

BIOTRONIK Lumax VR ICD, Lumax DR-T ICD, Lumax VR-T ICD, Lumax DR ICD, Lumax HF-T CRT-D Technical Manual
...Page 1

Lumax
Family of Implantable Cardioverter
Defibrillators and Cardiac
Resynchronization Therapy
Defibrillators
• VR ICD
• VR-T ICD
• DR ICD
• DR-T ICD
• HF CRT-D
• HF-T CRT-D
Technical Manual
Page 2

X-ray Identification
Lumax Family
Implantable Cardioverter Defibrillator and Cardiac
Resynchronization Therapy Defibrillators
Inside the housing:
X-Ray identification Year of manufacture
HR nn
Federal (U.S.A.) law restricts this device to sale by, or on the
order of, a physician.
CAUTION
2008 BIOTRONIK, Inc., all rights reserved.
Page 3

Lumax Technical Manual i
Contents
1.1 System Description .......................................................1
1.2 Indications and Usage...................................................3
1.3 Contraindications...........................................................4
1.4 Warnings and Precautions ............................................4
1.4.1 Sterilization, Storage, and Handling ......................7
1.4.2 Device Implantation and Programming .................7
1.4.3 Lead Evaluation and Connection ..........................9
1.4.4 Follow-up Testing ................................................11
1.4.5 Pulse Generator Explant and Disposal ...............11
1.4.6 Hospital and Medical Hazards.............................11
1.4.7 Home and Occupational Hazards .......................13
1.4.8 Cellular Phones ...................................................13
1.4.9 Electronic Article Surveillance (EAS) ..................14
1.4.10 Home Appliances ................................................14
1.4.11 Home Monitoring .................................................15
1.5 Potential/Observed Effects of the Device on Health ...16
1.5.1 Potential Adverse Events ....................................16
1.5.2 Observed Adverse Events...................................17
1.6 Clinical Studies............................................................27
1.6.1 Kronos LV-T Study ..............................................27
1.6.2 Tupos LV/ATx Study............................................29
1.7 Patient Selection and Treatment.................................48
1.7.1 Individualization of Treatment .............................48
1.7.2 Specific Patient Populations................................49
1.8 Patient Counseling Information ...................................50
1.9 Evaluating Prospective CRT-D/ICD Patients ..............50
2.1 Cardiac Resynchronization Therapy (CRT) ................53
2.2 Sensing (Automatic Sensitivity Control) ......................56
2.2.1 Right Ventricular Sensitivity Settings...................57
2.2.2 Minimum Right Ventricular Threshold .................59
2.2.3 Atrial Sensitivity Settings .....................................59
2.2.4 Minimum Atrial Threshold....................................60
2.2.5 Left Ventricular Sensitivity Settings .....................60
Page 4

ii Lumax Technical Manual
2.2.6 Minimum Left Ventricular Threshold....................61
2.2.7 Far Field Protection .............................................61
2.2.8 Additional Sensing Parameters ...........................62
2.3 Automatic Threshold Measurement (ATM) .................63
2.3.1 Functional Description.........................................63
2.4 Intra-Thoracic Impedance Measurement ....................65
2.4.1 Functional Description.........................................65
2.5 Ventricular Tachyarrhythmia Detection .......................66
2.5.1 VF Classifications ................................................67
2.5.2 VT Interval Counters............................................67
2.5.3 VT Classification..................................................67
2.5.4 SMART Detection™ ............................................68
2.5.5 Onset ...................................................................69
2.5.6 Stability ................................................................69
2.5.7 Sustained VT Timer.............................................70
2.5.8 VT Monitoring Zone .............................................70
2.5.9 Atrial Monitoring Zone .........................................71
2.6 Tachyarrhythmia Redetection......................................71
2.6.1 VT Redetection....................................................71
2.6.2 SMART Redetection............................................72
2.6.3 Forced Termination .............................................72
2.6.4 VF Redetection....................................................72
2.7 Tachyarrhythmia Termination ......................................72
2.8 Tachyarrhythmia Therapy............................................72
2.8.1 Therapy Options ..................................................73
2.8.2 Anti-Tachycardia Pacing (ATP) ...........................73
2.8.3 Shock Therapy ....................................................76
2.8.4 Progressive Course of Therapy...........................82
2.9 Bradycardia Therapy ...................................................83
2.9.1 Bradycardia Pacing Modes .................................83
2.9.2 Basic Rate ...........................................................84
2.9.3 Night Rate............................................................85
2.9.4 Rate Hysteresis ...................................................85
2.9.5 Dynamic AV Delay...............................................88
2.9.6 IOPT ....................................................................91
Page 5

Lumax Technical Manual iii
2.9.7 Upper Tracking Rate ...........................................91
2.9.8 Mode Switching ...................................................93
2.9.9 PMT Management ...............................................94
2.9.10 VES Discrimination after Atrial Sensed Events ...97
2.9.11 Rate Adaptive Pacing ..........................................98
2.9.12 Pulse Amplitude...................................................99
2.9.13 Pulse Width .........................................................99
2.9.14 Post Ventricular Atrial Refractory Period...........100
2.9.15 PVARP after VES ..............................................100
2.9.16 Auto PVARP ......................................................100
2.9.17 Noise Response ................................................100
2.9.18 Post Shock Pacing ............................................101
2.10 EP Test Functions .....................................................101
2.10.1 P and R-wave Amplitude Measurements ..........101
2.10.2 Pacing Impedance Measurements....................102
2.10.3 Shock Impedance Measurements.....................103
2.10.4 Testing for Retrograde Conduction ...................103
2.10.5 Pacing Threshold...............................................104
2.10.6 Arrhythmia Induction Features ..........................104
2.10.7 Manual Shock ....................................................105
2.10.8 Test Shock.........................................................105
2.10.9 Manual ATP .......................................................106
2.10.10 Emergency Shock .............................................106
2.11 Special Features........................................................107
2.11.1 ICD Therapy Status ...........................................107
2.11.2 Home Monitoring ...............................................107
2.11.3 Real-time IEGM Transmission ..........................117
2.11.4 Capacitor Reforming .........................................118
2.11.5 Patient and Implant Data ...................................118
2.11.6 System Status ...................................................119
2.11.7 HF Monitor Statistics .........................................119
2.11.8 Holter Memory ...................................................121
2.11.9 Timing Statistics ....................................................2
2.11.10 Atrial Arrhythmias ..................................................3
2.11.11 Ventricular Arrhythmias .........................................3
Page 6
iv Lumax Technical Manual
2.11.12 Sensor ...................................................................3
2.11.13 Sensing..................................................................3
2.11.14 Impedances ...........................................................4
2.11.15 Automatic Threshold..............................................4
4.1 Implant Preparation .......................................................7
4.2 Lead System Evaluation..............................................12
4.3 Opening the Sterile Container.....................................12
4.4 Pocket Preparation......................................................13
4.5 Lead to Device Connection .........................................13
4.6 Blind Plug Connection.................................................16
4.7 Program the ICD/CRT-D .............................................17
4.8 Implant the ICD/CRT-D ...............................................18
5.1 General Considerations ..............................................23
5.2 Longevity .....................................................................24
5.2.1 Lumax 300/340 Devices......................................25
5.2.2 Lumax 500/540 Devices......................................26
5.3 Explantation.................................................................29
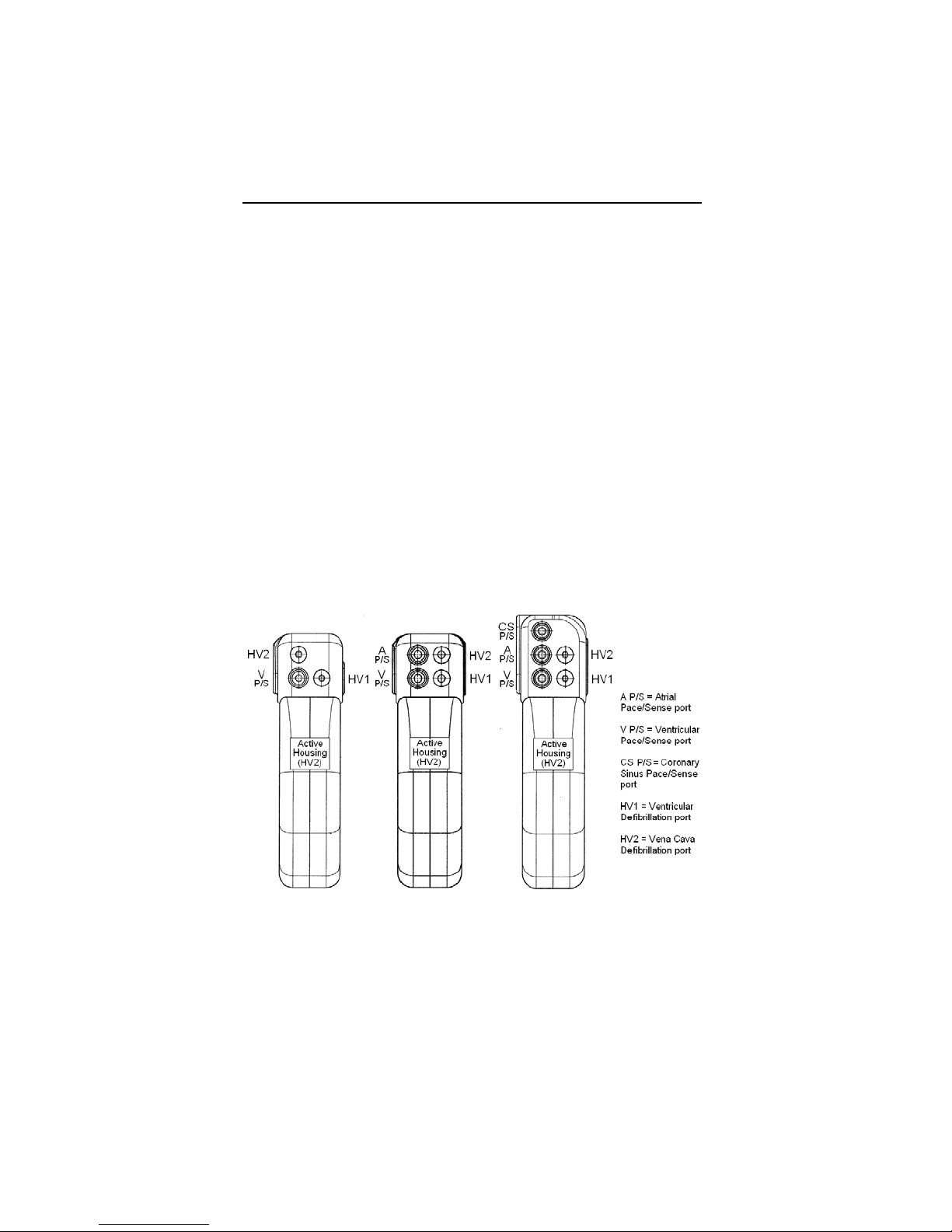
*Lumax VR (-T) and DR (-T) ICDs do not have coronary sinus
pace/sense ports
** Lumax VR (-T) ICDs do not have atrial pace/sense ports
Page 7

Lumax Technical Manual v
Lumax Specifications and Description
Battery Voltage: 3.2 Volts
300/500 models:
30 Joules programmed
Maximum Shock Energy:
340/540 models:
40 Joules programmed
Maximum Shock Energy:
Defibrillation Lead Ports Two DF-1 (3.2 mm)
Pacing Lead Ports Three IS-1 (3.2 mm)
(one for Lumax VR (-T)
and two for
Lumax DR (-T)s)
Dimensions:
Volume:
See Technical Details in
Section 6
Mass:
Housing Material: Titanium
Header Material: Epoxy Resin
Sealing Plug Material: Silicone
Page 8

vi Lumax Technical Manual
Page 9

Lumax Technical Manual 1
1. General
1.1 System Description
The Lumax family of Implantable Cardioverter Defibrillators
(ICDs) and Cardiac Resynchronization Therapy Defibrillators
(CRT-Ds) detect and treat ventricular tachyarrhythmias and
provide rate adaptive bradycardia pacing support. The HF and
HF-T versions of Lumax provide Cardiac Resynchronization
Therapy (CRT) through biventricular pacing. Both CRT-Ds and
ICDs detect and treat ventricular tachyarrhythmias and provide
rate adaptive bradycardia pacing support. They are designed to
collect diagnostic data to aid the physician’s assessment of a
patient’s condition and the performance of the implanted device.
The Lumax family of devices provides therapy for ventricular
tachyarrhythmias with a sophisticated range of programmable
anti-tachycardia pacing (ATP), and/or defibrillation therapy
features. The shock polarity and energy may be programmed to
tailor the therapy to appropriately treat each patient's
tachyarrhythmias. The ICDs/CRT-Ds provide shock therapies
with programmable energies from 5 to 40 joules.
The Lumax family of ICDs/CRT-Ds includes the following
members:
• Lumax HF - provides three chamber rate adaptive
bradycardia pacing support including biventricular pacing
via a left ventricular pacing lead. The CRT-D uses right
atrial and ventricular sensing/pacing leads to provide
enhanced atrial and ventricular tachyarrhythmia
discrimination through BIOTRONIK’s SMART
DetectionTM algorithm.
• Lumax HF-T - In addition to the functionality found with
HF model Lumax HF-T also has the added functionality of
BIOTRONIK’s Home Monitoring system. The Home
Monitoring System enables automatic exchange of
information about a patient’s cardiac status from the
implant to the physician remotely.
Page 10

2 Lumax Technical Manual
• Lumax DR - provides dual chamber rate adaptive
bradycardia pacing support. The ICD uses atrial and
ventricular sensing/pacing leads to provide enhanced
atrial and ventricular tachyarrhythmia discrimination
through BIOTRONIK’s SMART Detection
TM
algorithm.
• Lumax DR-T - In addition to the functionality found with
the DR model it also has the added functionality of
BIOTRONIK’s Home Monitoring system. The Home
Monitoring System enables automatic exchange of
information about a patient’s cardiac status from the
implant to the physician remotely.
• Lumax VR - provides single chamber rate adaptive
bradycardia pacing support as well as tachyarrhythmia
detection and therapy.
• Lumax VR-T - In addition to the functionality found with
standard VR model it also has the added functionality of
BIOTRONIK’s Home Monitoring system. The Home
Monitoring System enables automatic exchange of
information about a patient’s cardiac status from the
implant to the physician remotely.
The 300/500 and 340/540 designation for each of the abovedescribed models denote the maximum programmable shock
energy of 30 joules and 40 joules, respectively.
The Lumax 500/540 models also feature a third programmable
shock path for delivery of defibrillation/cardioversion shocks. The
shock path is programmable between the different shock coils
(SVC/RV) and/or the device housing. Section 2.8.3.6 provides
further details on the available shock configurations.
Additionally, the Lumax 500/540 models feature Automatic
Threshold Measurement (ATM) of ventricular pacing thresholds.
This feature is separately programmable for the right (RV) and left
(LV) ventricle. Section 2.3 provides further details.
Page 11

Lumax Technical Manual 3
The Lumax HF (-T) models have three IS-1 pacing/sensing
header ports and two DF-1 defibrillation/cardioversion ports. The
Lumax DR (-T) models have two IS-1 pacing/sensing header
ports. The Lumax VR (-T) models have one IS-1 pacing/sensing
header ports. IS-1 refers to the international standard whereby
leads and generators from different manufacturers are assured a
basic fit [Reference ISO 5841-3:1992]. DF-1 refers to the
international standard for defibrillation lead connectors
[Reference ISO 11318:1993].
External devices that interact with and test the implantable
devices are also part of the ICD/CRT-D System. These external
devices include the ICS 3000 Programming and Tachyarrhythmia
Monitoring System and the Implant Module System Analyzer or
Pacing System Analyzer for acute lead testing. The ICS 3000
programmer is used to interrogate and program the ICD/CRT-Ds.
1.2 Indications and Usage
The Lumax CRT-Ds are indicated for use in patients with all of the
following conditions:
• Indicated for ICD therapy
• Receiving optimized and stable Congestive Heart Failure
(CHF) drug therapy
• Symptomatic CHF (NYHA Class III/IV and LVEF ≤ 35%);
and
• Intraventricular conduction delay (QRS duration ≥130 ms)
The Lumax Implantable Cardioverter Defibrillators (ICDs) and
Cardiac Resynchronization Therapy Defibrillators (CRT-Ds) are
intended to provide ventricular anti-tachycardia pacing and
ventricular defibrillation, for automated treatment of lifethreatening ventricular arrhythmias.
Page 12

4 Lumax Technical Manual
1.3 Contraindications
The Lumax devices are contraindicated for use in patients with
the following conditions:
• Patients whose ventricular tachyarrhythmias may have
transient or reversible causes such as:
• Acute myocardial infarction
• Digitalis intoxication
• Drowning
• Electrocution
• Electrolyte imbalance
• Hypoxia
• Sepsis
• Patients with incessant ventricular fibrillation (VF) and
ventricular tachycardia (VT)
• Patients whose only disorder is brady arrhythmias or
atrial arrhythmias
1.4 Warnings and Precautions
MRI (Magnetic Resonance Imaging) - Do not expose a patient
to MRI device scanning. Strong magnetic fields may damage the
device and cause injury to the patient.
Electrical Isolation - To prevent inadvertent arrhythmia
induction, electrically isolate the patient during the implant
procedure from potentially hazardous leakage currents.
Left Ventricular Lead Systems – BIOTRONIK CRT-Ds may be
implanted with any legally marketed, compatible LV lead.
Compatibility is defined as:
• IS-1 pacing connector
• Active or passive fixation technology
• Insertion and withdrawal forces as specified by
ISO 5841-3 (IS-1)
The following LV leads were evaluated in the OPTION CRT/ATx
study with BIOTRONIK’s CRT-Ds:
• Guidant-Easytrak IS-1
• Guidant-Easytrak LV-1
Page 13

Lumax Technical Manual 5
• Guidant-Easytrak 2
• Guidant-Easytrak 3
• Medtronic-Attain
• St. Jude-Aescula
• St. Jude-Quicksite
• Biomec-Myopore Epicardial
• Medtronic-Epicardial 5071
• Medtronic-CapSure EPI
• Biotronik-ELC 54-UP
The following LV leads were bench tested for compatibility with
BIOTRONIK’s CRT-Ds:
• Guidant EasyTrak 4512 (unipolar)
• Guidant EasyTrak 4513 (bipolar)
• Guidant EasyTrak 3 4525 (bipolar)
• Medtronic Attain OTW 4193 (unipolar)
• Medtronic Attain OTW 4194 (bipolar)
• Medtronic Attain LV 2187 (unipolar)
• St. Jude Medical QuickSite 1056K (unipolar)
• ELA Situs OTW (unipolar)
• Biotronik Corox OTW 75-UP Steroid #346542 (unipolar)
• Biotronik Corox+ LV-H 75-BP #341885 (bipolar)
ICD Lead Systems – BIOTRONIK ICDs/CRT-Ds maybe
implanted with any legally marketed, compatible ICD lead.
Compatibility is defined as:
• IS-1 pacing and sensing connector(s)
• DF-1 shock coil connector(s)
• Integrated or dedicated bipolar pacing and sensing
configuration
• Active or passive fixation technology
• Single or dual defibrillation shock coil (s)
• High energy shock accommodation of at least 30 joules
• Insertion and withdrawal forces as specified by
ISO 5841-3 (IS-1) and ISO 11318:1993 (E) DF-1
Page 14

6 Lumax Technical Manual
The following leads were evaluated in a retrospective study with
BIOTRONIK’s ICDs/CRT-Ds:
• Medtronic Sprint 6932
• Medtronic Sprint 6943
• Medtronic Sprint Quattro 6944
• Medtronic Transvene RV 6936
• St. Jude (Ventritex) TVL- ADX 1559
• St. Jude SPL SP02
• Guidant Endotak DSP
• Guidant Endotak Endurance EZ, Endotak Reliance
• Guidant (Intermedics) 497-24.
The following leads were bench tested for compatibility with
BIOTRONIK’s ICDs/CRT-Ds:
• Guidant Endotak Endurance “CPI 0125”
• Guidant Endotak Reliance 0148
• Medtronic Sprint 6932
• Medtronic Sprint 6942
• Medtronic Sprint 6943
• Medtronic Sprint 6945
• Medtronic Sprint Quattro 6944
• St. Jude Riata 1571/65
• St. Jude SPL SPO1
Resuscitation Availability - Do not perform induction testing
unless an alternate source of patient defibrillation such as an
external defibrillator is readily available. In order to implant the
ICD/CRT-D system, it is necessary to induce and convert the
patient’s ventricular tachyarrhythmias.
Unwanted Shocks – Always program ICD Therapy to OFF prior
to handling the device to prevent the delivery of serious shocks to
the patient or the person handling the device during the implant
procedure.
Rate-Adaptive Pacing – Use rate-adaptive pacing with care in
patients unable to tolerate increased pacing rates.
Page 15

Lumax Technical Manual 7
1.4.1 Sterilization, Storage, and Handling
Device Packaging - Do not use the device if the device’s
packaging is wet, punctured, opened or damaged because the
integrity of the sterile packaging may be compromised. Return
the device to BIOTRONIK.
Re-sterilization - Do not re-sterilize and re-implant explanted
devices.
Storage (temperature) - Store the device between 5° to 45°C
(41° - 113° F) because temperatures outside this range could
damage the device.
Storage (magnets) - To avoid damage to the device, store the
device in a clean area, away from magnets, kits containing
magnets, and sources of electromagnetic interference (EMI).
Temperature Stabilization - Allow the device to reach room
temperature before programming or implanting the device
because temperature extremes may affect initial device function.
Use Before Date - Do not implant the device after the USE
BEFORE DATE because the device may have reduced longevity.
1.4.2 Device Implantation and Programming
Blind Plug - A blind plug must be inserted and firmly connected
into any unused header port to prevent chronic fluid influx and
possible shunting of high energy therapy.
Capacitor Reformation - Infrequent charging of the high voltage
capacitors may extend the charge times of the ICD/CRT-D. The
capacitors are reformed automatically at least every 85 days. For
further information, please refer to Section 2.11.4, Capacitor
Reforming.
Connector Compatibility – ICD/CRT-D and lead system
compatibility should be confirmed prior to the implant procedure.
Consult your BIOTRONIK representative regarding lead/pulse
generator compatibility prior to the implantation of an ICD/CRT-D
system. For further information, please refer to Appendix A.
Page 16

8 Lumax Technical Manual
ERI (Elective Replacement Indicator) - Upon reaching ERI, the
battery has sufficient energy remaining to continue monitoring for
at least three months and to deliver a minimum of six maximum
energy shocks. After this period (EOS), all tachyarrhythmia
detection and therapy is disabled. Bradycardia functions are still
active at programmed values until the battery voltage drops below
1.75 volts.
Magnets - Positioning of a magnet or the programming wand
over the ICD/CRT-D will suspend tachycardia detection and
treatment. The minimum magnet strength required to suspend
tachycardia treatment is 1.8 mT. When the magnet strength
decreases to less than 1 mT, the reed contact is reopened.
Programmed Parameters – Program the device parameters to
appropriate values based on the patient’s specific arrhythmias
and condition.
Programmers - Use only BIOTRONIK ICS 3000 programmers to
communicate with the device.
Sealing System - Failure to properly insert the torque wrench
into the perforation at an angle perpendicular to the connector
receptacle may result in damage to the sealing system and its
self-sealing properties.
Defibrillation Threshold - Be aware that the changes in the
patient’s condition, drug regimen, and other factors may change
the defibrillation threshold (DFT) which may result in nonconversion of the arrhythmia post-operatively. Successful
conversion of ventricular fibrillation or ventricular tachycardia
during arrhythmia conversion testing is no assurance that
conversion will occur post-operatively.
Manual Shocks – User-commanded shocks may be withheld if
the ICD/CRT-D is already busy processing a manual command or
the Battery Status is low.
Charge Time - When preparing a high energy shock the charge
circuit stops charging the capacitors after 20 seconds, and
delivers the stored energy as shock therapy. After the device
reaches ERI the stored energy may be less than the maximum
programmable energy for each shock.
Page 17

Lumax Technical Manual 9
Shipment Mode – The shipment mode is a factory set mode that
controls the charge current of automatic capacitor reformations.
This mode controls the charge current to avoid temporary low
battery readings. The shipment mode is automatically
deactivated as soon as electrophysiological tests (e.g.,
Impedance measurement) have been performed. To ensure
delivery of programmed shock energy, make sure shipment mode
is disabled prior to completion of implant procedure.
Shock Therapy Confirmation – Programming CONFIRMATION
to OFF may increase the incidence of the ICD/CRT-D delivering
inappropriate shocks.
Shock Impedance - If the shock impedance is less than twentyfive ohms, reposition the lead system to allow a greater distance
between the electrodes. Never implant the device with a lead
system that has measured shock impedance as less than twentyfive ohms. Damage to the device may result.
Negative AV Hysteresis – This feature insures ventricular
pacing, a technique which has been used in patients with
hypertrophic obstructive cardiomyopathy (HOCM) with normal AV
conduction in order to replace intrinsic ventricular activation. No
clinical study was conducted to evaluate this feature, and there is
conflicting evidence regarding the potential benefit of ventricular
pacing therapy for HOCM patients. In addition, there is evidence
with other patient groups to suggest that inhibiting the intrinsic
ventricular activation sequence by right ventricular pacing may
impair hemodynamic function and/or survival.
1.4.3 Lead Evaluation and Connection
Capping Leads - If a lead is abandoned rather than removed, it
must be capped to ensure that it is not a pathway for currents to
or from the heart.
Gripping Leads - Do not grip the lead with surgical instruments
or use excessive force or surgical instruments to insert a stylet
into a lead.
Kinking Leads - Do not kink leads. This may cause additional
stress on the leads that can result in damage to the lead.
Page 18

10 Lumax Technical Manual
Liquid Immersion - Do not immerse leads in mineral oil, silicone
oil, or any other liquid.
Short Circuit - Ensure that none of the lead electrodes are in
contact (a short circuit) during delivery of shock therapy as this
may cause current to bypass the heart or cause damage to the
ICD/CRT-D system.
Far-field sensing of signals from the atrium in the ventricular
channel or ventricular signals in the atrial channel should be
avoided by appropriate lead placement, programming of
pacing/sensing parameters, and maximum sensitivity settings. If
it is necessary to modify the Far Field Blanking parameter, the
parameter should be lengthened only long enough to eliminate
far-field sensing as evidenced on the IEGMs. Extending the
parameter unnecessarily may cause under sensing of actual atrial
or ventricular events.
Suturing Leads - Do not suture directly over the lead body as
this may cause structural damage. Use the appropriate suture
sleeve to immobilize the lead and protect it against damage from
ligatures.
Tricuspid Valve Bioprosthesis - Use ventricular transvenous
leads with caution in patients with a tricuspid valvular
bioprosthesis.
Setscrew Adjustment – Back-off the setscrew(s) prior to
insertion of lead connector(s) as failure to do so may result in
damage to the lead(s), and/or difficulty connecting lead(s).
Cross Threading Setscrew(s) – To prevent cross threading the
setscrew(s), do not back the setscrew(s) completely out of the
threaded hole. Leave the torque wrench in the slot of the
setscrew(s) while the lead is inserted.
Tightening Setscrew(s) – Do not overtighten the setscrew(s).
Use only the BIOTRONIK supplied torque wrench.
Sealing System – Be sure to properly insert the torque wrench
into the perforation at an angle perpendicular to the connector
receptacle. Failure to do so may result in damage to the plug and
its self-sealing properties.
Page 19

Lumax Technical Manual 11
1.4.4 Follow-up Testing
Defibrillation Threshold - Be aware that changes in the patient’s
condition, drug regimen, and other factors may change the
defibrillation threshold (DFT), which may result in non-conversion
of the arrhythmia post-operatively. Successful conversion of
ventricular fibrillation or ventricular tachycardia during arrhythmia
conversion testing is no assurance that conversion will occur
post-operatively.
Resuscitation Availability - Ensure that an external defibrillator
and medical personnel skilled in cardiopulmonary resuscitation
(CPR) are present during post-implant device testing should the
patient require external rescue.
Safe Program – Within the EP Test screen, pressing the “Safe
Program” key on the programmer head immediately sends the
safe program to the ICD/CRT-D.
1.4.5 Pulse Generator Explant and Disposal
Device Incineration – Never incinerate the ICD/CRT-D due to
the potential for explosion. The ICD/CRT-D must be explanted
prior to cremation.
Explanted Devices – Return all explanted devices to
BIOTRONIK.
Unwanted Shocks – Always program ICD Therapy to OFF prior
to handling the device to prevent the delivery of serious shocks to
the patient or the person handling the device during the implant
procedure.
1.4.6 Hospital and Medical Hazards
Electromagnetic interference (EMI) signals present in hospital and
medical environments may affect the function of any ICD/CRT-D
or pacemaker. The ICD/CRT-D is designed to selectively filter out
EMI noise. However, due to the variety of EMI signals, absolute
protection from EMI is not possible with this or any other
ICD/CRT-D.
Page 20

12 Lumax Technical Manual
The ICD/CRT-D system should have detection and therapy
disabled (OFF) prior to performing any of the following medical
procedures. In addition, the ICD/CRT-D should be checked after
the procedures to assure proper programming:
Diathermy - Diathermy therapy is not recommended for
ICD/CRT-D patients due to possible heating effects of the pulse
generator and at the implant site. If diathermy therapy must be
used, it should not be applied in the immediate vicinity of the
pulse generator or lead system.
Electrocautery - Electrosurgical cautery could induce ventricular
arrhythmias and/or fibrillation, or may cause device malfunction or
damage. If use of electrocautery is necessary, the current path
and ground plate should be kept as far away from the pulse
generator and leads as possible (at least 6 inches (15 cm)).
External Defibrillation - The device is protected against energy
normally encountered from external defibrillation. However, any
implanted device may be damaged by external defibrillation
procedures. In addition, external defibrillation may also result in
permanent myocardial damage at the electrode-tissue interface as
well as temporary or permanent elevated pacing thresholds. When
possible, observe the following precautions:
• Position the adhesive electrodes or defibrillation paddles
of the external defibrillator anterior-posterior or along a
line perpendicular to the axis formed by the implanted
device and the heart.
• Set the energy to a level not higher than is required to
achieve defibrillation.
• Place the paddles as far as possible away from the
implanted device and lead system.
• After delivery of an external defibrillation shock,
interrogate the ICD/CRT-D to confirm device status and
proper function.
Lithotripsy - Lithotripsy may damage the ICD/CRT-D. If
lithotripsy must be used, avoid focusing near the ICD/CRT-D
implant site.
MRI (Magnetic Resonance Imaging) - Do not expose a patient
to MRI device scanning. Strong magnetic fields may damage the
device and cause injury to the patient.
Page 21

Lumax Technical Manual 13
Radiation - High radiation sources such as cobalt 60 or gamma
radiation should not be directed at the pulse generator. If a
patient requires radiation therapy in the vicinity of the pulse
generator, place lead shielding over the device to prevent
radiation damage and confirm its function after treatment.
Radio Frequency Ablation - Prior to performing an ablation
procedure, deactivate the ICD/CRT-D during the procedure.
Avoid applying ablation energy near the implanted lead system
whenever possible.
1.4.7 Home and Occupational Hazards
Patients should be directed to avoid devices that generate strong
electromagnetic interference (EMI) or magnetic fields. EMI could
cause device malfunction or damage resulting in non-detection or
delivery of unneeded therapy. Moving away from the source or
turning it off will usually allow the ICD/CRT-D to return to its
normal mode of operation.
The following equipment (and similar devices) may affect normal
ICD/CRT-D operation: electric arc or resistance welders, electric
melting furnaces, radio/television and radar transmitters,
power-generating facilities, high-voltage transmission lines, and
electrical ignition systems (of gasoline-powered devices) if
protective hoods, shrouds, etc., are removed.
1.4.8 Cellular Phones
Testing has indicated there may be a potential interaction
between cellular phones and BIOTRONIK ICD/CRT-D systems.
Potential effects may be due to either the cellular phone signal or
the magnet within the telephone and may include inhibition of
therapy when the telephone is within 6 inches (15 centimeters) of
the ICD/CRT-D, when the ICD/CRT-D is programmed to standard
sensitivity.
Patients having an implanted BIOTRONIK ICD/CRT-D who
operate a cellular telephone should:
• Maintain a minimum separation of 6 inches
(15 centimeters) between a hand-held personal cellular
telephone and the implanted device.
Page 22

14 Lumax Technical Manual
• Set the telephone to the lowest available power setting, if
possible.
• Patients should hold the phone to the ear opposite the
side of the implanted device. Patients should not carry
the telephone in a breast pocket or on a belt over or
within 6 inches (15 centimeters) of the implanted device
as some telephones emit signals when they are turned
ON, but not in use (i.e., in the listen or stand-by mode).
Store the telephone in a location opposite the side of
implant.
Based on results to date, adverse effects resulting from
interactions between cellular telephones and implanted
ICDs/CRT-Ds have been transitory. The potential adverse effects
could include inhibition or delivery of additional therapies. If
electromagnetic interference (EMI) emitting from a telephone
does adversely affect an implanted ICD/CRT-D, moving the
telephone away from the immediate vicinity of the ICD/CRT-D
should restore normal operation. A recommendation to address
every specific interaction of EMI with implanted ICDs/CRT-Ds is
not possible due to the disparate nature of EMI.
1.4.9 Electronic Article Surveillance (EAS)
Equipment such as retail theft prevention systems may interact
with pulse generators. Patients should be advised to walk directly
through and not to remain near an EAS system longer than
necessary.
1.4.10 Home Appliances
Home appliances normally do not affect ICD/CRT-D operation if
the appliances are in proper working condition and correctly
grounded and shielded. There have been reports of the
interaction of electric tools or other external devices (e.g. electric
drills, older models of microwave ovens, electric razors, etc.) with
ICDs/CRT-Ds when they are placed in close proximity to the
device.
Page 23

Lumax Technical Manual 15
1.4.11 Home Monitoring
Patient’s Ability - Use of the Home Monitoring system requires
the patient and/or caregiver to follow the system instructions and
cooperate fully when transmitting data.
If the patient cannot understand or follow the instructions because
of physical or mental challenges, another adult who can follow the
instructions will be necessary for proper transmission.
Use in Cellular Phone Restricted Areas - The mobile patient
device (transmitter/receiver) should not be utilized in areas where
cellular phones are restricted or prohibited (i.e., commercial
aircraft).
Event-Triggered Report – A timely receipt of the event report
cannot be guaranteed. The receipt is also dependent on whether
the patient was physically situated in the required coverage range
of the patient device at the time the event information was sent.
Not for Diagnosis - The data transmitted by Home Monitoring
are not suitable for diagnosis, because not all information
available in the implant is being transmitted.
Follow-Ups - The use of Home Monitoring does not replace
regular follow-up examinations. Therefore, when using Home
Monitoring, the time period between follow-up visits may not be
extended.
Page 24

16 Lumax Technical Manual
1.5 Potential/Observed Effects of the
Device on Health
1.5.1 Potential Adverse Events
The following are possible adverse events that may occur relative
to the implant procedure and chronic implant of the CRT-D:
• Air embolism
• Allergic reactions to
contrast media
• Arrhythmias
• Bleeding
• Body rejection
phenomena
• Cardiac tamponade
• Chronic nerve damage
• Damage to heart valves
• Device migration
• Elevated pacing
thresholds
• Extrusion
• Fluid accumulation
• Hematoma
• Infection
• Keloid formation
• Lead dislodgment
• Lead fracture/ insulation
damage
• Lead-related thrombosis
• Local tissue reaction /
fibrotic tissue formation
• Muscle or nerve
stimulation
• Myocardial damage
• Myopotential sensing
• Pacemaker mediated
tachycardia
• Pneumothorax
• Pocket erosion
• Thromboembolism
• Undersensing of intrinsic
signals
• Venous occlusion
• Venous or cardiac
perforation
Page 25

Lumax Technical Manual 17
In addition, patients implanted with the ICD/CRT-D system may
have the following risks. These are the same risks relate with
implantation of any ICD/CRT-D system:
• Acceleration of
arrhythmias (speeding
up heart rhythm caused
by the CRT-D)
• Dependency
• Depression
• Fear of premature
battery depletion (fear
that battery will stop
working before predicted
time)
• Fear of shocking while
awake
• Fear that shocking
ability may be lost
There may be other risks associated with this device that are not
currently unforeseeable.
• Anxiety about the CRT-D
resulting from frequent
shocks
• Imagined shock (phantom
shock)
• Inappropriate detection of
ventricular arrhythmias
• Inappropriate shocks
• Potential death due to
inability to defibrillate or
pace
• Shunting current or
insulating myocardium
during defibrillation with
external or internal paddles
1.5.2 Observed Adverse Events
Reported Adverse Events are classified as either observations or
complications. Complications are defined as clinical events that
require additional invasive intervention to resolve. Observations
are defined as clinical events that do not require additional
invasive intervention to resolve.
1.5.2.1 Kronos LV-T Study
NOTE:
The Kronos LV-T CRT-D is an earlier generation of
BIOTRONIK devices. The Lumax CRT-Ds are based upon
the Kronos LV-T and other BIOTRONIK CRT-Ds and ICDs
(i.e., Tupos LV/ATx CRT-D, Lexos and Lumos families of
ICDs).
Page 26
18 Lumax Technical Manual
s
The HOME-CARE Observational study, conducted outside the US
on the Kronos LV-T cardiac resynchronization defibrillator
(CRT-D) in patients with congestive heart failure (CHF) involved
45 devices implanted with a cumulative implant duration of
202 months (mean implant duration of 4.5 months).
Of the 31 adverse events reported, there have been
26 observations in 23 patients and 5 complications in 3 patients
with a cumulative implant duration of 202 months (16.8 patientyears). 6.7% of the enrolled patients have experienced a
complication with two patients experiencing 2 separate
complications. The rate of complications per patient-year was
0.30. 51% of the enrolled study patients had a reported
observation with 3 patients having more than 1 observation. The
rate of observations per patient-year is 1.54. Complications and
observations for the patient group are summarized in Tab le 1 and
Table 2, respectively.
Table 1: Summary of Complications – Kronos LV-T
Category
Number
of
Patients
% of
Patient
Number
Per
patient-
year
Left Ventricular Lead Related
Dislodgement 1 2.2% 1
No Capture 1 2.2% 1
Total 2 4.4% 2
0.06
0.06
0.12
ICD Lead Related
Dislodgement 1 2.2% 1
Elevated Pacing
Threshold
12.2% 1
Total 2 4.4% 2
0.06
0.06
0.12
Unrelated to CRT-D or Leads
Hemathorax 1 2.2% 1
Total 1 2.2% 1
Overall
Complication Totals
Number of Patients = 45, Number of Patient-Years = 16.8
36.7% 5
0.06
0.06
0.30
Page 27

Lumax Technical Manual 19
Table 2: Summary of Observations – Kronos LV-T
Category
Unsuccessful LV
lead implant
Elevated LV pacing
threshold
Phrenic nerve
stimulation
Elevated DFT
measurement
T-wave oversensing
Worsening CHF
Elevated RV pacing
threshold
Hepatitis
Arrhythmias
Cardiac
Decompensation
Number
of
Patients
%of
Patients
Number
8 17.8% 8 0.48
5 11.1% 5 0.30
3 6.7% 3 0.18
2 4.4% 2 0.12
2 4.4% 2 0.12
2 4.4% 2 0.12
1 2.2% 1 0.06
1 2.2% 1 0.06
1 2.2% 1 0.06
1 2.2% 1 0.06
per
patient-
year
All Observations 23 51.1% 26 1.54
Number of Patients = 45, Number of Patient-Years = 16
Two patient deaths were reported during the HOME-CARE
Observational Study. One death resulted from worsening heart
failure and the second death resulted from cardiogenic shock due
to ischemic cardiomyopathy. None of the deaths were related to
the implanted CRT-D system. There were no device explants
during the HOME-CARE Observational Study.
Page 28

20 Lumax Technical Manual
1.5.2.2 Tupos LV/ATx Study
NOTE:
The clinical study information included in this section and in
Section 1.6.2 was performed with the Tupos LV/ATx CRT-D,
which is an earlier version of the Lumax CRT-D/ICD families.
The clinical study data presented here is applicable because
the Lumax family are downsized versions of the
Tupos LV/ATx CRT-D and Tachos ICD families. The Lumax
family is slightly different as compared to the Tupos LV/ATx
(and Tachos family) in the following areas:
• Reduced size from 50/48 cc to 40/35 cc
• Addition of Home Monitoring functionality
• Addition of triggered pacing for biventricular pacing
modes
• True three chamber pacing and sensing capabilities
(CRT-Ds)
The OPTION CRT/ATx study was a prospective, randomized,
multi-center study to demonstrate the safety and effectiveness of
the investigational Tupos LV/ATx Cardiac Resynchronization
Therapy Defibrillator (CRT-D) in patients with congestive heart
failure (CHF) and atrial tachyarrhythmias. All patients enrolled
into the clinical study were randomly assigned to either the study
group or the control group at a 2 to 1 ratio. Patients in the study
group were implanted with the Tupos LV/ATx. Patients in the
control group were implanted with a legally marketed ICD that
provides CRT.
Of the 278 adverse events reported in the Tupos LV/ATx study
group, there have been 210 observations in 104 patients and 68
complications in 50 patients with a cumulative implant duration of
1240.4 months (101.9 patient-years). 37.6% of the enrolled study
patients have experienced a complication. The rate of
complications per patient-year is 0.67. 78.2% of the enrolled
study patients have a reported observation. The rate of
observations per patient-year is 2.06.
Page 29

Lumax Technical Manual 21
s
Complications and observations for the Tupos LV/ATx study
group are summarized in Table 3 and Ta ble
4. The total number
of patients may not equal the sum of the number of patients listed
in each category, as an individual patient may have experienced
more than one complication or observation.
Table 3: Summary of Complications – Tupos LV/ATx
Number
Category
Hematoma 4 3.01% 4 0.04
Pneumothorax 2 1.50% 2 0.02
Total 6 4.51% 6 0.06
Dislodgement 3 2.26% 3 0.03
Total 3 2.26% 3 0.03
High threshold/ No
capture
Diaphragmatic/
Intercostal
stimulation (RV)
Total 3 2.26% 3 0.03
High threshold/
Intermittent
biventricular capture/
No capture
Unable to implant
lead via coronary
sinus
Dislodgement 4 3.01% 4 0.04
Diaphragmatic/
Intercostal
stimulation
Total 27 20.3% 29 0.28
of
Patients
Procedure Related
Atrial Lead Related
2 1.50% 2 0.02
1 0.75% 1 0.01
11 8.27% 12 0.12
11 8.27% 11 0.11
1 0.75% 2 0.02
% of
Patients
ICD Lead Related
LV Lead Related
Number of
omplications
Complication
per patient-
year
Page 30
22 Lumax Technical Manual
s
Table 3: Summary of Complications – Tupos LV/ATx
Number
Category
Infection 3 2.26% 7 0.07
Device migration 4 3.01% 4 0.04
Elective replacement
indicator reached
Inductions and
conversions
Unable to interrogate
device
Total 12 9.02% 17 0.17
Total Procedure
and Device Related
Non-CHF Cardiac
Symptoms
Ventricular
arrhythmias
Other medical 2 1.50% 2 0.02
Atrial arrhythmia 1 0.75% 1 0.01
Total 9 6.77% 10 0.10
Total – All Patients
and Categories
of
Patients
4 3.01% 4 0.04
1 0.75% 1 0.01
1 0.75% 1 0.01
43 32.33% 58 0.57
Other Medical Related
4 3.01% 4 0.04
2 1.50% 3 0.03
50 37.59% 68 0.67
% of
Patients
Device Related
Number of
omplications
Complication
per patient-
year
Number of Patients = 133, Number of Patient-Years = 101.9
* 1 Unanticipated Adverse Device Effect (UADE) occurred with a
Tupos LV/ATx CRT-D during the OPTION clinical study. The device was
explanted after it was unable to be interrogated with the programmer
software and no pacing output was evident. The analysis showed an
appropriately depleted battery and no anomalies with the IC module. The
battery depletion strongly suggests that the high voltage circuit was
activated over a prolonged period due to a single-bit execution path
failure. The current programmer software with Automatic Battery
Management (ABM) would have prevented the battery from becoming
completely depleted. There were no other instances of this failure
mechanism in Tupos LV/ATx devices.
Page 31

Lumax Technical Manual 23
For the Tupos LV/ATx study group, there were 210 observations
in 104 patients with cumulative implant duration of 1240.4 months
(101.9 patient years). 78.2% of the enrolled study patients have a
reported observation. The rate of observations per patient-year
was 2.06. Table 4 summarizes by category each type of
observation for the study group.
Table 4: Summary of Observations – Tupos LV/ATx
Number
Category
Hematoma 10 7.52% 10 0.10
Cardiac arrest 2 1.50% 2 0.02
Unable to implant
system
Total 13 9.77% 13 0.13
Dislodgement 1 0.75% 1 0.01
High threshold 1 0.75% 1 0.01
Total 2 1.50% 2 0.02
High threshold/No
capture
Total 1 0.75% 1 0.01
High threshold/
Intermittent
biventricular capture/
No capture
Diaphragmatic/
Intercostal stimulation
Total 30 22.56% 32 0.31
of
Patients
Procedure Related
1 0.75% 1 0.01
Atrial Lead Related
ICD Lead Related
1 0.75% 1 0.01
LV Lead Related
24 18.05% 24 0.24
8 6.02% 8 0.08
% of
Patients
Number
per patient-
year
Page 32

24 Lumax Technical Manual
Table 4: Summary of Observations – Tupos LV/ATx
Number
Category
Infection 1 0.75% 1 0.01
Inductions and
conversions
Inappropriate sensing 20 15.04% 20 0.20
Symptomatic with
biventricular pacing
Total 25 18.80% 29 0.28
Total Procedure,
Lead and Device
Related
Non-CHF Cardiac
Symptoms
Ventricular
arrhythmias
Other medical 26 19.55% 32 0.31
Atrial arrhythmia 14 10.53% 14 0.14
Dizziness 4 3.01% 4 0.04
Medication 5 3.76% 5 0.05
Worsening CHF 46 34.59% 46 0.45
Total 82 61.65% 133 1.31
Total – All Patients
and Categories
Number of Patients = 133 Number of Patient-Years = 101.9
of
Patients
Device Related
6 4.51% 6 0.06
2 1.50% 2 0.02
61 45.86% 77 0.76
Other Medical Related
21 15.79% 21 0.21
11 8.27% 11 0.11
104 78.20% 210 2.06
% of
Patients
Number
per patient-
year
Page 33

Lumax Technical Manual 25
There have been 4 patient deaths reported for the control group
(out of 67 total control patients) and 10 patient deaths have been
reported for the study group (out of 133 total study patients).
None of the deaths were related to the implanted CRT-D system.
One patient in the control group died prior to receiving a
biventricular device implant. There is no significant difference
between the number of deaths in the study group versus the
control group (p = 0.777, Fisher's Exact Test, 2 sided). Table 5
provides a summary of reported patient deaths and Table 6
provides survival percentages by follow-up interval during the first
12 months of study participation.
Table 5: Summary of Patient Deaths
Category of
Death
Study
(N = 133)
Control
(N = 67)
Number of Patients Number of Patients
Sudden Cardiac 1 1
Non-Sudden
52
Cardiac
Non-Cardiac 4 1
All Causes
10 4
Figure 1 shows the associated Kaplan-Meier survival curves for
the study and control group. The significance level for the
difference between the two study groups based on a Log Rank
test was p = 0.795.
Page 34

26 Lumax Technical Manual
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
Cumulative Survival
0.2
0.1
0.0
Log Rank = 0.795
Control
Study
211815129630
Survival Time (months)
Figure 1: Kaplan-Meier Survival Curves
Table 6 Survival Table
Study Group
(n = 133)
Control Group
(n = 66)
Number % Number %
Enrollment 133 100.00% 67 100.00%
3-month 131 98.50% 63 94.03%
6-month 127 95.49% 63 94.03%
12-month 123 92.48% 63 94.03%
Page 35

Lumax Technical Manual 27
1.6 Clinical Studies
The Kronos LV-T Clinical study (HOME-CARE, Section 1.6.1)
supports the safety of the Lumax CRT-D/ICD device family.
Additionally, because the Tupos LV/ATx and the Lumax CRT-D
devices have identical CRT and ventricular ICD therapy, the
effectiveness results from the OPTION CRT/ATx IDE Clinical
study (Tupos LV/ATx, Section 1.6.2) support the effectiveness of
the Lumax family.
1.6.1 Kronos LV-T Study
The purpose of the HOME-CARE Observational Study is to
demonstrate the safety of the CE-marked Kronos LV-T cardiac
resynchronization defibrillator (CRT-D) in patients with congestive
heart failure (CHF).
1.6.1.1 Methods
The multi-center, non-randomized observational study was
designed to evaluate the safety of the Kronos LV-T through an
analysis of the complication-free rate through three months.
The Home-CARE Observational Study Primary Endpoint was to
evaluate complications (adverse events that require additional
invasive intervention to resolve) related to the implanted CRT
system which includes the Kronos LV-T, the right atrial lead, the
right ventricular ICD lead, and the left ventricular lead
Inclusion Criteria
To support the objectives of this investigation, patients were
required to meet the following inclusion criteria prior to enrollment:
• Indication for Cardiac Resynchronization Therapy
• Sufficient GSM-network coverage in the patient’s area
• Age greater than or equal to 18 years
Page 36

28 Lumax Technical Manual
Exclusion Criteria
To support the objectives of this investigation, the exclusion
criteria at the time of patient enrollment included the following:
• Permanent atrial fibrillation
• Myocardial infarction or unstable angina pectoris within
the last 3 prior to enrollment
• Planned cardio-surgical intervention within 3 months after
enrollment (e.g. PTCA, CABG, HTX)
• Acute myocarditis
• Life expectancy less than 6 months
• Pregnant or breast-feeding woman
• Drug or Alcohol abuse
• The patient is mentally or physically unable to take part in
the observational study
• No signed declaration of consent for the patient
At the enrollment screening, the physician evaluated the patient
to verify that all inclusion/exclusion criteria were met in
accordance to the protocol and the patient signed the informed
consent. After successful enrollment, all patients were implanted
with the Kronos LV-T CRT-D. Evaluations at the One- and Threemonth follow-ups included resting ECG, NYHA classification,
medications, and activation of Home Monitoring.
1.6.1.2 Summary of Clinical Results
The study involved 45 patients (37 males, 82.2%, and 8 females,
17.8%), with a mean age of 64 years (range: 36-79), a left
ventricular ejection fraction of 26 % (range: 15-43), NYHA Class
III (NHYA Class 1 (2.3%), Class II (11.4%), Class III (79.5%),
Class IV (6.8%)) and QRS duration of 154 ms (range: 84-208).
The mean implant duration was 4.5 months with a cumulative
implant duration of 202 months. The patient follow-up compliance
rate was 95.9% out of 221 required follow-ups.
Page 37

Lumax Technical Manual 29
Primary Endpoint
The safety of the Kronos LV-T was evaluated based on
complications (adverse events that require additional invasive
intervention to resolve) related to the implanted CRT system
which includes the Kronos LV-T, the right atrial lead, the right
ventricular ICD lead, and the left ventricular lead. 5 complications
were seen in 3 patients with cumulative implant duration of 202
months (16.8 patient-years). 6.7% of the patients had a reported
complication. The rate of complications per patient-year is 0.30.
The freedom from Kronos LV-T system-related complications is
93.3% with a two sided lower 95% confidence bound of 83.8%.
The null hypothesis is rejected, and it is concluded that the
complication-free rate is equivalent to 85% within 10%.
1.6.2 Tupos LV/ATx Study
NOTE:
The clinical study information included in this section was
performed with the Tupos LV/ATx CRT-D, which is an earlier
version of the Lumax CRT-D/ICD families. The clinical study
data presented here is applicable because the Lumax family
are downsized versions of the Tupos LV/ATx CRT-D and
Tachos ICD families. The Lumax family is slightly different as
compared to the Tupos LV/ATx (and Tachos family) in the
following areas:
• Reduced size from 50/48 cc to 40/35 cc
• Addition of Home Monitoring functionality
• Addition of triggered pacing for biventricular pacing
modes
• True three chamber pacing and sensing capabilities
(CRT-Ds)
Page 38

30 Lumax Technical Manual
1.6.2.1 Study Overview
The purpose of the prospective, randomized, multi-center
OPTION CRT/ATx study was to demonstrate the safety and
effectiveness of the investigational Tupos LV/ATx Cardiac
Resynchronization Therapy Defibrillator (CRT-D) in patients with
congestive heart failure (CHF) and atrial tachyarrhythmias.
Patients in the study group were implanted with a BIOTRONIK
Tupos LV/ATx. Patients in the control group were implanted with
any legally marketed CRT-D. Patients in both the study and
control groups were implanted with a legally marketed left
ventricular lead.
1.6.2.2 Methods
Primarily, the study evaluates and compares the functional
benefits of CRT between the two randomized groups using a
composite endpoint consisting of a six-minute walk test (meters
walked) and quality of life measurement (assessed using the
Minnesota Living with Heart Failure Questionnaire). Relevant
measurements were completed twice for each patient: once at the
Baseline evaluation (two-week post implant follow-up) and again
at a six-month follow-up evaluation. The data collected during this
clinical study was used to demonstrate equivalent treatment of
CHF in both the study and control groups. This study also
evaluated other outcomes including: the effectiveness of atrial
therapy to automatically convert atrial tachyarrhythmias, the
percentage of time CRT is delivered, and other measures of CHF
status including NYHA classification, peak oxygen consumption
during metabolic exercise testing, and the rate of hospitalization
for CHF.
Inclusion Criteria
To support the objectives of this investigation, patients were
required to meet the following inclusion criteria prior to enrollment:
• Stable, symptomatic CHF status
• NYHA Class III or IV congestive heart failure
• Left ventricular ejection fraction ≤ 35% (measured within
Six-Months prior to enrollment)
• Intraventricular conduction delay (QRS duration greater
than or equal to 130 ms)
Page 39

Lumax Technical Manual 31
• For patients with an existing ICD/CRT-D, optimal and
stable CHF drug regimen including ACE-inhibitors and
beta-blockers unless contraindicated (stable is defined as
changes in dosages less than 50% during the last 30
days)
• Indicated for ICD therapy
• History or significant risk of atrial tachyarrhythmias
• Willing to receive possibly uncomfortable atrial shock
therapy for the treatment of atrial tachyarrhythmias
• Able to understand the nature of the study and give
informed consent
• Ability to tolerate the surgical procedure required for
implantation
• Ability to complete all required testing including the sixminute walk test and cardiopulmonary exercise testing
• Available for follow-up visits on a regular basis at the
investigational site
• Age greater than or equal to 18 years
Exclusion Criteria
To support the objectives of this investigation, the exclusion
criteria at the time of patient enrollment included the following:
• Previously implanted CRT device
• ACC/AHA/NASPE indication for bradycardia pacing
(sinus node dysfunction)
• Six-minute walk test distance greater than 450 meters
• Chronic atrial tachyarrhythmias refractory to
cardioversion shock therapy
• Receiving intermittent, unstable intravenous inotropic
drug therapy (patients on stable doses of positive
inotropic outpatient therapy for at least One-Month are
permitted)
• Enrolled in another cardiovascular or pharmacological
clinical investigation
• Expected to receive a heart transplant within 6 months
• Life expectancy less than 6 months
• Presence of another life-threatening, underlying illness
separate from their cardiac disorder
Page 40

32 Lumax Technical Manual
• Acute myocardial infarction, unstable angina or cardiac
revascularization within the last 30 days prior to
enrollment
• Conditions that prohibit placement of any of the lead
systems
1.6.2.3 Summary of Clinical Results
A total of 200 patients were enrolled in the OPTION CRT/ATx
clinical study at 25 sites:
There were 133 study patients and 67 active control patients in
this prospective, multi-center, randomized clinical study. For the
study group, there were 129 successful implants (91.4%) of the
Tupos LV/ATx CRT-D system. For the active control group, there
were 64 successful implants (92.2%) of the legally marketed
CRT-D systems.
Patient Accountability
After randomization and enrollment, 7 patients (4 in the study
group and 3 in the control group) did not receive an implant. The
reasons for patients not receiving an implant are outlined in
Figure 2.
Page 41

Enrolled and Randomized
Patients
Study 133
Control 67
Implant Attempted
Study 130
Control 65
Successful implant
Study 129
Control 64
Patients completed 6-Month
Follow-up
Study 100
Control 49
Lumax Technical Manual 33
No implant Attempted
Withdrawal of Consent
Study 2
Control 1
Not Meeting Inclusion Criteria
Study 1
Control 1
Unsuccessful implant
Withdrawal of IC before 2nd Attempt
Study 1
Control 0
Expired before Second Attempt
Study 0
Control 1
6-Month Follow-up Data
Patient Death before 6-Month
Study 7
Control 3
Withdrawal before 6-Month
Study 1
Control 2
Not Reached 6-Month FU
or Data Pending
Study 21
Control 10
Figure 2: Patient Accountability
Overall Results
• There were 192 endocardial and 19 epicardial leads
implanted in 193 patients. Investigators were allowed to
choose among any legally marketed LV lead according to
familiarity with the lead and patient anatomy. The
Tupos LV/ATx CRT-D was implanted with 7 endocardial
and 4 epicardial lead models from 6 different
manufacturers. There were no adverse events reported
attributable to lead-generator incompatibility.
• The cumulative implant duration was 1240.4 months with
a mean duration of 9.6 months for the study group. The
cumulative implant duration is 596.5 months with a mean
duration of 9.3 months for the control group.
Page 42

34 Lumax Technical Manual
• For the study group, there have been 278 adverse events
(210 observations in 104 patients and 68 complications in
50 patients). There has been one unanticipated adverse
device effect reported.
• For the control group, there have been 105 adverse
events (81 observations in 44 patients and 24
complications in 19 patients). There have been no
unanticipated adverse device effects reported.
• There have been 10 patient deaths reported in the study
group and 4 patient deaths reported in the control group.
The clinical investigators have determined that no deaths
were related to the study device.
1.6.2.4 Primary Endpoint 1: Six Minute Walk Test & QOL
(Effectiveness)
The purpose of Primary Endpoint 1 is to evaluate the
effectiveness of the Tupos LV/ATx system in providing CRT as
measured by the average composite rate of improvement in six
minute walk test and QOL.
Table 7 presents the average composite rate of improvement in
six minute walk test distance and QOL score, the average 6minute walk test distance and the average QOL score at Baseline
and at the Six-Month follow-up, as well as the average difference
in 6-minute walk test distance and QOL score between Baseline
and the Six-Month follow-up for the Study and Control Groups for
those patients with six minute walk test data and complete QOL
data at both Baseline and the Six-Month follow-up.
Page 43

Lumax Technical Manual 35
Table 7: Composite of Six Minute Walk Test and QOL
(Effectiveness)
Category
Distance Walked at
Baseline
Distance Walked at
Six-Months
∆ Distance Walked
QOL Score at
Baseline
QOL Score at Six-
Months
∆ in QOL Score**
Study
Group
(N = 74)
Mean ± SE
310.51 ±
10.89
340.77 ±
12.32
30.26 ±
10.40
17.27% ±
5.59%
44.39 ± 2.78 45.53 ± 4.13 0.817
28.68 ± 2.66 33.95 ± 4.35 0.279
15.72± 2.83
19.08% ±
12.21%
Control
Group
(N = 38)
Mean ± SE
288.76 ±
15.37
301.84 ±
17.02
13.08 ±
13.05
8.71% ±
5.26%
11.58 ± 3.45
-13.42% ±
34.54%
P-value*
0.249
0.067
0.322
0.326
0.376
0.281
Composite Rate***
*The calculated p-values are associated with a Student's t-test (2-sided)
of the equality of means in the two groups, except for the p-value of the
composite rate, which is associated with a test of equivalence (noninferiority).
**∆ in QOL Score is calculated as the average of the individual
differences between Baseline and Six-Months for each patient. Negative
values for mean ∆ QOL in percent are possible when positive mean
values for absolute changes in QOL are recorded. In some cases, small,
negative changes in absolute QOL scores resulted in relatively large
percentage changes.
***The Composite Rate (=(∆ Distance Walked (%) + ∆ QOL Score (%)) /
2) is calculated for each patient and then averaged to obtain the
Composite Rates. For all calculations, a positive number represents
improvement from Baseline to Six-Months.
18.18% ±
7.07%
-2.36% ±
17.73%
0.030
Page 44

36 Lumax Technical Manual
1.6.2.5 Effectiveness Endpoint Analysis and Conclusions
A composite rate of six minute walk test and QOL improvement
from Baseline to the Six-Month follow-up is evaluated as a
measure of CRT effectiveness. For this analysis both six minute
walk test and QOL are equally weighted at 50%.
The mean difference in the composite rate between study and
control group was 20.53% with an associated one-sided, 95%
confidence bound is (-6.10%). The p-value for non-inferiority
within 10% is 0.030. The analysis of the composite rate in six
minute walk test distance and QOL score demonstrates that the
study group is non-inferior to the control group and that the
primary effectiveness endpoint was met (p=0.030).
1.6.2.6 Primary Endpoint 2: Complication-Free Rate
(Safety)
The purpose of Primary Endpoint 2 was to evaluate complications
(adverse events that require additional invasive intervention to
resolve) related to the implanted CRT system which includes the
Tupos LV/ATx, the right atrial lead, the right ventricular ICD lead,
the left ventricular lead, and the implant procedure. The target
complication-free rate at Six-Months is 85%.
Table 8
provides the categorized complication rates at 6-months
for the study and the control group as well as a comparison
between the study and the control group.
Page 45

Lumax Technical Manual 37
Table 8: Complications at 6-Month – Study and Control
Study versus Control
Comparison
Delta 95% CI P-value
3.02%[-3.64%,
8.45%]
0.76%[-5.74%,
5.37%]
6.53%]
6.12%[-5.50%,
16.45%]
[-11.42%,
%
3.78
%
6.94%[-6.46%,
9.21%[-4.96%,
4.77%]
[-3.82%,
10.13%]
19.17%]
21.99%]
Category
Procedure
Related
Atrial Lead
Related
ICD Lead
Related
LV Lead
Related
Device
Related
Other
Medical
Related
Total
Procedure,
Lead and
Device
Related
Total
Study
N = 133
6 (4.51%) 1
3 (2.26%) 1
3 (2.26%) 0 (0%) 2.26%[-3.03%,
26
(19.55%)9(13.43%)
7 (5.26%) 5
9 (6.77%) 2
39
(29.32%)15(22.39%)
46
(34.59%)17(25.37%)
Control
N = 67
(1.49%)
(1.49%)
(7.46%)-2.20
(2.99%)
0.428
1.000
0.552
0.329
0.541
0.341
0.317
0.201
1.6.2.7 Primary Safety Enpoint Analysis and Conclusions
The observed procedure, lead and device related complicationfree rate at 6 months was 70.68%. The 95% confidence interval
for the complication-free rate was [62.16%, 78.25%]. The lower,
one-sided 95% confidence bound for the complication-free rate
was 63.50%. Therefore the procedure, lead and device related
complication-free rate at 6 months did not meet the pre-specified
acceptance criterion for this endpoint.
Page 46

38 Lumax Technical Manual
1.6.2.8 Post-hoc Safety Analysis
BIOTRONIK did not meet the pre-specified objective performance
criteria of 85% within 10% for the safety endpoint. Therefore, a
post-hoc safety analysis was conducted. It was noted that
79.80% (39 out of 49 events) of the complications were right atrial
lead, right ventricular ICD lead, left ventricular lead and procedure
related. The atrial, ICD and LV leads used during this study are
legally marketed devices.
This post-hoc analysis evaluated the LV lead complications that
were “related” or “possibly related” to the Tupos LV/ATx CRT-D,
but excludes the complications that were “not related” to the
Tupos LV/ATx device (see Table 9). There were 11 patients who
had an attempt to implant the LV lead, but the physician was
unsuccessful in either obtaining coronary sinus (CS) access or
unable to find a stable position for the LV lead. Additionally, there
were 4 patients with a documented LV lead dislodgement that has
no direct relationship to the implanted Tupos LV/ATx.
Table 9: Complications at 6-Months (Excluding LV Lead
Related) - Study versus Control
Difference
Study vs
Control
8.36%
11.39%
Category
Procedure Related
Atrial Lead Related
ICD Lead Related
LV Lead Related
Device Related
Other Medical Related
Total Procedure,
Lead and Device
Related
Total
Study
N=133
Control
N=67
6 (4.51%) 2 (2.99%) 1.53%
3 (2.26%) 1 (1.49%) 0.76%
3 (2.26%) 0 (0%) 2.26%
11 (8.27%) 1 (1.49%) 6.78%
7 (5.26%) 5 (7.46%) -2.20%
9 (6.77%) 2 (2.99%) 3.78%
27
(20.30%)8(11.94%)
35
(26.32%)10(14.93%)
Page 47

Lumax Technical Manual 39
The pulse generator related complication rate is higher in the
control group as compared to the study group. The complication
rates for procedure related, atrial lead related, ICD lead related,
LV lead related and other medical related are higher in the study
group as compared to the control group.
1.6.2.9 Post hoc Safety Analysis Conclusion
There are no clinically substantial differences in the total
complication rate or in the rates for the different complication rate
categories between the study and the control group.
Table 10 compares this post-hoc Safety Endpoint analysis to
previous CRT-D clinical studies:
Table 10 Safety Endpoint Comparisons
CRT-D Study
BIOTRONIK OPTION
(Original Analysis)
BIOTRONIK OPTION
(Post-hoc Analysis)
Estimated
freedom from
Complications
Lower 95%
CI
@ 6mos.
70.68% 63.5% 75%
78.95% 72.29% 75%
95%
lower
bound
criteria
Medtronic Insync ICD 81.1% 77.6% 67%
Guidant Contak CD N/A N/A 70%
St. Jude Medical Epic
HF
93.4% 90.6% 70%
This analysis confirms that the safety profile of the Tupos LV/ATx
is within a similar range determined during trials of other legally
marketed CRT-D devices.
Page 48

40 Lumax Technical Manual
1.6.2.10 Secondary Endpoint Results
1. The purpose of Secondary Endpoint 1 is to evaluate the
overall ability of the Tupos LV/ATx to appropriately convert
spontaneous AT (atrial tachycardia) and AF (atrial fibrillation).
The results from the OPTION study were compared to the
results from BIOTRONIK’s TACT study (P000009/S4, dated
09-09-2002) that demonstrated the effectiveness of these
atrial therapy features in the Tachos DR - Atrial Tx ICD.
Table 11 summarizes success rates for each individual atrial
tachyarrhythmia therapy type and overall success rate from
the OPTION study compared to the TACT study. The
number of episodes and patients receiving any therapy is less
than the total episodes of each therapy type, as episodes
may have included more than one type of therapy.
Table 11 Overall Atrial Conversion Rate
OPTION Study
Patients
Patients Success Episodes
Conversion
rate
ATP 3 3 5 60.0%
HF Burst 17 45 111 40.5%
Shock 12 30 34 88.2%
All
Therapies
25 78 129 60.5%
TACT Study
ATP 29 62 142 43.6 %
HF Burst 49 156 408 38.2 %
Shock 42 84 108 77.8 %
All
Therapies
66 302 542 55.7 %
The overall conversion rate and the conversion rates for each
therapy are comparable to the conversion rates observed in
the TACT study, demonstrating that the Tupos LV/ATx device
has similar atrial conversion capabilities as the legally
marketed Tachos DR – Atrial Tx ICD.
Page 49

Lumax Technical Manual 41
2. The purpose of Secondary Endpoint 2 is to evaluate VT
(ventricular tachycardia) and VF (ventricular fibrillation)
detection times of the Tupos LV/ATx. This is a measure of
the ability of the ventricular detection algorithm to detect VT
and VF in an appropriate timeframe. This endpoint was
evaluated based on the review of electrograms following
induced VT/VF episodes. A comparison of data from the
TACT study that utilized the legally marketed Tachos DR –
Atrial Tx ICD (P000009/S4, dated 09-09-2002) to data
collected during the OPTION study for the Tupos LV/ATx was
performed.
Table 12 summarizes and compares the results from these
two clinical studies.
Table 12: Summary of Detection Times
Detection
Time
Individual
Readings
Tachos DR -
Atrial Tx ICD
Mean (SE) / N
2.27 (0.06) / 52
By Patient 2.27 (0.07) / 26
Tupos
LV/ATx Mean
(SE) / N
2.26 (0.06) /
71
2.24 (0.06) /
35
Difference
0.01
0.03
The analysis demonstrates that the average detection times
of the Tupos LV/ATx are comparable to the detection times
observed with the legally marketed Tachos DR - Atrial Tx
ICD. Both devices utilize identical ventricular detection
algorithms and only sense with the right ventricular lead. This
clinical data demonstrates that the ventricular detection times
are similar in both devices.
3. The purpose of Secondary Endpoint 3 is to evaluate the
percentage of ventricular pacing (thus, CRT) as
demonstrated by the device diagnostics at required followups. This data was based on diagnostic data stored by the
Tupos LV/ATx.
Table 13 summarizes the percentage of ventricular pacing
between follow-ups as shown by device diagnostics for
patients in the study group.
Page 50

42 Lumax Technical Manual
Table 13: Percentage of Ventricular Pacing – 3-Month and
6-Month Follow-ups
Percentage of
Ventricular
Pacing
3-Months
Patients
(percentage)
6-Months Patients
(percentage)
<80% 9 (7.4%) 4 (4.0%)
81 – 85 % 4 (3.3%) 2 (2.0%)
86 – 90 % 13 (10.7%) 9 (9.1%)
91 – 95 % 19 (15.7%) 20 (20.2%)
96 – 100 % 76 (62.8%) 64 (64.7%)
Totals 121 (100%) 99 (100%)
The majority of the follow-ups (84.9%) show a percentage of
ventricular pacing of 91% or more at Six-Months.
4. The purpose of secondary endpoint 4 is to evaluate
improvement in functional capacity as measured by the six
minute walk test. The six minute walk test is a well-accepted
measure of functional capacity and exercise tolerance. Also,
this test more closely mimics the patient’s day-to-day
activities than maximal exercise testing.
Table 14 summarizes the six minute walk test distance at
Baseline and the Six-Month follow-up for patients in the study
group and the control group.
Page 51

Lumax Technical Manual 43
Table 14: Six Minute Walk Distance
Distance
(meters)
Study Control
Baseline
N
Mean ± SE
Range
Median
127
283.14 ± 9.27
23 to 511
302.00
61
269.43 ± 13.77
29 to 507
244.00
Six-Month
N
Mean ± SE
Range
Median
* Student's t-test, 2-sided
93
329.73 ± 10.82
78 to 596
335.00
44
310.70 ± 15.49
91 to 489
313.00
There are no clinically relevant differences in the six minute
walk test results between the study and the control group.
5. The purpose of Secondary Endpoint 5 is to evaluate the
improvement in the patient’s NYHA classification. Table 15
summarizes the average improvement in NYHA from
Baseline to Six-Months for 140 patients that were able to
complete both NYHA classification evaluations.
Table 15: Improvement in NYHA Classification at Six-Months
from Baseline
NYHA Change During OPTION Study
Change in NYHA
Class
Improved 2 classes
Improved 1 class
Total improved
No change
Worsened 1 class
Study Patients
(N=97)
(percentage)
Control Patients
(N=43)
(percentage)
10 (10.3%) 2 (4.7%)
47 (48.5%) 20 (46.5%)
57 (58.8%) 23 (51.2%)
39 (40.2%) 20 (46.5%)
1 (1.0%) 1 (2.3%)
Page 52

44 Lumax Technical Manual
The study and the control group have similar NYHA classes
and similar rates of improvement in NYHA class from
Baseline to the Six-Month follow-up.
6. The purpose of Secondary Endpoint 6 is to evaluate the rate
of hospitalization, for CHF and for all other causes. The
occurrence rate and reasons for hospitalization of the study
group were compared to the control group. To be consistent
with other large-scale clinical trials, clinical sites were
instructed to report hospitalizations for CHF using the
following definitions: 1) hospitalization for heart failure
management, 2) outpatient visit in which IV inotropes or
vasoactive infusion are administered continuously for at least
4 hours, or 3) emergency room (ER) visit of at least 12 hours
duration in which intravenous heart failure medications
including diuretics are administered.
Table 16 summarizes hospitalization, ER visits and outpatient
visits for enrolled patients.
Page 53

Lumax Technical Manual 45
Table 16: Hospitalization, ER Visits and Outpatient Visits
Medical Visits
Hospital
Study
(N=128)
CHF Related:
Control
(N=65)
CHF Related:
Admissions
Patients
Hospitalizations
Patients
Hospitalizations
20 (15.6%)
28
All causes:
68 (53.1%)
76
5 (7.7%)
9
All causes:
29 (44.6%)
46
Emergency
Room Visits
Patients
Patients
CHF Related:
Visits
Visits
1 (0.8%)
1
All causes:
13 (10.1%)
16
CHF Related:
0 (0.0%)
0
All causes:
2 (3.1%)
2
Outpatient Visits
Patients
Patients
CHF Related:
Visits
Visits
1 (0.8%)
1
All causes:
5 (3.9%)
5
CHF Related:
0 (0.0%)
0
All causes:
2 (3.1%)
2
A large percentage of All Cause hospitalizations can be
attributed to pacing lead revisions, device infections, or other
device-related interventions (e.g., pocket revision or device
replacements for ERI or device recall). The CHF
hospitalization rate for both the study and control groups is
clinically acceptable considering the enrollment CHF status of
the patients.
Page 54

46 Lumax Technical Manual
7. The purpose of Secondary Endpoint 7 is to evaluate the
observation rate. Observations are defined as clinical events
that do not require additional invasive intervention to resolve.
For the study group, there were 210 observations in 104
patients with cumulative implant duration of 1240.4 months
(101.9 patient years). 78.2% of the enrolled study patients
have a reported observation. The rate of observations per
patient-year is 2.06. For the control group, there were 81
observations in 44 patients with cumulative implant duration
of 596.5 months (49.0 patient years). 65.7% of the enrolled
control patients had a reported observation. The rate of
observations per patient-year was 1.65.
8. The purpose of Secondary Endpoint 8 is to evaluate peak
VO2 as a measure of effectiveness of the Tupos LV/ATx
system in providing CRT. The core lab was blinded to study
randomization assignments during evaluation of the results of
the cardiopulmonary exercise (CPX) testing in order to
minimize the potential for bias. According to the protocol, to
be included in the analysis, patients were required to attain a
respiratory exchange ratio (RER) of ≥ 1.
Table 17 provides a summary of peak VO
results for 42
2
patients with CPX testing completed at Baseline and the SixMonth follow-up and with an RER of ≥ 1.
Page 55

Lumax Technical Manual 47
Table 17: Peak VO2 Testing Results – Patients with RER ≥ 1
Results Study Control
Peak VO
(ml/kg/min)
2
N=32
Baseline:
Mean:
13.46 ± 0.57
Range:
6.9 to 21.1
Six-Month:
Mean:
13.39 ± 0.53
Range:
7.6 to 20.70
Difference:
Mean:
-0.06 ± 0.42
Range:
-7.9 to 4.9
N=10
Baseline:
Mean:
12.58 ± 0.75
Range:
8.0 to 14.8
Six-Month:
Mean:
12.89 ± 0.94
Range:
7.0 to 17.2
Difference:
Mean:
0.31 ± 0.67
Range:
-2.7 to 4.6
1.6.2.11 Multi-site Poolability and Gender Analysis
The OPTION CRT/ATx clinical report includes data from multiple
centers with centralized coordination, data processing, and
reporting at BIOTRONIK. All of the clinical centers followed the
requirements of an identical clinical protocol, and all of the clinical
centers used the same methods to collect and report the clinical
data. In order to justify pooling of the data from multiple centers,
several analyses were completed. All of the centers were divided
into two groups based on implant volume. Comparisons were
then made between the patient populations based on the results
of each of the endpoints. Additionally, analyses were performed
on the data collected in the OPTION CRT/ATx clinical
investigation in order to compare results between males and
females. The first type of analysis compared enrollment by patient
gender in each of the study and control groups. The second type
of analysis compared effectiveness outcomes in each gender.
The results of these analyses demonstrate poolability of the data
between sites. There were no significant differences in the
second primary endpoint or any of the secondary endpoints
between high and low volume implant centers.
Page 56

48 Lumax Technical Manual
The gender distribution in this clinical investigation is consistent
within the study groups and includes a representative proportion
of female participants. There were no significant differences in
any of the primary or secondary endpoints between the male and
female population.
1.6.2.12 Conclusions
The IDE Clinical study (OPTION LV/ATx) demonstrated that the
safety and effectiveness of the Tupos LV/ATx CRT-D device is
equivalent to that of similar legally marketed CRT-D devices.
Although the study missed its primary safety endpoint, additional
post hoc analyses were conducted to reassure that the safety
profile of the device is comparable to other legally marketed
CRT-D devices.
1.7 Patient Selection and Treatment
1.7.1 Individualization of Treatment
• Determine whether the expected device benefits
outweigh the possibility of early device replacement for
patients whose ventricular tachyarrhythmias require
frequent shocks.
• Determine if the device and programmable options are
appropriate for patients with drug-resistant
supraventricular tachyarrhythmias (SVTs), because drugresistant SVTs can initiate unwanted device therapy.
• Direct any questions regarding individualization of patient
therapy to your BIOTRONIK representative or
BIOTRONIK technical services at 1-800-547-0394.
The prospective patient’s size and activity level should be
evaluated to determine whether a pectoral or abdominal implant
is suitable. It is strongly recommended that candidates for an
ICD/CRT-D have a complete cardiac evaluation including EP
testing prior to device implant to gather electrophysiologic
information, including the rates and classifications of all the
patient’s cardiac rhythms. When gathering this information,
delineate all clinically significant ventricular and atrial arrhythmias,
whether they occur spontaneously or during EP testing.
Page 57

Lumax Technical Manual 49
If the patient’s condition permits, use exercise stress testing to do
the following:
• Determine the maximum rate of the patient’s normal
rhythm.
• Identify any supraventricular tachyarrhythmias.
• Identify exercise-induced tachyarrhythmias.
The maximum exercise rate or the presence of supraventricular
tachyarrhythmias may influence selection of programmable
parameters. Holter monitoring or other extended ECG monitoring
also may be helpful.
If the patient is being treated with antiarrhythmic or cardiac drugs,
the patient should be on a maintenance drug dose rather than a
loading dose at the time of pulse generator implantation. If
changes to drug therapy are made, repeated arrhythmia
inductions are recommended to verify pulse generator detection
and conversion. The pulse generator also may need to be
reprogrammed.
Changes in a patient’s antiarrhythmic drug or any other
medication that affect the patient’s normal cardiac rate or
conduction can affect the rate of tachyarrhythmias and/or efficacy
of therapy.
If another cardiac surgical procedure is performed prior to
implanting the pulse generator, it may be preferable to implant the
lead system at that time. This may prevent the need for an
additional thoracic operation.
1.7.2 Specific Patient Populations
Pregnancy - If there is a need to image the device, care should
be taken to minimize radiation exposure to the fetus and the
mother.
Nursing Mothers - Although appropriate biocompatibility testing
has been conducted for this implant device, there has been no
quantitative assessment of the presence of leachables in breast
milk.
Page 58

50 Lumax Technical Manual
Geriatric Patients - Most (about 71%) of the patients receiving a
CRT-D or ICD in the clinical studies detailed in this manual were
over the age of 60 years (see Clinical Studies).
Handicapped and Disabled Patients - Special care is needed in
using this device for patients using an electrical wheel chair or
other electrical (external or implanted) devices.
1.8 Patient Counseling Information
The implanted devices are subject to random component failure.
Such failure could cause inappropriate shocks, induction of
arrhythmias or inability to sense arrhythmias, and could lead to
the patient’s death.
Persons administering CPR may experience the presence of
voltage on the patient’s body surface (tingling) when the patient’s
CRT-D/ICD system delivers a shock.
A patient manual is available for the patient, patient’s relatives,
and other interested people. Discuss the information in the
manual with concerned individuals both before and after pulse
generator implantation so they are fully familiar with operation of
the device. (For additional copies of the patient manual, contact
the BIOTRONIK at the address listed in this manual.)
1.9 Evaluating Prospective CRT-D/ICD
Patients
The prospective ICD/CRT-D implant candidate should undergo a
cardiac evaluation to classify any and all tachyarrhythmias. In
addition, other patient specific cardiac information will help in
selecting the optimal device settings. This evaluation may
include, but is not limited to:
• an evaluation of the specific tachycardia rate(s)
• the confirmation and/or evaluation of any supraventricular
arrhythmias or bradyarrhythmias
• the evaluation of various ATP and cardioversion
therapies
• the presence of any post-shock arrhythmias, and
Page 59

Lumax Technical Manual 51
• an evaluation of the maximum sinus rate during exercise
If a patient’s drug regimen is changed or adjusted while the
CRT-D/ICD is implanted, additional EP testing may be required to
determine if detection or therapy parameter settings are relevant
and appropriate.
Empirical changes to the detection or therapy parameters should
be assessed based on patient safety. Some changes may
necessitate a re-assessment of sensing, pacing, or arrhythmia
conversion treatment. Thorough technical knowledge of
BIOTRONIK CRT-D/ICDs, additional CRT-D/ICD experience, and
individual medical judgment will aid in determining the need for
additional testing and follow-up.
Page 60

52 Lumax Technical Manual
Page 61

Lumax Technical Manual 53
2. Device Features
The Lumax family feature set is presented under the following
sub-headings: Tachyarrhythmia Detection, Tachyarrhythmia
Redetection / Acceleration, Tachyarrhythmia Therapy,
Tachyarrhythmia Termination, Bradycardia Therapy, EP Test
Functions and Special Features. The features apply to all
members of the Lumax family except where specifically
referenced differently.
CAUTION
Programmed Parameters – Program the device
parameters to appropriate values based on the patient’s
specific arrhythmias and condition.
2.1 Cardiac Resynchronization Therapy
(CRT)
HF versions only
For Cardiac Resynchronization Therapy (CRT), a sensing/pacing
lead is placed in the right atrium, while an ICD lead is placed in
the right ventricle. The third lead is positioned to pace the left
ventricle. When connected together, this system provides
coordinated, simultaneous stimulation of the right and left
ventricles. This resynchronization therapy is designed to
coordinate the contraction of both ventricles, which allows the
heart to contract more efficiently. As a result, patients with CHF
and intraventricular conduction delay may have a greater ability to
complete physical activities thus improving their quality of life.
Page 62

54 Lumax Technical Manual
As a result of the device design and header configuration,
ventricular pacing pulses can be delivered between the right / left
ventricular lead tip electrodes simultaneously (cathode) and the
ring of the right ventricular lead (anode). Ventricular sensing
primarily uses the poles of the right ventricular lead tip and ring.
This design avoids sensing of ventricular activity twice during a
single cardiac cycle (double counting) in patients with a wide QRS
complex. However, for diagnostic purposes and LV pacing
protection the Lumax HF devices can be programmed to sense in
the left ventricle.
Atrial Channel
The Lumax ICDs/CRT-Ds pace and sense in bipolar
configuration, between the atrial lead’s tip and ring electrodes. A
bipolar atrial lead must be used to ensure reliable sensing of atrial
activity.
Ventricular Channel
The Lumax HF devices can be programmed to pace in both the
right and left ventricle (as well as RV only). The Lumax HF
primarily senses in a bipolar configuration in the right ventricle.
However, for diagnostic purposes and LV pacing protection the
Lumax HF devices can be programmed to sense in the left
ventricle.
Potential Ventricular lead configurations are provided in Table 18.
Page 63

Sensing
Lumax Technical Manual 55
Table 18. Lead Configurations
Configuration Explanation
RV Only
Sensing takes place between
the tip and ring electrodes of
the right ventricular lead.
Pacing
Pacing
LV Only
RV & LV
Together (BiV)
RV Only
Sensing takes place between
the tip and ring electrodes
(bipolar) or the tip electrode of
the left ventricular lead and the
CRT-D housing (unipolar).
Pacing configuration is
programmable between the tip
and ring electrodes of the right
and left ventricular leads.
See Figure 3
Pacing takes place between
the tip and ring electrodes of
the right ventricular lead.
Figure 3 Programmable BiV Pacing Configurations
Page 64

56 Lumax Technical Manual
For CRT to be effective, ventricular pacing must occur.
Therefore, AV delays must be programmed short enough to
override intrinsic ventricular contractions. Additional information
to further optimize AV delays can be obtained with
echocardiography.
CRT can be programmed ON or OFF via the programmer using
the [Ventricular Pacing] parameter. Ventricular Pacing
Configuration allows either standard right ventricular [RV] (CRT =
OFF) pacing or Cardiac Resynchronization Therapy [BiV] (CRT =
ON).
The Lumax HF CRT-D can provide triggered biventricular pacing.
This is a functional expansion of the basic ventricular modes
(DDD(R); DDI(R); VDI(R); VDD(R); VVI(R)) used for biventricular
pacing. The “RVs triggering” was designed to ensure CRTis
delivered even when rapid intrinsic activation interferes with
pacing, such as in the case of conducted atrial fibrillation. This
function triggers LV pacing (Vp) after intrinsic sensing (RVs) in the
right ventricle. Triggered pacing can be programmed to react to
only normal RV sensed events or to right ventricular extrasystoles
as well as normal RV sensed events. The maximum Trigger rate
is normally limited by the programmed UTR, but can also be
programmed to function up to a separate and higher maximum
trigger rate.
2.2 Sensing (Automatic Sensitivity Control)
The Lumax ICDs/CRT-Ds use Automatic Sensitivity Control
(ASC) to adjust the input stage sensitivity threshold for each
channel to appropriately detect the various cardiac signals. The
characteristics of the sensing circuitry have been optimized to
ensure appropriate sensing during all potential cardiac rhythms.
Cardiac signals vary in amplitude; therefore detection thresholds
cannot be static. With the Automatic Sensitivity Control (ASC)
every sensed event is measured, and the upper and lower
thresholds are re-set accordingly (also known as beat-by-beat
adaptation). The ASC begins by tracking the cardiac signals (R
and P-waves) during the sensed refractory periods. The peak
values measured during this time are used to set the sensing
thresholds during the active detection periods.
Page 65

Lumax Technical Manual 57
2.2.1 Right Ventricular Sensitivity Settings
There are three programmable preset options for setting the
sensitivity of the right ventricular input stage. The sensitivity
selections are designed to adapt the parameters of the input
stage to various signal conditions. The predefined parameter
sets are described in Table 19.
Table 19: Sensitivity Settings
Setting Definition for Use
Standard This setting is recommended for most
patients, especially for those with
measured R-wave amplitude of ≥3 mV.
Enhanced
T Wave
Suppression
Enhanced
VF Sensitivity
This setting offers suppression of T-wave
oversensing. This mode should not to be
used on patients with the following
conditions:
• Sinus rhythms with small signal
amplitudes, R-waves <4 mV
• VF with highly fluctuating signal
amplitudes.
This setting enhances VF detection, in
cases of highly fluctuating signal
amplitudes. It is not to be used for patients
that have sinus rhythms containing large
amplitude T-waves.
Typically, the upper threshold is reset with each sensed R-wave,
but in order to ensure that pacing does not occur during an
episode of VF, the ASC behaves differently with paced events.
Each paced event is followed by a paced refractory period after
which the ventricular threshold is set to the minimum programmed
value.
Page 66

58 Lumax Technical Manual
For example, the upper threshold is set at 50% of the measured
R-wave for the Standard sensitivity setting following the 100 ms
sensed refractory period. The upper threshold decays 0.125 mV
every 250 ms through the T-wave discrimination period (hold of
upper threshold: 360 ms). After the T-wave discrimination period,
the threshold is decreased to the lower threshold. The lower
threshold is set to 25% of the measured peak R-wave. The lower
threshold then decreases 0.125 mV every 500 ms until the
Minimum Threshold is reach or until the next sensed (or paced)
event.
Figure 4 Automatic Sensitivity Control with Standard Setting
Figure 4 provides an illustration of Automatic Sensitivity Control
with the sensitivity programmed to Standard. The tracked R –
wave is measured to be 6.0 mV, following the sensed refractory
period the upper threshold is set to 3.0 mV. After the T-wave
discrimination period, the threshold is further reduced to 1.5 mV.
Both the Upper and Lower Thresholds decay over time, but the
Minimum Threshold is never violated. Nominally, the minimum
threshold is set to 0.8 mV, but it can be adjusted by the user.
The Enhanced VF Sensitivity setting is specifically designed to
improve VF detection when the VF signal is very small. Two
adjustments are made to ASC with this setting:
Page 67

Lumax Technical Manual 59
• The T-wave discrimination period (hold of upper
threshold) is decreased to 100 ms, thus eliminating the
Upper Threshold.
• The decay rate of the Lower Threshold is increased to
0.125 mV every 250 ms.
These adjustments ensure that the threshold reaches the lower
values more quickly in order to assure that all VF signals are
sensed appropriately.
The Enhanced T-Wave Suppression setting is specifically
designed to avoid double counting of each QRS-T complexes
during normal sinus rhythms. With sensitivity programmed to
Enhanced T-Wave Suppression:
• High pass filtering is increased to reduce low frequency
signal components such as T-waves and respiratory
artifacts.
• The Upper Threshold is increased to 75% of the
measured R-wave.
• The Upper Threshold may not retrigger with each sensed
event, it is only triggered when the new sensed R-wave
crosses the 50% point of the previous measured R-wave.
2.2.2 Minimum Right Ventricular Threshold
This parameter limits the minimum sensitivity of the ICD/CRT-D to
a programmable value. Nominally, the minimum threshold is set
to 0.8 mV, but it can be adjusted from 0.5 to 2.5 mV.
2.2.3 Atrial Sensitivity Settings
DR and HF versions only
The primary option for setting the sensitivity of the atrial input
stage is “Standard”. When atrial sensing is active, the sensitivity
is set to “Standard” for most patients, which is designed to adapt
the parameters of the input stage to various signal conditions.
The available settings are described in Table 20.
Page 68

60 Lumax Technical Manual
Table 20: Atrial Sensitivity Settings
Setting Definition for Use
Standard This setting is recommended for all
patients with a functioning atrial lead.
Inactive This setting deactivates the atrial channel
for sensing, EGM telemetry and Holter
recording and is typically used when no
atrial lead is implanted.
Typically, the upper threshold is reset with each sensed P-wave,
but in order to ensure that pacing does not occur during an
episode of AF/VF, the ASC behaves differently with paced events.
Each paced event is followed by a paced refractory period after
which the atrial threshold is set to the minimum programmed
value.
2.2.4 Minimum Atrial Threshold
This parameter limits the minimum sensitivity of the ICD/CRT-D to
a programmable value. Nominally, the minimum threshold is set
to 0.4 mV, but it can be adjusted from 0.2 to 2.0 mV.
2.2.5 Left Ventricular Sensitivity Settings
HF versions only
The primary option for setting the sensitivity of the left ventricular
input stage is “Standard”. When LV sensing is active, the
sensitivity is set to “Standard” for most patients, which is designed
to adapt the parameters of the input stage to various signal
conditions. The available settings are described in Table 20.
Page 69

Lumax Technical Manual 61
Table 21: Atrial Sensitivity Settings
Setting Definition for Use
Standard This setting is recommended for all
patients with a functioning left ventricular
lead.
Inactive This setting deactivates the left ventricular
channel for sensing, EGM telemetry and
Holter recording and is typically used when
no LV lead is implanted.
2.2.6 Minimum Left Ventricular Threshold
This parameter limits the minimum sensitivity of the CRT-D to a
programmable value. Nominally, the minimum threshold is set to
0.8 mV, but it can be adjusted from 0.5 to 2.5 mV.
2.2.7 Far Field Protection
DR and HF versions only
This parameter blanks the atrial channel of the ICD/CRT-D to the
period before and after each ventricular event. This blanking
period is programmable separately based on whether the
ventricular event is a paced or sensed event and is designed to
prevent sensing of ventricular signals with the atrial leads.
CAUTION
Far-field sensing of signals from the atrium in the
ventricular channel or ventricular signals in the atrial channel
should be avoided by appropriate lead placement,
programming of pacing/sensing parameters, and maximum
sensitivity settings. If it is necessary to modify the Far Field
Blanking parameter, the parameter should be lengthened
only long enough to eliminate far-field sensing as evidenced
on the IEGMs. Extending the parameter unnecessarily may
cause undersensing of actual atrial or ventricular events.
Page 70

62 Lumax Technical Manual
2.2.8 Additional Sensing Parameters
The parameters of the Additional Sensing Parameters menu are
to provide additional flexibility for physicians to non-invasively
correct over/undersensing situations. The ranges and nominal
values are located in table titled Additional Sensing Parameters in
Section 6.
Upper Threshold (A, RV & LV) - This feature allows the user to
change the upper sensing threshold level (UT in Figure 4) from
the nominal value of 50% of the sensed R-wave / P-wave
amplitude to either 75% or 87.5% of the R-wave / P-wave value.
This feature is used in the ventricle for example to eliminate
oversensing of large T-waves.
Hold of Upper Threshold (RV & LV) - This parameter
determines when the sensing decrement begins after an event
(small step-down on the 50% threshold before LT in the figure
above). This parameter “holds” the threshold at a constant value
(UT in Figure 4) for the programmed time. Maximum Hold Time
is programmable from 10 to 600 ms (T-Wave discrimination
period).
Lower Threshold (A, RV & LV)- This feature allows the user to
change the lower sensing threshold (labeled LT in Figure 4) from
the default value of 25% of the sensed R-wave / P-wave
amplitude to either 12.5 or 50% of the measured R-wave/ P-wave
value. This feature is also used in the ventricles to alleviate
T-wave oversensing and/or undersensing of small amplitude
events (e.g., fine VF).
Blank after atrial pacing (RV) - This feature is used to eliminate
sensing of artifacts after atrial paced events. Blank Post atrial
Pace in the RV is programmable from 50 to 100 ms. For the left
ventricle, this parameter is equal to the safety window time
(100 ms).
VES Discrimination after As - This feature is used to correctly
identify and classify ventricular extrasystoles (VES). With each
atrial sensed event (also with Ars falling into PVARP) a special
timing interval is started for the ventricle, if the subsequent
ventricular event does not fall within the AV delay or the
programmed VES discrimination interval, it is classified as a VES.
Page 71

Lumax Technical Manual 63
LV T-wave Protection - Used to eliminate unintended pacing in
the vulnerable period of the left ventricle. This feature is only
used when left ventricular sensing is active. HF versions only.
2.3 Automatic Threshold Measurement
(ATM)
Lumax 500/540 Models only
The Lumax 500/540 models have automatic threshold
measurement (ATM) feature for determining ventricular pacing
thresholds. This feature is separately programmable for the right
(RV) and left (LV) ventricles.
ATM is initiated once each day, typically during the nighttime while
the patient is normally sleeping. It can measure the right and the
left ventricular pacing thresholds and stores this information for
use in trends of daily threshold values. This information is
available on the programmer during in-office follow-ups and via
BIOTRONIK’s Home Monitoring system. The permanent pacing
amplitude is not adjusted by the ATM feature.
2.3.1 Functional Description
The ATM features of Lumax 500/540 are based on the evaluation
of the morphology of the ventricular evoked response (VER) to
determine if the pacing pulse has captured the myocardium. In
order to measure the pacing threshold, a sequence of two
algorithmic steps is carried out:
• Signal Quality Check (SQC)
• Threshold Measurement
Page 72

64 Lumax Technical Manual
The signal quality check determines if the morphology of the
evoked response of a captured beat is sufficiently different from
the morphology of the evoked response of a non-captured pace.
If the SQC is sufficient (i.e., if the algorithm can clearly distinguish
between capture and non-capture), then the automatic threshold
measurement is activated by initiating a series of pacing pulses.
The amplitude of these pulses is continuously decreased until a
loss of capture is detected. When loss of capture is detected a
back-up pulse is delivered (right ventricle only) to avoid
ventricular pauses caused by the threshold measurement.
2.3.1.1 Signal Quality Check
During the Signal Quality Check, five single effective pacing
pulses are delivered. These pulses are followed by five double
pulses. The second pulse of each double pulse is delivered into
the refractory phase of the ventricle (as a non-capturing pulse).
The VER morphology of the single pulses and the second pulse
of the double pulses are assessed. The signal quality check is
successful if the single pulses are recognized as capturing pulses
and the second pulses of the double pulses are classified as noncapturing pulses. The AV delay of the 2 x 5 pulses is shortened
to 50 ms in order to avoid ventricular fusion beats which would
otherwise disturb the SQC. If the SQC is successful, then the
threshold measurement is started, if not, the SQC is repeated
again after 30 minutes.
2.3.1.2 Threshold Measurement
The threshold measurement begins by delivering pulses at the
programmed pacing amplitude. The pulse amplitude is decreased
in steps of 0.6 Volts (V) each until loss of capture is detected.
During the loss of RV-capture sequence, a backup pulse is
issued. The back-up is set to a value of the programmed pacing
amplitude plus 0.6 V with a pulse width of 1.5 ms. There are no
backup paces for left ventricular ATM (LV-ATM).
The pulse amplitude is then set to the last effective value (0.6
Volts above the initial non-capture level) and then subsequent
pacing pulses are reduced in 0.1 V steps until loss of capture
reoccurs. The last effective pacing amplitude is then recorded as
the pacing threshold.
Page 73

Lumax Technical Manual 65
2.3.1.2.1 Loss of Capture Detection
In order to ensure against ATM loss of capture detection due to
an isolated non-capture event (i.e. as a result of VES), loss of
capture is only declared if two out of three consecutive cycles
show loss of capture.
2.3.1.2.2 ATM in Lumax HF-T Models
If the device is programmed to biventricular pacing (BiV), the RV
ATM functions in the RV mode as described above. The LV ATM,
if active, is initiated after the RV ATM is complete. For the LV
ATM, the left ventricle is stimulated first with a VV delay of 50 ms
and then completed in a similar manner.
2.4 Intra-Thoracic Impedance
Measurement
Lumax 500/540 Models only
2.4.1 Functional Description
The intra-thoracic impedance measurement of Lumax 500/540 is
based on the painless shock impedance feature of the
Lumax ICD/CRT-D Family. It utilizes 24 measurement windows
evenly distributed over the course of 24 hours to trend the data.
With a total of 1,024 impedance measurements per hour, there is
a significant amount of data being stored in the ICD/CRT-D.
The impedance is measured between RV tip and the distal shock
coil with a programmable excitation current. The device calculates
and stores the mean hourly impedance values and the
corresponding time points. All hourly values obtained within one
Home Monitoring interval are transmitted to the Home Monitoring
service center along with the daily message.
Upon interrogation, the programmer calculates and displays an
intra-thoracic impedance trend of the mean daily impedance
values. This feature is only available after inserting a special code
into the programmer.
Page 74

66 Lumax Technical Manual
2.5 Ventricular Tachyarrhythmia Detection
The Lumax ICDs/CRT-Ds detect and measure the rate of sensed
cardiac signals to discriminate ventricular tachyarrhythmias from
supraventricular tachycardias, sinus rhythm or sinus bradycardia.
This is accomplished through programmable rate detection
parameters in the device. When a tachyarrhythmia is present, the
ICD/CRT-D classifies the arrhythmia and delivers the appropriate
therapy. If a tachyarrhythmia continues following the first therapy
attempt, then the ICD/CRT-D will redetect the tachyarrhythmia
and deliver subsequent therapies as necessary.
WARNING
Unwanted Shocks – Always program ICD therapy to OFF
prior to handling the device to prevent the delivery of serious
shocks to the patient or the person handling the device
during the implant procedure.
Classification of cardiac signals is accomplished primarily by
measuring the cardiac cycle length (R-R, P-R and P-P). In
addition, the ICD/CRT-D can also utilize abrupt changes in rate or
irregularity of the cardiac signal to further differentiate ventricular
tachyarrhythmias. Each detected ventricular tachyarrhythmia is
classified into one of the following zones:
• VT-1 Lower rate ventricular tachycardia
• VT-2 Higher rate ventricular tachycardia
• VF Ventricular fibrillation
Each rhythm class is programmable to a separate rate with the
zone limit defining the lowest rate in each class. The upper rate
limit of each class is equal to the zone limit of the next higher
class, creating a continuous range of rate classes.
Page 75

Lumax Technical Manual 67
2.5.1 VF Classifications
Detection of ventricular fibrillation (VF) utilizes programmable X
out of Y criterion. Both X and Y are programmable. If X number
of intervals within the sliding window (defined by Y) are shorter
than the programmed VF rate interval (>bpm), VF is detected.
After fibrillation is detected, the programmed therapy sequence
for VF is initiated.
Nominal settings for classification of ventricular fibrillation (VF)
are 8 of 12 intervals; meaning that within a sample window of
12 intervals, 8 intervals must meet or exceed the VF zone rate
criteria.
2.5.2 VT Interval Counters
The VT Interval Counters are separately programmable for VT-1
and VT-2 rate classifications. The Counter: Detection is the
number of intervals required to declare a tachyarrhythmia as VT.
A tachyarrhythmia must meet both the rate/interval criteria and
the programmed Counter / Detection criteria, in addition to any
other detection enhancements to be declared a tachycardia.
2.5.3 VT Classification
Both VT-1 and VT-2 classification zones utilize separately
programmable detection parameters. Classification of VT-1 or
VT-2 is based on the last interval average preceding declaration
of tachyarrhythmia detection. If this average falls within the VT-1
zone, the programmed VT-1 therapy is delivered. If the average
falls within the VT-2 limits, the programmed VT-2 therapy is
delivered. If additional detection parameters are activated, each
of these supplemental criteria must also be satisfied before a VT
rhythm can be classified and treated.
Page 76

68 Lumax Technical Manual
The ICDs/CRT-Ds may be programmed to use ventricular-only
information, or both atrial and ventricular information for the
discrimination of ventricular tachycardias. With SMART
Detection™ turned ON, the Lumax ICDs/CRT-Ds use atrial and
ventricular signals for discrimination of fast heart rhythms. With
SMART Detection™ turned OFF, only the ventricular rate is used
to discriminate between ventricular rhythm classes. If SMART
Detection is enabled, this algorithm evaluates all cardiac signals
within the VT range and increments the VT Sample Count for all
intervals that are deemed VT. A full description of SMART
Detection™ is provided in the following text.
In addition, when the Lumax senses the programmed number of
consecutive intervals (termination count) within the sinus rate
zone, all tachyarrhythmia detection criteria, including the VT
sample counters are reset.
2.5.4 SMART Detection™
DR and HF versions only
This discrimination algorithm enhances VT-1 and VT-2 detection
by applying a series of tests to the sensed cardiac signal.
SMART Detection™ is intended to discriminate VT from a variety
of supraventricular arrhythmias that are conducted to the ventricle
and that would otherwise satisfy VT-1 or VT-2 rate detection
criteria.
First, the average ventricular rate is compared to the average
atrial rate. In the event that the measured ventricular rate is
faster than the atrial rate, the device immediately declares the
rhythm a VT and delivers programmed ventricular therapy for the
detected VT zone.
In the event that an atrial rate is faster compared to the ventricular
rate one of three tests are performed:
Ventricular rhythm stability, (see Stability on page 69) if the
ventricular signal is unstable, then the rhythm is declared a
supraventricular tachyarrhythmia, (SVT) and ventricular therapy is
typically withheld.
Page 77

Lumax Technical Manual 69
If the ventricular signal is stable, and the atrial rate is a multiple of
the ventricle rate, then the rhythm is declared a supraventricular
tachyarrhythmia (SVT) and ventricular therapy is typically
withheld.
If the ventricular rhythm is stable and the atrial rate is not a
multiple of the ventricular rate, then the rhythm is declared a VT
and ventricular tachycardia therapy is delivered.
In the event that both the atrial and ventricular signals are
detected at the same rate, a series of additional discrimination
tests are applied.
2.5.5 Onset
Another detection enhancement that may be used independently
(VT-1 or VT-2, when SMART Detection is active) or as an adjunct
to the SMART Detection™ algorithm is the Onset parameter.
This parameter measures abrupt changes in ventricular cycle
length to discriminate between sinus tachycardias and ventricular
and atrial tachyarrhythmias, which characteristically begin with an
abrupt change in cardiac rate.
This feature allows therapy to be withheld if a sinus tachycardia
rate crosses into one of the VT zones.
2.5.6 Stability
In VT-1 and VT-2 zones, the purpose of STABILITY is to assist in
discriminating between stable ventricular tachyarrhythmias and
supraventricular tachyarrhythmias that conduct irregularly to the
ventricles. STABILITY evaluates sudden changes in the
regularity of cardiac events (R-R and P-P intervals) on a beat by
beat basis. The STABILITY criterion compares the current
measured interval with the three preceding cardiac intervals. If a
difference between the current interval and each of the three
preceding intervals is less than the stability range, then the
current intervals are stable.
Page 78

70 Lumax Technical Manual
The SMART Detection™ algorithm utilizes both atrial and
ventricular STABILITY as integral parts of the discrimination
algorithm. Therefore, when SMART Detection™ is enabled, the
STABILITY parameter must also remain enabled and set to 12%.
2.5.7 Sustained VT Timer
The Sustained VT Timer can be programmed between
30 seconds and 30 minutes (or to OFF). When the timer expires,
therapy is initiated regardless of the detection enhancements.
The Sustained VT parameter is intended to force tachycardia
therapy in cases where a cardiac rhythm meets the VT rate
criteria but does not satisfy one or more detection enhancement
criterion (Onset, SMART Detection, or Stability) for an extended
duration. This “safety” timer is initiated within one of the VT
zones. If the programmed Sustained VT time period expires
without tachycardia detection, redetection is initiated without
utilizing the detection enhancements.
A simple up/down counter is used to initiate the safety timer. The
counter is incremented by one when an interval falls into a VT
zone and decrements by one when an interval falls into the sinus
zone. When the counter reaches a number equal to the
programmed VT detection counter, the safety timer is started.
The timer runs until the programmed time expires and therapy is
delivered or until the timer is reset. The timer is reset with initial
detection or VT termination.
The safety timer is not used in redetection. If initial detection was
due to the safety timeout and SMART Redetection is programmed
“ON”, then SMART Detection will not be used for redetection.
2.5.8 VT Monitoring Zone
The VT1 zone can be programmed to monitor for non-sustained
arrhythmias not requiring therapy. The monitoring zone can be
programmed with all the standard detection enhancements
including SMART DetectionTM to monitor for non-sustained
ventricular tachycardia or atrial tachyarrhythmias. Any
tachyarrhythmia meeting the Monitor Zone criteria will store an
IEGM.
Page 79

Lumax Technical Manual 71
2.5.9 Atrial Monitoring Zone
This feature allows the device to store an IEGM for atrial
tachyarrhythmias such as Atrial Fibrillation or Atrial Flutter. The
zone is programmed by rate with a range of 100 bpm to 250 bpm.
Any atrial tachyarrhythmia meeting the Atrial Arrhythmia Monitor
Zone criteria will store an IEGM.
2.6 Tachyarrhythmia Redetection
The Lumax ICDs/CRT-Ds offer independently programmable
settings for determining if tachyarrhythmias remain after therapy
has been delivered. The redetection routine allows the
ICDs/CRT-Ds to determine whether further therapy is required
when the initial therapy was unsuccessful in terminating the
arrhythmia.
Tachyarrhythmia redetection criteria are based on cardiac cycle
length and number of intervals. The number of intervals is distinct
and independent of the initial detection criteria.
2.6.1 VT Redetection
The Counter: Redetection parameter may be programmed
separately for each arrhythmia class, independent of the initial
detection parameters:
Redetection of an ongoing tachyarrhythmia is declared when the
Counter: Redetection is satisfied (based on individual cycles). If a
sensed cardiac signal meets any VT rate criteria, following
therapy, that signal is counted and compared to the programmed
Counter: Redetection setting.
Redetection functions are based exclusively on the VT criterion.
Page 80

72 Lumax Technical Manual
2.6.2 SMART Redetection
DR and HF versions only
With SMART Redetection programmed ON, both atrial and
ventricular signals are used for redetection after initial detection
and therapy for a VT. SMART Detection™ will function identically
as in initial VT detection.
2.6.3 Forced Termination
DR and HF versions only
With SMART Redetection programmed ON, this programmed
parameter sets a time after which the SMART Redetection will be
terminated even if the SVT is still ongoing. This forces the device
to terminate the episode and allow detection of a new VT or VF
episode.
2.6.4 VF Redetection
VF redetection uses the same X in Y algorithm as initial detection.
The X and Y values for initial detection are also used for
redetection to ensure consistent classification of VF.
2.7 Tachyarrhythmia Termination
Termination of a ventricular tachyarrhythmia episode is declared
when 12 out of 16 consecutive sensed intervals are longer than
the VT-interval parameter of the lowest VT class (sinus or
bradycardia rhythm).
2.8 Tachyarrhythmia Therapy
The Lumax ICDs/CRT-Ds offer a variety of therapy options that
can be tailored to meet a patient’s specific anti-tachycardia or
defibrillation therapy requirements. Anti-tachycardia pacing (ATP)
therapies can be combined with defibrillation therapies to provide
a broad spectrum of tachyarrhythmia treatment options.
Page 81

Lumax Technical Manual 73
2.8.1 Therapy Options
The Lumax ICDs/CRT-Ds offer multiple therapy options for each
tachyarrhythmia class (VT1, VT2, VF). Therapy options (up to 10
ATP sequences and 8 shocks) are available for the VT1 and VT2
zones, whereas ATP One Shot and up to 8 shock therapies are
available for the VF class. The specific characteristics of an ATP
and shock therapy are independently programmed for each
VT/VF zone.
The ATP and shock therapy options are discussed in detail in the
following sections.
2.8.2 Anti-Tachycardia Pacing (ATP)
Anti-tachycardia pacing therapy (ATP) is available in both VT
detection zones. Available modes of ATP include Burst, Ramp,
and Burst + PES (Programmed Extra Stimuli). In addition, the
Burst and Ramp modes allow interval scanning of the R-S1
Interval, the S1 Decrement, or both. The Attempts parameter
determines the number of burst schemes to be delivered before
the scan parameter is incremented.
Burst – This mode will deliver a series of pacing stimuli with user
defined duration of the burst (Number S1), coupling cycle length
(R-S1) and burst rate (S1-S1). The coupling interval and the start
interval are calculated from the intrinsic R-R average.
Ramp - This mode will deliver a series of pacing stimuli with the
above options including a parameter which decrements each
successive stimuli interval in the burst.
Burst + PES - This mode provides a pulse train followed by one
additional timed stimuli. The coupling cycle length of the burst
and each extra stimulus (S1-S2 Interval) is individually
programmed either as an adaptive value (as a percentage) or as
an absolute value (expressed in milliseconds).
Ventricular Pacing - This parameter defines the type of
ventricular pacing performed during delivery of ATP sequences in
CRT-Ds and is programmable to biventricular (VV delay = 0 ms)
or right ventricular only.
Page 82

74 Lumax Technical Manual
Number S1 - This parameter defines the number of stimuli for an
ATP. For Burst + PES, a single extra stimulus with a separate
parameter setting is coupled.
Add S1 - This feature can be programmed with any Burst, Ramp,
or Burst + PES scheme. When “Add S1” is “ON,” the number of
S1 intervals is incremented by one on each successive ATP
therapy. The next S1-S1 interval is dependent on the initial start
interval (S1 decrement) and the programmed Scan Decrement (if
activated).
R-S1 Interval - The R-S1 programmable coupling interval occurs
at the beginning of each ATP. It defines the interval between the
last R-wave signal and the first stimulus (S1). The second
stimulus always follows the first one with the same interval.
S1 Decrement - The S1 decrement continuously reduces the
pulse intervals of the ATP from the second pulse onward.
S1-S2 Interval - The S1-S2 programmable coupling interval
occurs between the Burst sequence and the extra stimuli (PES). It
defines the interval between the first stimulus (S1) and the extra
stimuli (S2).
Scan Decrement - The Scan decrement continuously reduces
the starting pulse intervals of each Burst or Scan.
Minimum ATP Interval - The programmed minimum interval
prevents ATPs from being given with stimulation values less than
the minimum interval. When the ATP interval reaches the value of
the minimum interval with the S1 decrement or scan decrement, it
then assumes this value and remains constant.
2.8.2.1 ATP Help
ATP help is a useful tool to assist the physician in choosing and
confirming appropriate ATP programming. The “ATP Help” button
is displayed in each ATP therapy option. When the ATP help
button is pressed, a histogram of the chosen therapy scheme is
shown. The histogram displays the intervals for the programmed
ATP scheme. When rate adaptive intervals are programmed, the
displayed intervals are based on the programmed R-R average.
Page 83

Lumax Technical Manual 75
2.8.2.2 ATP Optimization
In order to optimize future therapies, the ICD will store the
parameter configuration of the last successful ATP attempt in
each the VT1 and VT2 classes. The last successful stored ATP
attempt is then used as the starting point for the next detected
episode of the same arrhythmia class. If the stored parameter
configuration is not successful, it is deleted from the ATP
optimization memory of the respective arrhythmia class and
subsequent therapy sequences will begin with ATP1 for the next
detected episode.
ATP Optimization is programmable ON or OFF for all ATP
therapies and VT zones with one parameter.
NOTE:
In VT zones, the ICD/CRT-D stores successful ATP therapies
only. The stored information includes not only the number of
the ATP therapy (e.g., ATP2), but also the successful
configuration in detail (for example: Burst; R-S1 Interval:
320 ms, S1-S1 Interval: 320 ms; etc.).
2.8.2.3 ATP Timeout
ATP Timeout is a timer that decrements after the initial ventricular
ATP is delivered (VT-1 zone) and limits the additional ATP
therapies that may be delivered. Once the timer expires, all
further ATP therapies in the sequence are blocked. If further
therapy is required after the timer has expired, the system
advances to the first programmed shock therapy for the
applicable VT zone. Therapy continues until arrhythmia
termination or all programmed therapy (in the applicable zone)
has been delivered. The ATP Timeout is reset each time an
arrhythmia is terminated.
Page 84

76 Lumax Technical Manual
2.8.2.4 ATP One Shot
ATP One Shot offers the opportunity to treat fast VTs that are
detected in the VF zone with a single ATP sequence delivered
immediately before charging of the high energy capacitors. The
device performs a stability check (same as VT zones, criterion
12% fixed) to determine if the arrhythmia might be a fast VT and if
the rhythm is stable, the programmed ATP sequence is delivered
prior to charging. Charging starts without redetection and the
successful treatment of the arrhythmia will be confirmed prior to
shock delivery during shock charging. If the arrhythmia is
converted by the ATP, the shock is aborted. All ATP therapy
parameters are available for programming ATP One Shot and it
can only be used with shock therapy is programmed ON.
2.8.3 Shock Therapy
Shock Therapy is developed by internal circuitry that stores
energy across high energy capacitors. The voltage level for the
charging cycle is based on the programmed energy level. The
energy is then delivered over the connected ICD lead and through
the ICD/CRT-D housing utilizing a biphasic waveform. The first
and second shock energies in each shock module have
independently programmable Shock Energy.
A synchronization window is started at the end of the charging
period. During this window, the device will attempt to synchronize
the shock therapy to an R-wave. If no R-wave is detected and
Confirmation is programmed to OFF, the shock will be delivered
asynchronously at the end of the synchronization period (DF). If
no R-wave is detected and Confirmation is programmed to ON,
the shock will be aborted at the end of the synchronization period
(DFc = Cardio Version).
Page 85

Lumax Technical Manual 77
2.8.3.1 Confirmation
The Confirmation parameter is used to verify the presence of a
tachyarrhythmia during charging of the shock capacitors. This
function is designed to avoid delivery of shock therapy if a
tachyarrhythmia has spontaneously terminated. The
programmed shock will be delivered unless bradycardia or a
normal sinus rhythm is detected during the Confirmation period.
Confirmation may be programmed ON or OFF for each zone (VT1, VT-2 or VF). Although if a shock is aborted and redetection
occurs (even in a zone with confirmation programmed ON) the
subsequent shock will be delivered without confirmation (see
confirmation OFF).
Confirmation OFF
When Confirmation is programmed OFF, shock therapy will be
delivered to the patient during the synchronization period
regardless of the detected cardiac signal.
Confirmation ON
If the tachyarrhythmia spontaneously converts to bradycardia or a
normal sinus rhythm during the confirmation period, shock
therapy is aborted. If the device confirms the presence of the
tachyarrhythmia, the device will deliver the programmed shock
therapy synchronized to the R-wave.
2.8.3.2 Number of Shocks
The number of shocks defines the total number of shock attempts
per therapy zone (VT-1, VT-2 or VF). Up to 8 shocks are
available in each therapy zone. The first and second shock
energies are independently programmable, while the remaining
shocks are fixed at maximum energy (30 joules for 300 series
devices and 40 joules for 340 series devices).
2.8.3.3 Shock Waveform
Two waveforms of shock therapy are available with the Lumax
ICDs/CRT-Ds, Biphasic and Biphasic 2 (ms). The following
diagram describes each of the shock waveforms.
Page 86

78 Lumax Technical Manual
Figure 5. Biphasic Waveforms
Both waveforms start at the calculated voltage, based on the
programmed energy level. After an exponential discharge
through the lead system to 40% of the initial charge voltage, both
shock waveforms switch polarity. At the second phase the:
• Biphasic waveform discharges to 20% of the initial charge
voltage before the waveform is truncated
• Biphasic 2ms waveform discharges the remaining energy
for two milliseconds before the waveform is truncated
Figure 5 provides a pictorial representation of both biphasic
waveforms.
BIOTRONIK recommends use of the standard Biphasic shock
waveform for initial defibrillation threshold testing. If testing
demonstrates high defibrillation thresholds, testing with the
Biphasic 2ms waveform is offered as a therapeutic alternative to
the standard Biphasic shock.
2.8.3.4 Shock Energy
The Lumax ICDs/CRT-Ds are designed to charge to the energy
selected on the programmer screen, but similar to all other
commercially available ICDs/CRT-Ds, the actual therapy
delivered is somewhat less depending on several factors
including the shock lead impedance. The first two shock energies
in each therapy class are programmable between 1 joules and
maximum energy for the device type. The energy of the second
shock is always greater than the first shock. The remaining
shock energies will be delivered at maximum programmable
energy.
Page 87

Lumax Technical Manual 79
Actual energy delivered for each programmable shock energy is
approximately equal to the “Energy Delivered” for the high energy
Lumax 340/540 variants in Table 22, and for the Lumax 300/500
in Table 23.
Table 22 Delivered Shock Energy (340/540 Variants)
Programmed Energy
(joules)
Approximate Delivered
Energy (joules)
10.8
21.7
32.6
43.5
54.3
65.2
76.1
86.9
97.8
10 8.7
11 9.6
12 10.5
13 11.3
14 12.2
15 13.1
16 13.9
18 15.7
20 17.3
22 19.1
24 21.1
26 22.8
28 24.4
30 26.3
32 27.9
34 29.7
36 31.5
38 33.3
40 35.0
Page 88

80 Lumax Technical Manual
Table 23 Delivered Shock Energy (300/500 Variants)
Programmed Energy
(joules)
Approximate Delivered
Energy (joules)
10.8
21.7
32.5
43.3
54.3
65.1
76.0
87.0
97.8
10 8.6
11 9.6
12 10.6
13 11.4
14 12.3
15 13.1
16 14.2
18 16.0
20 17.7
22 19.5
24 21.4
26 23.2
28 24.9
30 27.0
Page 89

Lumax Technical Manual 81
CAUTION
Shock Impedance - If the shock impedance is less than
twenty-five ohms, reposition the lead system to allow a
greater distance between the electrodes. Never implant the
device with a lead system that has measured shock
impedance as less than twenty-five ohms. Damage to the
device may result.
Defibrillation Threshold - Be aware that changes in the
patient’s condition, drug regimen, and other factors may
change the defibrillation threshold (DFT), which may result in
non-conversion of the arrhythmia post-operatively.
Successful conversion of ventricular fibrillation or ventricular
tachycardia during arrhythmia conversion testing is no
assurance that conversion will occur post-operatively.
Shock Therapy Confirmation – Programming
CONFIRMATION to OFF may increase the incidence of the
ICD/CRT-D delivering inappropriate shocks.
2.8.3.5 Shock Polarity
The polarity of the shock therapy may be programmed and
changed non-invasively. The Normal polarity configures the
HV 1 connector port as the negative electrode and the HV 2
connector port and the outer housing of the ICD/CRT-D as the
positive electrode for the first phase of the shock. Reversed
polarity will switch the electrical polarity of the connector ports
and housing. The shock polarity is separately programmable for
each arrhythmia zone.
Alternating polarity is automatically employed when a maximum
energy shock with normal polarity fails to cardovert. The next
shock will have reversed polarity and alternates the polarity for all
subsequent shocks.
Page 90

82 Lumax Technical Manual
2.8.3.6 Third Programmable Shock Pathway
Lumax 500/540 Models only
The Lumax 500/540 models offer three programmable shock
delivery configurations. The defibrillation shock pathways include
a “non-active housing” shock path and a shock paths between the
ICD/CRT-D housing and either or both shock coils when
connected to a dual coil shock lead.
The Shock Configuration parameter operates independently of
Shock Polarity. So each of the pathways described below can be
reversed for delivered defibrillation/cardioversion shocks.
Table 24 outlines the current shock pathways that are available
with Lumax 500/540 models.
Table 24 Lumax 500/540 Programmable Shock Pathways
# Electrical Pathway
RVÎ SVC and Housing (can)*
RV Î SVC
RV Î Housing (can)
- 300/340 Models are limited to this configuration.
2.8.4 Progressive Course of Therapy
By design, the Lumax ICDs/CRT-Ds will deliver more aggressive
therapy for each successive attempt within a single detected
episode. Therefore, the device will not deliver ATP1 therapy
following ATP2 therapy, and will not deliver ATP therapy following
a high voltage defibrillation shock.
When Progressive Course of Therapy is turned ON, the ICD/CRTD will always deliver a maximum energy shock after re-detecting
in an arrhythmia class with programmed shock energy less than
or equal to the previously delivered therapy. In addition, the
ICD/CRT-D blocks all ATP therapy during the current episode if a
shock has already been delivered during the episode.
Page 91

Lumax Technical Manual 83
Furthermore, the ICD/CRT-D prevents therapies of different
arrhythmia classes from permanently retarding or accelerating a
VT in such a way that the cardiac rhythm fluctuates between the
different arrhythmia classes without achieving termination of the
arrhythmia regardless of the Progressive Course of Therapy
setting.
For example, a 10-joule defibrillation shock is delivered for an
arrhythmia detected in the VT-2 zone and results in a deceleration
of the VT so that it is subsequently redetected in the VT-1 zone.
At that point, the Lumax ICDs/CRT-Ds would continue with shock
therapy, but all shocks programmed at less than 10 joules would
be delivered at 10 joules.
If a defibrillation shock is delivered but does not terminate the
arrhythmia, the next shock will always have the same or higher
energy than the last delivered shock. Beginning with the third
shock, all shocks are delivered at maximum energy.
2.9 Bradycardia Therapy
The Lumax ICDs/CRT-Ds have independently programmable
single, dual and triple chamber and post-shock pacing functions.
The post-shock bradycardia parameters may be programmed to
higher rates or output values for the period following a delivered
shock, without significantly compromising the longevity of the
ICD/CRT-D for patients who require chronic bradycardia pacing.
The post-shock programmable values are presented in a
separate subsection from the normal bradycardia pacing support
values.
2.9.1 Bradycardia Pacing Modes
The available bradycardia pacing modes for each member of the
Lumax ICD/CRT-D family are listed in Table 25.
Page 92

84 Lumax Technical Manual
Table 25 Lumax Pacing Modes
Mode Lumax HF Lumax DR Lumax VR
DDDR X X N/A
DDIR X X N/A
VDDR X X N/A
VDIR X X N/A
AAIR X X N/A
VVIR X X X
DDD X X N/A
DDI X X N/A
VDD X X N/A
VDI X X N/A
AAI X X N/A
VVI X X X
OFF X X X
The basic rate timer is started by a sensed or paced event. A
sensed event outside of the refractory period inhibits pacing and
resets the lower rate time; in the absence of a sensed event, a
pacing pulse will be delivered at the end of the lower rate interval.
The pacing modes with an “R” indicate rate adaptive pacing
controlled by a motion based capacitive sensor. These modes
are functionally the same as the corresponding non-rate-adaptive
modes, except that the pacing rate is increased based on
physical activity.
2.9.2 Basic Rate
The basic rate is the pacing rate in the absence of a patient’s
intrinsic rhythm. This rate may be independently programmed for
normal and post-shock bradycardia pacing.
Page 93

Lumax Technical Manual 85
2.9.3 Night Rate
The night rate is the effective basic rate during the programmed
“sleep” period for the patient. This parameter provides a different
pacing rate during the patient’s normal sleep time in an attempt to
match for example the decreased metabolic needs during sleep.
When Night Mode is active, the basic rate automatically changes
to the programmed NIGHT RATE during the nighttime hours.
At the programmed start time (Begin of Night), the rate gradually
adapts to the night rate. When the internal clock reaches the
programmed end time (End of Night), the pacing rate gradually
changes to the programmed basic rate. The rate changes at the
same rate as the Sensor Gain decrease and increase
parameters.
NOTE:
The Night Mode time is based on the programmer clock.
Therefore, the programmer time should be checked prior to
device programming. If a patient travels across different time
zones, the Night Mode time may require adjustment.
2.9.4 Rate Hysteresis
The ability to decrease the effective lower rate through
Hysteresis is intended to preserve a spontaneous rhythm. The
pulse generator operates by waiting for a sensed event throughout
the effective lower rate interval (Hysteresis interval). If no sensed
event occurs, a pacing pulse is emitted following the Hysteresis
interval.
Hysteresis can be programmed OFF or to values as low as
-90 bpm of the basic rate. Hysteresis is initiated by a sensed
event. The resulting Hysteresis rate is always less than the lower
rate. The Hysteresis rate can only be programmed to provide a
basic rate that is 30 bpm or greater.
NOTES:
If rate adaptation is active, the Hysteresis rate is based on the
current sensor-indicated rate and the value of the
programmable parameter.
Page 94

86 Lumax Technical Manual
If Hysteresis is used in the DDI mode, the AV delay must be
programmed shorter than the spontaneous AV conduction
time. Otherwise, stimulation in the absence of spontaneous
activity occurs at the hysteresis rate instead of the lower rate.
Night Rate is the limit for the Hysteresis when Night Mode is
active. Programming conflicts arise when the total decrease
in rate is below 30 ppm. Care should be exercised to avoid
programming a Night Mode rate and hysteresis that are
below what is appropriate and may be tolerated by the
individual patient.
2.9.4.1 Repetitive Hysteresis
Repetitive hysteresis is expanded programmability of the
Hysteresis feature. Repetitive hysteresis searches for an intrinsic
cardiac rhythm, which may exist below the programmed lower
rate (or sensor-indicated rate) of the patient. Following 180
consecutive sensed events, this feature allows the intrinsic
rhythm to drop to or below the hysteresis rate. During the time
when the intrinsic rate is at or below the hysteresis rate, pacing
occurs at the hysteresis rate for the programmed number of beats
(up to 10). Should the number of programmed beats be
exceeded, the stimulation rate returns to the lower rate (or
sensor-indicated rate).
If an intrinsic cardiac rhythm is detected within the programmed
number of beats between the hysteresis rate and the lower rate,
the intrinsic rhythm is allowed and inhibits the pulse generator.
Page 95

Lumax Technical Manual 87
Figure 6. Repetitive Hysteresis
Repetitive hysteresis has been incorporated to promote
spontaneous cardiac rhythm and may reduce pulse generator
energy consumption.
NOTE:
Repetitive and Scan Hysteresis are only available when
Hysteresis is selected on.
There is one Standard Hysteresis interval which occurs
before the programmable number of Repetitive Hysteresis.
2.9.4.2 Scan Hysteresis
Scan hysteresis is expanded programmability of the Hysteresis
feature. Scan hysteresis searches for an intrinsic cardiac rhythm,
which may exist just below the programmed lower rate (or sensorindicated rate). Following 180 consecutive paced events, the
stimulation rate is temporarily decreased to the hysteresis rate for
a programmed number of beats. If a cardiac rhythm is not
detected within the programmed number of beats at the
hysteresis rate, the stimulation rate returns back to the original
lower rate (or sensor-indicated rate). Several programmable beat
intervals are available to allow a greater probability of detecting a
spontaneous rhythm.
Page 96

88 Lumax Technical Manual
If an intrinsic cardiac rhythm is detected within the programmed
number of beats between the hysteresis rate and the lower rate,
the intrinsic rhythm is allowed and the pulse generator inhibits.
Figure 7. Scan Hysteresis
Scan hysteresis has been incorporated to promote intrinsic
cardiac rhythm and may reduce pulse generator energy
consumption.
2.9.5 Dynamic AV Delay
DR and HF versions only
The AV Delay defines the interval between an atrial paced or
sensed event and the ventricular pacing pulse. If the pulse
generator is programmed to a dual chamber sensing mode, an
intrinsic ventricular event falling within the AV Delay will inhibit the
ventricular pacing pulse. If not contraindicated, a longer AV Delay
can be selected to preserve intrinsic AV conduction.
Dynamic AV Delay is where the AV Delay is varied depending on
the spontaneous atrial rate. Dynamic AV Delay provides a linear
change of AV Delays depending on current rate at preset lower
and upper AV Delay values.
Page 97

Lumax Technical Manual 89
In addition to selecting the preset values (Low, Medium, and
High) with the Dynamic AV Delay window, the Dynamic
AV Delays may be programmed individually (Individual) for each
rate zone or to a fixed AV Delay (Fixed).
The AV Delay feature includes an AV Delay shortening option
(Sense Compensation) for dual chamber pacing modes. When
enabled, the AV Delay is shortened by the programmed value
(5 to 60 ms) from the programmed AV Delay after an intrinsic
atrial sensed event.
The Dynamic AV Delay is intended to mimic physiologicshortening of the AV Delay with increasing heart rate. It also
serves for automatic prevention and termination of “circus
movement” pacemaker mediated tachycardia and for prevention
of reentrant supraventricular tachycardias. Dynamic AV Delay is
available with the Lumax DR-T and HF-T ICDs/CRT-Ds.
2.9.5.1 Positive AV Hysteresis
Positive AV Hysteresis allows a user-programmable change in AV
delay that is designed to encourage normal conduction of intrinsic
signals from the atrium into the ventricles. With Positive AV
hysteresis enabled, the AV delay is extended by a defined time
value after sensing a ventricular event (10 … (10) …150 ms).
The long AV interval is used as long as intrinsic ventricular activity
is detected. The programmed short AV delay interval resumes
after a ventricular paced event.
2.9.5.2 AV Repetitive Hysteresis
With AV Repetitive Hysteresis, the AV delay is extended by a
defined hysteresis value after sensing an intrinsic ventricular
event. When a ventricular paced event occurs, a long AV delay is
used for the programmed number of cycles. (OFF, 1 … 10). If an
intrinsic rhythm occurs during one of the repetitive cycles, the
long duration AV delay interval remains in effect. If an intrinsic
rhythm does not occur during the repetitive cycles, the original AV
delay interval resumes.
Page 98

90 Lumax Technical Manual
2.9.5.3 AV Scan Hysteresis
With AV Scan Hysteresis enabled, after 180 consecutive pacing
cycles, the AV delay is extended for the programmed number of
pacing cycles. (OFF, 1 … 10). If an intrinsic rhythm is detected
within the extended AV delay and the longer AV delay remains in
effect. If an intrinsic rhythm is not detected within the number of
scan cycles, the original AV delay value resumes.
2.9.5.4 Negative AV Hysteresis
With Negative AV Hysteresis, the AV delay is decreased by a
programmable value (10 … (10) …150 ms) after a ventricular
event is sensed, thereby promoting ventricular pacing. The
shorten AV interval is used for one time.
2.9.5.5 Negative AV Repetitive Hysteresis
With AV Repetitive Hysteresis and negative AV Hysteresis, the AV
delay is shortened by a defined hysteresis value after sensing an
intrinsic ventricular event as described above. The short AV Delay
will retain until the programmed number of cycles has elapsed.
(OFF,1 … 180). The normal AV delay resumes after the
programmed number of consecutive ventricular paced events
(Repetitive Negative AV Hysteresis) elapses.
CAUTION
Negative AV Hysteresis – This feature insures ventricular
pacing, a technique which has been used in patients with
hypertrophic obstructive cardiomyopathy (HOCM) with normal
AV conduction in order to replace intrinsic ventricular
activation. No clinical study was conducted to evaluate this
feature, and there is conflicting evidence regarding the
potential benefit of ventricular pacing therapy for HOCM
patients. In addition, there is evidence with other patient
groups to suggest that inhibiting the intrinsic ventricular
activation sequence by right ventricular pacing may impair
hemodynamic function and/or survival.
Page 99

Lumax Technical Manual 91
2.9.6 IOPT
DR versions only
The IOPT function serves to support the patient’s intrinsic rhythm
and avoid excessive ventricular pacing. This feature simply
activates all of the AV hysteresis parameters with a single
selection. Table 26 details the settings that are preset when IOPT
is turned ON:
Table 26 IOPT Parameters
Parameter IOPT
AV Hysteresis (max AV Delay) 400 ms
AV Scan Hysteresis 5
Repetitive AV Hysteresis 5
2.9.7 Upper Tracking Rate
DR and HF versions only
In the atrial tracking modes (DDDR, VDDR, DDD, and VDD)
ventricular pacing tracks atrial pace/sense events. The maximum
tracking rate (ventricular pacing rate) is limited by the Upper Rate
parameter.
The UTR response will automatically toggle between 2:1 and
WKB (Wenkebach) depending on the relative programmed values
for upper rate and atrial refractory period (AV Delay + PVARP).
Page 100

92 Lumax Technical Manual
If the UTR is less than the maximum sensed atrial rate, defined
by the atrial refractory period (60,000/ARP), the WKB response is
utilized. Atrial rates exceeding the selected upper rate will result
in a Wenckebach-type pacing pattern. This is accomplished by
progressively lengthening the AV delay to keep the ventricular
pacing rate at the upper rate. Lengthening of the AV interval is
interrupted as soon as: 1) a P-wave falls within the atrial blanking
period and is not detected; or 2) a succeeding P-wave is detected
before the end of the AV delay previously started. In the second
case, the corresponding ventricular pacing pulse is suppressed.
If the atrial rate is just above the upper rate, a low degree (i.e.
6:5) block results. Higher atrial rates result in higher degrees of
AV block until the intrinsic atrial cycle length violates the
programmed atrial refractory period causing a 2:1 or greater
block.
The 2:1 response is utilized when the rate defined by the atrial
refractory period is less than the upper rate. In such a case, the
maximum pacing rate is regulated by the inability to respond to
P-waves falling within the atrial refractory period.
If the resulting length of the spontaneous atrial cycle is shorter
than the atrial refractory period in a rate-adaptive mode, the
resulting pacing rate will depend on whether the 2:1 rate has
been exceeded. If this is the case, the pulse generator will use
the sensor rate as the pacing rate. If the 2:1 rate is not exceeded,
the pulse generator will use a rate that lies between the sensor
rate and the rate determined by the atrial refractory period.
Atrial Upper Rate is designed to prevent pacing in the vulnerable
period after an atrial sensed event during PVARP. It ensures that
the next atrial pace is emitted outside of the patient’s normal sinus
atrial refractory period. Atrial Upper Rate is limited to 240 ms or
OFF.
NOTE:
Lumax DR ICDs and Lumax HF CRT-Ds allow the UTR to be
programmed within the VT-1 zone. This feature is for patients
that are active and have exercise and VT rates that overlap.
This may be desirable in young active patients. Also, with
RVsense Triggering programming the UTR in the VT-1 zone
is blocked.
 Loading...
Loading...