για ανοικτή χειρουργική αποκατάσταση
• Ανατομική καταλληλότητα του ασθενούς για ενδαγγειακή αποκατάσταση
• Ο κίνδυνος ρήξης ανευρύσματος σε σύγκριση με τον κίνδυνο της θεραπείας με
το ενδαγγειακό μόσχευμα AAA Zenith Flex
• Ικανότητα ανοχής γενικής, περιοχικής ή τοπικής αναισθησίας
• Το μέγεθος και η μορφολογία του αγγείου λαγονομηριαίας προσπέλασης
(ελάχιστος θρόμβος, ασβέστιο ή/και ελίκωση) πρέπει να είναι συμβατά με τις
τεχνικές αγγειακής προσπέλασης και τα παρελκόμενα του προφίλ τοποθέτησης
ενός θηκαριού αγγειακού εισαγωγέα 14 έως 22 French
• Μη ανευρυσματικό, υπονεφρικό αορτικό τμήμα (αυχένας) εγγύς προς το
ανεύρυσμα:
• με μήκος τουλάχιστον 15 mm,
• με διάμετρο που μετράται από εξωτερικό τοίχωμα σε εξωτερικό τοίχωμα το
πολύ 32 mm και τουλάχιστον 18 mm,
• με γωνία μικρότερη από 60 μοίρες σε σχέση με τον μακρύ άξονα του
ανευρύσματος και
• με γωνία μικρότερη από 45 μοίρες σε σχέση με τον άξονα της επινεφρικής
αορτής
• Περιφερική θέση καθήλωσης λαγόνιας αρτηρίας μεγαλύτερη από 10 mm σε
μήκος και 7,5 έως 20 mm σε διάμετρο (μετρημένη από εξωτερικό τοίχωμα σε
εξωτερικό τοίχωμα)
• Απουσία σημαντικής αποφρακτικής νόσου μηριαίας/λαγόνιας αρτηρίας, η
οποία θα ήταν δυνατόν να παρεμποδίσει τη ροή μέσω του ενδαγγειακού
μοσχεύματος
Η τελική απόφαση θεραπείας είναι στην κρίση του ιατρού και του ασθενούς.
8 ΠΛΗΡΟΦΟΡΙΕΣ ΓΙΑ ΤΙΣ ΣΥΣΤΑΣΕΙΣ ΠΡΟΣ ΤΟΝ ΑΣΘΕΝΗ
Κατά τη συζήτηση αυτής της ενδαγγειακής συσκευής και της διαδικασίας, ο
ιατρός και ο ασθενής (ή/και τα μέλη της οικογένειας) πρέπει να ανασκοπήσουν
τους κινδύνους και τα οφέλη, που περιλαμβάνουν:
• Κινδύνους και διαφορές μεταξύ ενδαγγειακής αποκατάστασης και χειρουργικής
αποκατάστασης
• Δυνητικά πλεονεκτήματα της κλασσικής ανοικτής χειρουργικής αποκατάστασης
• Δυνητικά πλεονεκτήματα της ενδαγγειακής αποκατάστασης
• Η πιθανότητα ανάγκης επακόλουθης επεμβατικής ή ανοικτής χειρουργικής
αποκατάστασης του ανευρύσματος μετά την αρχική ενδαγγειακή
αποκατάσταση
Επιπρόσθετα των κινδύνων και των ωφελειών μιας ενδαγγειακής
αποκατάστασης, ο ιατρός πρέπει να εκτιμήσει τη δέσμευση και τη συμμόρφωση
του ασθενούς στη μετεγχειρητική παρακολούθηση, όπως είναι απαραίτητο για
τη διασφάλιση ασφαλών και αποτελεσματικών αποτελεσμάτων. Παρακάτω
παρατίθενται επιπλέον θέματα προς συζήτηση με τον ασθενή, όσον αφορά τις
προσδοκίες μετά από μια ενδαγγειακή αποκατάσταση:
• Η μακροχρόνια απόδοση και ασφάλεια των ενδαγγειακών μοσχευμάτων
δεν έχει ακόμα επιβεβαιωθεί. Όλοι οι ασθενείς πρέπει να ενημερώνονται
ότι η ενδαγγειακή θεραπεία απαιτεί μακροχρόνια, τακτική
παρακολούθηση για την εκτίμηση της υγείας τους και της απόδοσης του
ενδαγγειακού μοσχεύματός τους. Ασθενείς με ειδικά κλινικά ευρήματα (π.χ.
ενδοδιαφυγές, διευρυνόμενα ανευρύσματα ή μεταβολές της δομής ή της θέσης
του ενδαγγειακού μοσχεύματος) πρέπει να τελούν υπό αυξημένη
παρακολούθηση. Ειδικές κατευθυντήριες οδηγίες παρακολούθησης
περιγράφονται στην ενότητα 12, ΚΑΤΕΥΘΥΝΤΉΡΙΕΣ ΟΔΗΓΊΕΣ ΑΠΕΙΚΌΝΙΣΗΣ
ΚΑΙ ΜΕΤΕΓΧΕΙΡΗΤΙΚΉ ΠΑΡΑΚΟΛΟΎΘΗΣΗ.
• Πρέπει να παρέχονται συμβουλές στους ασθενείς σχετικά με τη σημασία της
τήρησης του προγράμματος παρακολούθησης, τόσο κατά τη διάρκεια του
πρώτου έτους όσο και σε ετήσια διαστήματα στη συνέχεια. Οι ασθενείς πρέπει
να ενημερώνονται ότι η τακτική και συνεπής παρακολούθηση αποτελεί κρίσιμο
μέρος της διασφάλισης της συνεχούς ασφάλειας και αποτελεσματικότητας της
ενδαγγειακής θεραπείας των AAA. Ως ελάχιστο, απαιτείται ετήσια απεικόνιση
και τήρηση των απαιτήσεων μετεγχειρητικής παρακολούθησης ρουτίνας και
πρέπει να θεωρείται δια βίου δέσμευση στην υγεία και στην καλή κατάσταση
του ασθενούς.
• Ο ασθενής θα πρέπει να ενημερωθεί ότι η επιτυχής αποκατάσταση του
ανευρύσματος δεν αναστέλλει τη διαδικασία της νόσου. Εξακολουθεί να
υπάρχει η πιθανότητα παρουσίας σχετιζόμενης εκφύλισης των αγγείων.
• Οι ιατροί πρέπει να ενημερώσουν όλους τους ασθενείς ότι είναι σημαντικό να
αναζητήσουν άμεση ιατρική φροντίδα σε περίπτωση που παρουσιάσουν
σημεία απόφραξης του μέλους, διεύρυνσης ή ρήξης του ανευρύσματος. Τα
σημεία απόφραξης του μέλους του μοσχεύματος περιλαμβάνουν πόνο στο(α)
ισχίο(α) ή στην(ις) κνήμη(ες) κατά τη διάρκεια του βαδίσματος ή κατά την
ανάπαυση ή αποχρωματισμό ή ψυχρότητα της κνήμης. Η ρήξη του
ανευρύσματος ενδέχεται να είναι ασυμπτωματική, αλλά συνήθως
παρουσιάζεται ως: πόνος, μούδιασμα, αδυναμία στις κνήμες, οποιοσδήποτε
πόνος στη ράχη, στο θώρακα, στην κοιλιά ή στο βουβώνα, ζάλη, λιποθυμία,
ταχύς καρδιακός παλμός ή αιφνίδια αδυναμία.
• Λόγω της απεικόνισης που απαιτείται για την επιτυχή τοποθέτηση και
παρακολούθηση των ενδαγγειακών συσκευών, οι κίνδυνοι που προκαλούνται
Χειρουργικός τυπικός
κίνδυνος Τιμή P
σε αναπτυσσόμενο ιστό λόγω της έκθεσης σε ακτινοβολία θα πρέπει να
συζητώνται με γυναίκες οι οποίες είναι ή έχουν υποψία ότι είναι έγκυες. Οι
άντρες που υποβάλλονται σε ενδαγγειακή ή ανοικτή χειρουργική
αποκατάσταση μπορεί να εκδηλώσουν ανικανότητα.
Οι ιατροί πρέπει να παραπέμπουν τον ασθενή στον Οδηγό ασθενούς σχετικά με
τους κινδύνους που παρουσιάζονται κατά τη διάρκεια ή μετά την εμφύτευση της
συσκευής. Οι κίνδυνοι που σχετίζονται με τη διαδικασία περιλαμβάνουν
καρδιακές, πνευμονικές, νευρολογικές, εντερικές και αιμορραγικές επιπλοκές.
Οι κίνδυνοι που σχετίζονται με τη συσκευή περιλαμβάνουν απόφραξη,
ενδοδιαφυγή, διεύρυνση ανευρύσματος, θραύση, ενδεχόμενο για επανεπέμβαση
και μετατροπή σε ανοικτή χειρουργική επέμβαση, ρήξη και θάνατο (δείτε την
ενότητα 5.1, Παρατηρούμενες ανεπιθύμητες ενέργειες και την ενότητα 5.2,
Δυνητικές ανεπιθύμητες ενέργειες). Ο ιατρός πρέπει να συμπληρώσει την
Κάρτα ταυτοποίησης ασθενούς και να τη δώσει στον ασθενή, έτσι ώστε να μπορεί
να τη φέρει μαζί του συνεχώς. Ο ασθενής πρέπει να ανατρέχει στην κάρτα
οποιαδήποτε στιγμή επισκέπτεται επιπλέον επαγγελματίες υγείας, ιδιαίτερα για
τυχόν επιπλέον διαγνωστικές διαδικασίες (π.χ. MRI).
9 ΤΡΟΠΟΣ ΔΙΑΘΕΣΗΣ
• Το ενδαγγειακό μόσχευμα AAA Zenith Flex παρέχεται στείρο και
προτοποθετημένο σε αποκολλούμενες συσκευασίες.
• Η συσκευή προορίζεται για μία χρήση μόνο. Μην επαναποστειρώνετε τη
συσκευή.
• Επιθεωρήστε τη συσκευή και τη συσκευασία, έτσι ώστε να επαληθεύσετε ότι
δεν έχει συμβεί καμία ζημιά ως αποτέλεσμα της αποστολής. Μη χρησιμοποιείτε
τη συσκευή αυτή εάν έχει συμβεί ζημιά ή εάν ο φραγμός αποστείρωσης έχει
υποστεί ζημιά ή έχει σπάσει. Εάν έχει συμβεί ζημιά, μη χρησιμοποιείτε το προϊόν
και επιστρέψτε το στην COOK.
• Πριν από τη χρήση, επαληθεύστε ότι έχουν παρασχεθεί οι σωστές συσκευές
(ποσότητα και μέγεθος) για τον ασθενή, ελέγχοντας τη συσκευή με βάση την
παραγγελία που έχει συνταγογραφηθεί από τον ιατρό για τον συγκεκριμένο
ασθενή.
• Η συσκευή κύριου σώματος τοποθετείται πάνω σε ένα θηκάρι εισαγωγέα
Flexor® 18, 20 ή 22 French. Η επιφάνεια του θηκαριού φέρει υδρόφιλη
επικάλυψη, η οποία όταν ενυδατωθεί, ενισχύει τη στρεπτικότητα. Για την
ενεργοποίηση της υδρόφιλης επικάλυψης, η επιφάνεια πρέπει να σκουπίζεται
με στείρο επίθεμα γάζας εμποτισμένο σε φυσιολογικό ορό.
• Μη χρησιμοποιείτε μετά την ημερομηνία (λήξης) “ΧΡΗΣΗ ΕΩΣ” που
αναγράφεται στην ετικέτα.
• Φυλάσσετε σε δροσερό και ξηρό χώρο.
Υψηλός κίνδυνος από το
μόσχευμα Zenith
Εισαγωγική περίοδος με
το μόσχευμα Zenith
10 ΠΛΗΡΟΦΟΡΙΕΣ ΓΙΑ ΤΗΝ ΚΛΙΝΙΚΗ ΧΡΗΣΗ
10.1 Εκπαίδευση ιατρού
ΠΡΟΣΟΧΗ: Να έχετε πάντοτε διαθέσιμη μια ομάδα αγγειοχειρουργικής κατά
τη διάρκεια των διαδικασιών εμφύτευσης ή επανεπέμβασης, σε περίπτωση
που είναι απαραίτητη η μετατροπή σε ανοικτή χειρουργική αποκατάσταση.
ΠΡΟΣΟΧΗ: Το ενδαγγειακό μόσχευμα AAA Zenith Flex με το σύστημα
εισαγωγής H&L-B One-Shot πρέπει να χρησιμοποιείται μόνον από ιατρούς
και ομάδες που έχουν εκπαιδευτεί σε αγγειακές επεμβατικές τεχνικές και
στη χρήση της συσκευής αυτής. Οι συνιστώμενες απαιτήσεις δεξιοτήτων/
γνώσεων για τους ιατρούς που χρησιμοποιούν το ενδαγγειακό μόσχευμα
AAA Zenith Flex με το σύστημα εισαγωγής H&L-B One-Shot περιγράφονται
παρακάτω:
Επιλογή ασθενούς:
• Γνώση του φυσικού ιστορικού των ανευρυσμάτων κοιλιακής αορτής (AAA) και
συνυπάρχουσες νόσους που σχετίζονται με την αποκατάσταση AAA.
• Γνώση της ερμηνείας των ακτινογραφικών εικόνων, επιλογής και
προσδιορισμού μεγέθους συσκευής.
Μια διεπιστημονική ομάδα που έχει συνδυασμένη εμπειρία της
διαδικασίας με:
• Μηριαία τομή, αρτηριοτομή και αποκατάσταση
• Τεχνικές διαδερμικής προσπέλασης και σύγκλεισης
• Μη εκλεκτικές και εκλεκτικές τεχνικές συρμάτινου οδηγού και καθετήρα
• Ερμηνεία ακτινοσκοπικών και αγγειογραφικών εικόνων
• Εμβολισμός
• Αγγειοπλαστική
• Τοποθέτηση ενδαγγειακής ενδοπρόσθεσης
• Τεχνικές βρόχου
• Κατάλληλη χρήση ακτινογραφικού σκιαγραφικού υλικού
• Τεχνικές για την ελαχιστοποίηση της έκθεσης σε ακτινοβολία
• Ειδίκευση στις απαραίτητες μεθόδους παρακολούθησης ασθενούς
10.2 Επιθεώρηση πριν από τη χρήση
Επιθεωρήστε τη συσκευή και τη συσκευασία, έτσι ώστε να επαληθεύσετε ότι δεν
έχει συμβεί καμία ζημιά ως αποτέλεσμα της αποστολής. Μη χρησιμοποιείτε τη
συσκευή αυτή εάν έχει συμβεί ζημιά ή εάν ο φραγμός αποστείρωσης έχει
υποστεί ζημιά ή έχει σπάσει. Εάν έχει συμβεί ζημιά, μη χρησιμοποιείτε το προϊόν
και επιστρέψτε το στην COOK. Πριν από τη χρήση, επαληθεύστε ότι έχουν
παρασχεθεί οι σωστές συσκευές (ποσότητα και μέγεθος) για τον ασθενή,
ελέγχοντας τη συσκευή με βάση την παραγγελία που έχει συνταγογραφηθεί από
τον ιατρό για τον συγκεκριμένο ασθενή.
10.3 Απαιτούμενα υλικά
(Δεν περιλαμβάνονται σε αρθρωτό σύστημα 3 τεμαχίων)
• Βοηθητικό κιτ ενδαγγειακού μοσχεύματος AAA Zenith
• Ακτινοσκόπιο με δυνατότητες ψηφιακής ακτινογραφίας (βραχίονας C ή
σταθερή μονάδα)
• Σκιαγραφικό μέσο
• Σύριγγα
• Ηπαρινισμένο αλατούχο διάλυμα
• Στείρα επιθέματα γάζας
10.4 Συνιστώμενα υλικά
(Δεν περιλαμβάνονται σε αρθρωτό σύστημα 3 τεμαχίων)
Τα ακόλουθα προϊόντα συνιστώνται για την εμφύτευση οποιουδήποτε
εξαρτήματος στη σειρά προϊόντων Zenith. Για πληροφορίες σχετικά με τη χρήση
86

των προϊόντων αυτών, ανατρέξτε στις προτεινόμενες οδηγίες χρήσης του
εκάστοτε προϊόντος.
• Υπεράκαμπτος συρμάτινος οδηγός 0,035” (0,89 mm), 260 cm, για παράδειγμα:
• Υπεράκαμπτοι συρμάτινοι οδηγοί Cook Lunderquist (LES)
• Τυπικός συρμάτινος οδηγός 0,035” (0,89 mm), για παράδειγμα:
• Συρμάτινοι οδηγοί 0,035” (0,89 mm) Cook
• Συρμάτινοι οδηγοί Cook Nimble™
• Μπαλόνια διαμόρφωσης, για παράδειγμα:
• Καθετήρας με μπαλόνι Cook CODA®
• Σετ εισαγωγέα, για παράδειγμα:
• Σετ εισαγωγέα Cook Check-Flo®
• Σετ πολύ μεγάλου εισαγωγέα Cook Check-Flo®
• Ετερόπλευροι εισαγωγείς Cook Flexor® Balkin Up & Over®
• Καθετήρας προσδιορισμού μεγέθους, για παράδειγμα:
• Καθετήρες προσδιορισμού μεγέθους σε εκατοστά Cook Aurous®
• Αγγειογραφικοί καθετήρες με ακτινοσκιερό άκρο, για παράδειγμα:
• Αγγειογραφικοί καθετήρες με άκρο Beacon® της Cook
• Καθετήρες Royal Flush με άκρο Beacon® της Cook
• Βελόνες εισαγωγής, για παράδειγμα:
• Βελόνες εισόδου μονού τοιχώματος Cook
10.5 Κατευθυντήριες οδηγίες προσδιορισμού μεγέθους διαμέτρου
συσκευής
Η επιλογή της διαμέτρου πρέπει να προσδιορίζεται από τη διάμετρο του αγγείου
από εξωτερικό τοίχωμα σε εξωτερικό τοίχωμα και όχι από τη διάμετρο του
αυλού. Ο προσδιορισμός ανεπαρκούς ή υπερβολικού μεγέθους ενδέχεται να έχει
ως αποτέλεσμα ατελή στεγανοποίηση ή περιορισμένη ροή.
Πίνακας 10.5.1 Οδηγός προσδιορισμού μεγέθους διαμέτρου
Προοριζόμενη
διάμετρος
αορτικού
αγγείου
(mm)
18-19 22 82/112, 96/126, 111/141,
20-21 24 82/112, 96/126, 111/141,
22 26 82/112, 96/126, 111/141,
23-24 28 82/112, 96/126, 111/141,
25-26 30 82/112, 96/126, 111/141,
27-28 32 82/112, 96/126, 111/141,
29-32 36 95/125, 113/143,
1
Μέγιστη διάμετρος κατά μήκος της εγγύς θέσης καθήλωσης.
2
Στρογγυλοποιήστε τη μετρημένη διάμετρο αορτής στο πλησιέστερο mm.
3
Επιπλέον ζητήματα ενδέχεται να επηρεάσουν την επιλογή της διαμέτρου.
*Όλες οι διαστάσεις είναι ονομαστικές.
Πίνακας 10.5.2 Οδηγός προσδιορισμού μεγέθους διαμέτρου
Διάμετρος
προοριζόμενου
λαγόνιου
αγγείου
(mm)
<8 8 37, 54, 71, 88, 105, 122 14
8-9 10 37, 54, 71, 88, 105, 122 14
10-11 12 37, 54, 71, 88, 105, 122 14
12-13 14 37, 54, 71, 88 14
14-15 16 37, 54, 71, 88 14
16-17 18 37, 54, 71, 88 16
18 20 37, 54, 71, 88 16
19 22 37, 54, 71, 88 16
20 24 37, 54, 71, 88 16
1
Μέγιστη διάμετρος κατά μήκος της περιφερικής θέσης καθήλωσης.
2
Στρογγυλοποιήστε τη μετρημένη λαγόνια διάμετρο στο πλησιέστερο mm.
3
Επιπλέον ζητήματα ενδέχεται να επηρεάσουν την επιλογή της διαμέτρου.
4
Συνολικό μήκος σκέλους = μήκος εργασίας + 22 mm ενδοπρόσθεσης σύνδεσης.
*Όλες οι διαστάσεις είναι ονομαστικές.
11 ΟΔΗΓΙΕΣ ΧΡΗΣΗΣ
Ανατομικές απαιτήσεις
• Το μέ γεθος και η μορφολογία του αγγείου λαγονομηριαίας προσπέλασης
(ελάχιστος θρόμβος, ασβέστιο ή/και ελίκωση) πρέπει να είναι συμβατά με τις
τεχνικές αγγειακής προσπέλασης και τα παρελκόμενα. Ενδέχεται να
απαιτούνται τεχνικές αρτηριακού αγωγού.
• Το μήκος του εγγύς αορτικού αυχένα θα πρέπει να είναι τουλάχιστον 15 mm με
διάμετρο που μετράται από εξωτερικό τοίχωμα σε εξωτερικό τοίχωμα
18-32 mm.
• Περιφερική θέση καθήλωσης λαγόνιας αρτηρίας μεγαλύτερη από 10 mm σε
μήκος και 7,5-20 mm σε διάμετρο (μετρημένη από εξωτερικό τοίχωμα σε
εξωτερικό τοίχωμα).
Πριν από τη χρήση του ενδαγγειακού μοσχεύματος AAA Zenith Flex με το
σύστημα εισαγωγής H&L-B One-Shot, ανασκοπήστε αυτό το φυλλάδιο των
προτεινόμενων οδηγιών χρήσης. Οι ακόλουθες οδηγίες ενσωματώνουν μια
βασική κατευθυντήρια οδηγία για την τοποθέτηση της συσκευής. Ενδέχεται να
είναι απαραίτητες παραλλαγές στις ακόλουθες διαδικασίες. Οι οδηγίες αυτές
προορίζονται για βοήθεια στην καθοδήγηση του ιατρού και δεν αντικαθιστούν
την κρίση του ιατρού.
μοσχεύματος κύριου σώματος*
Διάμετρος
1,2
σώματος
Συνολικό μήκος έως ετερόπλευρο
κύριου
3
(mm)
μοσχεύματος λαγόνιου σκέλους*
Διάμετρος
λαγόνιου
1,2
σκέλους
(mm)
3
μέλος/Συνολικό
μήκος έως σύστοιχο μέλος
(mm)
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
125/155, 140/170
131/161, 149/179
Μήκος εργασίας
λαγόνιου σκέλους
(mm)
4
Θηκάρι
εισαγωγέα
(French)
18
18
18
20
20
20
22
Θηκάρι
εισαγωγέα
(French)
Γενικές πληροφορίες χρήσης
• Κατά τη διάρκεια της χρήσης του ενδαγγειακού μοσχεύματος AAA Zenith Flex
με το σύστημα εισαγωγής H&L-B One-Shot, πρέπει να χρησιμοποιούνται
τυπικές τεχνικές για την τοποθέτηση θηκαριών αρτηριακής προσπέλασης,
οδηγών καθετήρων, αγγειογραφικών καθετήρων και συρμάτινων οδηγών. Το
ενδαγγειακό μόσχευμα AAA Zenith Flex με το σύστημα εισαγωγής H&L-B OneShot είναι συμβατό με συρμάτινους οδηγούς διαμέτρου 0,035” (0,89 mm).
• Η τοποθέτηση ενδαγγειακού μοσχεύματος είναι μία χειρουργική επέμβαση,
και μπορεί να παρουσιαστεί απώλεια αίματος από διάφορες αιτίες, η οποία
σε σπάνιες περιπτώσεις απαιτεί παρέμβαση (συμπεριλαμβανομένης της
μετάγγισης αίματος) προκειμένου να αποτραπούν οι ανεπιθύμητες εκβάσεις.
Είναι σημαντικό να παρακολουθείται η απώλεια αίματος από την
αιμοστατική βαλβίδα καθόλη τη διάρκεια της επέμβασης, αλλά αυτό είναι
ιδιαίτερα σημαντικό κατά τη διάρκεια και μετά από τον χειρισμό της γκρι
διάταξης τοποθέτησης. Μετά από την αφαίρεση της γκρι διάταξης
τοποθέτησης, εάν η απώλεια αίματος είναι υπερβολική, εξετάστε το
ενδεχόμενο τοποθέτησης ενός μη πληρωμένου μπαλονιού διαμόρφωσης ή
ενός διαστολέα συστήματος εισαγωγής εντός της βαλβίδας, περιορίζοντας
τη ροή.
Προσδιοριστικοί παράγοντες προ της εμφύτευσης
Επαληθεύστε από το σχεδιασμό προ της εμφύτευσης ότι έχει επιλεγεί η σωστή
συσκευή. Οι προσδιοριστικοί παράγοντες περιλαμβάνουν:
1. Ε πιλογή μηριαίας αρτηρίας για εισαγωγή του συστήματος κύριου σώματος
(δηλ. καθορισμός αντίστοιχων ετερόπλευρων και σύστοιχων λαγόνιων
αρτηριών).
2. Γωνίωση αυχένα αορτής, ανευρύσματος και λαγόνιων αρτηριών.
3. Ποιότητα του αυχένα αορτής.
4. Διάμετροι αυχένα υπονεφρικής αορτής και περιφερικών λαγόνιων αρτηριών.
5. Απόσ ταση από τις νεφρικές αρτηρίες έως το διχασμό της αορτής.
6. Μήκος από το διχασμό της αορτής έως τις έσω λαγόνιες αρτηρίες/θέση(εις)
πρόσφυσης.
7. Ανεύρυσμα(τα) που προεκτείνεται(ονται) εντός των λαγόνιων αρτηριών
ενδέχεται να απαιτήσει(ουν) ειδική εξέταση στην επιλογή κατάλληλης θέσης
επιφάνειας επαφής μοσχεύματος/αρτηρίας.
8. Εξετάσ τε το βαθμό αγγειακής αποτιτάνωσης.
Προετοιμασία ασθενούς
1. Ανατρέξτε στα πρωτόκολλα του ιδρύματος που σχετίζονται με την
αναισθησία, την αντιπηκτική αγωγή και την παρακολούθηση των ζωτικών
σημείων.
2. Τοποθετήστε τον ασθενή επάνω στο τραπέζι απεικόνισης, επιτρέποντας την
ακτινοσκοπική απεικόνιση από το αορτικό τόξο έως το διχασμό των μηριαίων
αρτηριών.
3. Αποκαλύψτε αμφότερες τις κοινές μηριαίες αρτηρίες με χρήση πρότυπης
χειρουργικής τεχνικής.
4. Επιτύχετε επαρκή εγγύς και περιφερικό αγγειακό έλεγχο αμφότερων των
μηριαίων αγγείων.
11.1 Διχαλωτό σύστημα (Εικ. 2)
11.1.1 Προετοιμασία/Έκπλυση διχαλωτού κύριου σώματος
1. Αφαιρέστε το στειλεό αποστολής με τον μαύρο ομφαλό (από την εσωτερική
κάνουλα), τον προστατευτικό σωλήνα της κάνουλας (από την εσωτερική
κάνουλα) και το προστατευτικό του άκρου διαστολέα (από το άκρο του
διαστολέα). Αφαιρέστε το θηκάρι Peel-Away® από την πίσω πλευρά της
αιμοστατικής βαλβίδας. (Εικ. 5) Ανασηκώστε το περιφερικό άκρο του
συστήματος και εκπλύνετε μέσω της στρόφιγγας στην αιμοστατική βαλβίδα,
έως ότου εξέλθει το υγρό από την πλευρική θύρα κοντά στο άκρο του
θηκαριού εισαγωγέα. (Εικ. 6) Συνεχίστε την έγχυση διαλύματος έκπλυσης
όγκου 20 ml μέσω της συσκευής. Διακόψτε την έγχυση και κλείστε τη
στρόφιγγα στο σωλήνα σύνδεσης.
ΣΗΜΕΙΩΣΗ: Συχνά χρησιμοποιείται διάλυμα έκπλυσης μοσχεύματος
ηπαρινισμένου αλατούχου διαλύματος.
2. Προσαρτήσ τε τη σύριγγα με ηπαρινισμένο φυσιολογικό ορό στον ομφαλό
της εσωτερικής κάνουλας. Εκπλύνετε έως ότου εξέλθει υγρό από το άκρο του
διαστολέα. (Εικ. 7)
ΣΗΜΕΙΩΣΗ: Κατά την έκπλυση του συστήματος, ανασηκώστε το περιφερικό του
άκρο για τη διευκόλυνση της αφαίρεσης του αέρα.
3. Εμποτίσ τε στείρα επιθέματα γάζας σε φυσιολογικό ορό και χρησιμοποιήστε
τα για να σκουπίσετε το θηκάρι εισαγωγέα Flexor®, για την ενεργοποίηση
της υδρόφιλης επικάλυψης. Ενυδατώστε άφθονα τόσο το θηκάρι όσο και το
διαστολέα.
11.1.2 Προετοιμασία/Έκπλυση ετερόπλευρου λαγόνιου σκέλους
1. Αφαιρέστε τον εσωτερικό στειλεό με τον μαύρο ομφαλό (από την εσωτερική
κάνουλα), τον προστατευτικό σωλήνα της κάνουλας (από την εσωτερική
κάνουλα) και το προστατευτικό του άκρου διαστολέα (από το άκρο του
διαστολέα). Αφαιρέστε το θηκάρι Peel-Away® από την πίσω πλευρά της
αιμοστατικής βαλβίδας. (Εικ. 8) Ανασηκώστε το περιφερικό άκρο του
συστήματος και εκπλύνετε μέσω της στρόφιγγας στην αιμοστατική βαλβίδα,
έως ότου εξέλθει το υγρό από την πλευρική θύρα κοντά στο άκρο του
θηκαριού εισαγωγέα. (Εικ. 9) Συνεχίστε την έγχυση διαλύματος έκπλυσης
όγκου 20 ml μέσω της συσκευής. Διακόψτε την έγχυση και κλείστε τη
στρόφιγγα στο σωλήνα σύνδεσης.
ΣΗΜΕΙΩΣΗ: Συχνά χρησιμοποιείται διάλυμα έκπλυσης μοσχεύματος
ηπαρινισμένου αλατούχου διαλύματος.
2. Προσαρτήσ τε τη σύριγγα με ηπαρινισμένο αλατούχο διάλυμα στον ομφαλό
της περιφερικής εσωτερικής κάνουλας. Εκπλύνετε έως ότου εξέλθει υγρό από
το περιφερικό άκρο του διαστολέα. (Εικ. 7)
ΣΗΜΕΙΩΣΗ: Κατά την έκπλυση του συστήματος, ανασηκώστε το περιφερικό του
άκρο για τη διευκόλυνση της αφαίρεσης του αέρα.
11.1.3 Προετοιμασία/Έκπλυση σύστοιχου λαγόνιου σκέλους
Ακολουθήστε τις οδηγίες που αναφέρονται στην προηγούμενη ενότητα 11.1.2,
Προετοιμασία/Έκπλυση ετερόπλευρου λαγόνιου σκέλους, έτσι ώστε να
διασφαλιστεί η σωστή έκπλυση του μοσχεύματος σύστοιχου λαγόνιου σκέλους.
11.1.4 Αγγειακή προσπέλαση και αγγειογραφία
1. Παρακεν τήστε τις επιλεγμένες κοινές μηριαίες αρτηρίες με χρήση τυπικής
τεχνικής με (υπέρλεπτη) αρτηριακή βελόνα 18UT ή 19UT gauge. Κατά την
είσοδο στο αγγείο, εισαγάγετε:
• Συρμάτινους οδηγούς – τυπικούς, διαμέτρου 0,035” (0,89 mm), μήκους
145 cm, άκρου σχήματος J ή συρμάτινο οδηγό Bentson
• Θηκάρια κατάλληλου μεγέθους (π.χ. 6,0 ή 8,0 French)
• Καθετήρα έκπλυσης (συχνά ακτινοσκιεροί καθετήρες προσδιορισμού
μεγέθους – π.χ. καθετήρας προσδιορισμού μεγέθους σε εκατοστά ή ευθύς
καθετήρας έκπλυσης)
2. Εκτελέστε αγγειογραφία για την αναγνώριση του(ων) επιπέδου(ων) των
νεφρικών αρτηριών, του διχασμού της αορτής και του διχασμού των
λαγόνιων αρτηριών.
ΣΗΜΕΙΩΣΗ: Εάν χρησιμοποιείται γωνίωση ακτινοσκοπίου με γωνιωτό αυχένα,
ενδέχεται να είναι απαραίτητη η εκτέλεση αγγειογραμμάτων με χρήση διαφόρων
προβολών.
11.1.5 Τοποθέτηση κύριου σώματος
1. Βεβαιωθείτε ότι το σύστημα τοποθέτησης έχει εκπλυθεί με ηπαρινισμένο
αλατούχο διάλυμα και ότι έχει αφαιρεθεί όλος ο αέρας από το σύστημα.
2. Χορηγήστε συστηματική ηπαρίνη και ελέγξτε τα διαλύματα έκπλυσης.
Εκπλύνετε μετά από κάθε εναλλαγή καθετήρα ή/και συρμάτινου οδηγού.
ΣΗΜΕΙΩΣΗ: Παρακολουθείτε την κατάσταση της πήξης του αίματος του
ασθενούς καθόλη τη διάρκεια της διαδικασίας.
3. Στη σύστοιχη πλευρά, αντικαταστήστε το σύρμα σχήματος J με τον άκαμπτο
συρμάτινο οδηγό (LES) 0,035” (0,89 mm), μήκους 260 cm και προωθήστε
μέσω του καθετήρα και έως τη θωρακική αορτή. Αφαιρέστε τον καθετήρα
έκπλυσης και το θηκάρι. Διατηρήστε τη θέση του συρμάτινου οδηγού.
4. Πριν από την εισαγωγή, τοποθετήστε το σύστημα τοποθέτησης κύριου
σώματος στην κοιλία του ασθενούς υπό ακτινοσκόπηση, για τον
προσδιορισμό του προσανατολισμού του ακτινοσκιερού δείκτη του
ετερόπλευρου μέλους. Ο πλευρικός βραχίονας της αιμοστατικής βαλβίδας
μπορεί να χρησιμεύσει ως εξωτερική αναφορά στον ακτινοσκιερό δείκτη του
ετερόπλευρου μέλους.
5. Εισαγάγετε το σύστημα τοποθέτησης κύριου σώματος πάνω από το σύρμα,
εντός της μηριαίας αρτηρίας προσέχοντας την αναφορά του πλευρικού
βραχίονα.
ΠΡΟΣΟΧΗ: Διατηρείτε τη θέση του συρμάτινου οδηγού κατά τη διάρκεια
της εισαγωγής του συστήματος τοποθέτησης.
ΠΡΟΣΟΧΗ: Για να α ποφύγετε τυχόν συστροφή του ενδαγγειακού
μοσχεύματος, κατά τη διάρκεια οποιασδήποτε περιστροφής του
συστήματος τοποθέτησης, προσέχετε έτσι ώστε να περιστρέφετε όλα τα
87

εξαρτήματα του συστήματος ως ενιαία μονάδα (από το εξωτερικό θηκάρι
έως την εσωτερική κάνουλα).
6. Προωθήστε το σύστημα τοποθέτησης έως ότου οι τέσσερις χρυσοί
ακτινοσκιεροί δείκτες (οι οποίοι είναι τοποθετημένοι σε απόσταση 2 mm από
το πλέον εγγύς τμήμα του υλικού μοσχεύματος) (Εικ. 10, απεικόνιση 1) είναι
μόλις κάτω από το πλέον κατώτερο νεφρικό στόμιο.
7. Επαληθεύστε τη θέση του συρμάτινου οδηγού στη θωρακική αορτή.
Βεβαιωθείτε ότι το σύστημα μοσχεύματος είναι προσανατολισμένο, έτσι
ώστε να τοποθετηθεί το ετερόπλευρο μέλος επάνω από και πρόσθια προς
την έκφυση της ετερόπλευρης λαγόνιας αρτηρίας. Εάν ο ακτινοσκιερός
δείκτης του ετερόπλευρου μέλους δεν ευθυγραμμιστεί σωστά, περιστρέψτε
ολόκληρο το σύστημα έως ότου τοποθετηθεί σωστά στο μέσον της
απόστασης μεταξύ μιας εξωτερικής και μιας πρόσθιας θέσης στην
ετερόπλευρη πλευρά.
• Ένας σχηματισμός δείκτη ✓ υποδεικνύει πρόσθια θέση του βραχέος
(ετερόπλευρου) μέλους. (Εικ. 10, απεικόνιση 4)
• Ένας σχηματισμός δείκτη υποδεικνύει οπίσθια θέση του βραχέος
(ετερόπλευρου) μέλους. (Εικ. 10, απεικόνιση 5)
• Ένας σχηματισμός δείκτη | υποδεικνύει πλευρική θέση του βραχέος
(ετερόπλευρου) μέλους. (Εικ. 10, απεικόνιση 6)
ΣΗΜΕΙΩΣΗ: Ακτινοσκιερές κάνουλες σε κάθε ενδοπρόσθεση μεταξύ των
νεφρικών αρτηριών και του ετερόπλευρου μέλους ευθυγραμμίζονται με τον
ακτινοσκιερό δείκτη του ετερόπλευρου μέλους.
8. Επαναλάβετε το αγγειόγραμμα, έτσι ώστε να επαληθεύσετε ότι οι τέσσερις
χρυσοί ακτινοσκιεροί δείκτες απέχουν 2 mm ή περισσότερο κάτω από το
πλέον κατώτερο νεφρικό στόμιο.
9. Βεβαιωθείτε ότι η αιμοστατική βαλβίδα Captor στο θηκάρι εισαγωγέα Flexor®
είναι στραμμένη στην ανοικτή θέση. (Εικ. 11)
10. Σταθεροποιήστε τη γκρι διάταξη τοποθέτησης (το στέλεχος του συστήματος
τοποθέτησης) ενώ αποσύρετε το θηκάρι. Εκπτύξτε τις πρώτες δύο (2)
καλυμμένες ενδοπροσθέσεις αποσύροντας το θηκάρι, ενώ παρακολουθείτε
τη θέση της συσκευής.
11. Χωρίς να μετακινήσετε το τραπέζι, μειώστε τη μεγέθυνση για να ελέγξετε
τη θέση του ακτινοσκιερού δείκτη του ετερόπλευρου μέλους και τη θέση
των νεφρικών αρτηριών. Προχωρήστε με την έκπτυξη έως ότου εκπτυχθεί
πλήρως το ετερόπλευρο μέλος. (Εικ. 12) Σταματήστε την απόσυρση του
θηκαριού.
ΣΗΜΕΙΩΣΗ: Επαληθεύστε ότι το ετερόπλευρο μέλος απέχει τουλάχιστον 5 mm
πάνω από το διχασμό της αορτής και είναι στην επιθυμητή θέση για
καθετηριασμό.
11.1.6 Τοποθέτηση του ετερόπλευρου λαγόνιου συρμάτινου οδηγού
1. Χειρισ τείτε τον καθετήρα και τον συρμάτινο οδηγό μέσω του ανοικτού
άκρου του ετερόπλευρου μέλους εντός του σώματος του μοσχεύματος.
Προωθήστε τον συρμάτινο οδηγό έως ότου σχηματίσει καμπύλη εντός του
σώματος του μοσχεύματος. Οι προσθοπίσθιες και οι λοξές ακτινοσκοπικές
προβολές μπορούν να βοηθήσουν στην επαλήθευση του καθετηριασμού της
συσκευής.
2. Μετά τον καθετηριασμό, προωθήστε τον αγγειογραφικό καθετήρα πάνω από
το σύρμα εντός του σώματος του ενδαγγειακού μοσχεύματος. Αφαιρέστε
το σύρμα και εκτελέστε αγγειογραφία για την επιβεβαίωση της θέσης.
Προωθήστε τον συρμάτινο οδηγό έως ότου σχηματίσει καμπύλη εντός του
σώματος του μοσχεύματος. Αφαιρέστε τον αγγειογραφικό καθετήρα. (Εικ. 13)
11.1.7 Εγγύς (άνω) απελευθέρωση του κύριου σώματος
1. Εκτελέστε αγγειογραφία μέσω αγγειογραφικού καθετήρα για την
επαλήθευση της θέσης του ενδαγγειακού μοσχεύματος σε σχέση με τις
νεφρικές αρτηρίες. Εάν είναι απαραίτητο, επανατοποθετήστε προσεκτικά
το καλυμμένο τμήμα του ενδαγγειακού μοσχεύματος σε σχέση με τις
νεφρικές αρτηρίες. (Η επανατοποθέτηση μπορεί να λάβει χώρα μόνον
πάνω από ένα μικρό εύρος απόστασης στο στάδιο αυτό.)
ΣΗΜΕΙΩΣΗ: Διασφαλίστε τη βατότητα των νεφρικών αρτηριών
επιβεβαιώνοντας ότι οι εγγύς δείκτες του μοσχεύματος απέχουν 2 mm ή
περισσότερο κάτω από την κατώτατη βατή νεφρική αρτηρία.
ΠΡΟΣΟΧΗ: Κατά την αφαίρεση του περιφερικού σύρματος
ενεργοποίησης, την προώθηση του άνω πώματος και την επακόλουθη
απελευθέρωση της επινεφρικής ενδοπρόσθεσης, επαληθεύστε ότι η
θέση του συρμάτινου οδηγού του κυρίου σώματος προεκτείνεται μόλις
περιφερικά προς το αορτικό τόξο και ότι μεγιστοποιείται η υποστήριξη
του συστήματος.
2. Αφαιρέστε την ασφάλεια από τον μαύρο μηχανισμό απελευθέρωσης
του σύρματος ενεργοποίησης. Υπό ακτινοσκόπηση, αποσύρετε και
αφαιρέστε το σύρμα ενεργοποίησης σύροντας τον μαύρο μηχανισμό
απελευθέρωσης σύρματος ενεργοποίησης εκτός της λαβής και κατόπιν
αφαιρέστε τον μέσω της σχισμής του πάνω από την εσωτερική κάνουλα.
(Εικ. 14)
Εάν αισθανθείτε αντίσταση ή παρατηρήσετε κάμψη του συστήματος, το
σύρμα ενεργοποίησης βρίσκεται υπό τάση. Η υπερβολική ισχύς μπορεί να
προκαλέσει αλλαγή της θέσης του μοσχεύματος. Εάν παρατηρήσετε
υπερβολική αντίσταση ή μετακίνηση του συστήματος τοποθέτησης, διακόψτε
και αξιολογήστε την κατάσταση. Εάν δεν μπορείτε να αφαιρέσετε το μαύρο
μηχανισμό απελευθέρωσης του σύρματος ενεργοποίησης από το άνω πώμα,
διενεργήστε τα ακόλουθα βήματα υπό ακτινοσκόπηση:
a. Εκτονώσ τε την τάση από το σύρμα ενεργοποίησης χαλαρώνοντας τη
μέγγενη ακίδας και τραβώντας λίγο την εσωτερική κάνουλα για να
μετακινήσετε το άνω πώμα προς τα κάτω επάνω από την επινεφρική
ενδοπρόσθεση. Αποφύγετε τη συμπίεση του κυρίου σώματος του
μοσχεύματος Zenith Flex.
b. Ξανασφίξτε τη μέγγενη ακίδας.
c. Αφαιρέστε το μαύρο μηχανισμό απελευθέρωσης του σύρματος
ενεργοποίησης.
d. Συνεχίστε με το βήμα 3 της ενότητας 11.1.7, Εγγύς (άνω)
απελευθέρωση του κύριου σώματος.
ΣΗΜΕΙΩΣΗ: Εάν και πάλι δεν μπορείτε να αφαιρέσετε τον μηχανισμό
απελευθέρωσης του σύρματος ενεργοποίησης της άνω ενδοπρόσθεσης από
το άνω πώμα, δείτε την ενότητα 14.1, Αντιμετώπιση προβλημάτων με την
απελευθέρωση του σύρματος ενεργοποίησης.
3. Ξεσφίξτε τη μέγγενη ακίδας. (Εικ. 15) Ελέγξτε τη θέση του μοσχεύματος
σταθεροποιώντας την γκρι διάταξη τοποθέτησης του εισαγωγέα.
4. Εκπτύξτε την επινεφρική ενδοπρόσθεση προωθώντας την εσωτερική
κάνουλα του άνω πώματος 1 έως 2 mm κάθε φορά, ενώ ελέγχετε τη θέση
του κύριου σώματος έως ότου εκπτυχθεί πλήρως η άνω ενδοπρόσθεση.
(Εικ. 16 και 17) Προωθήστε την κάνουλα του άνω πώματος 1 έως
2 cm επιπλέον και κατόπιν σφίξτε πάλι τη μέγγενη ακίδας, έτσι ώστε να
αποφύγετε την επαφή με την εκπτυγμένη επινεφρική ενδοπρόσθεση.
ΣΗΜΕΙΩΣΗ: Εάν δεν μπορείτε να απελευθερώσετε πλήρως την επινεφρική
ενδοπρόσθεση με την προώθηση της εσωτερικής κάνουλας του άνω πώματος,
δείτε την ενότητα 14.2, Αντιμετώπιση προβλημάτων απελευθέρωσης της
επινεφρικής ενδοπρόσθεσης.
ΣΗΜΕΙΩΣΗ: Μόλις εκπτυχθεί η ακιδωτή επινεφρική ενδοπρόσθεση, δε
συνιστώνται περαιτέρω απόπειρες για την επανατοποθέτηση του
μοσχεύματος.
ΠΡΟΕΙΔΟΠΟΙΗΣΗ: Το ενδαγγειακό μόσχευμα AAA Zenith Flex φέρει
ενσωματωμένη μια επινεφρική ενδοπρόσθεση με ακίδες καθήλωσης.
Πρέπει να δίνετε ιδιαίτερη προσοχή κατά το χειρισμό επεμβατικών
συσκευών στην περιοχή της επινεφρικής ενδοπρόσθεσης.
5. Προωθήστε τον ετερόπλευρο συρμάτινο οδηγό μέσα στη θωρακική
αορτή.
11.1.8 Τοποθέτηση και απελευθέρωση του ετερόπλευρου λαγόνιου σκέλους
ΠΡΟΣΟΧΗ: Επαληθεύστε ότι επιλέγεται το προκαθορισμένο ετερόπλευρο
λαγόνιο σκέλος για εισαγωγή στην ετερόπλευρη πλευρά του ασθενούς
πριν από την εμφύτευση.
1. Τοποθετήστε τον ενισχυτή εικόνας για την εμφάνιση τόσο της ετερόπλευρης
έσω λαγόνιας αρτηρίας όσο και της ετερόπλευρης κοινής λαγόνιας αρτηρίας.
2. Πριν από την εισαγωγή του συστήματος τοποθέτησης ετερόπλευρου
λαγόνιου σκέλους, εγχύστε σκιαγραφικό μέσο μέσω του ετερόπλευρου
μηριαίου θηκαριού για τον εντοπισμό της ετερόπλευρης έσω λαγόνιας
αρτηρίας.
3. Εισαγάγετε το σύστημα τοποθέτησης ετερόπλευρου λαγόνιου σκέλους
εντός της αρτηρίας. Προωθήστε αργά έως ότου το μόσχευμα λαγόνιου
σκέλους επικαλύψει τουλάχιστον μία πλήρη ενδοπρόσθεση λαγόνιου
σκέλους (δηλ. εγγύς ενδοπρόσθεση του μοσχεύματος λαγόνιου σκέλους)
εντός του ετερόπλευρου μέλους του κύριου σώματος. (Εικ. 18) Εάν υπάρχει
οποιαδήποτε τάση μετακίνησης του μοσχεύματος κύριου σώματος κατά τη
διάρκεια του χειρισμού αυτού, κρατήστε το στη θέση του σταθεροποιώντας
την γκρι διάταξη τοποθέτησης στη σύστοιχη πλευρά.
ΣΗΜΕΙΩΣΗ: Εάν παρουσιαστεί δυσκολία κατά την προώθηση του συστήματος
τοποθέτησης λαγόνιου σκέλους, εναλλάξτε το με έναν πιο υποστηρικτικό
συρμάτινο οδηγό. Σε ελικοειδή αγγεία, η ανατομία ενδέχεται να μεταβληθεί
σημαντικά με την εισαγωγή των άκαμπτων συρμάτων και των συστημάτων
θηκαριού.
4. Ε πιβεβαιώστε τη θέση του περιφερικού άκρου του μοσχεύματος λαγόνιου
σκέλους. Επανατοποθετήστε το μόσχευμα λαγόνιου σκέλους εάν είναι
απαραίτητο, έτσι ώστε να διασφαλιστεί τόσο η βατότητα της έσω λαγόνιας
αρτηρίας όσο και μια ελάχιστη επικάλυψη μιας πλήρους ενδοπρόσθεσης
λαγόνιου σκέλους (δηλ. εγγύς ενδοπρόσθεση του μοσχεύματος λαγόνιου
σκέλους, μέγιστη επικάλυψη 1,5 ενδοπροσθέσεων) εντός του ενδαγγειακού
μοσχεύματος του κύριου σώματος.
5. Για να εκπτύξετε, κρατήστε το μόσχευμα του λαγόνιου σκέλους στη θέση του
με τη γκρι διάταξη τοποθέτησης, ενώ αποσύρετε το θηκάρι. (Εικ. 19 και 20)
Βεβαιωθείτε ότι διατηρείται επικάλυψη ίση με το μήκος μίας ενδοπρόσθεσης.
6. Σταματήστε την απόσυρση του θηκαριού αμέσως μόλις απελευθερωθεί το
περιφερικό άκρο του μοσχεύματος λαγόνιου σκέλους.
7. Υπό ακτινοσκόπηση και μετά από επαλήθευση της θέσης του μοσχεύματος
λαγόνιου σκέλους, ξεσφίξτε τη μέγγενη ακίδας, αποσύρετε την εσωτερική
κάνουλα για τη σύνδεση του κωνικού διαστολέα με τη γκρι διάταξη
τοποθέτησης. Σφίξτε τη μέγγενη ακίδας. Διατηρήστε τη θέση του θηκαριού
ενώ αποσύρετε τη γκρι διάταξη τοποθέτησης με στερεωμένη την εσωτερική
κάνουλα. (Εικ. 21)
8. Ε λέγξτε πάλι τη θέση του συρμάτινου οδηγού.
11.1.9 Περιφερική (κάτω) απελευθέρωση του κύριου σώματος
1. Ε πανέλθετε στη σύστοιχη πλευρά.
2. Εκπτύξτε πλήρως το σύστοιχο μέλος του κύριου σώματος αποσύροντας το
θηκάρι, έως ότου επεκταθεί η πλέον περιφερική ενδοπρόσθεση. (Εικ. 22 και
23) Σταματήστε την απόσυρση του θηκαριού.
ΣΗΜΕΙΩΣΗ: Η περιφερική ενδοπρόσθεση είναι ακόμα στερεωμένη από το
σύρμα ενεργοποίησης.
3. Αφαιρέστε την ασφάλεια από τον λευκό μηχανισμό απελευθέρωσης
σύρματος ενεργοποίησης. Αποσύρετε και αφαιρέστε το σύρμα
ενεργοποίησης σύροντας τον λευκό μηχανισμό απελευθέρωσης σύρματος
ενεργοποίησης εκτός της λαβής και κατόπιν αφαιρέστε το μέσω της σχισμής
του, επάνω από την εσωτερική κάνουλα της συσκευής. (Εικ. 24)
11.1.10 Σύνδεση του άνω πώματος
1. Ξ εσφίξτε τη μέγγενη ακίδας. (Εικ. 25)
2. Στερεώστε το θηκάρι και την εσωτερική κάνουλα, έτσι ώστε να αποφύγετε
τυχόν κίνηση των εξαρτημάτων αυτών.
3. Προωθήστε τη γκρι διάταξη τοποθέτησης πάνω από την εσωτερική κάνουλα
έως ότου συνδεθεί με το άνω πώμα. (Εικ. 26, 27 και 28)
ΣΗΜΕΙΩΣΗ: Εάν παρουσιαστεί αντίσταση, περιστρέψτε ελαφρά τη γκρι διάταξη
τοποθέτησης και συνεχίστε να προωθείτε απαλά.
4. Σφίξτε πάλι τη μέγγενη ακίδας και αποσύρετε ολόκληρο το άνω πώμα και
τη γκρι διάταξη τοποθέτησης μέσω του μοσχεύματος και του θηκαριού,
έλκοντας την εσωτερική κάνουλα. (Εικ. 29) Αφήστε το θηκάρι και τον
συρμάτινο οδηγό στη θέση τους.
ΣΗΜΕΙΩΣΗ: Διατηρήστε τη θέση του θηκαριού και του συρμάτινου οδηγού.
5. Κ λείστε την αιμοστατική βαλβίδα Captor® στο θηκάρι εισαγωγέα Flexor®
στρέφοντας την δεξιόστροφα, έως ότου σταματήσει. (Εικ. 30)
11.1.11 Τοποθέτηση και απελευθέρωση του σύστοιχου λαγόνιου σκέλους
ΣΗΜΕΙΩΣΗ: Βεβαιωθείτε ότι η αιμοστατική βαλβίδα Captor στο θηκάρι
εισαγωγέα είναι στραμμένη στην ανοικτή θέση. (Εικ. 31)
1. Χρησιμοποιήσ τε τη διάταξη σύρματος του μοσχεύματος κύριου σώματος και
θηκαριού για την εισαγωγή του μοσχεύματος σύστοιχου λαγόνιου σκέλους.
Προωθήστε τη διάταξη διαστολέα και θηκαριού εντός του θηκαριού κύριου
σώματος.
ΣΗΜΕΙΩΣΗ: Σε ελικοειδή αγγεία, η θέση των έσω λαγόνιων αρτηριών ενδέχεται
να μεταβληθεί σημαντικά με την εισαγωγή των άκαμπτων συρμάτων και των
συστημάτων θηκαριού.
2. Προωθήστε αργά έως ότου το μόσχευμα του σύστοιχου λαγόνιου σκέλους
επικαλύψει τουλάχιστον μία πλήρη ενδοπρόσθεση λαγόνιου σκέλους
(δηλ. εγγύς ενδοπρόσθεση του μοσχεύματος λαγόνιου σκέλους) εντός του
σύστοιχου μέλους του κύριου σώματος. (Εικ. 32)
ΣΗΜΕΙΩΣΗ: Εάν απαιτείται επικάλυψη μεγαλύτερη από 3 ενδοπροσθέσεις
λαγόνιου σκέλους (μεγαλύτερη από 2 ενδοπροσθέσεις λαγόνιου σκέλους για
μήκη σκέλους 37 mm και 54 mm), ενδέχεται να είναι απαραίτητο να εξετάσετε το
ενδεχόμενο χρήσης μιας προέκτασης σκέλους στην περιοχή διχασμού της
αντίθετης πλευράς.
3. Ε πιβεβαιώστε τη θέση του περιφερικού άκρου του μοσχεύματος λαγόνιου
σκέλους. Επανατοποθετήστε το μόσχευμα λαγόνιου σκέλους εάν είναι
απαραίτητο, έτσι ώστε να διασφαλίσετε τη βατότητα της έσω λαγόνιας
αρτηρίας.
4. Για να εκπτύξετε, σταθεροποιήστε το μόσχευμα του λαγόνιου σκέλους με τη
γκρι διάταξη τοποθέτησης, ενώ αποσύρετε το θηκάρι λαγόνιου σκέλους.
(Εικ. 33 και 34) Εάν είναι απαραίτητο, αποσύρετε το θηκάρι κύριου σώματος.
5. Υπό ακτινοσκόπηση και μετά από επαλήθευση της θέσης του μοσχεύματος
λαγόνιου σκέλους, ξεσφίξτε τη μέγγενη ακίδας, αποσύρετε την εσωτερική
κάνουλα για τη σύνδεση του κωνικού διαστολέα με τη γκρι διάταξη
τοποθέτησης. Σφίξτε τη μέγγενη ακίδας. Διατηρήστε τη θέση του θηκαριού
ενώ αποσύρετε τη γκρι διάταξη τοποθέτησης με στερεωμένη την εσωτερική
κάνουλα. (Εικ. 35)
6. Κ λείστε την αιμοστατική βαλβίδα Captor στο θηκάρι εισαγωγέα Flexor®
στρέφοντας την δεξιόστροφα, έως ότου σταματήσει.
7. Ε λέγξτε πάλι τη θέση των συρμάτινων οδηγών. Αφήστε το θηκάρι και τους
συρμάτινους οδηγούς στη θέση τους.
11.1.12 Εισαγωγή μπαλονιού διαμόρφωσης
1. Προετοιμάσ τε το μπαλόνι διαμόρφωσης ως εξής:
• Εκπλύνετε τον αυλό του σύρματος με ηπαρινισμένο αλατούχο διάλυμα.
88

• Αφαιρέστε όλο τον αέρα από το μπαλόνι.
2. Σε προετοιμασία για την εισαγωγή του μπαλονιού διαμόρφωσης, ανοίξτε την
αιμοστατική βαλβίδα Captor στρέφοντάς την αριστερόστροφα.
3. Προωθήστε το μπαλόνι διαμόρφωσης πάνω από τον συρμάτινο οδηγό και
μέσω της αιμοστατικής βαλβίδας Captor του συστήματος εισαγωγής του
κύριου σώματος έως το επίπεδο των νεφρικών αρτηριών. Διατηρήστε τη
σωστή θέση του θηκαριού.
4. Σφίξτε την αιμοστατική βαλβίδα Captor γύρω από το μπαλόνι διαμόρφωσης
με απαλή πίεση, στρέφοντάς το δεξιόστροφα.
ΠΡΟΣΟΧΗ: Μη φουσκώνετε το μπαλόνι σε αγγείο εκτός του μοσχεύματος.
5. Διασ τείλετε το μπαλόνι διαμόρφωσης με αραιωμένο σκιαγραφικό μέσο
(σύμφωνα με τις οδηγίες του κατασκευαστή) στην περιοχή της υπερνεφρικής
επικαλυμμένης ενδοπρόσθεσης και του υπονεφρικού αυχένα, αρχίζοντας
εγγύς και προχωρώντας προς την περιφερική κατεύθυνση. (Εικ. 36)
ΠΡΟΣΟΧΗ: Επιβεβαιώστε το πλήρες ξεφούσκωμα του μπαλονιού πριν
από την επανατοποθέτηση.
ΠΡΟΣΟΧΗ: Η αιμοστατική βαλβίδα Captor πρέπει να είναι ανοικτή πριν
από την επανατοποθέτηση του μπαλονιού διαμόρφωσης.
6. Αποσύρετε το μπαλόνι διαμόρφωσης προς την επικάλυψη του σύστοιχου
μέλους και διαστείλετέ το.
ΠΡΟΣΟΧΗ: Η αιμοστατική βαλβίδα Captor πρέπει να είναι ανοικτή πριν
από την επανατοποθέτηση του μπαλονιού διαμόρφωσης.
7. Αποσύρετε το μπαλόνι διαμόρφωσης έως την περιφερική θέση καθήλωσης
του σύστοιχου μέλους και διαστείλετέ το.
ΠΡΟΣΟΧΗ: Μη φουσκώνετε το μπαλόνι σε αγγείο εκτός του μοσχεύματος.
8. Ξ εφουσκώστε και αφαιρέστε το μπαλόνι διαμόρφωσης. Μεταφέρετε
το μπαλόνι διαμόρφωσης πάνω στον ετερόπλευρο συρμάτινο οδηγό
και εντός του συστήματος εισαγωγής του ετερόπλευρου λαγόνιου
σκέλους. Προωθήστε το μπαλόνι διαμόρφωσης προς την επικάλυψη του
ετερόπλευρου μέλους και διαστείλετέ το.
ΠΡΟΣΟΧΗ: Επιβεβαιώστε το πλήρες ξεφούσκωμα του μπαλονιού πριν
από την επανατοποθέτηση.
9. Αποσύρετε το μπαλόνι διαμόρφωσης προς το ετερόπλευρο λαγόνιο σκέλος/
περιφερική θέση καθήλωσης αγγείου και διαστείλετέ το. (Εικ. 36)
ΠΡΟΣΟΧΗ: Μη φουσκώνετε το μπαλόνι σε αγγείο εκτός του μοσχεύματος.
10. Αφαιρέστε το μπαλόνι διαμόρφωσης και αντικαταστήστε το με έναν
αγγειογραφικό καθετήρα, για την εκτέλεση των αγγειογραμμάτων
ολοκλήρωσης.
11. Αφαιρέστε ή αντικαταστήστε όλους τους άκαμπτους συρμάτινους οδηγούς,
έτσι ώστε να επιτραπεί στις λαγόνιες αρτηρίες να επανέλθουν στη φυσική
τους θέση.
Τελικό αγγειόγραμμα
1. Τοποθετήστε τον αγγειογραφικό καθετήρα ακριβώς επάνω από το
επίπεδο των νεφρικών αρτηριών. Εκτελέστε αγγειογραφία, έτσι ώστε να
επαληθεύσετε τη βατότητα των νεφρικών αρτηριών και την απουσία
ενδοδιαφυγών. Επαληθεύστε τη βατότητα των έσω λαγόνιων αρτηριών.
2. Ε πιβεβαιώστε ότι δεν υπάρχουν ενδοδιαφυγές ή στρεβλώσεις και
επαληθεύστε τη θέση των εγγύς χρυσών ακτινοσκιερών δεικτών. Αφαιρέστε
τα θηκάρια, τα σύρματα και τους καθετήρες.
ΣΗΜΕΙΩΣΗ: Εάν παρατηρηθούν ενδοδιαφυγές ή άλλα προβλήματα, ανατρέξτε
στις προτεινόμενες οδηγίες χρήσης για τα βοηθητικά εξαρτήματα του
ενδαγγειακού μοσχεύματος AAA Zenith.
3. Αποκαταστήσ τε τα αγγεία και συγκλείστε με πρότυπο χειρουργικό τρόπο.
12 ΚΑΤΕΥΘΥΝΤΗΡΙΕΣ ΟΔΗΓΙΕΣ ΑΠΕΙΚΟΝΙΣΗΣ ΚΑΙ ΜΕΤΕΓΧΕΙΡΗΤΙΚΗ
ΠΑΡΑΚΟΛΟΥΘΗΣΗ
12.1 Γενικά
• Η μακροχρόνια απόδοση και ασφάλεια των ενδαγγειακών μοσχευμάτων
δεν έχει ακόμα επιβεβαιωθεί. Όλοι οι ασθενείς πρέπει να ενημερώνονται
ότι η ενδαγγειακή θεραπεία απαιτεί μακροχρόνια, τακτική
παρακολούθηση για την εκτίμηση της υγείας τους και της απόδοσης του
ενδαγγειακού μοσχεύματός τους. Ασθενείς με ειδικά κλινικά ευρήματα (π.χ.
ενδοδιαφυγές, διευρυνόμενα ανευρύσματα ή μεταβολές της δομής ή της θέσης
του ενδαγγειακού μοσχεύματος) πρέπει να τελούν υπό πρόσθετη
παρακολούθηση.
• Πρέπει να παρέχονται συμβουλές στους ασθενείς σχετικά με τη σημασία της
τήρησης του προγράμματος παρακολούθησης, τόσο κατά τη διάρκεια του
πρώτου έτους όσο και σε ετήσια διαστήματα στη συνέχεια. Οι ασθενείς πρέπει
να ενημερώνονται ότι η τακτική και συνεπής παρακολούθηση αποτελεί κρίσιμο
μέρος της διασφάλισης της συνεχούς ασφάλειας και αποτελεσματικότητας της
ενδαγγειακής θεραπείας των AAA.
• Οι ιατροί πρέπει να αξιολογούν τους ασθενείς σε εξατομικευμένη βάση και να
δίνουν οδηγίες για την παρακολούθησή τους σε σχέση με τις ανάγκες και τις
καταστάσεις του εκάστοτε ασθενούς. Το συνιστώμενο πρόγραμμα απεικόνισης
παρουσιάζεται στον πίνακα 12.1. Το πρόγραμμα αυτό συνεχίζει να είναι η
ελάχιστη απαίτηση για την παρακολούθηση των ασθενών και πρέπει να
διατηρείται ακόμα και απουσία κλινικών συμπτωμάτων (π.χ. πόνος, μούδιασμα,
αδυναμία). Ασθενείς με ειδικά κλινικά ευρήματα (π.χ. ενδοδιαφυγοές,
διευρυνόμενα ανευρύσματα ή μεταβολές της δομής ή της θέσης του
μοσχεύματος ενδοπρόσθεσης) πρέπει να τελούν υπό παρακολούθηση σε πιο
συχνά διαστήματα.
• Η ετήσια παρακολούθηση με απεικόνιση πρέπει να περιλαμβάνει κοιλιακές
ακτινογραφίες, καθώς και εξετάσεις αξονικής τομογραφίας τόσο με όσο και
χωρίς σκιαγραφικό μέσο. Σε περίπτωση που αποκλείεται η χρήση σκιαγραφικού
μέσου απεικόνισης λόγω νεφρικών επιπλοκών ή άλλων παραγόντων, μπορούν
να χρησιμοποιηθούν κοιλιακές ακτινογραφίες, αξονική τομογραφία χωρίς
σκιαγραφικό μέσο και υπέρηχος duplex.
• Ο συνδυασμός απεικόνισης αξονικής τομογραφίας με και χωρίς σκιαγραφικό
μέσο παρέχει πληροφορίες σχετικά με τη μεταβολή της διαμέτρου του
ανευρύσματος, την ενδοδιαφυγή, τη βατότητα, την ελίκωση των αγγείων, την
προοδευτική νόσο, το μήκος καθήλωσης και άλλες μορφολογικές μεταβολές.
• Οι κοιλιακές ακτινογραφίες παρέχουν πληροφορίες σχετικά με την ακεραιότητα
της συσκευής (διαχωρισμός μεταξύ εξαρτημάτων, θραύση της ενδοπρόσθεσης
και διαχωρισμός ακίδων).
• Η απεικόνιση με υπέρηχο duplex μπορεί να παρέχει πληροφορίες σχετικά με τη
μεταβολή της διαμέτρου του ανευρύσματος, την ενδοδιαφυγή, τη βατότητα,
την ελίκωση των αγγείων και την προοδευτική νόσο. Στην περίπτωση αυτή,
πρέπει να εκτελείται αξονική τομογραφία χωρίς σκιαγραφικό μέσο, για χρήση
σε συνδυασμό με τον υπέρηχο. Ο υπέρηχος μπορεί να είναι μια λιγότερο
αξιόπιστη και ευαίσθητη διαγνωστική μέθοδος σε σύγκριση με την αξονική
τομογραφία.
Στον πίνακα 12.1 παρατίθεται η ελάχιστη παρακολούθηση με απεικόνιση για
ασθενείς με το ενδαγγειακό μόσχευμα AAA Zenith Flex. Οι ασθενείς που
χρειάζονται αυξημένη παρακολούθηση πρέπει να υποβάλλονται σε ενδιάμεσες
αξιολογήσεις.
Πίνακας 12.1 Συνιστώμενο πρόγραμμα απεικόνισης για
ασθενείς με ενδαγγειακό μόσχευμα
Αγγειόγραμμα
Πριν από τη διαδικασία X
Κατά τη διαδικασία X
Πριν από το εξιτήριο
(εντός 7 ημερών)
1 μήνας X
3 μήνες X
6 μήνες X
12 μήνες (στη συνέχεια
ετησίως)
1
Η απεικόνιση πρέπει να εκτελείται εντός 6 μηνών πριν από τη διαδικασία.
2
Ο υπέρηχος duplex μπορεί να χρησιμοποιηθεί για εκείνους τους ασθενείς που
παρουσιάζουν νεφρική βλάβη ή οι οποίοι κατά τα άλλα δεν είναι σε θέση να υποβληθούν
σε ενισχυμένη με σκιαγραφικό μέσο αξονική τομογραφία. Με τον υπέρηχο, συνιστάται
ακόμα η χρήση αξονικής τομογραφίας χωρίς σκιαγραφικό μέσο.
3
Συνιστάται η εκτέλεση αξονικής τομογραφίας είτε πριν από το εξιτήριο είτε μετά από
1 μήνα.
4
Εάν υπάρχει ενδοδιαφυγή τύπου I ή III, συνιστάται άμεση επέμβαση και επιπλέον
παρακολούθηση μετά την επέμβαση. Δείτε την ενότητα 12.6, Επιπλέον παρακολούθηση
και θεραπεία.
5
Συνιστάται εάν αναφέρεται ενδοδιαφυγή προ του εξιτηρίου ή στον 1 μήνα.
12.2 Συστάσεις αξονικής τομογραφίας με και χωρίς σκιαγραφικό μέσο
• Τα σετ φιλμ πρέπει να περιλαμβάνουν όλες τις διαδοχικές εικόνες στο κατώτατο
δυνατό πάχος τομής (≤3 mm). ΜΗΝ εκτελείτε σετ εικόνων/φιλμ αξονικής
τομογραφίας μεγάλου πάχους τομής (>3 mm) ή/και μην παραλείπετε διαδοχικά
σετ εικόνων/φιλμ αξονικής τομογραφίας, καθώς αυτό αποτρέπει τις ακριβείς
συγκρίσεις ανατομίας και συσκευής σε συνάρτηση με το χρόνο.
• Όλες οι εικόνες πρέπει να περιλαμβάνουν μια κλίμακα για κάθε φιλμ/εικόνα. Εάν
χρησιμοποιείται φιλμ, οι εικόνες πρέπει να είναι σε διάταξη τουλάχιστον 20:1
εικόνες, σε φύλλα 35,5 x 43,2 cm (14” x 17”).
• Απαιτούνται λήψεις τόσο χωρίς όσο και με σκιαγραφικό μέσο, με ταιριαστές ή
αντίστοιχες θέσεις τραπεζιού.
• Το πάχος τομής και το διάστημα των λήψεων προ της χρήσης σκιαγραφικού
μέσου και με χρήση σκιαγραφικού μέσου πρέπει να συμφωνούν.
• ΜΗΝ αλλάξετε τον προσανατολισμό και την ανατομική θέση λήψης των
μετρήσεων του ασθενούς μεταξύ των λήψεων χωρίς και με σκιαγραφικό μέσο.
Η απεικόνιση γραμμής βάσης και παρακολούθησης χωρίς σκιαγραφικό μέσο
και με ενίσχυση με σκιαγραφικό μέσο είναι σημαντική για τη βέλτιστη
παρακολούθηση του ασθενούς. Είναι σημαντικό να ακολουθείτε αποδεκτά
πρωτόκολλα απεικόνισης κατά τη διάρκεια της εξέτασης αξονικής τομογραφίας.
Στον πίνακα 12.2 παρατίθενται παραδείγματα αποδεκτών πρωτοκόλλων
απεικόνισης.
Αξονική
τομογραφία
(με και χωρίς
σκιαγραφικό
1
μέσο)
X
2,3,4
X
2,3,4
2,4,5
X
1
2,4
2,4
Κοιλιακές
ακτινογραφίες
Πίνακας 12.2 Αποδεκτά πρωτόκολλα απεικόνισης
Χωρίς σκιαγραφικό μέσο Με σκιαγραφικό μέσο
ΕΦ σκιαγραφικό μέσο Όχι Ναι
Αποδεκτά μηχανήματα
Όγκος έγχυσης μη διαθ. 150 ml
Ρυθμός έγχυσης μη διαθ. >2,5 ml/sec
Τρόπος έγχυσης μη διαθ. Μηχανική
Χρονισμός bolus μη διαθ.
Κάλυψη - έναρξη Διάφραγμα
Κάλυψη - τέλος Εγγύς μηριαίο οσ τό
Διαφραγματοποίηση <3 mm <3 mm
Ανακατασκευή
Αξονικό διπλό οπτικό
πεδίο
Λήψεις μετά την έγχυση Καμία Καμία
12.3 Κοιλιακές ακτινογραφίες
Απαιτούνται οι ακόλουθες προβολές:
• Τέσσερα φιλμ: ύπτια-μετωπιαία (ΠΟ) προβολή, εγκάρσια προς το τραπέζι πλάγια
προβολή, αριστερή οπίσθια λοξή προβολή 30 μοιρών και δεξιά οπίσθια λοξή
προβολή 30 μοιρών, κεντραρισμένες στον ομφαλό.
• Καταγράψτε την απόσταση από το τραπέζι έως το φιλμ και χρησιμοποιήστε την
ίδια απόσταση σε κάθε επακόλουθη εξέταση.
Βεβαιωθείτε ότι καταγράφεται ολόκληρη η συσκευή σε κάθε μορφή μονής
εικόνας κατά μήκος.
Εάν υπάρχει οποιαδήποτε ανησυχία σχετικά με την ακεραιότητα της
συσκευής (π.χ. στρέβλωση, ρήξεις ενδοπροσθέσεων, διαχωρισμός
ακίδων, σχετική μετανάστευση εξαρτημάτων), συνιστάται η χρήση
μεγεθυσμένων προβολών. Ο θεράπων ιατρός πρέπει να αξιολογεί
τα φιλμ για την ακεραιότητα της συσκευής (ολόκληρο το μήκος της
συσκευής, συμπεριλαμβανομένων των εξαρτημάτων) με χρήση οπτικού
βοηθήματος μεγέθυνσης 2-4X.
12.4 Υπέρηχος
Όταν παράγοντες του ασθενούς αποκλείουν τη χρήση απεικονιστικού
σκιαγραφικού μέσου, μπορεί να εκτελεστεί υπερηχητική απεικόνιση αντί της
αξονικής τομογραφίας με σκιαγραφικό μέσο. Ο υπέρηχος μπορεί να συνδυαστεί
με αξονική τομογραφία χωρίς σκιαγραφικό μέσο. Πρέπει να βιντεοσκοπείται ένα
πλήρες αορτικό duplex για μέγιστη διάμετρο ανευρύσματος, ενδοδιαφυγές,
βατότητα της ενδοπρόσθεσης και στένωση. Στη βιντεοταινία πρέπει να
συμπεριλαμβάνονται οι ακόλουθες πληροφορίες, όπως παρουσιάζεται
παρακάτω:
• Πρέπει να λαμβάνεται εγκάρσια και διαμήκης απεικόνιση από το επίπεδο της
εγγύς αορτής, στην οποία να εμφανίζονται οι μεσεντέριες και νεφρικές αρτηρίες
έως το διχασμό των λαγονίων αρτηριών, έτσι ώστε να προσδιοριστεί εάν
υπάρχουν ενδοδιαφυγές με χρήση αγγειογραφίας έγχρωμης απεικόνισης ροής
και μεθόδου ισχύος (Color Flow και Color Power), εάν είναι προσπελάσιμη.
• Πρέπει να εκτελείται επιβεβαίωση με φασματική ανάλυση για τυχόν
πιθανολογούμενες ενδοδιαφυγές.
• Πρέπει να λαμβάνεται εγκάρσια και διαμήκης απεικόνιση του μέγιστου
ανευρύσματος.
12.5 Ασφάλεια και συμβατότητα με μαγνητική τομογραφία (MRI)
Μη κλινικές δοκιμές έχουν καταδείξει ότι το ενδαγγειακό μόσχευμα AAA Zenith
είναι ασφαλές για μαγνητική τομογραφία υπό προϋποθέσεις. Μπορεί να σαρωθεί
με ασφάλεια υπό τις ακόλουθες προϋποθέσεις:
Συστήματα 1,5 Tesla:
• Στατικό μαγνητικό πεδίο 1,5 Tesla
Ελικοειδής αξονική
τομογραφία με
δυνατότητα
>40 δευτερόλεπτα
2,5 mm πλήρης –
αλγόριθμος λογισμικού
32 cm 32 cm
Ελικοειδής αξονική
τομογραφία με
δυνατότητα
>40 δευτερόλεπτα
Bolus δοκιμασίας: SmartPrep,
C.A.R.E. ή ισοδύναμο
1 cm άνω από τον κοιλιακό
άξονα
Έκφυση εν τω βάθει μηριαίας
αρτηρίας
2,5 mm πλήρης –
αλγόριθμος λογισμικού
89
X
X
X
X

• Πεδίο χωρικής βαθμίδωσης 450 Gauss/cm
• Μέγιστος μεσοτιμημένος ρυθμός ειδικής ολοσωματικής απορρόφησης (SAR)
2,0 W/kg για 15 λεπτά σάρωσης
Σε μη κλινικές δοκιμές, το ενδαγγειακό μόσχευμα AAA Zenith προκάλεσε αύξηση
θερμοκρασίας χαμηλότερη από ή ίση με 1,4 °C σε μέγιστο μεσοτιμημένο ρυθμό
ειδικής ολοσωματικής απορρόφησης (SAR) 2,8 W/kg, όπως υπολογίστηκε βάσει
θερμιδομετρίας για 15 λεπτά σάρωσης μαγνητικής τομογραφίας σε μαγνητικό
τομογράφο 1,5 Tesla, Siemens Medical Magnetom, με λογισμικό Numaris/4,
έκδοσης Syngo MR 2002B DHHS. Ο μέγιστος μεσοτιμημένος ρυθμός ειδικής
ολοσωματικής απορρόφησης (SAR) ήταν 2,8 W/kg, που αντιστοιχεί σε τιμή
μετρούμενη με θερμιδομετρία ίση με 1,5 W/kg.
Συστήματα 3,0 Tesla:
• Στατικό μαγνητικό πεδίο 3,0 Tesla
• Πεδίο χωρικής βαθμίδωσης 720 Gauss/cm
• Μέγιστος μεσοτιμημένος ρυθμός ειδικής ολοσωματικής απορρόφησης (SAR)
2,0 W/kg για 15 λεπτά σάρωσης
Σε μη κλινικές δοκιμές, το ενδαγγειακό μόσχευμα AAA Zenith προκάλεσε αύξηση
θερμοκρασίας χαμηλότερη από ή ίση με 1,9 °C σε μέγιστο μεσοτιμημένο ρυθμό
ειδικής ολοσωματικής απορρόφησης (SAR) 3,0 W/kg, όπως υπολογίστηκε βάσει
θερμιδομετρίας για 15 λεπτά σάρωσης μαγνητικής τομογραφίας σε μαγνητικό
τομογράφο 3,0 Tesla Excite, GE Electric Healthcare, με λογισμικό G3.0-052B. Ο
μέγιστος μεσοτιμημένος ρυθμός ειδικής ολοσωματικής απορρόφησης (SAR) ήταν
3,0 W/kg, που αντιστοιχεί σε τιμή μετρούμενη με θερμιδομετρία ίση με 2,8 W/kg.
Το τέχνημα εικόνας εκτείνεται σε όλη την ανατομική περιοχή που περιέχει τη
συσκευή, ασαφοποιώντας τις παρακείμενες ανατομικές δομές σε ακτίνα περίπου
20 cm περιμετρικά της συσκευής, καθώς επίσης και ολόκληρη τη συσκευή και
τον αυλό της, κατά τη διενέργεια σάρωσης σε μη κλινικές δοκιμές με χρήση της
ακολουθίας: Ταχείας στροφορμικής ηχούς, σε σύστημα μαγνητικής τομογραφίας
(MR) 3,0 Tesla, Excite, GE Electric Healthcare, με λογισμικό G3.0-052B, με πηνίο
ραδιοσυχνοτήτων σώματος.
Για όλους τους τομογράφους, το τέχνημα εικόνας εξασθενεί όσο μεγαλώνει η
απόσταση της συσκευής από την περιοχή ενδιαφέροντος. Μαγνητικές
τομογραφίες της κεφαλής, του αυχένα και των κάτω άκρων είναι δυνατόν να
ληφθούν χωρίς τέχνημα εικόνας. Τέχνημα εικόνας μπορεί να υπάρχει σε
τομογραφίες της κοιλιακής χώρας και των άνω άκρων, ανάλογα με την απόσταση
της συσκευής από την περιοχή ενδιαφέροντος.
Διατίθενται κλινικές πληροφορίες για δεκαεπτά ασθενείς οι οποίοι υποβλήθηκαν
σε μαγνητική τομογραφία μετά από την εμφύτευση του μοσχεύματος
ενδοπρόσθεσης. Δεν έχει αναφερθεί κανένα ανεπιθύμητο συμβάν ή πρόβλημα
με τη συσκευή σε οποιονδήποτε από αυτούς τους ασθενείς λόγω της διενέργειας
μαγνητικής τομογραφίας. Επιπλέον, έχουν εμφυτευθεί πολύ περισσότερα από
50.000 ενδαγγειακά μοσχεύματα AAA Zenith παγκοσμίως, στα οποία δεν έχει
αναφερθεί κανένα ανεπιθύμητο συμβάν ή πρόβλημα με τη συσκευή λόγω
διενέργειας μαγνητικής τομογραφίας.
Η Cook συνιστά την καταχώρηση εκ μέρους του ασθενούς των όρων μαγνητικής
τομογραφίας που δίνονται σε αυτές τις οδηγίες χρήσης στο MedicAlert
Foundation. Τα σ τοιχεία επικοινωνίας με το MedicAlert Foundation είναι τα εξής:
Ταχυδρομική διεύθυνση: MedicAlert Foundation International
Τηλέφωνο: +1 888-633-4298 (γραμμή χωρίς χρέωση)
Φαξ: +1 209-669-2450
Ιστοσελίδα: www.medicalert.org
2323 Colorado Avenue
Turlock, CA 95382 Η.Π.Α.
+1 209-668-3333 για κλήσεις εκτός των Η.Π.Α.
12.6 Επιπλέον παρακολούθηση και θεραπεία
Επιπλέον παρακολούθηση και πιθανή θεραπεία συνιστάται για:
• Ανευρύσματα με ενδοδιαφυγή τύπου Ι
• Ανευρύσματα με ενδοδιαφυγή τύπου ΙIΙ
• Διεύρυνση ανευρύσματος, μέγιστης διαμέτρου ≥5 mm (ανεξάρτητα από την
κατάσταση της ενδοδιαφυγής)
• Μετανάστευση
• Ανεπαρκές μήκος στεγανοποίησης
Η εξέταση για επανεπέμβαση ή μετατροπή σε ανοικτή αποκατάσταση πρέπει να
περιλαμβάνει την εκτίμηση του θεράποντος ιατρού για τις συνυπάρχουσες
νόσους του εκάστοτε ασθενούς, το προσδόκιμο ζωής και τις προσωπικές
επιλογές του ασθενούς. Οι ασθενείς πρέπει να ενημερώνονται για την πιθανότητα
επακόλουθων επανεπεμβάσεων, συμπεριλαμβανομένης της επέμβασης με βάση
καθετήρα και της μετατροπής σε ανοικτή χειρουργική επέμβαση, μετά από
τοποθέτηση ενδαγγειακού μοσχεύματος.
13 ΠΛΗΡΟΦΟΡΙΕΣ ΠΑΡΑΚΟΛΟΥΘΗΣΗΣ ΑΣΘΕΝΩΝ
Επιπλέον αυτών των οδηγιών χρήσης, το ενδαγγειακό μόσχευμα ΑΑΑ Zenith Flex
με το σύστημα εισαγωγής H&L-B One-Shot συσκευάζεται με ένα Έντυπο
παρακολούθησης συσκευής, το οποίο απαιτείται να συμπληρώσει το
νοσοκομειακό προσωπικό και να το προωθήσει στην COOK για τους σκοπούς
παρακολούθησης όλων των ασθενών που λαμβάνουν το ενδαγγειακό μόσχευμα
ΑΑA Zenith Flex (όπως απαιτείται από τον ομοσπονδιακό κανονισμό των Η.Π.Α.).
14 ΑΝΤΙΜΕΤΩΠΙΣΗ ΠΡΟΒΛΗΜΑΤΩΝ
ΣΗΜΕΙΩΣΗ: Μπορείτε να λάβετε τεχνική βοήθεια από έναν ειδικό προϊόντων
της Cook επικοινωνώντας με τον τοπικό σας αντιπρόσωπο της Cook.
14.1 Αντιμετώπιση προβλημάτων με την απελευθέρωση του σύρματος
ενεργοποίησης
ΠΡΟΣΟΧΗ: Θα πρέπει να εκτελείτε τα παρακάτω βήματα μόνον εάν δεν είστε
σε θέση να αφαιρέσετε το εγγύς σύρμα ενεργοποίησης με τον τρόπο που
περιγράφεται στην ενότητα 11.1.7.
14.1.1 Εγγύς (άνω) απελευθέρωση του κύριου σώματος
1. Εάν ο μαύρος (άνω πώμα) μηχανισμός απελευθέρωσης του σύρματος
ενεργοποίησης δεν μπορεί να αφαιρεθεί από τη λαβή, κόψτε το σύρμα
κοντά στο μηχανισμό απελευθέρωσης (Εικ. 37) και αφαιρέστε το μηχανισμό
απελευθέρωσης από τη λαβή.
2. Σταθεροποιήστε τη γκρι διάταξη τοποθέτησης, ενώ αποσύρετε το θηκάρι για
να εκπτύξετε πλήρως το σύστοιχο μέλος.
3. Αφαιρέστε την ασφάλεια από το λευκό μηχανισμό απελευθέρωσης σύρματος
ενεργοποίησης.
4. Αποσύρετε και αφαιρέστε το σύρμα ενεργοποίησης σύροντας το λευκό
μηχανισμό απελευθέρωσης σύρματος ενεργοποίησης εκτός της λαβής και
κατόπιν αφαιρέστε το μέσω της σχισμής του, επάνω από την εσωτερική
κάνουλα της συσκευής.
5. Χρησιμοποιώντας λαβίδα ασφάλισης, συσφίξτε και ασφαλίστε το άκρο κοπής
του σύρματος ενεργοποίησης του άνω πώματος. (Εικ. 38)
6. Χαλαρώστε τη μέγ γενη ακίδας, ενώ διατηρείτε την εσωτερική κάνουλα και
το σύρμα ενεργοποίησης στη θέση τους, και προωθήστε τη γκρι διάταξη
τοποθέτησης και το θηκάρι μέσα στο μόσχευμα μέχρι να βρεθεί η άκρη της
γκρι διάταξης τοποθέτησης σε απόσταση περίπου 2 cm από τους χρυσούς
δείκτες. (Εικ. 39) Η προώθηση της γκρι διάταξης τοποθέτησης προσφέρει
πρόσθετη υποστήριξη στην εσωτερική κάνουλα.
ΣΗΜΕΙΩΣΗ: Διατηρήστε ήπια τάση στο σύρμα ενεργοποίησης για να αφαιρέσετε
τυχόν περίσσεια σύρματος, καθώς προωθούνται η γκρι διάταξη τοποθέτησης και
το θηκάρι.
ΣΗΜΕΙΩΣΗ: Βεβαιωθείτε ότι δεν προωθείται το άκρο της γκρι διάταξης
τοποθέτησης μέσα στο άνω πώμα.
7. Ασφαλίστε τη μέγ γενη ακίδας. Επιβεβαιώστε ότι το σύρμα ενεργοποίησης
έχει ασφαλίσει στη λαβίδα.
8. Σταθεροποιήστε τη γκρι διάταξη τοποθέτησης και προωθήστε αργά το
θηκάρι μέχρι να βρεθεί το άκρο του θηκαριού σε απόσταση 2 mm από τους
χρυσούς δείκτες. (Εικ. 40)
ΣΗΜΕΙΩΣΗ: Φροντίστε να μην προωθήσετε το ίδιο το μόσχευμα κατά τη
διάρκεια της προώθησης του θηκαριού.
9. Σταθεροποιήστε το θηκάρι και αποσύρετε τη γκρι διάταξη τοποθέτησης μαζί
με την εσωτερική κάνουλα για να τραβήξετε το άνω πώμα επάνω από την
επινεφριδιακή ενδοπρόσθεση. (Εικ. 41)
10. Επιβεβαιώστε ότι οι χρυσοί δείκτες έχουν τοποθετηθεί χαμηλότερα από τις
νεφρικές αρτηρίες.
11. Αφαιρέστε το σύρμα ενεργοποίησης.
12. Αποσύρετε το θηκάρι μέχρι να αποκαλυφθεί το κωνικό άκρο της γκρι
διάταξης τοποθέτησης.
13. Προωθήσ τε ένα μπαλόνι διαμόρφωσης διαμέσου του ετερόπλευρου μέλους
του κυρίου σώματος και τοποθετήστε το λίγο υψηλότερα από το διχασμό του
μοσχεύματος.
14. Φουσκώ στε το μπαλόνι έως την πλήρη διάμετρο του μοσχεύματος. (Εικ. 42)
15. Ξ εσφίξτε τη μέγγενη ακίδας.
16. Σταθεροποιήστε τη γκρι διάταξη τοποθέτησης και τον καθετήρα με μπαλόνι
και προωθήστε την εσωτερική κάνουλα για να εκπτύξετε το άνω πώμα.
17. Σφίξτε τη μέγγενη ακίδας.
18. Ξ εφοσυκώστε το μπαλόνι και προωθήστε τον ετερόπλευρο συρμάτινο οδηγό
στη θωρακική αορτή.
ΣΗΜΕΙΩΣΗ: Λόγω της πρόωρης έκπτυξης του σύστοιχου σκέλους και της
απελευθέρωσης του σύρματος ενεροποίησης, προτείνεται να αφήσετε το
μπαλόνι διαμόρφωσης μέσα ή λίγο επάνω από το ετερόπλευρο μέλος για τη
σταθεροποίηση του μοσχεύματος κατά τη διάρκεια της τοποθέτησης του
σύστοιχου μέλους.
14.1.2 Σύνδεση του άνω πώματος
1. Ξεσφί ξτε τη μέγγενη ακίδας. (Εικ. 25)
2. Στερεώστε το θηκάρι και την εσωτερική κάνουλα, έτσι ώστε να αποφύγετε
τυχόν κίνηση των εξαρτημάτων αυτών.
3. Προωθήσ τε τη γκρι διάταξη τοποθέτησης πάνω από την εσωτερική κάνουλα
έως ότου συνδεθεί με το άνω πώμα. (Εικ. 43, 27, 44)
ΣΗΜΕΙΩΣΗ: Εάν παρουσιαστεί αντίσταση, περιστρέψτε ελαφρά τη γκρι διάταξη
τοποθέτησης και συνεχίστε να προωθείτε απαλά.
4. Σφίξτε πάλι τη μέγγενη ακίδας και αποσύρετε ολόκληρο το άνω πώμα και
τη γκρι διάταξη τοποθέτησης μέσω του μοσχεύματος και του θηκαριού,
έλκοντας την εσωτερική κάνουλα. (Εικ. 45) Αφήστε το θηκάρι και τον
συρμάτινο οδηγό στη θέση τους.
5. Κλείστε την αιμοστατική βαλβίδα Captor® στο θηκάρι εισαγωγέα Flexor®
στρέφοντας τη δεξιόστροφα, έως ότου σταματήσει. (Εικ. 30)
14.1.3 Τοποθέτηση και απελευθέρωση του σύστοιχου λαγόνιου σκέλους
ΣΗΜΕΙΩΣΗ: Βεβαιωθείτε ότι η αιμοστατική βαλβίδα Captor στο θηκάρι
εισαγωγέα είναι στραμμένη στην ανοικτή θέση. (Εικ. 31)
1. Χρησιμοποιήστε τη διάταξη σύρματος του μοσχεύματος κύριου σώματος και
θηκαριού για την εισαγωγή του μοσχεύματος σύστοιχου σκέλους.
ΣΗΜΕΙΩΣΗ: Λόγω της τροποποίησης της απελευθέρωσης του άνω πώματος, η
διάταξη θηκαριού κυρίου σώματος πρέπει να αποσύρεται έως ένα σημείο που
βρίσκεται σε απόσταση 1-2 cm στο εσωτερικό του εγγύς σύστοιχου μέλους.
Προωθήστε τη διάταξη θηκαριού διαστολέα του σύστοιχου μέλους μέσα στο
θηκάρι του κυρίου σώματος.
ΣΗΜΕΙΩΣΗ: Μπορείτε να φουσκώσετε το μπαλόνι διαμόρφωσης στο
ετερόπλευρο μέλος του μοσχεύματος κυρίου σώματος εάν απαιτείται πρόσθετη
σταθεροποίηση του μοσχέυματος.
ΠΡΟΣΟΧΗ: Μη φουσκώνετε το μπαλόνι εκτός του μοσχεύματος.
ΣΗΜΕΙΩΣΗ: Σε ελικοειδή αγγεία, η θέση των έσω λαγόνιων αρτηριών ενδέχεται
να μεταβληθεί σημαντικά με την εισαγωγή των άκαμπτων συρμάτων και των
συστημάτων θηκαριού.
2. Προωθήσ τε αργά έως ότου το μόσχευμα του σύστοιχου λαγόνιου σκέλους
επικαλύψει τουλάχιστον μία πλήρη ενδοπρόσθεση λαγόνιου σκέλους
(δηλ. εγγύς ενδοπρόσθεση του μοσχεύματος λαγόνιου σκέλους) εντός του
σύστοιχου μέλους του κύριου σώματος. (Εικ. 46)
ΣΗΜΕΙΩΣΗ: Εάν απαιτείται αλληλεπικάλυψη μεγαλύτερη από τρεις
ενδοπροσθέσεις λαγόνιου σκέλους (μεγαλύτερη από δύο ενδοπροσθέσεις
λαγόνιου σκέλους για σκέλη με μήκος 37 και 54 mm), ενδέχεται να είναι
απαραίτητο να εξετάσετε το ενδεχόμενο χρήσης μιας προέκτασης σκέλους στην
περιοχή διχασμού της αντίθετης πλευράς.
3. Επιβεβαιώσ τε τη θέση του περιφερικού άκρου του μοσχεύματος λαγόνιου
σκέλους. Επανατοποθετήστε το μόσχευμα λαγόνιου σκέλους εάν είναι
απαραίτητο, έτσι ώστε να διασφαλίσετε τη βατότητα της έσω λαγόνιας
αρτηρίας.
4. Για να εκπτύξετε, σταθεροποιήστε το μόσχευμα του λαγόνιου σκέλους με τη
γκρι διάταξη τοποθέτησης, ενώ αποσύρετε ταυτόχρονα το θηκάρι λαγόνιου
σκέλους και το θηκάρι κύριου σώματος. (Εικ. 33 και 47) Εάν είναι απαραίτητο,
αποσύρετε το θηκάρι κύριου σώματος.
5. Υπό ακτινοσκόπηση και μετά από επαλήθευση της θέσης του μοσχεύματος
λαγόνιου σκέλους, ξεσφίξτε τη μέγγενη ακίδας και αποσύρετε την
εσωτερική κάνουλα για τη σύνδεση του κωνικού διαστολέα στη γκρι διάταξη
τοποθέτησης. Σφίξτε τη μέγγενη ακίδας. Διατηρήστε τη θέση του θηκαριού
ενώ αποσύρετε τη γκρι διάταξη τοποθέτησης με στερεωμένη την εσωτερική
κάνουλα. (Εικ. 48)
6. Κλείστε την αιμοστατική βαλβίδα Captor στο θηκάρι εισαγωγέα Flexor
στρέφοντας τη δεξιόστροφα, έως ότου σταματήσει.
7. Ελέγξτε πάλι τη θέση των συρμάτινων οδηγών. Αφήστε το θηκάρι και τους
συρμάτινους οδηγούς στη θέση τους.
8. Αφαιρέστε το ξεφουσκωμένο μπαλόνι από την ετερόπλευρη μεριά.
14.1.4 Τοποθέτηση και απελευθέρωση του ετερόπλευρου λαγόνιου
σκέλους
ΠΡΟΣΟΧΗ: Επαληθεύστε ότι επιλέγεται το προκαθορισμένο ετερόπλευρο
λαγόνιο σκέλος για εισαγωγή στην ετερόπλευρη πλευρά του ασθενούς πριν
από την εμφύτευση.
1. Τοποθετήστε τον ενισχυτή εικόνας για την εμφάνιση τόσο της ετερόπλευρης
έσω λαγόνιας αρτηρίας όσο και της ετερόπλευρης κοινής λαγόνιας αρτηρίας.
2. Πριν από την εισαγωγή του συστήματος τοποθέτησης ετερόπλευρου
λαγονίου σκέλους, εγχύστε σκιαγραφικό μέσο μέσω του ετερόπλευρου
μηριαίου θηκαριού για τον εντοπισμό της ετερόπλευρης έσω λαγόνιας
αρτηρίας.
3. Εισαγάγετε το σύστημα τοποθέτησης ετερόπλευρου λαγόνιου σκέλους εντός
της αρτηρίας. Προωθήστε αργά έως ότου το μόσχευμα λαγόνιου σκέλους
επικαλύψει τουλάχιστον μία πλήρη ενδοπρόσθεση λαγόνιου σκέλους
(δηλ. εγγύς ενδοπρόσθεση του μοσχεύματος λαγόνιου σκέλους) εντός του
ετερόπλευρου μέλους του κύριου σώματος. (Εικ. 49)
ΣΗΜΕΙΩΣΗ: Εάν παρουσιαστεί δυσκολία κατά την προώθηση του συστήματος
τοποθέτησης λαγόνιου σκέλους, εναλλάξτε το με έναν πιο υποστηρικτικό
συρμάτινο οδηγό. Σε ελικοειδή αγγεία, η ανατομία ενδέχεται να μεταβληθεί
σημαντικά με την εισαγωγή των άκαμπτων συρμάτων και των συστημάτων
θηκαριού.
90

4. Επιβεβαιώσ τε τη θέση του περιφερικού άκρου του μοσχεύματος λαγόνιου
σκέλους. Επανατοποθετήστε το μόσχευμα λαγόνιου σκέλους εάν είναι
απαραίτητο, έτσι ώστε να διασφαλιστεί τόσο η βατότητα της έσω λαγόνιας
αρτηρίας όσο και ελάχιστη επικάλυψη μιας πλήρους ενδοπρόσθεσης
λαγόνιου σκέλους (δηλ. εγγύς ενδοπρόσθεση του μοσχεύματος λαγόνιου
σκέλους, μέγιστη επικάλυψη 1,5 ενδοπροσθέσεων) εντός του ενδαγγειακού
μοσχεύματος του κύριου σώματος.
5. Για να εκπτύξετε, κρατήστε το μόσχευμα του λαγόνιου σκέλους στη θέση του
με τη γκρι διάταξη τοποθέτησης, ενώ αποσύρετε το θηκάρι. (Εικ. 19 και 50)
Βεβαιωθείτε ότι διατηρείται επικάλυψη ίση με το μήκος μίας ενδοπρόσθεσης.
6. Σταματήστε την απόσυρση του θηκαριού αμέσως μόλις απελευθερωθεί το
περιφερικό άκρο του μοσχεύματος λαγόνιου σκέλους.
7. Υπό ακτινοσκόπηση και μετά από επιβεβαίωση της θέσης του μοσχεύματος
λαγονίου σκέλους, ξεσφίξτε τη μέγγενη ακίδας και αποσύρετε την εσωτερική
κάνουλα για να συνδέσετε τον κωνικό διαστολέα στη γκρι διάταξη
τοποθέτησης. Σφίξτε τη μέγγενη ακίδας. Διατηρήστε τη θέση του θηκαριού
ενώ αποσύρετε τη γκρι διάταξη τοποθέτησης με στερεωμένη την εσωτερική
κάνουλα. (Εικ. 51)
8. Ελέγξτε πάλι τη θέση των συρμάτινων οδηγών.
9. Διενεργήστε την εισαγωγή του μπαλονιού διαμόρφωσης και μια τελική
αγγειογραφία, όπως περιγράφεται στην ενότητα 11.1.12 Εισαγωγή
μπαλονιού διαμόρφωσης.
14.2 Αντιμετώπιση προβλημάτων απελευθέρωσης της επινεφρικής
ενδοπρόσθεσης
ΠΡΟΣΟΧΗ: Θα πρέπει να εκτελείτε τα παρακάτω βήματα μόνον εάν
δεν είστε σε θέση να απελευθερώσετε το άνω πώμα με τον τρόπο που
περιγράφεται στην ενότητα 11.1.7.
14.2.1 Εγγύς (άνω) απελευθέρωση του κύριου σώματος
1. Εάν δεν μπορεί να απελευθερωθεί πλήρως η επινεφρική ενδοπρόσθεση
με την προώθηση της εσωτερικής κάνουλας του άνω πώματος,
προωθήστε ένα μπαλόνι διαμόρφωσης διαμέσου του ετερόπλευρου
μέλους του κύριου σώματος και τοποθετήστε το λίγο ψηλότερα από τον
διχασμό του μοσχεύματος ενδοπρόσθεσης.
2. Για να υποστηρίξετε περαιτέρω την εσωτερική κάνουλα, πληρώστε το
μπαλόνι έως τη μέγιστη διάμετρο του μοσχεύματος.
3. Ξεσφίξτε τη μέγγενη ακίδας. (Εικ. 15)
4. Σταθεροποιήστε τη γκρι διάταξη τοποθέτησης και τον καθετήρα με
μπαλόνι και προωθήστε την εσωτερική κάνουλα για να απελευθερώσετε
την επινεφρική ενδοπρόσθεση.
Εάν η επινεφρική ενδοπρόσθεση έχει απελευθερωθεί πλήρως:
5. Σφίξτε τη μέγγενη ακίδας. Ξεφουσκώσ τε και αποσύρετε το μπαλόνι.
6. Προωθήστε τον ετερόπλευρο συρμάτινο οδηγό στη θωρακική αορτή
και επιστρέψτε στο κατάλληλο βήμα των οδηγιών χρήσης για να
ολοκληρώσετε τη διαδικασία.
Εάν εξακολουθείτε να μην μπορείτε να απελευθερώσετε πλήρως την
επινεφρική ενδοπρόσθεση:
5. Σφίξτε τη μέγγενη ακίδας και ξεφουσκώστε το μπαλόνι. Διατηρώντας το
μπαλόνι στη θέση του, σταθεροποιήστε τη γκρι διάταξη τοποθέτησης και
αποσύρετε το θηκάρι για να απελευθερώσετε πλήρως το σύστοιχο μέλος.
6. Αφαιρέστε την ασφάλεια από τον μηχανισμό απελευθέρωσης σύρματος
ενεργοποίησης του σύστοιχου μέλους.
7. Αποσύρετε και αφαιρέστε το σύρμα ενεργοποίησης σύροντας τον
μηχανισμό απελευθέρωσης σύρματος ενεργοποίησης σύστοιχου μέλους
εκτός της λαβής και αφαιρέστε το μέσω της σχισμής του επάνω από την
εσωτερική κάνουλα της συσκευής.
8. Χαλαρώστε την μέγγενη ακίδας (Εικ. 25) και, ενόσω διατηρείτε
την εσωτερική κάνουλα στη θέση της, προωθήστε τη γκρι διάταξη
τοποθέτησης και το θηκάρι μέσα στο μόσχευμα μέχρι να βρεθεί το
άκρο της γκρι διάταξης τοποθέτησης σε απόσταση περίπου 2 cm από
τους εγγύς χρυσούς δείκτες. (Εικ. 52) Η προώθηση της γκρι διάταξης
τοποθέτησης προσφέρει πρόσθετη υποστήριξη στην εσωτερική κάνουλα.
ΣΗΜΕΙΩΣΗ: Φροντίστε να μην προωθήσετε το μόσχευμα κατά τη
διάρκεια της προώθησης της γκρι διάταξης τοποθέτησης και του
θηκαριού.
ΣΗΜΕΙΩΣΗ: Βεβαιωθείτε ότι δεν προωθείται το άκρο της γκρι διάταξης
τοποθέτησης μέσα στο άνω πώμα.
9. Ασφαλίστε τη μέγγενη ακίδας.
10. Επιβεβαιώστε ότι οι χρυσοί δείκτες έχουν τοποθετηθεί χαμηλότερα από
τις νεφρικές αρτηρίες.
11. Επανατοποθετήστε το μπαλόνι διαμόρφωσης ώστε να εφαρμόσει επί του
διχασμού.
12. Πληρώστε το μπαλόνι έως την πλήρη διάμετρο του μοσχεύματος.
(Εικ. 42)
13. Ξεσφίξτε τη μέγγενη ακίδας.
14. Σταθεροποιήστε τη γκρι διάταξη τοποθέτησης και τον καθετήρα με
μπαλόνι και προωθήστε την εσωτερική κάνουλα για να απελευθερώσετε
την επινεφρική ενδοπρόσθεση.
15. Σφίξτε τη μέγγενη ακίδας.
16. Ξεφουσκώστε το μπαλόνι και προωθήστε τον ετερόπλευρο συρμάτινο
οδηγό στη θωρακική αορτή.
ΣΗΜΕΙΩΣΗ: Λόγω της πρόωρης απελευθέρωσης του σύστοιχου σκέλους
και της απελευθέρωσης του σύρματος ενεργοποίησης, προτείνεται
να αφήσετε το μπαλόνι διαμόρφωσης μέσα ή λίγο επάνω από το
ετερόπλευρο μέλος που χρησιμοποιείτε για τη σταθεροποίηση του
μοσχεύματος κατά τη διάρκεια της τοποθέτησης του σύστοιχου μέλους.
14.2.2 Σύνδεση του άνω πώματος
1. Ξεσφίξτε τη μέγγενη ακίδας. (Εικ. 25)
2. Στερεώστε το θηκάρι και την εσωτερική κάνουλα, έτσι ώστε να αποφύγετε
τυχόν κίνηση των εξαρτημάτων αυτών.
3. Προωθήστε τη γκρι διάταξη τοποθέτησης πάνω από την εσωτερική
κάνουλα έως ότου συνδεθεί με το άνω πώμα. (Εικ. 43, 27 και 44)
ΣΗΜΕΙΩΣΗ: Εάν παρουσιαστεί αντίσταση, περιστρέψτε ελαφρά τη γκρι
διάταξη τοποθέτησης και συνεχίστε να προωθείτε απαλά.
4. Σφίξτε πάλι τη μέγγενη ακίδας και αποσύρετε ολόκληρο το άνω πώμα και
τη γκρι διάταξη τοποθέτησης μέσω του μοσχεύματος και του θηκαριού,
έλκοντας την εσωτερική κάνουλα. (Εικ. 45) Αφήστε το θηκάρι και τον
συρμάτινο οδηγό στη θέση τους.
5. Κλείστε την αιμοστατική βαλβίδα Captor® στο θηκάρι εισαγωγέα Flexor®,
στρέφοντάς τη δεξιόστροφα, έως ότου σταματήσει. (Εικ. 30)
14.2.3 Τοποθέτηση και απελευθέρωση του σύστοιχου λαγόνιου σκέλους
ΣΗΜΕΙΩΣΗ: Βεβαιωθείτε ότι η αιμοστατική βαλβίδα Captor στο θηκάρι
εισαγωγέα είναι στραμμένη στην ανοικτή θέση. (Εικ. 31)
1. Τοποθετήστε τον ενισχυτή εικόνας έτσι ώστε να δείχνει και την σύστοιχη
έσω λαγόνια αρτηρία και τη σύστοιχη κοινή λαγόνια αρτηρία.
2. Μετά από την εισαγωγή του συστήματος τοποθέτησης του σύστοιχου
λαγόνιου σκέλους, χορηγήστε σκιαγραφικό μέσο δια μέσου του θηκαριού
του κυρίου σώματος για να εντοπίσετε τη σύστοιχη έσω λαγόνια αρτηρία.
3. Χρησιμοποιήστε τη διάταξη σύρματος του μοσχεύματος κύριου σώματος
και θηκαριού για την εισαγωγή του μοσχεύματος σύστοιχου σκέλους.
ΣΗΜΕΙΩΣΗ: Λόγω της τροποποίησης της απελευθέρωσης του άνω
πώματος, η διάταξη θηκαριού κυρίου σώματος πρέπει να αποσύρεται
έως ένα σημείο που βρίσκεται σε απόσταση 1-2 cm στο εσωτερικό του
εγγύς σύστοιχου μέλους. Προωθήστε τη διάταξη θηκαριού διαστολέα του
σύστοιχου μέλους μέσα στο θηκάρι του κυρίου σώματος.
ΣΗΜΕΙΩΣΗ: Μπορείτε να πληρώσετε το μπαλόνι διαμόρφωσης στο
ετερόπλευρο μέλος του μοσχεύματος κυρίου σώματος εάν απαιτείται
πρόσθετη σταθεροποίηση του μοσχεύματος.
ΠΡΟΣΟΧΗ: Μην πληρώνετε το μπαλόνι εκτός του μοσχεύματος.
ΣΗΜΕΙΩΣΗ: Σε ελικοειδή αγγεία, η θέση των έσω λαγόνιων αρτηριών
ενδέχεται να μεταβληθεί σημαντικά με την εισαγωγή των άκαμπτων
συρμάτων και των συστημάτων θηκαριού.
4. Προωθήστε αργά το σύστημα τοποθέτησης σύστοιχου λαγονίου
σκέλους έως ότου το μόσχευμα σύστοιχου λαγόνιου σκέλους επικαλύψει
τουλάχιστον μία πλήρη ενδοπρόσθεση λαγόνιου σκέλους (δηλ. εγγύς
ενδοπρόσθεση του μοσχεύματος λαγόνιου σκέλους) εντός του σύστοιχου
μέλους του κύριου σώματος. (Εικ. 46)
ΣΗΜΕΙΩΣΗ: Εάν απαιτείται αλληλεπικάλυψη μεγαλύτερη από τρεις
ενδοπροσθέσεις λαγόνιου σκέλους (μεγαλύτερη από δύο ενδοπροσθέσεις
λαγόνιου σκέλους για σκέλη με μήκος 37 και 54 mm), ενδέχεται να
είναι απαραίτητο να εξετάσετε το ενδεχόμενο χρήσης μιας προέκτασης
σκέλους στην περιοχή διχασμού της αντίθετης πλευράς.
5. Επιβεβαιώστε τη θέση του περιφερικού άκρου του μοσχεύματος λαγόνιου
σκέλους. Επανατοποθετήστε το μόσχευμα λαγόνιου σκέλους εάν είναι
απαραίτητο, έτσι ώστε να διασφαλίσετε τη βατότητα της έσω λαγόνιας
αρτηρίας.
6. Για να απελευθερώσετε, σταθεροποιήστε το μόσχευμα του λαγόνιου
σκέλους με τη γκρι διάταξη τοποθέτησης, ενόσω αποσύρετε ταυτόχρονα
το θηκάρι λαγόνιου σκέλους και το θηκάρι κύριου σώματος.
(Εικ. 33 και 47)
7. Υπό ακτινοσκόπηση και μετά από επιβεβαίωση της θέσης του
μοσχεύματος λαγονίου σκέλους, ξεσφίξτε τη μέγγενη ακίδας και
αποσύρετε την εσωτερική κάνουλα για να συνδέσετε τον κωνικό
διαστολέα στη γκρι διάταξη τοποθέτησης. Σφίξτε τη μέγγενη ακίδας.
Διατηρήστε τη θέση του θηκαριού ενώ αποσύρετε τη γκρι διάταξη
τοποθέτησης με στερεωμένη την εσωτερική κάνουλα. (Εικ. 48)
8. Κλείστε την αιμοστατική βαλβίδα Captor στο θηκάρι εισαγωγέα Flexor
στρέφοντας τη δεξιόστροφα, έως ότου σταματήσει.
9. Ελέγξτε πάλι τη θέση των συρμάτινων οδηγών. Αφήστε το θηκάρι και τους
συρμάτινους οδηγούς στη θέση τους.
10. Αφαιρέστε το ξεφουσκωμένο μπαλόνι από την ετερόπλευρη μεριά.
14.2.4 Τοποθέτηση και απελευθέρωση ετερόπλευρου λαγόνιου σκέλους
ΠΡΟΣΟΧΗ: Επαληθεύστε ότι επιλέγεται το προκαθορισμένο ετερόπλευρο
λαγόνιο σκέλος για εισαγωγή στην ετερόπλευρη πλευρά του ασθενούς
πριν από την εμφύτευση.
1. Τοποθετήστε τον ενισχυτή εικόνας έτσι ώστε να δείχνει και την
ετερόπλευρη έσω λαγόνια αρτηρία και την ετερόπλευρη κοινή λαγόνια
αρτηρία.
2. Πριν από την εισαγωγή του συστήματος τοποθέτησης ετερόπλευρου
λαγονίου σκέλους, εγχύστε σκιαγραφικό μέσο μέσω του ετερόπλευρου
μηριαίου θηκαριού για τον εντοπισμό της ετερόπλευρης έσω λαγόνιας
αρτηρίας.
3. Εισάγετε το σύστημα τοποθέτησης του ετερόπλευρου λαγόνιου σκέλους
μέσα στην αρτηρία. Προωθήστε αργά έως ότου το μόσχευμα λαγόνιου
σκέλους επικαλύψει τουλάχιστον μία πλήρη ενδοπρόσθεση λαγόνιου
σκέλους (δηλ. εγγύς ενδοπρόσθεση του μοσχεύματος λαγόνιου σκέλους)
εντός του ετερόπλευρου μέλους του κύριου σώματος. (Εικ. 49)
ΣΗΜΕΙΩΣΗ: Εάν παρουσιαστεί δυσκολία κατά την προώθηση του
συστήματος τοποθέτησης λαγόνιου σκέλους, εναλλάξτε το με έναν
πιο υποστηρικτικό συρμάτινο οδηγό. Στα ελικοειδή αγγεία, η ανατομία
ενδέχεται να αλλοιωθεί σημαντικά με την εισαγωγή άκαμπτων συρμάτων
και θηκαριών.
4. Επιβεβαιώστε τη θέση του περιφερικού άκρου του μοσχεύματος
λαγόνιου σκέλους. Επανατοποθετήστε το μόσχευμα λαγόνιου σκέλους,
εάν είναι απαραίτητο, έτσι ώστε να διασφαλιστεί τόσο η βατότητα της
έσω λαγόνιας αρτηρίας όσο και ελάχιστη επικάλυψη μιας πλήρους
ενδοπρόσθεσης λαγόνιου σκέλους (δηλ. εγγύς ενδοπρόσθεση
του μοσχεύματος λαγόνιου σκέλους, μέγιστη επικάλυψη 1,5
ενδοπροσθέσεων) εντός του ενδαγγειακού μοσχεύματος του κύριου
σώματος.
5. Για να απελευθερώσετε, κρατήστε το μόσχευμα του λαγόνιου σκέλους
στη θέση του με τη γκρι διάταξη τοποθέτησης, ενόσω αποσύρετε το
θηκάρι. (Εικ. 19 και 50) Φροντίστε να διατηρείται επικάλυψη ίση με το
μήκος μίας ενδοπρόσθεσης.
6. Σταματήστε την απόσυρση του θηκαριού αμέσως μόλις απελευθερωθεί
το περιφερικό άκρο του μοσχεύματος λαγόνιου σκέλους.
7. Υπό ακτινοσκόπηση και μετά από επιβεβαίωση της θέσης του
μοσχεύματος λαγονίου σκέλους, ξεσφίξτε τη μέγγενη ακίδας και
αποσύρετε την εσωτερική κάνουλα για να συνδέσετε τον κωνικό
διαστολέα στη γκρι διάταξη τοποθέτησης. Σφίξτε τη μέγγενη ακίδας.
Διατηρήστε τη θέση του θηκαριού ενώ αποσύρετε τη γκρι διάταξη
τοποθέτησης με στερεωμένη την εσωτερική κάνουλα. (Εικ. 51)
8. Ελέγξτε πάλι τη θέση των συρμάτινων οδηγών.
9. Διενεργήστε την εισαγωγή του μπαλονιού διαμόρφωσης και τελική
αγγειογραφία, όπως περιγράφεται στην ενότητα Εισαγωγή μπαλονιού
διαμόρφωσης των οδηγιών χρήσης του προϊόντος.
91

ESPAÑOL
ENDOPRÓTESIS VASCULAR PARA AAA ZENITH FLEX® CON EL
SISTEMA DE INTRODUCCIÓN H&L-B ONE-SHOT™
Lea atentamente todas las instrucciones. Si no se siguen correctamente las
instrucciones, las advertencias y las precauciones, el paciente puede sufrir
consecuencias quirúrgicas o lesiones graves.
AVISO: Las leyes federales estadounidenses restringen la venta de este
dispositivo a médicos o por prescripción facultativa.
AVISO: Todo el contenido de la bolsa exterior (incluidos el sistema de
introducción y las endoprótesis vasculares) se suministra estéril y es para
un solo uso.
Hay cuatro documentos de instrucciones de uso recomendadas relacionados
con la línea de productos Zenith. Este documento describe las instrucciones de
uso recomendadas de la endoprótesis vascular para AAA Zenith Flex (cuerpo
principal y ramas ilíacas). Para obtener información sobre otros componentes
Zenith, consulte las instrucciones de uso recomendadas siguientes:
• Endoprótesis vascular para AAA Zenith (cuerpo principal y ramas ilíacas de la
endoprótesis vascular para AAA Zenith);
• Componentes auxiliares para la endoprótesis vascular para AAA Zenith
(extensión de cuerpo principal, extensión de rama ilíaca, convertidor y tapón
ilíaco);
• Endoprótesis vascular auxiliar para AAA Zenith® Renu™ (extensión de cuerpo
principal y configuraciones del convertidor);
• Catéter balón CODA®.
1 DESCRIPCIÓN DEL DISPOSITIVO
1.1 Cuerpo principal aórtico y ramas ilíacas
La endoprótesis vascular para AAA Zenith Flex es un sistema modular compuesto
de tres componentes, un cuerpo principal aórtico bifurcado y dos ramas ilíacas.
(Fig. 1) Los módulos de la endoprótesis vascular están fabricados de tela de
poliéster tejido de espesor total cosida a stents Cook-Z® autoexpandibles
de acero inoxidable con hilo de sutura de poliéster trenzado y polipropileno
monofilamento. Estos módulos tienen stents a todo lo largo para ofrecer
estabilidad y la fuerza de expansión necesaria para abrir la luz de la endoprótesis
vascular durante el despliegue. Los stents Cook-Z también proporcionan el
necesario acoplamiento y sellado de la endoprótesis vascular a la pared vascular.
El stent suprarrenal descubierto del extremo proximal de la endoprótesis
vascular tiene púas colocadas a 3 mm una de otra para reforzar la fijación
del dispositivo. Para facilitar la visualización fluoroscópica de la endoprótesis
vascular, hay marcadores radiopacos de oro colocados de la forma siguiente:
uno en la superficie lateral del stent más distal de la ramificación contralateral
de la parte bifurcada del cuerpo principal y cuatro orientados en circunferencia
a menos de 2 mm de la superficie más superior del material de la endoprótesis
vascular.
1.2 Sistema de implantación del cuerpo principal
El cuerpo principal de la endoprótesis vascular para AAA Zenith Flex se
suministra precargado sobre el sistema de introducción H&L-B One-Shot. (Fig. 2)
Tiene un método de despliegue secuencial con características integradas
que permiten el control continuo de la endoprótesis vascular durante todo el
procedimiento de despliegue. El sistema de introducción H&L-B One-Shot está
diseñado para su colocación precisa y permite el reajuste de la posición final de
la endoprótesis vascular antes del despliegue del stent suprarrenal con púas.
El sistema de implantación del cuerpo principal de la endoprótesis vascular
utiliza un sistema de introducción H&L-B One-Shot de 18, 20 ó 22 Fr. Unos
mecanismos de liberación de alambre disparador doble inmovilizan la
endoprótesis vascular sobre el sistema de implantación hasta que es liberado por
el médico. Todos los sistemas son compatibles con una guía de 0,035 pulgadas
(0,89 mm).
Para potenciar la hemostasia, la válvula hemostática Captor® puede aflojarse o
apretarse para introducir los dispositivos auxiliares en la vaina y para extraerlos
de ésta. Los sistemas de implantación del cuerpo principal incluyen una vaina
introductora Flexor® resistente a la plicatura y revestida con una sustancia
hidrofílica. Ambas características están concebidas para facilitar el control del
desplazamiento en las arterias ilíacas y en la aorta abdominal.
1.3 Sistema de implantación de la rama ilíaca
Las ramas ilíacas de la endoprótesis vascular para AAA Zenith se suministran
precargadas sobre el sistema de introducción H&L-B One-Shot. (Fig. 3) El sistema
de implantación está diseñado para que sea fácil de utilizar con una preparación
mínima. El sistema de implantación de las ramas ilíacas utiliza un sistema de
introducción H&L-B One-Shot de 14 ó 16 Fr. Todos los sistemas son compatibles
con una guía de 0,035 pulgadas (0,89 mm).
1.4 Componentes auxiliares de la endoprótesis vascular para AAA Zenith
También se comercializan componentes auxiliares endovasculares adicionales
(extensiones de cuerpo principal, extensiones de rama ilíaca, convertidores y
tapones ilíacos). (Fig. 4) Para obtener más información, consulte las instrucciones
de uso de los componentes auxiliares de la endoprótesis vascular para AAA
Zenith.
2 INDICACIONES
La endoprótesis vascular para AAA Zenith Flex con el sistema de introducción
H&L-B One-Shot está indicada para el tratamiento endovascular de pacientes
con aneurismas aórticos abdominales o aortoilíacos cuyas morfologías permitan
la reparación endovascular, lo que incluye:
• Acceso ilíaco/femoral adecuado compatible con los sistemas de introducción
requeridos,
• Segmento (cuello) aórtico infrarrenal no aneurismático proximal al aneurisma:
• con una longitud de al menos 15 mm,
• con un diámetro medido de pared exterior a pared exterior no superior a
32 mm y no inferior a 18 mm,
• con un ángulo respecto al eje largo del aneurisma de menos de 60 grados, y
• con un ángulo respecto al eje de la aorta suprarrenal de menos de
45 grados.
• Lugar de fijación distal en la arteria ilíaca de más de 10 mm de longitud y de
7,5-20 mm de diámetro (medido de pared exterior a pared exterior).
3 CONTRAINDICACIONES
Estos dispositivos no tienen ninguna contraindicación conocida.
4 ADVERTENCIAS Y PRECAUCIONES
4.1 Generales
• Lea atentamente todas las instrucciones. Si no se siguen correctamente las
instrucciones, las advertencias y las precauciones, el paciente puede sufrir
consecuencias o lesiones graves.
• Durante los procedimientos de implantación o reintervención deberá haber un
equipo quirúrgico cualificado disponible en todo momento para el caso de
que sea necesaria una conversión a reparación quirúrgica abierta.
• La endoprótesis vascular para AAA Zenith Flex con el sistema de introducción
H&L-B One-Shot sólo deben utilizarlo médicos y equipos que hayan recibido
formación en técnicas intervencionistas vasculares (basadas en catéter y
quirúrgicas) y en el uso de este dispositivo. La formación específica necesaria
se describe en el apartado 10.1, Formación de médicos.
• En los pacientes que presenten agrandamiento del aneurisma, disminución
inaceptable de la longitud de fijación (solapamiento de vaso y componente) o
endofugas, deben considerarse otras intervenciones endovasculares o de una
conversión a reparación quirúrgica abierta convencional después de la
reparación endovascular inicial. El aumento del tamaño del aneurisma o una
endofuga persistente o la migración pueden producir la rotura del aneurisma.
• Es posible que los pacientes que presenten fugas o reducción del flujo
sanguíneo a través de la ramificación de la endoprótesis vascular tengan que
someterse a intervenciones o procedimientos quirúrgicos secundarios.
4.2 Selección, tratamiento y seguimiento de los pacientes
• La endoprótesis vascular para AAA Zenith Flex está diseñada para tratar
cuellos aórticos de diámetros no inferiores a 18 mm y no superiores a 32 mm.
La endoprótesis vascular para AAA Zenith Flex está diseñada para tratar
cuellos aórticos proximales (distales a la arteria renal más inferior) de al menos
15 mm de longitud. Se requiere un lugar de fijación distal en la arteria ilíaca de
más de 10 mm de longitud y de 7,5-20 mm de diámetro (medido de pared
exterior a pared exterior). Estas medidas de tamaño son esenciales para la
eficacia de la reparación endovascular.
• Los principales elementos anatómicos que pueden dificultar la exclusión
correcta del aneurisma incluyen: exceso de angulación del cuello proximal
(>60 grados en el caso del cuello infrarrenal respecto al eje del AAA o
>45 grados en el del cuello suprarrenal respecto al cuello infrarrenal
inmediato); cuello aórtico proximal corto (<15 mm); forma de embudo
invertido (con un incremento de diámetro de más de un 10% en 15 mm de
longitud del cuello aórtico proximal); y trombo o calcificación circunferenciales
en los lugares de la implantación arterial, concretamente en el punto de
encuentro del cuello aórtico proximal y la arteria ilíaca distal. En presencia de
limitaciones anatómicas, puede ser necesario un cuello más largo para obtener
un sellado y una fijación adecuados. Las irregularidades en la calcificación o la
placa pueden comprometer la fijación y el sellado en los lugares de la fijación.
Los cuellos que presenten estos elementos anatómicos clave pueden conllevar
un mayor riesgo de migración de la endoprótesis vascular o endofuga.
• La introducción del dispositivo en la vasculatura requiere un acceso ilíaco o
femoral adecuado. El diámetro (medido de pared interior a pared interior) y la
morfología (tortuosidad mínima, enfermedad oclusiva o calcificación) del vaso
de acceso deben ser compatibles con las técnicas de acceso vascular y los
sistemas de implantación de una vaina introductora vascular de entre 16 y
22 Fr. Los vasos que muestren un exceso de calcificación, oclusión, tortuosidad
o trombos pueden ser inadecuados para la colocación de la endoprótesis
vascular o presentar un mayor riesgo de embolización. El tratamiento
satisfactorio de algunos pacientes puede requerir una técnica de conducto
vascular.
• La endoprótesis vascular para AAA Zenith Flex con el sistema de introducción
H&L-B One-Shot no está recomendada para los pacientes que no puedan
tolerar los medios de contraste necesarios para los estudios de imagen
intraoperatorios y los estudios de imagen de seguimiento posoperatorios.
Todos los pacientes deben vigilarse estrechamente y examinarse
periódicamente para comprobar si presentan cambios en el estado de su
enfermedad y para evaluar la integridad de la endoprótesis.
• La endoprótesis vascular para AAA Zenith Flex con el sistema de introducción
H&L-B One-Shot no está recomendada para pacientes que superen los límites
de peso o tamaño que comprometen o impiden el cumplimiento de los
requisitos necesarios de los estudios de imagen.
• La incapacidad para mantener la permeabilidad de al menos una arteria ilíaca
interna o la oclusión de una arteria mesentérica inferior indispensable pueden
aumentar el riesgo de isquemia pélvica o intestinal.
• La existencia de varias arterias lumbares grandes permeables, trombos
murales y una arteria mesentérica inferior permeable puede predisponer a un
paciente a endofugas de tipo II. Los pacientes con coagulopatía incorregible
también pueden tener mayor riesgo de endofugas de tipo II o de
complicaciones hemorrágicas.
• La seguridad y la eficacia de la endoprótesis vascular para AAA Zenith Flex con
el sistema de introducción H&L-B One-Shot no se han evaluado en las
siguientes poblaciones de pacientes:
• lesión aórtica traumática;
• aneurismas con fugas, rotura o rotura inminente;
• aneurismas micóticos;
• pseudoaneurismas producidos por la colocación previa de endoprótesis
vasculares;
• revisión de endoprótesis vasculares colocadas con anterioridad;
• coagulopatía incorregible;
• arteria mesentérica indispensable;
• trastornos genéticos del tejido conjuntivo (p. ej., síndromes de Marfan o de
Ehlers-Danlos);
• aneurismas aórticos torácicos o toracoabdominales concomitantes;
• pacientes con infecciones generalizadas activas;
• mujeres embarazadas o lactantes;
• pacientes con obesidad mórbida;
• pacientes de menos de 18 años de edad;
• pacientes con cuellos aórticos proximales de menos de 15 mm de longitud
o de más de 60 grados de angulación respecto al eje largo del aneurisma.
• La selección satisfactoria de los pacientes requiere estudios de imagen
específicos y mediciones precisas; consulte el apartado 4.3, Técnicas de
medición y estudios de imagen previos al procedimiento.
• El médico debe disponer de unidades de todas las longitudes y los diámetros
de los dispositivos necesarios para realizar el procedimiento, sobre todo
cuando las medidas (diámetros y longitudes de tratamiento) preoperatorias de
planificación del caso no sean precisas. Esto permitirá una mayor flexibilidad
intraoperatoria para conseguir resultados óptimos.
4.3 Técnicas de medición y estudios de imagen previos al procedimiento
• La falta de TAC sin contraste puede impedir apreciar la calcificación ilíaca o
aórtica, que puede imposibilitar el acceso o la fijación y el sellado apropiados
del dispositivo.
• Si en las TAC previas al procedimiento se utilizan espesores de reconstrucción
de más de 3 mm, es posible que el tamaño del dispositivo no sea el óptimo o
que no se aprecien estenosis focales.
• La experiencia clínica indica que la angiografía tomográfica computarizada
(ATC) espiral con contraste y reconstrucción tridimensional es la modalidad de
estudio de imagen que se recomienda encarecidamente para evaluar con
exactitud la configuración anatómica del paciente antes del tratamiento con la
endoprótesis vascular para AAA Zenith Flex. Si no se dispone de equipo de
angiografía tomográfica computarizada (ATC) espiral con contraste y
reconstrucción tridimensional, debe remitirse al paciente a un centro que
disponga de dicho equipo.
• Los clínicos recomiendan colocar el brazo en C radiográfico durante la
angiografía del procedimiento de tal forma que los orígenes de las arterias
renales, y especialmente la arteria renal permeable más inferior, estén
claramente visibles antes del despliegue del borde proximal del material de la
endoprótesis vascular (stent de sellado) del cuerpo principal. Además, antes
del despliegue de los componentes de rama ilíaca, la angiografía debe mostrar
las bifurcaciones de las arterias ilíacas de tal forma que las arterias ilíacas
primitivas distales estén bien definidas respecto al origen de las arterias ilíacas
internas bilateralmente.
Diámetros:
Los diámetros deben determinarse mediante TAC y medirse de pared exterior a
pared exterior del vaso (y no de su luz) para facilitar la determinación correcta
de los tamaños de los dispositivos y la selección adecuada de los dispositivos. El
barrido de la tomografía computarizada espiral con contraste debe comenzar
en un punto 1 cm superior al eje celíaco y continuar a través de las cabezas
femorales con un espesor de corte axial de 3 mm o menos.
Longitudes:
Las longitudes deben determinarse mediante TAC para evaluar con exactitud
la longitud del cuello proximal infrarrenal y para planificar los tamaños de los
cuerpos principales y los componentes de ramas de la endoprótesis vascular
para AAA Zenith Flex. Estas reconstrucciones deben realizarse en proyecciones
sagitales, coronales y tridimensionales.
• La eficacia a largo plazo de las endoprótesis vasculares no se ha
determinado aún. Todos los pacientes deben ser informados de que el
tratamiento endovascular requiere un seguimiento periódico durante el
resto de la vida para evaluar el estado de salud y la eficacia de las
endoprótesis vasculares. Los pacientes que presenten signos clínicos
específicos (p. ej., endofugas, aneurisma en crecimiento o cambios en la
estructura o posición de la endoprótesis vascular) deben someterse a un
seguimiento más exhaustivo. Las pautas específicas del seguimiento se
describen en el apartado 12, PAUTAS PARA LOS ESTUDIOS DE IMAGEN Y
SEGUIMIENTO POSOPERATORIO.
92

• La endoprótesis vascular para AAA Zenith Flex con el sistema de introducción
H&L-B One Shot no está recomendada para pacientes que no puedan o no
estén dispuestos a someterse a los estudios de imagen y de implantación
preoperatorios y posoperatorios necesarios descritos en el apartado 12,
PAUTAS PARA LOS ESTUDIOS DE IMAGEN Y SEGUIMIENTO
POSOPERATORIO.
• Tras la colocación de la endoprótesis vascular, los pacientes deben ser
examinados periódicamente para determinar el flujo alrededor de la
endoprótesis vascular, el crecimiento del aneurisma y los cambios en la
estructura o posición de la endoprótesis vascular. Como mínimo, es necesario
realizar estudios de imagen anuales que incluyan: 1) radiografías abdominales
para examinar la integridad del dispositivo (separación entre componentes,
fisura en el stent o separación de las púas) y 2) TAC con contraste y sin él para
determinar los cambios del aneurisma, el flujo alrededor de la endoprótesis
vascular, la permeabilidad, la tortuosidad y la evolución de la patología. Si hay
complicaciones renales u otros factores que impidan el uso de medios de
contraste en los estudios de imagen, las radiografías abdominales y las
ecografías dúplex pueden ofrecer información similar.
4.4 Selección de los dispositivos
• Al seleccionar el tamaño de dispositivo adecuado, se recomienda
encarecidamente seguir al pie de la letra la guía para la selección del tamaño
de las instrucciones de uso de la endoprótesis vascular para AAA Zenith Flex
(tablas 10.5.1 y 10.5.2). La guía para la selección del tamaño de las
instrucciones de uso incorpora los sobredimensionamientos adecuados de los
dispositivos. Si se seleccionan tamaños que estén fuera de este intervalo,
pueden producirse endofugas, fracturas, migración, doblamiento de los
dispositivos hacia el interior y compresión de los dispositivos.
4.5 Procedimiento de implantación
(Consulte el apartado 11, MODO DE EMPLEO)
• Durante el procedimiento, es necesario mantener una visualización adecuada
mediante estudios de imagen para colocar satisfactoriamente la endoprótesis
vascular para AAA Zenith Flex y asegurar la yuxtaposición adecuada a la pared
aórtica.
• No doble ni retuerza el sistema de implantación. Si lo hace, podría dañar el
sistema de implantación y la endoprótesis vascular para AAA Zenith Flex.
• Para evitar el retorcimiento de la prótesis endovascular, al hacer girar el sistema
de implantación se debe tener cuidado para que giren conjuntamente todos
los componentes del sistema (desde la vaina exterior hasta la cánula interior).
• No siga haciendo avanzar ninguna parte del sistema de implantación si siente
resistencia durante el avance de la guía o el sistema de implantación.
Deténgase y determine la causa de la resistencia; el vaso, el catéter o la
endoprótesis vascular pueden resultar dañados. Tenga especial cuidado en
zonas de estenosis, trombosis intravascular o en vasos calcificados o tortuosos.
• El despliegue parcial o la migración inadvertidos de la endoprótesis pueden
requerir la extracción quirúrgica.
• A menos que lo indique un médico, no despliegue la endoprótesis vascular
para AAA Zenith Flex en lugares que ocluyan arterias necesarias para
suministrar flujo sanguíneo a órganos o extremidades. No cubra arterias
renales o mesentéricas importantes (con excepción de la arteria mesentérica
inferior) con la endoprótesis. Es posible que se ocluya el vaso. Durante el
estudio clínico, este dispositivo no se estudió en pacientes con dos arterias
ilíacas internas ocluidas.
• No intente reenvainar la endoprótesis vascular después de haberla desplegado
parcialmente o totalmente.
• Si la endoprótesis vascular se cambia de posición distalmente después del
despliegue parcial del stent proximal cubierto, la endoprótesis vascular o el
vaso pueden resultar dañados.
• La colocación incorrecta o el sellado incompleto de la endoprótesis vascular
para AAA Zenith Flex dentro del vaso pueden aumentar el riesgo de
endofugas, migración u oclusión inadvertida de las arterias renales o ilíacas
internas. La arteria renal debe mantenerse permeable para evitar o reducir el
riesgo de fallo renal y complicaciones posteriores.
• La fijación inapropiada de la endoprótesis vascular para AAA Zenith Flex
puede aumentar el riesgo de migración de la endoprótesis vascular. El
despliegue incorrecto o la migración de la endoprótesis pueden requerir una
intervención quirúrgica.
• Durante el procedimiento de implantación debe utilizarse anticoagulación
sistémica administrada según el protocolo habitual del hospital y el protocolo
preferido del médico. Si la heparina está contraindicada, deberá considerarse
otro anticoagulante.
• Para activar el revestimiento hidrofílico del exterior de la vaina introductora
Flexor, la superficie debe limpiarse con paños de gasa estériles empapados
con solución salina. Para conseguir una eficacia óptima, mantenga la vaina
hidratada en todo momento.
• Para disminuir el riesgo de contaminación e infección de la endoprótesis
contenida en el sistema de implantación, manipúlela lo menos posible durante
la preparación y la introducción.
• Mantenga la posición de la guía durante la introducción del sistema de
implantación.
• Debe utilizarse fluoroscopia durante la introducción y el despliegue para
confirmar el funcionamiento adecuado de los componentes del sistema de
implantación, la colocación adecuada de la endoprótesis vascular y el
resultado deseado del procedimiento.
• El uso de la endoprótesis vascular para AAA Zenith Flex con el sistema de
introducción H&L-B One-Shot requiere la administración de contraste
intravascular. Los pacientes con insuficiencia renal preexistente pueden tener
mayor riesgo de fallo renal en el período posoperatorio. Se debe procurar
limitar la cantidad de medios de contraste utilizados durante el procedimiento
y emplear métodos preventivos de tratamiento para disminuir el grado de
afectación renal (p. ej., hidratación adecuada).
• Al retirar la vaina o la guía, la configuración anatómica y la posición de la
endoprótesis vascular pueden cambiar. Vigile constantemente la posición de la
endoprótesis vascular y realice una angiografía para comprobar la posición
según sea necesario.
• La endoprótesis vascular para AAA Zenith Flex incorpora un stent suprarrenal
con púas de fijación. Tenga mucho cuidado al manipular dispositivos de
intervención y angiográficos en la región del stent suprarrenal.
• Tenga cuidado al manipular catéteres, guías y vainas en el interior de un
aneurisma. Las alteraciones excesivas pueden desplazar fragmentos de
trombo que pueden causar embolización distal o rotura del aneurisma.
• Si es necesario volver a intervenir con instrumental (intervención secundaria) a
través de la endoprótesis vascular, evite dañarla o alterar su posición.
• Antes del despliegue del stent suprarrenal, compruebe que la guía de acceso
se extiende hasta quedar en posición ligeramente distal al cayado aórtico.
• Compruebe que la rama ilíaca contralateral predeterminada se selecciona para
la introducción en el lado contralateral del paciente antes de la implantación.
4.6 Uso del balón moldeador
• No hinche el balón en el vaso fuera de la endoprótesis vascular, ya que el vaso
podría resultar dañado. Utilice el balón de acuerdo con lo indicado en su
documentación.
• Tenga cuidado al hinchar el balón dentro de la endoprótesis vascular en
presencia de calcificación, ya que el hinchado excesivo podría dañar el vaso.
• Asegúrese de que el balón esté deshinchado por completo antes de cambiarlo
de posición.
• Para potenciar la hemostasia, la válvula hemostática Captor puede aflojarse o
apretarse para permitir la introducción y extracción posterior de un balón
moldeador.
4.7 Seguridad y compatibilidad con MRI
Las pruebas no clínicas han demostrado que la endoprótesis vascular para AAA
Zenith es «MR Conditional» (esto es, segura bajo ciertas condiciones de la MRI),
según la clasificación de la American Society for Testing and Materials. El stent
puede someterse a MRI de manera segura en las siguientes condiciones:
Sistemas de 1,5 teslas:
• Campo magnético estático de 1,5 teslas
• Campo de gradiente espacial de 450 gauss/cm
• Promedio de índice de absorción específica (SAR, según sus siglas en inglés)
de cuerpo entero máximo de 2,0 W/kg durante 15 minutos de exploración
En las pruebas no clínicas, la endoprótesis vascular para AAA Zenith produjo un
aumento de temperatura inferior o igual a 1,4 °C con un promedio de índice de
absorción específica (SAR, según sus siglas en inglés) de cuerpo entero máximo
informado de 2,8 W/kg, evaluado mediante calorimetría, durante 15 minutos de
MRI en un escáner de MRI Siemens Medical Magnetom de 1,5 teslas con software
Numaris/4, versión Syngo MR 2002B DHHS. El promedio de índice de absorción
específica (SAR, según sus siglas en inglés) de cuerpo entero máximo fue de
2,8 W/kg, que corresponde a un valor medido por calorimetría de 1,5 W/kg.
Sistemas de 3,0 teslas:
• Campo magnético estático de 3,0 teslas
• Campo de gradiente espacial de 720 gauss/cm
• Promedio de índice de absorción específica (SAR, según sus siglas en inglés)
de cuerpo entero máximo de 2,0 W/kg durante 15 minutos de exploración
En las pruebas no clínicas, la endoprótesis vascular para AAA Zenith produjo un
aumento de temperatura inferior o igual a 1,9 °C con un promedio de índice de
absorción específica (SAR, según sus siglas en inglés) de cuerpo entero máximo
de 3,0 W/kg, evaluado mediante calorimetría, durante 15 minutos de MRI en
un escáner de MRI GE Electric Healthcare Excite de 3,0 teslas con software
G3.0-052B. El promedio de índice de absorción específica (SAR, según sus siglas
en inglés) de cuerpo entero máximo fue de 3,0 W/kg, que corresponde a un valor
medido por calorimetría de 2,8 W/kg.
El artefacto de la imagen se extiende por toda la región anatómica que contiene
el dispositivo, y oscurece la vista de las estructuras anatómicas inmediatamente
adyacentes situadas en un radio aproximado de 20 cm del dispositivo, así como
todo el dispositivo y su luz, cuando la imagen se obtiene en pruebas no clínicas
utilizando la siguiente secuencia: Secuencia spin eco rápida en un sistema de
MRI GE Electric Healthcare Excite de 3,0 teslas con software G3.0-052B y bobina
de radiofrecuencia de cuerpo.
En todos los escáneres, el artefacto de la imagen se disipa a medida que
aumenta la distancia entre el dispositivo y la zona de interés. Las imágenes
de MRI de la cabeza, el cuello y las extremidades inferiores pueden obtenerse
sin artefactos en las imágenes. Las imágenes pueden presentar artefactos en
barridos de la región abdominal y de las extremidades superiores, dependiendo
de la distancia entre el dispositivo y la zona de interés.
Se dispone de información clínica sobre diecisiete pacientes que se sometieron
a MRI después de la implantación de endoprótesis vasculares. En estos pacientes
no se ha informado de ninguna reacción adversa ni de ningún problema con
el dispositivo debidos a la MRI. Además, se han implantado bastante más de
50.000 endoprótesis vasculares para AAA Zenith en todo el mundo, y no se
ha informado de ninguna reacción adversa ni de ningún problema con el
dispositivo debidos a la MRI.
Cook recomienda que el paciente ponga en conocimiento de MedicAlert
Foundation las condiciones de la MRI reveladas en estas instrucciones de uso.
Para ponerse en contacto con la MedicAlert Foundation, utilice uno de los
medios siguientes:
Correo: MedicAlert Foundation International
2323 Colorado Avenue
Teléfono: +1 888-633-4298 llamada gratuita
Turlock, CA 95382 EE.UU.
+1 209-668-3333 desde fuera de EE.UU.
Fax: +1 209-669-2450
Web: www.medicalert.org
5 REACCIONES ADVERSAS
5.1 Reacciones adversas observadas
La tabla 5.1.1 enumera los índices de reacciones adversas observados en un
estudio prospectivo multicéntrico estadounidense de una versión anterior del
dispositivo (la endoprótesis vascular para AAA Zenith), realizado en 15 centros y
que incluyó a 352 pacientes endovasculares (200 de riesgo normal, 100 de alto
riesgo y 52 de prueba [pacientes realizados durante la curva de aprendizaje]) y
80 pacientes de control. Los pacientes se incluyeron en el grupo de riesgo
normal si eran fisiológicamente capaces de soportar una reparación abierta o
endovascular y tenían una configuración anatómica adecuada para el
tratamiento con la endoprótesis vascular para AAA Zenith con el sistema de
introducción H&L-B One-Shot. Los pacientes que tenían una configuración
anatómica adecuada pero un mayor riesgo de morbilidad o mortalidad con la
reparación abierta se incluyeron en el grupo de alto riesgo. Los pacientes
iniciales tratados en el estudio se incluyeron en el grupo de prueba. El grupo de
control incluyó pacientes cuya configuración anatómica vascular podía no ser
adecuada para la reparación endovascular de AAA.
Tabla 5.1.1 Muertes y roturas observadas en el estudio clínico
Muerte y rotura Riesgo normal, Zenith
Todas las muertes
2
(0-30 días)
3
(31-365 días)
Relacionadas con el AAA 0,0% (0/198) 1,3% (1/78) 0,29 3,1% (3/98) 0,0% (0/51)
No relacionadas con el AAA 3,0% (6/198) 0,0% (0/78) 0,19 4,1% (4/98) 9,8% (5/51)
2,3,4
(0-365 días)
Relacionadas con el AAA 0,5% (1/199) 3,8% (3/80) 0,07 5,0% (5/100) 1,9% (1/52)
No relacionadas con el AAA 3,0% (6/199) 0,0% (0/80) 0,19 4,0% (4/100) 9,6% (5/52)
Rotura
(0-30 días) 0,0% (0/199) -- -- 0,0% (0/100) 0,0% (0/52)
(31-365 días) 0,0% (0/198) -- -- 1,0% (1/98) 0,0% (0/51)
(0-365 días) 0,0% (0/199) -- -- 1,0% (1/100) 0,0% (0/52)
1
El denominador es 199 porque a un paciente de riesgo normal no se le implantó un dispositivo.
2
Todas las muertes (0-30 días) se consideraron relacionadas con el AAA y con el procedimiento.
3
De las muertes (31-365 días), cuatro se consideraron relacionadas con el AAA: 1 quirúrgica (shock séptico derivado de colitis isquémica) y 3 de alto riesgo (pancreatitis con fallo y
septicemia renales, hemorragia de aneurisma abdominal superior [AAA no tratado] y fallo multiorgánico).
4
De las muertes (0-365 días), diez se consideraron relacionadas con el AAA: 1 de riesgo normal (fallo cardíaco), 3 quirúrgicas (hemorragia masiva, isquemia mesentérica y choque séptico
derivado de colitis isquémica), 5 de alto riesgo (fallo respiratorio, fallo cardíaco con embolia pulmonar, pancreatitis con fallo y septicemia renales, hemorragia de aneurisma abdominal
superior [AAA no tratado] y fallo multiorgánico) y 1 del grupo de prueba (supuesto fallo cardíaco).
0,5% (1/199) 2,5% (2/80) 0,20 2,0% (2/100) 1,9% (1/52)
3,0% (6/198) 1,3% (1/78) 0,68 7,1% (7/98) 9,8% (5/51)
3,5% (7/199) 3,8% (3/80) >0,99 9,0% (9/100) 11,5% (6/52)
1
Riesgo normal, cirugía Valor P Alto riesgo, Zenith Prueba, Zenith
93

Tabla 5.1.2 Reacciones adversas1 observadas en el estudio clínico
Riesgo normal, Zenith Riesgo normal, cirugía Valor P Alto riesgo, Zenith Prueba, Zenith
Ausencia de morbilidad
(0-30 días)
Cardiovasculares
Pulmonares
Renales
Intestino
Incisión quirúrgica
Neurológicas
Vasculares
Ausencia de morbilidad
(31-365 días)
Cardiovasculares
Pulmonares
Renales
Intestino
Incisión quirúrgica
Neurológicas
Vasculares
Ausencia de morbilidad
(0-365 días)
Cardiovasculares
Pulmonares
Renales
Intestino
Incisión quirúrgica
Neurológicas
Vasculares
1
Del índice de morbilidad.
2
Las reacciones adversas cardiovasculares incluyeron: infartos de miocardio con onda Q y sin ella, fallo cardíaco congestivo, arritmias que requieren una medicación o un tratamiento
nuevos, isquemia cardíaca que requiere intervención, necesidad de apoyo inotrópico e hipertensión resistente al tratamiento médico.
3
Las reacciones adversas pulmonares incluyeron: reintubación o ventilación >24 horas, neumonía que requirió antibióticos, y oxígeno suplementario en el alta hospitalaria.
4
Las reacciones adversas renales incluyeron: diálisis en pacientes con función renal preoperatoria normal, y aumento de creatinina >30% del nivel basal en dos o más pruebas de
seguimiento.
5
Las reacciones adversas intestinales incluyeron: obstrucción intestinal, isquemia intestinal, fístula aortoentérica e íleo paralítico >4 días.
6
Las reacciones adversas de la incisión quirúrgica incluyeron: infección que requirió tratamiento con antibióticos, hernia, fístula linfática, dehiscencia y necrosis que requirió
desbridamiento.
7
Las reacciones adversas neurológicas incluyeron: infarto cerebral, accidente isquémico transitorio y parálisis o isquemia espinales.
8
Las reacciones adversas vasculares incluyeron: trombosis de ramificación, embolización distal que produce pérdida tisular o requirió intervención, transfusión después del
procedimiento (por pseudoaneurisma, lesión vascular, fuga aneurismática u otras causas relacionadas con el procedimiento), pseudoaneurisma, lesión vascular (como oclusión o
disección involuntarias u otras causas relacionadas con el procedimiento), fuga o rotura aneurismáticas, aumento de más de 0,5 cm del tamaño del aneurisma respecto a la menor de
las medidas previas.
9
Los investigadores informaron de otros dos pacientes de alto riesgo, uno que presentó oclusión de una arteria renal accesoria y otro que presentó insuficiencia renal crónica, que se
clasificaron como «otras» reacciones adversas.
10
Los investigadores informaron de otros dos pacientes, uno de prueba y otro de cirugía, que presentaron insuficiencia renal y se clasificaron como «otras» reacciones adversas.
11
Los investigadores informaron de otro paciente de prueba que presentó rotura intraoperatoria de la placa aórtica que produjo oclusión de la arteria renal, que se clasificó como «otra»
reacción adversa.
5.2 Reacciones adversas posibles
Las reacciones adversas posibles que pueden requerir intervención incluyen,
entre otras:
• Agrandamiento del aneurisma
• Amputación
• Claudicación (p. ej., en nalga o extremidad inferior)
• Complicaciones cardíacas y problemas asociados posteriores (p. ej., arritmia,
infarto de miocardio, insuficiencia cardíaca congestiva, hipotensión e
hipertensión)
• Complicaciones de la anestesia y problemas asociados posteriores (p. ej.,
aspiración)
• Complicaciones de la herida quirúrgica y problemas asociados posteriores (p.
ej., dehiscencia e infección)
• Complicaciones del lugar de acceso vascular, que incluyen infección, dolor,
hematoma, pseudoaneurisma y fístula arteriovenosa
• Complicaciones genitourinarias y problemas asociados posteriores (p. ej.,
isquemia, erosión, fístula, incontinencia, hematuria e infección)
• Complicaciones intestinales (p. ej., íleo, isquemia transitoria, infarto y necrosis)
• Complicaciones linfáticas y problemas asociados posteriores (p. ej., fístula
linfática)
• Complicaciones neurológicas locales o generalizadas y problemas asociados
posteriores (p. ej., infarto cerebral, accidente isquémico transitorio, paraplejia,
paraparesia y parálisis)
• Complicaciones pulmonares o respiratorias y problemas asociados posteriores
(p. ej., neumonía, insuficiencia respiratoria e intubación prolongada)
• Complicaciones renales y problemas asociados posteriores (p. ej., oclusión de
la arteria, toxicidad del contraste, insuficiencia y fallo)
• Conversión quirúrgica a reparación abierta
• Daño aórtico, que incluye perforación, disección, hemorragia, rotura y muerte
• Daño vascular
• Edema
• Embolización (micro y macro) con isquemia transitoria o permanente o infarto
• Endofuga
• Endoprótesis: colocación incorrecta de componentes; despliegue incompleto
de componentes; migración de componentes; rotura del hilo de sutura;
oclusión; infección; fisura en el stent; desgaste del material de la endoprótesis
vascular; dilatación; erosión; punción; flujo alrededor de la endoprótesis
vascular; separación y corrosión de las púas
• Espasmo vascular o traumatismo vascular (p. ej., disección del vaso
iliofemoral, hemorragia, rotura y muerte)
• Fiebre e inflamación localizada
• Fístula arteriovenosa
• Hemorragia, hematoma o coagulopatía
• Impotencia
• Infección del aneurisma, el dispositivo o el lugar de acceso, lo que incluye
formación de abscesos, fiebre transitoria y dolor
• Insuficiencia hepática
• Muerte
• Oclusión de la endoprótesis vascular o del vaso nativo
• Rotura del aneurisma y muerte
• Trombosis y pseudoaneurisma arteriales o venosos
Informes de reacciones adversas relacionadas con el dispositivo
Cualquier reacción adversa (incidente clínico) relacionada con la endoprótesis
vascular auxiliar para AAA Zenith Flex debe comunicarse inmediatamente a
COOK. Clientes de Estados Unidos: para informar sobre incidentes, llamen al
Departamento de Relaciones con los Clientes al número 1-800-457-4500
(24 horas) o 1-812-339-2235. Clientes de fuera de Estados Unidos: pónganse
en contacto con su distribuidor.
4, 9
4, 10
4, 9,10
2
3
5
6
7
8,11
2
3
5
6
7
8
2
3
5
6
7
8,11
80% (160/200) 58% (46/80) <0,001 68% (68/100) 73% (38/52)
3,0% (6/200) 11% (9/80) 0,02 14% (14/100) 1,9% (1/52)
1,0% (2/200) 15% (12/80) <0,001 2,0% (2/100) 0,0% (0/52)
2,5% (5/200) 10% (8/80) 0,01 6,0% (6/100) 5,8% (3/52)
1,0% (2/200) 3,8% (3/80) 0,14 1,0% (1/100) 1,9% (1/52)
4,5% (9/200) 7,5% (6/80) 0,38 2,0% (2/100) 3,8% (2/52)
0,0% (0/200) 2,5% (2/80) 0,08 0,0% (0/100) 0,0% (0/52)
11% (21/200) 31% (25/80) <0,001 20% (20/100) 19% (10/52)
91% (181/198) 86% (67/78) 0,25 79% (77/98) 86% (44/51)
2,5% (5/198) 3,8% (3/78) 0,69 5,1% (5/98) 2,0% (1/51)
0,5% (1/198) 1,3% (1/78) 0,49 4,1% (4/98) 0,0% (0/51)
0,5% (1/198) 0,0% (0/78) >0,99 3,1% (3/98) 0,0% (0/51)
0,5% (1/198) 1,3% (1/78) 0,49 0,0% (0/98) 0,0% (0/51)
2,0% (4/198) 5,1% (4/78) 0,23 3,1% (3/98) 2,0% (1/51)
1,0% (2/198) 0,0% (0/78) >0,99 1,0% (1/98) 3,9% (2/51)
3,0% (6/198) 3,8% (3/78) 0,72 8,2% (8/98) 5,9% (3/51)
76% (151/200) 49% (39/80) <0,001 55% (55/100) 62% (32/52)
5,0% (10/200) 14% (11/80) 0,02 19% (19/100) 3,8% (2/52)
1,5% (3/200) 16% (13/80) <0,001 6,0% (6/100) 0,0% (0/52)
2,5% (5/200) 10% (8/80) 0,01 9,0% (9/100) 5,8% (3/52)
1,5% (3/200) 3,8% (3/80) 0,36 1,0% (1/100) 1,9% (1/52)
5,5% (11/200) 13% (10/80) 0,08 5,0% (5/100) 5,8% (3/52)
1,0% (2/200) 2,5% (2/80) 0,32 1,0% (1/100) 3,8% (2/52)
12% (24/200) 33% (26/80) <0,001 25% (25/100) 23% (12/52)
mejores medidas de utilidad clínica en comparación con los pacientes de
control tratados mediante cirugía. La seguridad se determinó evaluando si los
sujetos tratados con la endoprótesis vascular para AAA Zenith presentaban
morbilidad reducida a los 30 días, y resultados no inferiores de la supervivencia
a 30 días y a 12 meses y del éxito del tratamiento a 12 meses, en comparación
con los sujetos tratados mediante cirugía abierta. La eficacia se basó en la
exclusión del aneurisma, lo que incluye ausencia de endofugas, ausencia de
agrandamiento (≥5 mm) del aneurisma y ausencia de reacciones adversas
importantes al dispositivo evaluadas durante un año de seguimiento. Los
objetivos secundarios incluyeron una evaluación de las ventajas clínicas y
medidas de la calidad de vida.
6.2 Diseño del estudio
El estudio clínico estadounidense fue un estudio multicéntrico no aleatorio que
comparó a los pacientes de riesgo médico normal a los que se implantó una
endoprótesis vascular con un grupo de control de pacientes tratados con cirugía
abierta. El estudio también incluyó otros dos grupos: uno de alto riesgo médico
y uno de prueba. El estudio se realizó en 15 centros con 200 pacientes de riesgo
normal, 80 de control de cirugía, 100 de alto riesgo y 52 de prueba. El grupo de
control incluyó pacientes cuya configuración anatómica vascular podía no ser
adecuada para la reparación endovascular de AAA. Las evaluaciones del
seguimiento se programaron para los siguientes momentos: una previa al alta y
otras a 1 mes, 6 meses, 12 meses y 24 meses. El seguimiento y la disponibilidad
de los pacientes a 1 mes, 12 meses y 24 meses se presentan en la tabla 6.2.1, ya
que éstos fueron los momentos principales del análisis de los datos. Los datos
de los estudios de imagen de este resumen se basan en los resultados de un
laboratorio de análisis de imagen centralizado independiente (laboratorio
central), que revisó las TAC y las radiografías abdominales para evaluar los
cambios del diámetro del aneurisma, la migración del dispositivo y de los
componentes relacionados, la integridad del dispositivo (guía y endoprótesis
vascular) y la presencia y el tipo de endofugas. Las reacciones clínicas fueron
clasificadas por un comité de reacciones clínicas independiente, y la seguridad
fue vigilada por un comité de vigilancia de datos y seguridad.
Los pacientes de riesgo normal del grupo de cirugía y del grupo de la
endoprótesis vascular Zenith cumplieron criterios idénticos de riesgo
fisiopatológico. Los criterios de exclusión de los grupos endovasculares
incluyeron: trombo circunferencial en el cuello proximal, cuello proximal de
menos de 15 mm de longitud, cuello proximal con diámetro de pared exterior a
pared exterior de menos de 18 mm o de más de 28 mm, cuello proximal
demasiado angulado, arteria ilíaca con diámetro de pared exterior a pared
exterior de menos de 7,5 mm o de más de 20 mm en el lugar de fijación distal y
arteria ilíaca con lugares de fijación distal de menos de 10 mm de longitud.
Se consideró que los pacientes tenían un mayor riesgo de reparación quirúrgica
si tenían más de 80 años de edad, concentraciones basales de creatinina de
>2,0 mg/dl, tratamiento domiciliario con oxígeno, volumen espiratorio forzado
en 1 segundo <1 litro, fracción de eyección <25%, enfermedad pulmonar
obstructiva crónica (EPOC) incapacitante, grado 3 ó 4 de la New York Heart
Association, abdomen hostil, diálisis, infarto de miocardio en los 6 meses
anteriores, hipertensión resistente al tratamiento médico, infarto cerebral previo
con déficit residual, objeción cultural a las transfusiones de sangre o
hemoderivados, cirugía de derivación renal anterior o aneurisma inflamatorio.
Antes de incluir a los pacientes en el estudio fundamental, se exigió que los
centros que no tenían experiencia con la endoprótesis vascular para AAA Zenith
trataran a pacientes iniciales bajo la supervisión de un monitor. Estos pacientes
de prueba incluyeron pacientes de riesgo normal y de alto riesgo, y fueron
seguidos de acuerdo con el mismo programa que los pacientes del estudio
fundamental.
6 RESUMEN DE ESTUDIOS CLÍNICOS
6.1 Objetivos
El objetivo principal del estudio clínico fue evaluar la seguridad y eficacia de una
versión anterior del dispositivo (la endoprótesis vascular para AAA Zenith) como
alternativa a la reparación quirúrgica abierta en el tratamiento primario de
aneurismas aórticos abdominales infrarrenales. Las hipótesis del estudio
examinaron si los pacientes de riesgo normal experimentaron menor morbilidad
a los 30 días, resultados no inferiores de los índices de supervivencia a 30 días,
los índices de supervivencia a 12 meses y el éxito del tratamiento a 12 meses, y
94

Tabla 6.2.1 Seguimiento y disponibilidad de los pacientes
1
Tratamiento Riesgo normal, Zenith Riesgo normal, cirugía
Intervalo
Sin dispositivo 1 1
Conversión a reparación abierta 0 2
1 mes 12 meses 24 meses 1 mes 12 meses 24 meses
2
2
2
1
2
2
0 0 --
-- -- -Fallecido 1 7 17 2 3 -Retirado o perdido para el seguimiento 0 0 2 0 4 -Disponible 198 190 178 78 73 -TAC en el centro clínico 191 168 110 69 58 -TAC en el laboratorio central 190 165 99 69 59 -Estudios de imagen de riñones, uréteres y vejiga en el centro clínico 179 153 108 -- -- -Estudios de imagen de riñones, uréteres y vejiga en el laboratorio central 178 149 93 -- -- -Evaluado por el centro clínico para comprobar la presencia de endofugas 187 163 107 -- -- -Evaluado por el laboratorio central para comprobar la presencia de endofugas 161 148 92 -- -- -Evaluado por el centro clínico para comprobar la presencia de agrandamiento
del aneurisma
Evaluado por el laboratorio central para comprobar la presencia de agran-
damiento del aneurisma
1
El tamaño de la muestra del análisis de datos varió en cada uno de los momentos indicados en la tabla anterior y en las tablas siguientes. Esta variabilidad se debe a la disponibilidad
del paciente para el seguimiento, así como a la cantidad y a la calidad de las imágenes diagnósticas disponibles para su evaluación en momentos específicos. Por ejemplo, el número y
la calidad de las imágenes diagnósticas disponibles para la evaluación de endofugas a los 12 meses es diferente del número y la calidad de las imágenes diagnósticas disponibles a los
24 meses, debido a la variación del número de exámenes de imágenes realizados, del número de imágenes diagnósticas suministradas por el centro clínico al laboratorio central y del
número de imágenes diagnósticas de calidad aceptable para la evaluación. Los totales de los distintos momentos no son acumulativos a menos que se indique.
2
Los totales de los distintos momentos son acumulativos.
-- 149 104 -- -- --
-- 151 94 -- -- --
6.3 Datos demográficos de los pacientes
Las tablas 6.3.1 y 6.3.2 comparan las características de los sujetos y el diámetro inicial de los aneurismas de las poblaciones de la endoprótesis vascular para
AAA Zenith y de la cirugía abierta, respectivamente.
Tabla 6.3.1 Comparación de las características de los sujetos
Artículo Riesgo normal, Zenith Riesgo normal, cirugía Valor P Alto riesgo, Zenith Prueba, Zenith
Edad (años) 71 ± 7 69 ± 7 0,03 77 ± 7 74 ± 8
Sexo masculino 94% (187/200) 89% (71/80) 0,22 92% (92/100) 90% (47/52)
Afecciones médicas actuales
Enfermedad vascular periférica
Hipertensión 64% (127/200) 83% (65/78) 0,001 68% (67/99) 67% (35/52)
16% (31/195) 25% (19/76) 0,12 24% (23/96) 9,6% (5/52)
Fallo renal 0,0% (0/197) 0,0% (0/79) >0,99 5,2% (5/97) 1,9% (1/52)
EPOC 20% (39/199) 18% (14/78) 0,87 34% (33/98) 22% (11/51)
Reacción tromboembólica 4,5% (9/199) 7,7% (6/78) 0,38 7,1% (7/99) 1,9% (1/52)
Enfermedad hepática 2,1% (4/192) 5,1% (4/79) 0,24 1,0% (1/99) 1,9% (1/52)
Diabetes mellitus 12% (24/199) 15% (12/79) 0,55 17% (17/99) 14% (7/51)
Insulinodependiente 17% (4/24) 8,3% (1/12) 0,65 24% (4/17) 43% (3/7)
Afecciones médicas anteriores
Infarto de miocardio
39% (74/192) 29% (23/80) 0,13 35% (34/98) 35% (18/52)
Fallo cardíaco congestivo 5,0% (10/199) 12% (9/78) 0,07 16% (16/100) 10% (5/50)
Angina 49% (98/198) 39% (31/79) 0,14 45% (44/98) 44% (23/52)
Arritmia 20% (40/197) 22% (17/78) 0,87 28% (27/98) 24% (12/51)
Enfermedad cerebrovascular 9,5% (19/199) 16% (13/79) 0,14 20% (20/99) 9,8% (5/51)
Infección generalizada 1,0% (2/196) 0,0% (0/78) >0,99 3,1% (3/97) 0,0% (0/49)
Cáncer 22% (43/200) 19% (15/80) 0,74 31% (31/99) 29% (15/51)
Antecedentes familiares de enfermedad
aneurismática
16% (24/150) 27% (17/63) 0,09 14% (11/77) 26% (10/38)
Cirugía previa en el centro 10% (20/200) 15% (12/79) 0,22 10% (10/99) 14% (7/51)
Radiación previa en el centro 0,5% (1/197) 0,0% (0/79) >0,99 2,0% (2/100) 2,0% (1/51)
Consumo excesivo de alcohol 3,6% (7/193) 10% (8/77) 0,04 3,1% (3/96) 4,0% (2/50)
Tabaquismo
Nunca ha fumado
Ex fumador 69% (133/193) 60% (48/80) 0,03 69% (66/96) 57% (28/49)
Fumador actual 21% (40/193) 35% (28/80) 18% (17/96) 18% (9/49)
Debido a los criterios de inclusión, los pacientes de alto riesgo tuvieron más edad (P<0,001) y presentaron más fallos renales (P=0,004), EPOC (P=0,01), fallos cardíacos congestivos
(P=0,004) y más enfermedades cerebrovasculares (P=0,02) que los pacientes de riesgo normal.
10% (20/193) 5,0% (4/80)
14% (13/96) 24% (12/49)
Tabla 6.3.2 Distribución de los diámetros de los aneurismas
Intervalo de diámetros Riesgo normal, Zenith Riesgo normal, cirugía Alto riesgo, Zenith Prueba, Zenith
<30 mm
30-39 mm
40-49 mm
50-59 mm
60-69 mm
70-79 mm
80-89 mm
≥90 mm
La distribución de los diámetros de los aneurismas no se evaluó en tres pacientes de alto riesgo y en uno de prueba.
6.4 Resultados
Los datos de las tablas 6.4.1 a 6.4.3 fueron recopilados por los centros del estudio clínico y por el laboratorio central. Los datos de los 24 meses se suministran
donde se dispone de ellos. A los pacientes de control no se les realizó seguimiento después de los 12 meses, y algunos datos posteriores a los 12 meses no se
han clasificado aún. Por lo tanto, en este apartado algunos resultados se presentan hasta los 12 meses, y otros hasta los 24 meses. La tabla 6.4.1 describe los
dispositivos implantados en los pacientes del estudio clínico. La tabla 6.4.2 describe los resultados primarios del estudio clínico. Las figuras 6.4.1 y 6.4.2 son
las curvas de Kaplan-Meier de la supervivencia relacionada con todas las causas y con el AAA hasta los 24 meses, respectivamente. Un comité de reacciones
clínicas independiente clasificó todas las muertes según su posible relación con la reparación aneurismática. Todas las muertes precoces (0-30 días) se
consideraron relacionadas con el AAA. Las muertes después de los 30 días se consideraron relacionadas con el AAA si se confirmó enfermedad de AAA o
implicación del dispositivo. La tabla 6.4.3 presenta las medidas de éxito, y la figura 6.4.3 es la curva de Kaplan-Meier de ausencia de morbilidad.
0,0% (0/199) 0,0% (0/78) 0,0% (0/100) 0,0% (0/52)
0,5% (1/199) 0,0% (0/78) 1,0% (1/100) 0,0% (0/52)
23% (45/199) 7,7% (6/78) 15% (15/100) 13% (7/52)
48% (95/199) 33% (26/78) 47% (47/100) 40% (21/52)
24% (47/199) 29% (23/78) 27% (27/100) 42% (22/52)
3,0% (6/199) 21% (16/78) 5,0% (5/100) 1,9% (1/52)
2,5% (5/199) 6,4% (5/78) 1,0% (1/100) 0,0% (0/52)
0,0% (0/199) 2,6% (2/78) 1,0% (1/100) 0,0% (0/52)
95

Tabla 6.4.1 Dispositivos implantados
Artículo Riesgo normal, Zenith Alto riesgo, Zenith Prueba, Zenith
Cuerpo principal y ramas 99,5% (199/200)* 100% (100/100)
Extensión de cuerpo principal
Extensión de rama ilíaca ipsilateral
Extensión de rama ilíaca contralateral
Convertidor 0,5% (1/199)** 0,0% (0/100) 0,0% (0/52)
Oclusor 0,0% (0/199) 0,0% (0/100) 0,0% (0/52)
*A un paciente de riesgo normal no se le implantó un dispositivo debido a la tortuosidad y la calcificación del vaso de acceso.
**Se utilizó convertidor sin oclusor.
1
Un dispositivo fue personalizado.
2
A dos pacientes de riesgo normal y a uno de alto riesgo se les implantaron extensiones de cuerpo principal después del procedimiento; a un paciente de riesgo normal se le
implantaron dos extensiones de cuerpo principal.
3
A dos pacientes de riesgo normal y a uno de alto riesgo se les implantaron extensiones de rama ipsilateral después del procedimiento.
4
A cuatro pacientes de riesgo normal, a dos de alto riesgo y a uno de prueba se les implantaron extensiones de rama contralateral después del procedimiento.
5
A tres pacientes de riesgo normal y a tres de alto riesgo se les implantaron extensiones tanto ipsilaterales como contralaterales durante el procedimiento; a uno de riesgo normal se le
implantaron extensiones tanto ipsilaterales como contralaterales después del procedimiento.
2
3,5
4,5
1,5% (3/199) 1,0% (1/100) 5,8% (3/52)
9,5% (19/199) 11% (11/100) 0,0% (0/52)
11% (21/199) 11% (11/100) 13,5% (7/52)
1
100% (52/52)
Tabla 6.4.2 Resultados primarios
Artículo Riesgo normal, Zenith
Todas las muertes
2
(0-30 días)
Todas las muertes
(31-365 días)
2, 3
0,5% (1/199) 2,5% (2/80) 0,20 2,0% (2/100) 1,9% (1/52)
3,0% (6/199) 1,3% (1/80) 0,68 7,0% (7/100) 9,6% (5/52)
1
Riesgo normal, cirugía Valor P Alto riesgo, Zenith Prueba, Zenith
Relacionadas con el AAA 0,0% (0/199) 1,3% (1/80) 0,29 3,0% (3/100) 0,0% (0/52)
No relacionadas con el AAA 3,0% (6/199) 0,0% (0/80) 0,19 4,0% (4/100) 9,6% (5/52)
Todas las muertes
(0-365 días)
2,3,4
3,5% (7/199) 3,8% (3/80) >0,99 9,0% (9/100) 11,5% (6/52)
Relacionadas con el AAA 0,5% (1/199) 3,8% (3/80) 0,07 5,0% (5/100) 1,9% (1/52)
No relacionadas con el AAA 3,0% (6/199) 0,0% (0/80) 0,19 4,0% (4/100) 9,6% (5/52)
Rotura
(0-30 días) 0,0% (0/199) -- -- 0,0% (0/100) 0,0% (0/52)
(31-365 días) 0,0% (0/199) -- -- 1,0% (1/100) 0,0% (0/52)
(0-365 días) 0,0% (0/199) -- -- 1,0% (1/100) 0,0% (0/52)
Conversión
(0-30 días) 0,0% (0/199) -- -- 0,0% (0/100) 0,0% (0/52)
5
(31-365 días)
5
(0-365 días)
Reacciones adversas
6
(0-30 días)
6
(31-365 días)
6
(0-365 días)
1
El denominador es 199 porque a un paciente de riesgo normal no se le implantó un dispositivo.
2
Todas las muertes (0-30 días) se consideraron relacionadas con el AAA y con el procedimiento.
3
De las muertes (31-365 días), cuatro se consideraron relacionadas con el AAA: 1 quirúrgica (shock séptico derivado de colitis isquémica) y 3 de alto riesgo (pancreatitis con fallo y
septicemia renales, hemorragia de aneurisma abdominal superior [AAA no tratado] y fallo multiorgánico).
4
De las muertes (0-365 días), diez se consideraron relacionadas con el AAA: 1 de riesgo normal (fallo cardíaco), 3 quirúrgicas (hemorragia masiva, isquemia mesentérica y choque
séptico derivado de colitis isquémica), 5 de alto riesgo (fallo respiratorio, fallo cardíaco con embolia pulmonar, pancreatitis con fallo y septicemia renales, hemorragia de aneurisma
abdominal superior [AAA no tratado] y fallo multiorgánico) y 1 del grupo de prueba (supuesto fallo cardíaco).
5
Algunos pacientes de riesgo normal fueron sometidos a conversiones debido a endofuga proximal de tipo I persistente y a un nuevo aneurisma aórtico suprarrenal. Tres pacientes
de cirugía sufrieron hemorragias masivas; de ellos, dos requirieron una nueva intervención y uno murió.
6
Reacciones adversas incluidas en el índice de morbilidad.
1,0% (2/199) -- -- 1,0% (1/100) 0,0% (0/52)
1,0% (2/199) -- -- 1,0% (1/100) 0,0% (0/52)
20% (40/200) 43% (34/80) <0,001 32% (32/100) 27% (14/52)
8,6% (17/199) 14% (11/78) 0,25 21% (21/98) 14% (7/51)
25% (49/200) 51% (41/80) <0,001 45% (45/100) 38% (20/52)
Los pacientes varones y mujeres a los que se implantó la endoprótesis
vascular para AAA Zenith no presentaron diferencias significativas en cuanto
a supervivencia y ausencia de reacciones adversas importantes. (Las barras de
error de las figuras 6.4.1, 6.4.2 y 6.4.3 representan límites de confianza del
95%). La figura 6.4.1 presenta la supervivencia de todas las causas hasta los
24 meses. La tabla adjunta presenta el análisis de Kaplan-Meier a 1, 6, 12 y
24 meses.
Figura 6.4.1 Supervivencia a los 24 meses
Riesgo normal, Zenith
(N=200, 1 muer te a los 30 días
4 muertes a los 6 meses
7 muertes a los 12 meses
17 muertes a los 24 meses)
Riesgo normal, cirugía
(N=80, 2 muertes a los 30 días
3 muertes a los 6 meses
3 muertes a los 12 meses)
Porcentaje de supervivencia
Meses después del procedimiento
1 mes 6 meses 12 meses 24 meses
% de super-
n
vivencia n
Riesgo
normal,
198* 99,5 194 98,0 190 96,5 97 90,9
Zenith
Riesgo
normal,
cirugía
n=Pacientes vivos y disponibles para el seguimiento al final del intervalo
P = 0,81
*Un paciente falleció antes de un mes y a otro no se le implantó ningún dispositivo.
78 97,5 73 96,2 67 96,2 -- --
% de super-
vivencia n
% de super-
vivencia n
% de super-
vivencia
La figura 6.4.2 presenta la supervivencia relacionada con el AAA
(determinada por el comité de reacciones clínicas) a los 24 meses. La tabla
adjunta presenta el análisis de Kaplan-Meier a 1, 6, 12 y 24 meses.
Figura 6.4.2 Supervivencia relacionada con el AAA a los 24 meses
Riesgo normal, Zenith
(N=200, 1 muer te a los 30 días
1 muerte a los 6 meses
1 muerte a los 12 meses
2 muertes a los 24 meses)
Riesgo normal, cirugía
(N=80, 2 muertes a los 30 días
3 muertes a los 6 meses
murieron por el aneurisma
Porcentaje de pacientes que no
3 muertes a los 12 meses)
Reacción: muerte relacionada con el aneurisma
Meses después del procedimiento
1 mes 6 meses 12 meses 24 meses
% de super-
n
vivencia n
Riesgo
normal,
198* 99,5 194 99,5 190 99,5 97 99,0
Zenith
Riesgo
normal,
cirugía
n=Pacientes vivos y disponibles para el seguimiento al final del intervalo
P = 0,04
*Un paciente falleció antes de un mes y a otro no se le implantó ningún dispositivo.
78 97,5 73 96,2 67 96,2 -- --
% de super-
vivencia n
% de super-
vivencia n
% de super-
vivencia
96

Tabla 6.4.3 Medidas de éxito
Artículo Riesgo normal, Zenith Riesgo normal, cirugía Alto riesgo, Zenith Prueba, Zenith
1
Éxito técnico
Éxito del procedimiento a los
2
30 días
Éxito del tratamiento a los
3
12 meses
1
Endoprótesis vascular permeable después del despliegue.
2
Éxito técnico sin complicaciones importantes, endoprótesis vascular permeable y ausencia de endofugas de tipos I o III a los 30 días.
3
El éxito del procedimiento se prolongó hasta los 12 meses sin ningún agrandamiento del aneurisma (>5 mm).
La figura 6.4.3 presenta la ausencia de morbilidad (reacciones del índice de morbilidad) hasta los 12 meses. La tabla adjunta presenta el análisis de
Kaplan-Meier a 1, 6 y 12 meses.
99,5% (199/200) 98,8% (79/80) 100% (100/100) 100% (52/52)
95,1% (155/163) 88% (60/68) 86% (70/81) 91% (30/33)
89% (122/137) 85% (52/61) 70% (44/63) 87% (26/30)
Figura 6.4.3 Ausencia de morbilidad (0-365 días)
Riesgo normal, Zenith
(N=200, 38 pacientes con reacciones a los 30 días
43 pacientes con reacciones a los 6 meses
45 pacientes con reacciones a los 12 meses)
Riesgo normal, cirugía
(N=80, 34 pacientes con reacciones a los 30 días
38 pacientes con reacciones a los 6 meses
41 pacientes con reacciones a los 12 meses)
Porcentaje de pacientes sin morbilidad
Meses después del procedimiento
1 mes 6 meses 12 meses
n % n % n %
Riesgo normal, Zenith 162 81,0 154 78,5 133 77,4
Riesgo normal, cirugía 45 57,1 39 52,0 33 47,8
n=Pacientes vivos y sin morbilidad al final del intervalo
P = <0,001
Las tablas 6.4.4 a 6.4.7 describen los resultados de los sujetos a los que se implantó la endoprótesis vascular para AAA Zenith documentados por el
laboratorio central. Los factores relacionados con la eficacia del dispositivo analizados por el laboratorio central incluyeron la integridad del dispositivo (tabla
6.4.4), la permeabilidad del dispositivo (tabla 6.4.5), la migración (tabla 6.4.6) y la separación de las ramificaciones (tabla 6.4.7).
Tabla 6.4.4 Resultados radiográficos abdominales: integridad del dispositivo
Artículo Riesgo normal, Zenith Alto riesgo, Zenith Prueba, Zenith
Fisuras en los stents
1
Antes del alta hospitalaria 0,0% (0/172) 0,0% (0/81) 0,0% (0/39)
30 días 0,0% (0/172) 0,0% (0/83) 0,0% (0/43)
6 meses 0,0% (0/166) 0,0% (0/78) 0,0% (0/35)
12 meses 0,0% (0/148) 0,0% (0/60) 0,0% (0/28)
24 meses 0,0% (0/93) 0,0% (0/42) 0,0% (0/19)
Separación de las púas
2
Antes del alta hospitalaria 0,0% (0/176) 0,0% (0/86) 0,0% (0/39)
30 días 0,0% (0/178) 0,0% (0/86) 0,0% (0/43)
6 meses 1,2% (2/167) 2,5% (2/80) 0,0% (0/35)
12 meses 2,0% (3/149) 1,7% (1/60) 0,0% (0/28)
24 meses 1,1% (1/93) 0,0% (0/42) 0,0% (0/19)
Rotura del material de la endoprótesis
vascular
Antes del alta hospitalaria 0,0% (0/176) 0,0% (0/86) 0,0% (0/39)
30 días 0,0% (0/178) 0,0% (0/86) 0,0% (0/43)
6 meses 0,0% (0/167) 0,0% (0/80) 0,0% (0/35)
12 meses 0,0% (0/149) 0,0% (0/60) 0,0% (0/28)
24 meses 0,0% (0/93) 0,0% (0/42) 0,0% (0/19)
1
Los porcentajes de fisuras en los stents se refieren al cuerpo principal. Además, el laboratorio central no observó ninguna fisura en las ramas ilíacas derechas, las ramas ilíacas
izquierdas, los oclusores, los convertidores, las extensiones ilíacas izquierdas, las extensiones ilíacas derechas ni las extensiones de cuerpo principal.
2
Pacientes con separación de una o dos púas (de un total de 10 ó 12); ninguna secuela adversa clínica.
Tabla 6.4.5 Resultados de las TAC: permeabilidad de la endoprótesis vascular
Artículo Riesgo normal, Zenith Alto riesgo, Zenith Prueba, Zenith
Permeabilidad de la endoprótesis vascular
30 días 100% (185/185) 99% (85/86) 100% (47/47)
6 meses 99% (183/184) 100% (74/74) 100% (39/39)
12 meses 99% (153/155) 100% (62/62) 100% (30/30)
24 meses 100% (96/96) 100% (33/33) 100% (25/25)
Tabla 6.4.6 Resultados de las TAC: migración de la endoprótesis vascular (cuerpo principal)
Artículo Riesgo normal, Zenith Alto riesgo, Zenith Prueba, Zenith
Migración de la endoprótesis vascular (>5 mm) a los 12 meses
con secuelas clínicas1 o intervención 0,0% (0/162) 0,0% (0/71) 0,0% (0/34)
sin secuelas clínicas1 ni intervención 2,5% (4/162) 2,8% (2/71) 0,0% (0/34)
Migración de la endoprótesis vascular (>10 mm) 0,0% (0/162) 0,0% (0/71) 0,0% (0/34)
1
La migración con secuelas clínicas incluía endofuga, conversión, rotura o muerte relacionada con el AAA.
97

Tabla 6.4.7 Resultados radiográficos abdominales: separación de las ramificaciones
Artículo Riesgo normal, Zenith Alto riesgo, Zenith Prueba, Zenith
Separación de las ramificaciones
Antes del alta hospitalaria 0,0% (0/176) 0,0% (0/86) 0,0% (0/39)
30 días 0,0% (0/178) 0,0% (0/86) 0,0% (0/43)
6 meses 0,0% (0/167) 0,0% (0/80) 0,0% (0/35)
12 meses 0,0% (0/149) 0,0% (0/60) 0,0% (0/28)
24 meses 0,0% (0/93) 0,0% (0/42) 0,0% (0/19)
6.5 Tratamiento de las endofugas
En el estudio clínico, las endofugas de tipo I se trataron durante el procedimiento inicial mediante el uso de angioplastia con balón adicional o, si no se tenía
éxito, con prótesis adicionales. Las endofugas de tipo II se observaron durante un período de uno a seis meses para determinar si trombosaban
espontáneamente o, en ausencia de agrandamiento de los aneurismas, se trataron con técnicas endovasculares a discreción del médico. Si hubo
agrandamiento del aneurisma, se consideró la conveniencia del tratamiento con embolización o ligadura y, en algunos casos, se llevó a cabo dicho
tratamiento. Las endofugas de tipo III causadas por defectos de las endoprótesis vasculares, el sellado inadecuado o la desconexión de los componentes
modulares se trataron mediante el hinchado de otro balón o con prótesis adicionales. Según el laboratorio central angiográfico, no hubo endofugas de tipo IV
durante el estudio clínico estadounidense. El material de la endoprótesis vascular utilizado para fabricar la endoprótesis vascular para AAA Zenith es de
espesor estándar y es el mismo material utilizado en intervenciones quirúrgicas abiertas. La tabla 6.5.1 presenta, por intervalo de evaluación, la incidencia de
endofugas identificadas por el laboratorio central en pacientes de riesgo normal, de alto riesgo y de prueba, respectivamente.
Tabla 6.5.1 Endofugas (todos los tipos, nuevas y persistentes)
Artículo Riesgo normal, Zenith Alto riesgo, Zenith Prueba, Zenith
Endofugas
Antes del alta hospitalaria 15% (23/153) 14% (11/78) 12% (3/26)
1
30 días
1
6 meses
1
12 meses
1
Incluye endofugas persistentes y nuevas observaciones.
Las tablas 6.5.2–6.5.4 presentan, por intervalo de evaluación, la incidencia de la primera aparición de endofugas identificadas por el laboratorio central en los
exámenes realizados a los 30 días, los 6 meses y los 12 meses, o antes de dichos exámenes, en pacientes de riesgo normal, de alto riesgo y de prueba,
respectivamente. También se indica el número de pacientes que no presentaron fugas a partir de entonces.
9,9% (16/161) 12% (9/75) 6,3% (2/32)
8,7% (15/172) 11% (8/70) 8,6% (3/35)
7,4% (11/148) 8,8% (5/57) 3,4% (1/29)
Tabla 6.5.2 Primera aparición de una endofuga1 en pacientes de riesgo normal
Artículo
Hasta el examen realizado al mes
N=179
% Endofuga
Ausencia de
a partir de
1
entonces
fugas
Examen realizado a los seis meses
2
% Endofuga
N=172
Ausencia de
a partir de
1
entonces
fugas
Examen realizado a los doce meses
2
% Endofuga
N=148
Ausencia de
a partir de
1
entonces
Endofugas 17 31 17 2,3 4 3 3,4 5 2
De tipo I proximales 2,8 5 4 0,0 0 0 0,0 0 0
Distales
De tipo I 1,7 3 1 0,0 0 0 0,7 1 1
De tipo II 9,5 17 9 2,3 4 3 1,4 2 1
De tipo III 1,1 2 2 0,0 0 0 0,7 1 0
De tipo IV 0,0 0 0 0,0 0 0 0,0 0 0
Múltiples 1,1 2 1 0,0 0 0 0,0 0 0
Desconocidas 1,1 2 0 0,0 0 0 0,7 1 0
1
Identificadas por el laboratorio central.
2
Las endofugas posteriores pueden ser de un tipo diferente del de la original.
3
Sólo dos pacientes presentaron endofugas nuevas después de 12 meses; no se dispone de datos de seguimiento posteriores a los 24 meses.
Tabla 6.5.3 Primera aparición de una endofuga1 en pacientes de alto riesgo
Artículo
Hasta el examen realizado al mes
% Endofuga
N=88
1
Ausencia de
fugas
a partir de
entonces
Examen realizado a los seis meses
2
% Endofuga
N=70
1
Ausencia de
fugas
a partir de
entonces
Examen realizado a los doce meses
2
% Endofuga
N=57
Ausencia de
1
Endofugas 18 16 6 2,9 2 0 3,5 2 1
De tipo I proximales 2,3 2 1 0,0 0 0 0,0 0 0
Distales
De tipo I 1,1 1 1 0,0 0 0 0,0 0 0
De tipo II 9,1 8 3 1,4 1 0 1,8 1 0
De tipo III 0,0 0 0 1,4 1 0 1,8 1 1
De tipo IV 0,0 0 0 0,0 0 0 0,0 0 0
Múltiples 4,5 4 0 0,0 0 0 0,0 0 0
Desconocidas 1,1 1 1 0,0 0 0 0,0 0 0
1
Identificadas por el laboratorio central.
2
Las endofugas posteriores pueden ser de un tipo diferente del de la original.
3
Ausencia de endofugas después de 12 meses; no se dispone de datos de seguimiento posteriores a los 24 meses.
3
fugas
3
fugas
a partir de
entonces
2
2
Tabla 6.5.4 Primera aparición de una endofuga1 en pacientes de prueba
Artículo
Hasta el examen realizado al mes
% Endofuga
N=36
1
Ausencia de
fugas
a partir de
entonces
Examen realizado a los seis meses
2
% Endofuga
N=35
1
Ausencia de
fugas
a partir de
entonces
Examen realizado a los doce meses
2
% Endofuga
N=29
Ausencia de
1
Endofugas 11 4 2 2,9 1 0 0,0 0 0
De tipo I proximales 0,0 0 0 0,0 0 0 0,0 0 0
Distales
De tipo I 0,0 0 0 0,0 0 0 0,0 0 0
De tipo II 5,6 2 1 2,9 1 0 0,0 0 0
De tipo III 2,8 1 0 0,0 0 0 0,0 0 0
De tipo IV 0,0 0 0 0,0 0 0 0,0 0 0
Múltiples 0,0 0 0 0,0 0 0 0,0 0 0
Desconocidas 2,8 1 1 0,0 0 0 0,0 0 0
1
Identificadas por el laboratorio central.
2
Las endofugas posteriores pueden ser de un tipo diferente del de la original.
3
Ausencia de endofugas después de 12 meses; no se dispone de datos de seguimiento posteriores a los 24 meses.
98
3
fugas
a partir de
entonces
2

6.6 Cambio del aneurisma
Las tablas 6.6.1-6.6.3 presentan el cambio del diámetro de los aneurismas identificados por el laboratorio central en los pacientes endovasculares. La
tabla 6.6.1 presenta el cambio del diámetro máximo del aneurisma por intervalos. Las tablas 6.6.2 y 6.6.3 presentan el cambio del aneurisma y la endofuga a
los 12 y los 24 meses, respectivamente.
Tabla 6.6.1 Cambio del diámetro máximo del aneurisma por intervalos*
Artículo Riesgo normal, Zenith Alto riesgo, Zenith Prueba, Zenith
Desde antes del alta a los 30 días
Disminución >5 mm 1,7% (3/180) 4,8% (4/84) 0,0% (0/40)
Sin cambios 97% (174/180) 94% (79/84) 97,5% (39/40)
Aumento >5 mm 1,7% (3/180) 1,2% (1/84) 2,5% (1/40)
Cambio desde antes del alta a los 6 meses
Disminución >5 mm 36% (63/173) 41% (30/73) 49% (18/37)
Sin cambios 62% (108/173) 59% (43/73) 51% (19/37)
Aumento >5 mm 1,2% (2/173) 0,0% (0/73) 0,0% (0/37)
Cambio desde antes del alta a los 12 meses
Disminución >5 mm 68% (102/151) 63% (39/62) 67% (20/30)
Sin cambios 31% (47/151) 35% (22/62) 33% (10/30)
Aumento >5 mm 1,3% (2/151) 1,6% (1/62) 0,0% (0/30)
Cambio desde antes del alta a los 24 meses
Disminución >5 mm 78% (74/94) 75% (27/36) 71% (17/24)
Sin cambios 19% (18/94) 25% (9/36) 25% (6/24)
Aumento >5 mm 2,1% (2/94) 0,0% (0/36) 4,2% (1/24)
*Sólo incluye sujetos con películas de imágenes interpretables y medidas del cambio del aneurisma desde el seguimiento realizado al mes hasta el realizado a los 24 meses.
Tabla 6.6.2 Cambio del tamaño del aneurisma y endofugas a los 12 meses
Riesgo normal, Zenith
N=140
Alto riesgo, Zenith
N=53
Prueba, Zenith
N=27
Endofuga Endofuga Endofuga
Artículo
Cambio del tamaño del aneurisma desde antes del alta hasta los 12 meses
N n % N n % N n %
Disminución >5 mm 96 3 3,0 35 3 9,0 20 0 0,0
Sin cambios 42 8 19 17 2 12 7 1 14
Aumento >5 mm 2 0 0,0 1 0 0,0 0 0 0,0
Tabla 6.6.3 Cambio del tamaño del aneurisma y endofugas a los 24 meses
Riesgo normal, Zenith
N=90
Alto riesgo, Zenith
N=31
Prueba, Zenith
N=21
Endofuga Endofuga Endofuga
Artículo
Cambio del tamaño del aneurisma desde antes del alta hasta los 24 meses
N n % N n % N n %
Disminución >5 mm 71 3 4,0 24 1 4,0 16 0 0,0
Sin cambios 18 1 6,0 7 3 43 5 0 0,0
Aumento >5 mm 1 1 100 1 0 0,0 0 0 0,0
6.7 Intervenciones secundarias relacionadas con el AAA
En los sujetos tratados con la endoprótesis vascular Zenith, durante el primer año se realizaron intervenciones secundarias relacionadas con el AAA en el 11%
de los de riesgo normal, en el 13% de los de alto riesgo y en el 5,8% de los de prueba, como muestra la tabla 6.7.1. Más del 50% de las intervenciones
secundarias incluyeron cateterismo para el tratamiento de una endofuga. En los sujetos tratados con la endoprótesis vascular Zenith, durante el segundo año
se realizaron intervenciones secundarias relacionadas con el AAA en el 4,2% de los de riesgo normal, en el 2,2% de los de alto riesgo y en el 2,3% de los de
prueba, como muestra la tabla 6.7.2.
Tabla 6.7.1 Intervenciones secundarias (hasta los 12 meses)
Intervención
Conversión a reparación abierta 2 1,0 1 1,0 0 0,0
Riesgo normal, Zenith
N=199
Alto riesgo, Zenith
N=100
Prueba, Zenith
N=52
n % n % n %
Sujetos con ≥1 intervención 21 11 13 13 3 5,8
Tratamiento de una endofuga
Embolización 5 2,5 3 3,0 1 2,0
Componente auxiliar 6 3,0 4 4,0 1 2,0
Stent 1 0,5 0 0,0 0 0,0
Angioplastia 0 0,0 1 1,0 0 0,0
Tratamiento de aumento del aneurisma
Embolización 0 0,0 0 0,0 0 0,0
Tratamiento de oclusión de una ramificación 1 0,5 3 3,0 1 2,0
Tratamiento de estenosis de una ramificación 1 0,5 0 0,0 0 0,0
Tratamiento de arteria renal 5 2,5 0 0,0 0 0,0
Tratamiento de isquemia infrainguinal 1 0,5 1 1,0 0 0,0
Tratamiento de varias reacciones 1 0,5 1 1,0 0 0,0
Tabla 6.7.2 Intervenciones secundarias (desde los 12 hasta los 24 meses)
Intervención
Conversión a reparación abierta 1 0,5 1 1,1 0 0,0
Riesgo normal, Zenith
N=190
Alto riesgo, Zenith
N=90
Prueba, Zenith
N=44
n % n % n %
Sujetos con ≥1 intervención 8 4,2 2 2,2 1 2,3
Tratamiento de una endofuga
Embolización 3
1
1,6 0 0,0 1
2
Componente auxiliar 1 0,5 0 0,0 0 0,0
Stent 0 0,0 1 1,1 0 0,0
Angioplastia 0 0,0 0 0,0 0 0,0
Tratamiento de aumento del aneurisma
Embolización 1 0,5 0 0,0 1
2
Tratamiento de oclusión de una ramificación 1 0,5 0 0,0 0 0,0
Tratamiento de estenosis de una ramificación 0 0,0 0 0,0 0 0,0
Tratamiento de arteria renal 0 0,0 1 1,1 0 0,0
Tratamiento de isquemia infrainguinal 1 0,5 0 0,0 0 0,0
Tratamiento de varias reacciones 1 0,5 0 0,0 0 0,0
1
Al paciente también se le implantó un componente auxiliar.
2
El paciente se sometió a una intervención para el tratamiento de un aumento del aneurisma y una endofuga.
99
2,3
2,3

6.8 Medidas de los resultados secundarios
Como se describe en la tabla 6.8.1, al comparar el tratamiento de AAA con la endoprótesis vascular para AAA Zenith con el grupo de control de cirugía se
observó que el primero mostraba ventajas considerables en las medidas de recuperación y de calidad de vida.
Tabla 6.8.1 Resultados de las intervenciones secundarias por grupo de tratamiento
Artículo Riesgo normal, Zenith R iesgo normal, cirugía Valor P Alto riesgo, Zenith Prueba, Zenith
Duración de la anestesia (min) 221,6 ± 67,3 304,5 ± 102,7 <0,001 218,9 ± 69,6 213,9 ± 57,7
Duración del procedimiento (min) 153,2 ± 56,3 238,7 ± 92,2 <0,001 153,5 ± 58,6 155,9 ± 43,2
Productos del banco de sangre recibidos 5,0% (10/200) 84% (67/80) <0,001 12% (12/100) 3,8% (2/52)
Pérdida de sangre (ml) 299 ± 324 1676 ± 1676 <0,001 356 ± 514 265 ± 226
Días en la UCI 0,4 ± 0,9 3,4 ± 4,6 <0,001 0,5 ± 1,2 0,5 ± 0,9
Días hasta el alta 2,6 ± 1,7 8,8 ± 5,6 <0,001 3,0 ± 2,8 2,7 ± 1,5
Días hasta los líquidos orales 0,5 ± 0,8 3,9 ± 2,5 <0,001 0,5 ± 0,6 0,7 ± 0,5
Días hasta la dieta normal 1,3 ± 1,2 6,6 ± 4,9 <0,001 1,3 ± 0,8 1,1 ± 0,7
Días hasta la función intestinal normal 2,6 ± 1,4 4,2 ± 2,1 <0,001 2,6 ± 1,5 2,0 ± 1,2
Días hasta la ambulación 1,2 ± 0,7 3,5 ± 3,4 <0,001 1,2 ± 0,7 1,2 ± 0,6
Horas de intubación 1,9 ± 2,2 11,7 ± 13,6 <0,001 1,2 ± 1,7 2,6 ± 4,6
Temperatura máxima (°F) 101,1 ± 1,3 100,7 ± 1,2 0,06 100,8 ± 1,1 101,0 ± 1,2
7 SELECCIÓN Y TRATAMIENTO DE LOS PACIENTES
Véase el apartado 4, Advertencias y precauciones
Individualización del tratamiento
Cook recomienda que la selección de los diámetros de los componentes de la
endoprótesis vascular para AAA Zenith Flex se haga según lo descrito en las
tablas
10.5.1 y 10.5.2. La longitud de la endoprótesis vascular para AAA Zenith
Flex debe extenderse desde la arteria renal más inferior hasta un punto
ligeramente por encima de la bifurcación de la arteria ilíaca interna
(hipogástrica). El médico debe disponer de unidades de todas las longitudes y
los diámetros de los dispositivos necesarios para realizar el procedimiento, sobre
todo cuando las medidas (diámetros y longitudes de tratamiento) preoperatorias
de planificación del caso no sean precisas. Esto permitirá una mayor flexibilidad
intraoperatoria para conseguir resultados óptimos. Los riesgos y las ventajas
descritos en el
considerarse atentamente en cada paciente antes de utilizar la endoprótesis
vascular para AAA Zenith Flex. Las consideraciones adicionales que deben
tenerse en cuenta para la selección de los pacientes incluyen, entre otras:
• La edad y la esperanza de vida del paciente
• Las comorbilidades (p. ej., insuficiencia cardíaca, pulmonar o renal antes de la
intervención quirúrgica, y obesidad mórbida)
• La idoneidad del paciente para la reparación quirúrgica abierta
• La idoneidad anatómica del paciente para la reparación endovascular
• El riesgo de rotura del aneurisma comparado con el riesgo del tratamiento con
la endoprótesis vascular para AAA Zenith Flex
• La capacidad para tolerar la anestesia general, regional o local
• El tamaño y la morfología (trombo mínimo, calcio y tortuosidad) del vaso de
acceso iliofemoral deben ser compatibles con las técnicas de acceso vascular y
los accesorios del perfil de implantación de una vaina introductora vascular de
entre 14 y 22 Fr.
• Segmento (cuello) aórtico infrarrenal no aneurismático proximal al aneurisma:
• Lugar de fijación distal en la arteria ilíaca de más de 10 mm de longitud y de
7,5 a 20 mm de diámetro (medido de pared exterior a pared exterior)
• La ausencia de enfermedad oclusiva importante de la arteria femoral/ilíaca que
pueda impedir el flujo a través de la endoprótesis vascular
apartado 6, RESUMEN DE ESTUDIOS CLÍNICOS
• con una longitud de al menos 15 mm,
• con un diámetro medido de pared exterior a pared exterior no superior a
32 mm y no inferior a 18 mm,
• con un ángulo respecto al eje largo del aneurisma de menos de 60 grados, y
• con un ángulo respecto al eje de la aorta suprarrenal de menos de 45 grados
, deben
La decisión final del tratamiento deben tomarla el médico y el paciente.
8 INFORMACIÓN PARA EL ASESORAMIENTO DE LOS PACIENTES
El médico y el paciente (y los miembros de la familia) deben considerar los
riesgos y las ventajas al deliberar sobre la conveniencia de este dispositivo
endovascular y del procedimiento, lo que incluye:
• Los riesgos y las diferencias entre la reparación endovascular y la reparación
quirúrgica
• Las ventajas posibles de la reparación quirúrgica abierta tradicional
• Las ventajas posibles de la reparación endovascular
• La posibilidad de que sea necesaria la reparación intervencionista o quirúrgica
abierta posterior del aneurisma después de la reparación endovascular
inicial
Además de los riesgos y las ventajas de la reparación endovascular, el médico
debe evaluar el compromiso del paciente con el seguimiento posoperatorio y
su cumplimiento de los requisitos de éste, ya que son necesarios para garantizar
la seguridad y la eficacia continuadas del tratamiento. A continuación se
enumeran otros temas relacionados con las expectativas posteriores a una
reparación endovascular que deben discutirse con el paciente:
• La eficacia a largo plazo de las endoprótesis vasculares no se ha
determinado aún. Todos los pacientes deben ser informados de que el
tratamiento endovascular requiere un seguimiento periódico durante
el resto de la vida para evaluar el estado de salud y la eficacia de las
endoprótesis vasculares. Los pacientes que presenten signos clínicos
específicos (p. ej., endofugas, aneurismas en crecimiento o cambios en la
estructura o posición de la endoprótesis vascular) deben someterse a un
seguimiento más exhaustivo. Las pautas específicas del seguimiento se
describen en el apartado 12, PAUTAS PARA LOS ESTUDIOS DE IMAGEN Y
SEGUIMIENTO POSOPERATORIO.
• Los pacientes deben ser informados de la importancia del cumplimiento del
programa de seguimiento, tanto durante el primer año como a intervalos
anuales de entonces en adelante. También debe decírseles que la regularidad y
la constancia del seguimiento son fundamentales para garantizar la seguridad
y la eficacia continuadas del tratamiento endovascular de los AAA. Como
mínimo, es necesario realizar estudios de imagen anuales y cumplir los
requisitos del seguimiento posoperatorio periódico, lo que debe considerarse
un compromiso de por vida con la salud y el bienestar del paciente.
• El paciente debe ser informado de que la reparación satisfactoria de
aneurismas no detiene el proceso de la enfermedad. Aún es posible presentar
una degeneración vascular asociada.
• Los médicos deben advertir a todos los pacientes de la importancia de buscar
atención médica inmediata en caso de que se experimenten signos de
oclusión de ramificación o de agrandamiento o rotura del aneurisma. Los
signos de oclusión de ramificación de la endoprótesis vascular incluyen dolor
en las caderas o las piernas al caminar o en reposo, decoloración de las piernas
y sensación de frío en las piernas. La rotura del aneurisma puede ser
asintomática, pero normalmente se presenta con: dolor; entumecimiento;
debilidad en las piernas; dolor en la espalda, el tórax, el abdomen o la ingle;
mareos; desmayos; aumento del ritmo cardíaco o debilidad repentina.
• Debido a los estudios de imagen requeridos para la colocación y el
seguimiento satisfactorios de dispositivos endovasculares, las mujeres que
estén embarazadas o sospechen estarlo deben ser informadas de los riesgos
que tiene la exposición a la radiación para los tejidos en desarrollo. Los varones
que se sometan a reparación endovascular o quirúrgica abierta pueden
experimentar impotencia.
Los médicos deben aconsejar al paciente que consulte la Guía del paciente para
informarse sobre los riesgos posibles durante la implantación del dispositivo y
después de ella. Los riesgos relacionados con el procedimiento incluyen
complicaciones cardíacas, pulmonares, neurológicas, intestinales y
hemorrágicas. Los riesgos relacionados con el dispositivo incluyen oclusión,
endofuga, agrandamiento del aneurisma, fractura, posibilidad de que sea
necesario volver a intervenir y realizar una conversión quirúrgica abierta, rotura
y muerte (véase el apartado 5.1, Reacciones adversas observadas, y el apartado
5.2, Reacciones adversas posibles). El médico debe rellenar la Tarjeta de
identificación del paciente y dársela al paciente para que éste pueda llevarla
consigo en todo momento. El paciente debe hacer referencia a la tarjeta
siempre que visite a otros profesionales sanitarios, sobre todo en caso de que
tenga que someterse a otros procedimientos diagnósticos (p. ej., MRI).
9 PRESENTACIÓN
• La endoprótesis vascular para AAA Zenith Flex se suministra estéril y
precargada en envases de apertura pelable.
• El dispositivo está indicado para un solo uso. No reesterilice el dispositivo.
• Inspeccione el dispositivo y el envase para comprobar que no han resultado
dañados durante el transporte. No utilice este dispositivo si está dañado o si la
barrera estéril está dañada o rota. Si se ha producido algún daño, no utilice el
producto y devuélvalo a COOK.
• Antes del uso, asegúrese de que se dispone de los dispositivos apropiados
(cantidad y tamaño) para el paciente, comprobando que los dispositivos se
corresponden con los prescritos por el médico para ese paciente particular.
• El dispositivo del cuerpo principal está cargado en una vaina introductora
Flexor® de 18, 20 ó 22 Fr. La superficie de la vaina está tratada con un
revestimiento hidrofílico que, al hidratarse, mejora el control del
desplazamiento. Para activar el revestimiento hidrofílico, la superficie debe
limpiarse con un paño de gasa empapado con solución salina.
• No utilice el dispositivo después de la fecha «USE BY» (fecha de caducidad)
impresa en la etiqueta.
• Almacénelo en un lugar fresco y seco.
10 INFORMACIÓN SOBRE EL USO CLÍNICO
10.1 Formación de médicos
AVISO: Durante los procedimientos de implantación o reintervención
deberá haber un equipo quirúrgico cualificado disponible en todo
momento para el caso de que sea necesaria una conversión a reparación
quirúrgica abierta.
AVISO: La endoprótesis vascular para AAA Zenith Flex con el sistema de
introducción H&L-B One-Shot sólo deben utilizarlo médicos y equipos
que hayan recibido formación en técnicas intervencionistas vasculares
y en el uso de este dispositivo. A continuación se resumen los requisitos
teóricos y técnicos recomendados que deben cumplir los médicos que
utilicen la endoprótesis vascular para AAA Zenith Flex con el sistema de
introducción H&L-B One-Shot.
Selección de los pacientes:
• Conocimiento de la historia natural de los aneurismas aórticos abdominales
(AAA) y las comorbilidades asociadas a la reparación de AAA.
• Conocimiento de la interpretación de imágenes radiográficas, la selección de
los dispositivos y la selección de tamaños.
Un equipo multidisciplinar que tenga experiencia conjunta en la
realización de procedimientos tales como:
• Incisión, arteriotomía y reparación femorales
• Técnicas de acceso y cierre percutáneos
• Técnicas selectivas y no selectivas con guías y catéteres
• Interpretación de imágenes fluoroscópicas y angiográficas
• Embolización
• Angioplastia
• Colocación de stents endovasculares
• Técnicas con dispositivo de recuperación (lazos)
• Uso adecuado de material de contraste radiográfico
• Técnicas para reducir al mínimo la exposición a radiación
• Las modalidades necesarias para el seguimiento de los pacientes
10.2 Inspección previa al uso
Inspeccione el dispositivo y el envase para comprobar que no han resultado
dañados durante el transporte. No utilice este dispositivo si está dañado o si la
barrera estéril está dañada o rota. Si se ha producido algún daño, no utilice el
producto y devuélvalo a COOK. Antes del uso, asegúrese de que se dispone de
los dispositivos apropiados (cantidad y tamaño) para el paciente, comprobando
que los dispositivos se corresponden con los prescritos por el médico para ese
paciente particular.
10.3 Material necesario
(No incluido en el sistema modular de tres piezas)
• Kit de dispositivos auxiliares de la endoprótesis vascular para AAA Zenith
• Fluoroscopio con funciones de angiografía digital (brazo en C o unidad fija)
• Medios de contraste
• Jeringa
• Solución salina heparinizada
• Paños de gasa estériles
10.4 Material recomendado
(No incluido en el sistema modular de tres piezas)
Para la implantación de cualquier componente de la línea de productos Zenith
se recomiendan los productos siguientes. Para obtener información sobre el uso
de estos productos, consulte las Instrucciones de uso sugeridas del producto en
cuestión.
• Guía extrarrígida de 0,035 pulgadas (0,89 mm) y 260 cm de longitud; por ejemplo:
• Guías extrarrígidas Cook Lunderquist (LES)
• Guía estándar de 0,035 pulgadas (0,89 mm); por ejemplo:
• Guías Cook de 0,035 pulgadas (0,89 mm)
• Guías Cook Nimble
100
™
 Loading...
Loading...