

Page 1

Das Agilent Metabolomics
Labor: Alle Werkzeuge für eine
erfolgreiche Metabolomforschung
ÜBERBLICK
Die Metabolomics-Forschung ist eine
logische Ergänzung groß angelegter
Studien auf den Gebieten Expressionsprofiling und Proteomics
und liefert wertvolle Einblicke in die
Biochemie von Organismen. Sie stellt
Wissenschaftler aber gleichzeitig
auch vor analytische Herausforderungen. Agilent Technologies
unterstützt mit Fachwissen, Systemen
und Werkzeugen für die Datenanalyse,
damit diese Herausforderungen
bewältigt werden und eine erfolgreiche Metabolomforschung
betrieben werden kann.
Page 2

2
Ein leistungsstarker Ansatz für die Erforschung biologischer Systeme
Metaboliten spielen in biologischen Systemen eine wichtige Rolle. Mit Metabolomics, d. h. der vergleichenden Analyse von Metaboliten, die in ähnlichen biologischen Proben gefunden wurden, lassen sich
potenzielle Biomarker, Auswirkungen von Medikamenten oder Krankheiten auf bekannten oder unerwarteten biologischen Pathways erkennen.
Metaboliten als wesentliche
Komponenten in biologischen
Systemen
Metaboliten spielen in biologischen
Systemen eine wichtige Rolle. Sie
transportieren Energie, ermöglichen die
zelluläre Kommunikation und Steuerung
und funktionieren als Bausteine für
andere Prozesse. Metabolitänderungen
liefern wertvolle Einblicke in die zugrundeliegenden biochemischen Prozesse. Sie
dienen als Marker für Krankheiten und
weisen auf Zusammenhänge zwischen
Genen und Funktionen hin.
Die Metabolomics-Forschung dient dem
Verständnis biologischer Systeme. Sie
kann als leistungsstarke Ergänzung der
Genom- und Proteonomanalyse eingesetzt
werden.
Metabolomics – eine analytische
Herausforderung
Metaboliten weisen eine enorme
chemische Vielfalt mit beträchtlichen
Variationen in Bezug auf Struktur, funktionale Gruppen und physikochemische
Eigenschaften auf. Aus diesem Grund
und wegen der großen Häufigkeitsunterschiede sind Metabolomanalysen sehr
schwierig. Darüber hinaus wird die
Aufgabe durch die unterschiedlichen
analytischen Anforderungen erschwert,
da das Ziel entweder darin besteht, Substanzen zu finden, diese zu identifizieren,
zu quantifizieren oder alle diese drei Ziele
zusammen.
Es gibt kein Gerät und keine Technologie
mit Eignung für alle Metabolomanalysen. Die gemeinsam mit einem Gasoder Flüssigchromatographiesystem eingesetzte Massenspektrometrie (GC/MS
bzw. LC/MS) ist die am häufigsten verwendete Analysenmethode. Die Kombination
aus Kapillarelektrophorese und Massenspektrometrie (CE/MS) ist eine alternative
Lösung für hydrophile Substanzen. Metabolomuntersuchungen erfordern in der Regel
eine große Probenanzahl und komplexe
Datenverarbeitungsvorgänge. Daher
werden zudem leistungsstarke
Datenanalysenfunktionen benötigt.
Herausragende Werkzeuge für
die Metabolomforschung
Agilent Technologies liefert ein umfassendes Angebot an Werkzeugen für die
Metabolomforschung: Es beinhaltet GC-,
LC-, CE-, GC/MS-, LC/MS- und CE/MSSysteme sowie leistungsstarke Softwareapplikationen zur Identifizierung von
Metaboliten, zur Quantifizierung und
statistischen Analyse.
Bei Agilent erhalten Sie Hardware,
Software, Verbrauchsmaterialien,
Service und Support verlässlich aus
einer Hand. Außerdem können Sie auf
ein sachkundiges Team aus Applikationsspezialisten zurückgreifen, die verstehen,
worum es bei Ihrer Arbeit geht, und die
Sie unterstützen, aus allen Experimenten
biologisch aussagekräftige Daten
abzuleiten.
Agilent bietet die umfassendste
Palette an Geräten und Software
für Metabolomics, einschließlich
vollständiger GC/MS- und
LC/MS-Systeme.
Page 3
www.agilent.com/chem/metabolomics
3
Das Agilent Metabolomics Labor
Der gesamte Prozess der Metabolomanalyse wird in verschiedene Schritte
aufgeteilt. Je nach untersuchten Proben
und Metaboliten weichen die Details
voneinander ab - der Prozess ist aber
immer gleich:
1. Profiling: Suche nach Metaboliten mit
statistisch signifikanten Abweichungen
in der Häufigkeit innerhalb einer kleinen
Probenmenge aus experimentellen und
Kontrollproben.
2. Identifizierung: Identifizierung der im
Profiling markierten Metaboliten.
3. Validierung: Validierung der statistischen Signifikanz der identifizierten
Metaboliten in den ursprünglichen Proben,
gefolgt von der Validierung gegen größere
Probenmengen, um die Auswirkungen
natürlicher Abweichungen auszuschließen.
4. Interpretation: Auswertung der
gefundenen metabolomischen Marker
im Kontext des entsprechenden
biologischen Systems.
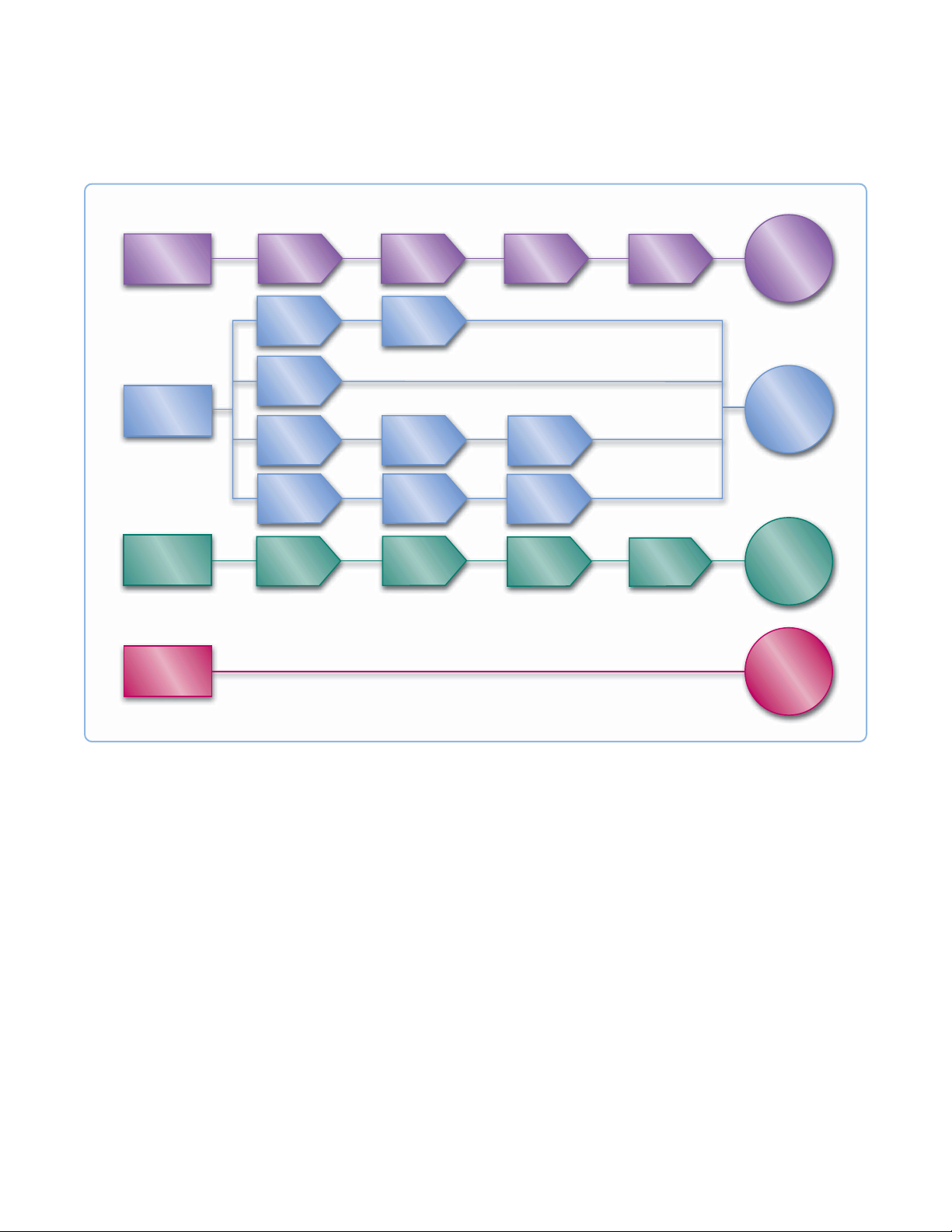
Der Metabolomics-Workflow: Von der Probe über Daten zur biologischen Bedeutung
1. Profiling
Probenabhängige
Vorbereitung
Aufreinigung
Bibliothekssuche
nach Fragmenten
(GC/MS)
Datenbanksuche
(LC/MS)
Probenabhängige
Vorbereitung
Probenabhängige
Analyse
Standards
erfassen und
analysieren
Standards
und Proben
vergleichen
De-Novo-
Spektrenauswertung
Standards
erfassen und
analysieren
Standards
und Proben
vergleichen
Gezielte
Quantifizierung
Statistische
Analyse
Validierte
Metaboliten
Pathways
und Durchsatz/
Umsatz
NMR-Analyse
Probenabhängige
Analyse
Merkmalerkennung
Datennormalisierung und
statistische
Analyse
Signifikante
Metaboliten
MetabolitIdentitäten
2. Identifizierung
3. Validierung
4. Interpretation
Der Workflow für die Metabolomanalyse ist ein mehrstufiger
Prozess mit unterschiedlichen
analytischen Herausforderungen
und Wahlmöglichkeiten bei jedem
Schritt.
Page 4
4
Profiling zur Suche nach statistisch signifikanten Metaboliten
Beim Profiling wird nach Metaboliten mit
statistisch signifikanten Abweichungen
in der Häufigkeit zwischen der experimentellen und der Kontrollgruppe gesucht.
Das Profiling kann sehr umfangreich
ausfallen, wenn Sie klare Ziele, aber nur
ein begrenztes Wissen über das untersuchte System haben. Wenn Sie jedoch
mit einem gut untersuchten System
arbeiten, kann das Profiling sehr zielgerichtet durchgeführt werden. Häufig
ist die beste Vorgehensweise, beide Ansätze zu verfolgen, d. h. ein gezieltes
Profiling bekannter Metaboliten und eine
umfangreiche Merkmalerkennung zur
Suche nach unerwarteten Metaboliten.
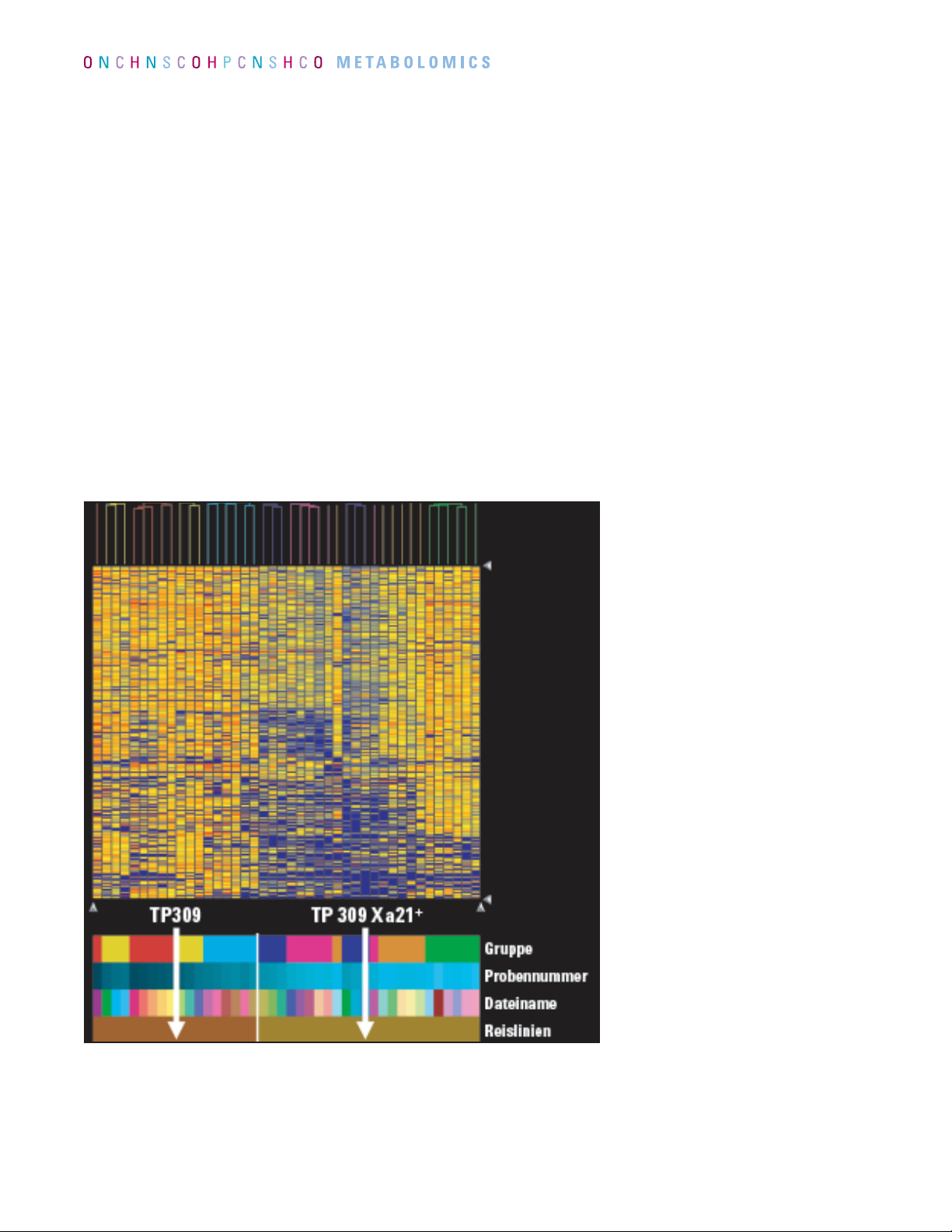
Durch hierarchische Clusterbildung werden Proben
basierend auf der Ähnlichkeit
ihrer Massenhäufigkeitsprofile
gruppiert. Diese Vorgehensweise
eignet sich zur Bewertung der
Datenqualität. Replizierte Proben
aus derselben experimentellen
Gruppe sollten enger beieinander
liegen als Proben aus unterschiedlichen Gruppen. In diesem Beispiel
erlaubt es die hierarchische
Clusterbildung bei Reisdaten
sofort zwischen der Wildreislinie
(TP309) und einer transgenen
Linie (TP309 Xa21
+
) zu unter-
scheiden.
Das Profiling setzt sich üblicherweise
aus folgenden Schritte zusammen:
• Methode auswählen: Auswertung der
Kenntnisse über das System und Ziele,
gefolgt von der Auswahl der besten
Methode für das Profiling
• Probenvorbereitung: Probenabhängige
Extraktion, Proteinpräzipitation und
Vorfraktionierung
• Analyse: Trennung und Nachweis von
Metaboliten, i. d. R. durch GC/MS oder
LC/MS
• Merkmalerkennung: Suche nach und
Quantifizierung aller Metaboliten in
der Probe
• Datennormalisierung: Korrektur der
durch Retentionszeiten oder Response
verursachten Drift
• Statistische Analyse: Erkennung
statistisch signifikanter Unterschiede
zwischen Probenmengen
Die analytische Reproduzierbarkeit ist
für das Expression Profiling von immenser
Bedeutung. Die Kombination aus normalen
Probenabweichungen und analytischen
Abweichungen bestimmt die Anzahl
an notwendigen Wiederholversuchen,
um festzustellen, ob die Unterschiede
zwischen Proben oder Probenmengen
statistisch signifikant sind. Je kleiner
die analytischen Abweichungen, desto
weniger Wiederholversuche sind
erforderlich.
Page 5
www.agilent.com/chem/metabolomics
5
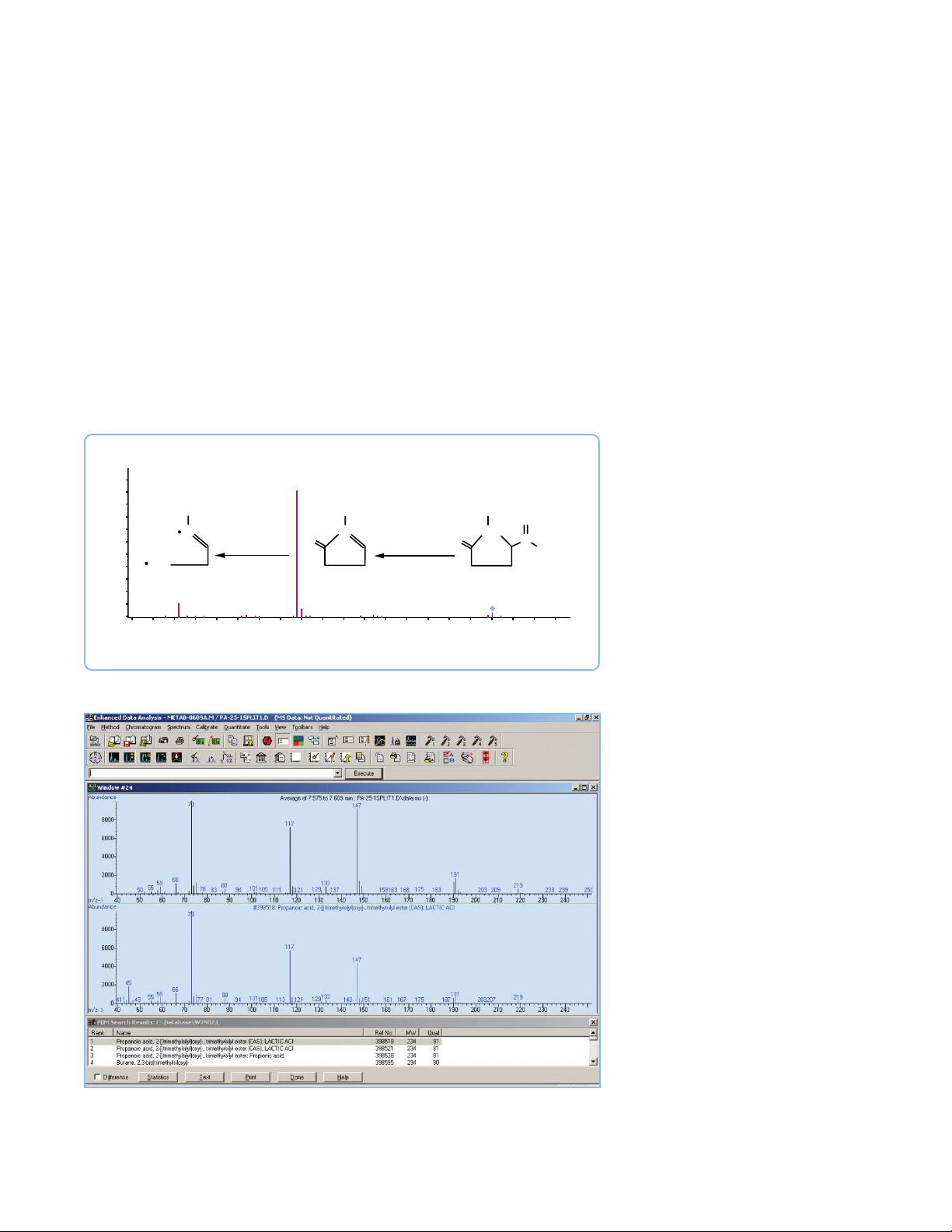
Eine Suche in GC/MSSpektrenbibliotheken kann
häufig zu einer schnellen
positiven Identifizierung
führen. In diesem Beispiel
wurde die Substanz positiv
als eine bestimmte Propionsäure identifiziert.
Eine Suche in der METLIN
Metabolite Database mit
TOF-Daten bekannter Masse
ergab für ein Metabolit mit
einer Masse von 130,0494
(Molekularion) sechs
mögliche Identitäten. Durch
das in der nachfolgenden
Q-TOF-Analyse erzeugte
MS/MS-Spektrum ließ sich
die Identität auf eines von
zwei Enantiomeren reduzieren: Pyroglutaminsäure
oder Pyrrolidoncarbonsäure.
x10
3
0
1
2
3
4
5
Verhältnis von Masse zu Ladung (m/z)
50 60 70 80 90 100 110 120 130 140
84,04484
56,05052
130,05320
(gemessen)
Häufigkeit
MS/MS-Spektrum von
Vorläuferion m/z 130,0532
NH
C
O
OH
H
+
O
N
H
+
O
-CH2O
2
N
H
+
CH
2
-CO
Das Agilent Metabolomics Labor
Identifizierung signifikanter
Metaboliten
Nach dem Profiling müssen die statistisch signifikanten Metaboliten identifiziert werden. Es gibt vier übliche Vorgehensweisen:
• Suche in Spektrenbibliotheken:
Kann eine erneute Analyse überflüssig
machen und liefert eine positive
Identifizierung. Diese Methode wird
am häufigsten mit reproduzierbaren
EI-Spektren aus GC/MS-Analysen
verwendet.
• Datenbanksuche: Wird häufig mit
LC/MS-Daten verwendet, um nach
wahrscheinlichen Identitäten zu
suchen. Mit einer guten Datenbank
ist die Suche relativ einfach. Zur
positiven Identifizierung müssen
jedoch Standards erfasst und
analysiert werden.
• De-novo-Spektrenauswertung: Ist vor
allem bei der Erforschung gut untersuchter Systeme möglich, erfordert
aber hervorragende Fachkenntnisse.
Für eine positive Identifizierung müssen
Standards erfasst und analysiert werden.
• Aufreinigung und NMR-Analyse:
Hoher Zuverlässigkeitsgrad, kann
aber schwierig, zeitaufwändig und
kostenintensiv sein.
Page 6
6
Validierung bestätigt Signifikanz
Um natürliche Abweichungen zwischen
einzelnen Organismen und exogenes
chemisches Rauschen zu berücksichtigen,
müssen die Schlussfolgerungen aus den
vorherigen Schritten validiert werden.
In der Regel handelt es sich dabei um
einen zweistufigen Prozess:
• Gezielte quantitative Neuanalyse der
ursprünglichen Proben zur Überprüfung
der statistischen Signifikanz der
während des Profilings markierten
Metaboliten
• Quantitative Analyse viel größerer
Probenmengen zur Validierung des
ursprünglichen Profilings und Eliminierung natürlicher Abweichungen.
Auch wenn bei der Validierungsanalyse
mehr Routinearbeiten anfallen als beim
Profiling, gilt es dennoch, hunderte
oder tausende Proben zu verarbeiten.
Zur Reduzierung der Analysenkosten
müssen die Protokolle optimiert werden.
Im Allgemeinen erfolgen diese quantitativen Analysen per Selected Ion
Monitoring (SIM) mit einem GC/MSSystem oder durch ein gezieltes
MS/MS-Verfahren (MRM) mit einem
Triple-Quadrupol-LC/MS-System. Zur
Gewährleistung einer genauen Quantifizierung werden in der Regel interne
Standards verwendet.
Interpretation im biologischen
Kontext
Nach der Suche, Identifizierung und
Verifizierung signifikanter Metaboliten
müssen diese noch im untersuchten
biologischen Kontext ausgewertet
werden. Dies umfasst ggf. eine PathwayAnalyse, Auswertung des Umsatzes
oder die Korrelation zwischen Genen
und dem funktionalen Phänotyp des
Organismus. Möglicherweise müssen
Metabolomdaten mit Genom- oder
Proteonomdaten integriert werden.
Die quantitative Validierung kann Konzentrationsvergleiche (Kalibrierkurven) umfassen, wenn Standards
der Zielsubstanz verfügbar sind, oder Flächenvergleiche, wenn keine Probenstandards verfügbar sind.
Page 7

www.agilent.com/chem/metabolomics
7
Erforderliche Werkzeuge
Metabolome weisen bereits in normalen Organismen signifikante natürliche Abweichungen auf. Wirkliche
Unterschiede lassen sich nur durch die Analyse großer Probenmengen nachweisen. Daher werden Analyseninstrumente mit geringen Fehlerraten und hohem Durchsatz bevorzugt. Die leicht zu bedienenden Agilent
GC/MS- und LC/MS-Systeme sind zuverlässig und bieten eine hervorragende Analysenleistung. Damit sind
sie bestens für Metabolomanalysen mit hohem Durchsatz geeignet.
Die GC/MS-Systeme
ermöglichen empfindliche und
wirtschaftliche Profiling- und
Identifizierungsverfahren.
GC/MS-Geräte bieten eine hohe Trennleistung und sind empfindlich genug für
flüchtige Substanzen und Substanzen,
die in flüchtige Derivate verwandelt
werden können. Sie ermöglichen die
Probenidentifizierung mit Hilfe einer
Suche in EI-Spektrenbibliotheken.
Außerdem sind sie bei weitem die
wirtschaftlichsten Geräte für
Metabolomanalysen.
Agilent ist der weltweit führende
Anbieter von GC/MS-Systemen und
verfügt über das entsprechende Fachwissen. Die Kombination aus dem
Verkaufsschlager Agilent 6890N Gaschromatograph und dem innovativen
Quadrupol-Massenspektrometer der
Agilent Serie 5975B ist in Bezug auf
Leistung und Zuverlässigkeit unschlagbar:
• Branchenweit höchste Empfindlichkeit
• Reproduzierbare und in Bibliotheken
durchsuchbare EI-Spektren
• Automatisierung, mit der die optionale
chemische Ionisierung genauso einfach
wie die Elektronenionisierung ist
• Bis zu 6 Ordnungen dynamischer Bereiche zur Analyse von Metaboliten
mit stark variierender Häufigkeit
• Schnelles Scannen (10.000 U/s), das
mit der schnellen Chromatographie für
einen hohen Durchsatz mithalten kann
• Hervorragende Stabilität der Massenachse verringert Anzahl der Gerätefehler
• Chemisch inerte Ionenquelle verhindert
den Zerfall der Metaboliten
Die technisch ausgereiften GC-Säulen
der Agilent Marke J&W Scientific bieten
auch bei den schwierigsten Probentypen
eine herausragende und reproduzierbare
Leistung. Diese Säulen weisen die geringsten Ausblutungswerte, die beste Inertheit
für Säuren/Basen/gemischt-funktionale
Substanzen und die höchste verfügbare
Reproduzierbarkeit bei anderen Säulen
des gleichen Typs auf.
Agilent GC/MSSysteme weisen eine
hohe Trennleistung
und Empfindlichkeit für
Metabolomanalysen auf.
Als Bibliothek durchsuchbare EI-Spektren
vereinfachen die
Identifizierung.
Das Agilent Metabolomics Labor
Page 8

8
LC/MS-Systeme als Lösung
vieler Probleme der
Metabolomanalyse
Mit der Flüssigchromatographie/
Massenspektrometrie (LC/MS) lässt
sich eine große Menge an Metaboliten
analysieren. Sie eignet sich vor allem für
Metabolitklassen, die nicht flüchtig sind
und nicht derivatisiert werden können.
Die LC/MS bietet eine hervorragende
Selektivität und Empfindlichkeit. Außerdem erzeugt sie Molekularionen- und
Strukturdaten.
Agilent hat eine große Anzahl zuverlässiger HPLC/MS-Systeme im Angebot.
Jedes System eignet sich für eine andere
Phase des Metabolomics-Workflows und
ist eine Kombination aus hervorragender
Leistung mit der für Agilent charakteristischen Zuverlässigkeit und
Benutzerfreundlichkeit.
In einem Metabolomics-Labor mit mehreren Geräten ist eine gleichbleibende,
plattformübergreifende Leistung erforderlich, um den Durchsatz zu maximieren
und analytische Abweichungen möglichst
gering zu halten. Der Einsatz gleicher
Technologien, z. B. der Kollisionszelle im
Q-TOF und der Triple-Quadrupol-Geräte
sowie der TOF-Komponenten in den
TOF- und Q-TOF-Geräten, erleichtert den
Gerätewechsel bei Experimenten und
sorgt für gleichbleibende Ergebnisse.
Auswechselbare LC/MS-Ionenquellen
stellen bei allen Workflow-Schritten
eine gleichbleibende Ionisierung sicher,
wodurch sich eine Quelle erheblicher
analytischer Abweichungen entfernen
lässt.
Schnelle, reproduzierbare Trennungen
mit dem Rapid Resolution LC-System
der Serie 1200
Eine gute Probentrennung ist bei der
Metabolomanalyse unverzichtbar. Das
Rapid Resolution LC-System der Agilent
Serie 1200 eignet sich besonders gut für
die Metabolomanalyse. Der hohe maximale
Betriebsdruck ermöglicht den Einsatz
längerer Säulen mit kleineren Partikeln
für eine optimale chromatographische
Trennung.
Durch den hohen Druck lässt sich außerdem in der mobilen Phase Methanol
anstelle von Acetonitril verwenden.
Acetonitril führt zu Problemen mit der
APCI, während Methanol diese erleichtert.
Die APCI, entweder mit einer Singlemodeoder Multimode-Quelle, kann entscheidend für die Erzielung eines hohen
Metabolitumfangs sein. Eigenschaften
der Serie 1200:
• Umfassende Systeme für Analyse- und
Vorbereitungsaufgaben
• Automatische Probenflaschen- und
Wellplate-Probengeber
• Temperaturgesteuerte Säulenöfen und
automatische Probengeber
• Fraktionssammler
ZORBAX RRHT-Säulen für eine
optimale Substanztrennung
Agilent ZORBAX Rapid Resolution High
Throughput (RRHT) Säulen wurden speziell
für das Rapid Resolution LC-System der
Serie 1200 entwickelt, um ultraschnelle
HPLC-Trennungen mit einer hohen Auflösung durchführen zu können. ZORBAX
RRHT-Säulen verwenden 1,8-µm-Partikel
zur Optimierung der Auflösung. Kurze
Säulen (15-50 mm) werden für Hochgeschwindigkeitsanalysen und lange
Säulen (100 oder 150 mm) für eine
maximale Auflösung eingesetzt.
Die schnellen, hochauflösenden
Trennungen mit dem Rapid Resolution
LC-System der Serie 1200 verbessern
den Durchsatz und die Anzahl
gefundener Metaboliten.
Page 9

www.agilent.com/chem/metabolomics
9
Aufgrund der hervorragenden
Reproduzierbarkeit und
Massengenauigkeit ist das
6210 TOF bestens für
das Profiling geeignet.
Das 6510 Q-TOF vereint eine
genaue Massenbestimmung
mit MS/MS-Spektren und
ermöglicht so eine zuverlässige
Identifizierung der Metaboliten.
6210 Time-of-Flight-LC/MS-Systeme
für schnelles und genaues Profiling
Das Agilent 6210 TOF-LC/MS-System ist
ein benutzerfreundliches und sehr stabiles
System, das ideal für das MetabolomProfiling ist. Die fortlaufende Zuführung
einer Referenzmasse optimiert die
Massengenauigkeit und gewährleistet
die Reproduzierbarkeit. Gleichzeitig
werden weniger Proben benötigt, um
statistisch zuverlässige Ergebnisse zu
erzielen. Eigenschaften des 6210:
• Massengenauigkeit von 2 ppm für den
genauen Vergleich von Metaboliten aus
unterschiedlichen Proben
• TOF-Tischgerät mit unübertroffener
Empfindlichkeit
• Auflösung größer als 13.000 zur
Unterscheidung von Metaboliten
mit ähnlicher Masse
• Schnelle Spektrenerfassung für genaue
Peakflächenbestimmungen und eine
sehr genaue Quantifizierung
• Herausragende Stabilität und
Reproduzierbarkeit
Das 6210 TOF kann mit den unterschiedlichsten Ionisierungsquellen ausgestattet
werden, z. B. der extrem vielseitigen
Multimode-Quelle.
6510 Q-TOF-LC/MS-Systeme für die
zuverlässige Identifizierung von
Metaboliten
Das Agilent 6510 Quadrupol-Time-ofFlight-LC/MS-System ist ein extrem
vielseitiges Gerät, das perfekt auf das
Metaboliten-Profiling und die Identifizierung abgestimmt ist. Es kombiniert
die ergiebigen Strukturdaten der
MS/MS-Analyse mit sehr genauen
Massenbestimmungen für eine hochzuverlässige Identifizierung der
Metaboliten. Eigenschaften des 6510:
• Schnelle Spektrenerfassung: 1 MSSpektrum und 5 MS/MS-Spektren pro
Sekunde - dadurch stehen mehr Daten
für die Identifizierung zur Verfügung
• Fortschrittliche Kollisionszelltechnologie
eliminiert chemische Crosstalk-Effekte
• Routinemäßige Massengenauigkeit von
2 ppm (MS) bzw. 5 ppm (MS/MS) für
eine zuverlässige Identifizierung der
Metaboliten
• Auflösung größer als 13.000 zur Unter-
scheidung von Metaboliten mit ähnlicher
Masse
• Automatisches Tuning und automatische
Massenkalibrierung für optimale Leistung
bei geringstmöglichem Aufwand
Das Agilent Metabolomics Labor
Page 10

10
Maximale Flexibilität und
Beständigkeit durch
auswechselbare LC/MSIonenquellen
Die hohe chemische Vielfalt der Metaboliten ist der Grund dafür, dass es
keinen Ionisierungsmodus gibt, der für
alle Analysen geeignet wäre. Agilent
bietet in der Branche die größte Auswahl
an auswechselbaren Ionenquellen. Der
Ionisierungsmodus kann nach Bedarf an
das Gerät angepasst werden. Mögliche
Ionisierungsverfahren:
• Electrospray (ESI) mit StandardFlussraten (Mikroliter und Nanoliter)
• Chemische Ionisierung unter
Atmosphärendruck (APCI)
• Multimode-ESI/APCI
• Photoionisierung unter
Atmosphärendruck (APPI)
Agilent LC/MS-Ionenquellen arbeiten mit
orthogonaler Spraytechnik und hohen
Temperaturen, Gegenstrom-Trocknungsgas zur Maximierung von Leistung und
Reproduzierbarkeit sowie zur Minimierung
des chemischen Rauschens.
Besonders zu beachten ist die
Multimode-Quelle, die Ionen gleichzeitig
per Electrospray und APCI erzeugen kann,
ohne signifikante Empfindlichkeitsverluste
gegenüber Singlemode-Quellen hinnehmen
zu müssen. Auf diese Weise lässt sich
der Probendurchsatz in Profiling- und
Validierungsstudien verdoppeln.
Auswechselbare LC/MS-Ionenquellen
ermöglichen es Ihnen, den Ionisierungsmodus an die erwarteten
Metaboliten anzupassen, und
gewährleisten eine gleichmäßige
Ionisierung über den gesamten
Metabolomics-Workflow hinweg.
6410 Triple Quadrupole LC/MS-System
für eine durchsatzstarke Validierung
In Validierungsstudien werden hunderte
oder tausende Proben untersucht, sodass
eine genaue, durchsatzstarke Quantifizierung unerlässlich ist. Triple-QuadrupolMRM ist der Standard für die gezielte
Quantifizierung. Das Agilent 6410 Triple
Quadrupole LC/MS-System bietet Ihnen
Detektionsstufen im Femtogramm-Bereich
und auch im Dauereinsatz eine hohe
Zuverlässigkeit. Die Quantifizierungssoftware ist durch Funktionen wie die
substanz- oder probenbasierte Navigation
und die parameterlose Integration sehr
einfach zu bedienen.
• Empfindlichkeit bis in den FemtogrammBereich
• Durch schnelle MRM-Wechsel können
in einem einzelnen Analysenlauf mehr
Proben analysiert werden.
• Verringerung chemischer CrosstalkEffekte durch fortschrittliche Kollisionszelltechnologie zur Verbesserung
von Empfindlichkeit und Linearität
• Zeitersparnis durch einfaches Einrichten von Methoden und parameterlose
Integration
• Automatisches Tuning für optimale
Leistung bei geringstmöglichem
Aufwand
Das 6410 Triple Quad System liefert
zuverlässige MRM-Quantifizierungsdaten
für die Validierung großer Probenmengen.
Page 11

www.agilent.com/chem/metabolomics
11
Das Agilent CE-System bietet eine hervorragende Trennleistung
und eine einfache Verbindung mit einem Massenspektrometer.
Vor allem bei der Analyse hydrophiler Metaboliten kann die
CE/MS eine wertvolle Alternative zu GC/MS- und LC/MSSystemen sein.
CE/MS - hervorragende
Trenneigenschaften in
Kombination mit einer hohen
Analysengeschwindigkeit
Die Kapillarelektrophorese (CE) bildet
eine Alternative mit Flüssigphase zur
Flüssigchromatographie. Sie bietet eine
wechselnde Selektivität und hervorragende Trennleistung. Die Kombination
aus Kapillarelektrophorese und Massenspektrometrie (CE/MS) wird für
Metabolomanalysen weniger häufig
eingesetzt als GC/MS- und LC/MSSysteme, kann sich aber vor allem bei
der Analyse hydrophiler Metaboliten als
wertvoll erweisen. CE/MS kombiniert die
kurze Analysendauer und die exzellente
Trennleistung der Kapillarelektrophorese
mit dem Molekulargewicht und den
Strukturdaten der Massenspektrometrie.
Das Agilent CE-System kann als
Branchenführer mit verschiedenen
Massenspektrometern kombiniert
werden. Der gemeinsame Einsatz der
Kapillarelektrophorese und des Agilent
6210 Time-of-Flight-LC/MS-Systems
verbindet eine hervorragende Trennleistung
mit einer exzellenten Massengenauigkeit
und Massenauflösung und garantiert so
herausragende Lösungen für das Profiling
von Metaboliten.
Durch die in Agilent Ionenquellen eingesetzten, geerdeten Zerstäuber sind die
elektrischen CE-Einstellungen von den
Einstellungen der MS-Ionenquelle unabhängig. Damit wurde ein großer Nachteil
vieler anderer CE/MS-Systeme behoben.
Das Agilent Metabolomics Labor
Page 12

Software, die Daten in Informationen verwandelt
Das Ziel der Metabolomforschung ist ein besseres Verständnis der komplexen Funktionsweise biologischer
Systeme. Zur Umwandlung qualitativ extrem hochwertiger Daten in biologisch aussagekräftige Informationen
werden entsprechende Software-Tools benötigt. Genau dies gewährleistet die Agilent Software für
Metabolomics-Applikationen.
Methodenspezifische
Dekonvolution zur Auffindung
aller Metaboliten
Ein nicht erkanntes Metabolit stellt eine
ungenutzte Gelegenheit dar. Daher ist es
wichtig, in einer Probe so viele Metaboliten
wie möglich zu finden. Es reicht daher
nicht aus, einfach nur nach chromatographischen Peaks zu suchen, da sich
ein Peak auch nach der besten Trennung
noch aus mehreren Substanzen zusammensetzen kann. Die Lösung für die
Suche nach koeluierenden Metaboliten
wird als Dekonvolution bezeichnet. Mit der
Dekonvolution lassen sich Ionen finden,
deren individuelle Häufigkeiten über die
Zeit betrachtet gemeinsam ansteigen
und fallen, was darauf hindeutet, dass
sie zur selben Substanz gehören. Bei der
Dekonvolution werden diese Ionen für
jedes Metabolit zu einem Einzelsubstanzspektrum rekonstruiert.
Die Ionisierungsprozesse und
Massenanalysen von GC/MS- und
LC/MS-Spektren weisen einzigartige
chemische und physikalische Eigenschaften auf. Aus diesem Grund ist ein
einzelnes Dekonvolutionsprogramm
nicht für alle Applikationen optimal.
Agilent bietet verschiedene Dekonvolutionsprogramme, die diese einzigartigen
Eigenschaften von GC/MS- und LC/MSDaten berücksichtigen.
AMDIS für eine nützliche GC/MSDekonvolution
Als Teil des NIST-Bibliothekensuchpakets
bietet Agilent zur Dekonvolution von
GC/MS-Spektren die Automated Mass
Spectral Deconvolution and Identification
Software (AMDIS) an. AMDIS extrahiert
reine Substanzspektren aus komplexen
GC/MS-Daten und hilft Ihnen bei der
Bestimmung von Ionen/Peak-Verknüpfungen. AMDIS erzeugt als Bibliothek
durchsuchbare Spektren und exportiert
automatisch eine Merkmalsliste.
MassHunter Workstation für eine
bessere LC/MS-Dekonvolution
Die Agilent MassHunter WorkstationSoftware für LC/MS enthält einen
proprietären Algorithmus zur Merkmalerkennung und -korrelation. Der Algorithmus
ist für Time-of-Flight-LC/MS-Daten optimiert und erkennt kovariante Ionen, die
mit einer einzelnen Substanz verknüpft
sind. Das Erkennen und Gruppieren derartiger Ionen verbessert die Genauigkeit
der daraus bestimmten empirischen
Formel und der nachfolgenden Suche
in einer Metabolit-Datenbank.
12
Bei der Dekonvolution werden Metaboliten gefunden, die sich chromatographisch gar nicht
oder nur schlecht bestimmen lassen. Für jeden Metaboliten werden ein extrahiertes Ionenchromatogramm und ein rekonstruiertes Einzelsubstanzspektrum erzeugt.
TIC-Peak
Metabolit 1
Metabolit 2
Metabolit 3
Metabolit 1
Metabolit 2
Metabolit 3
Dekonvolution
Massenspektrum
Page 13

www.agilent.com/chem/metabolomics
13
Die Auswertung eines
einzelnen Peaks aus einem
Totalionen-Chromatogramm
(oben) durch die AMDISDekonvolutionssoftware ergibt
kovariante Ionen (Mitte), die
zu einer einzelnen Substanz
gehören. Der untere Bereich
zeigt das rekonstruierte
Massenspektrum des Peaks.
Das Agilent Metabolomics Labor
Der Algorithmus für die molekulare
Merkmalerkennung und -korrelation
der MassHunter-Software findet
nicht nur alle chromatographischen
Peaks, sondern alle Komponenten
in einem Chromatogramm. In diesem
Beispiel findet er eine reale Komponente in einem Segment des
Totalionen-Chromatogramms
(oben), das hauptsächlich aus
Rauschen und einem Anstieg der
Basislinie zu bestehen scheint.
Page 14

14
Vereinfachte Identifizierung mit
der METLIN Metabolite Database
Es gibt mehrere Vorgehensweisen zur
Identifizierung von Metaboliten.
EI-Spektren aus der GC/MS-Analyse
eignen sich für die Suche in Spektrenbibliotheken. In der LC/MS kann die
Suche in einer Datenbank mit Metabolitdaten dabei helfen, die Liste möglicher
Kandidaten einzuschränken. Durch
genaue Massendaten wird die Suche
in der Datenbank effektiver, da das zu
durchsuchende Spektrum der Massewerte
kleiner ist, wodurch sich die Anzahl
möglicher Identitäten verringert.
Die METLIN Metabolite Database wurde
vom Center for Mass Spectrometry am
The Scripps Research Institute
entwickelt und ist derzeit vermutlich
die bekannteste und umfassendste
Metabolitdatenbank weltweit. Sie enthält mit Anmerkungen versehene Listen
von mehr als 15.000 endogenen und
exogenen Metaboliten sowie Di- und Tripeptiden. Die Einträge umfassen
folgende Informationen:
• Masse
• Chemische Formel
• Struktur
Die METLIN-Datenbank ist öffentlich
zugänglich. Über das Internet kann
jeweils nach einer einzelnen Substanz
gesucht werden. Es ist jetzt jedoch auch
möglich, eine eigene Kopie der METLINDatenbank lokal zu speichern. Vorteile
einer lokalen Kopie:
• Private Suche
• Möglichkeit, eigene Substanzen zur
Datenbank hinzuzufügen
• Automatisierte Suche anhand einer
Massenliste
Agilent Technologies ist der einzige
Zulieferer der METLIN Personal
Metabolite Database. Die Agilent
MassHunter Workstation-Software und
die GeneSpring MS-Software enthalten
Links zur METLIN Personal-Datenbank,
um den Durchsatz zu steigern und die
Benutzerfreundlichkeit zu erhöhen.
Mit den Angaben zu
Massen, chemischen
Formeln und Strukturdaten für mehr als
15.000 endogene und
exogene Metaboliten
sowie Di- und Tripeptiden ist die
METLIN-Datenbank
ein leistungsstarkes
Werkzeug für die
Identifizierung von
Metaboliten.
Page 15
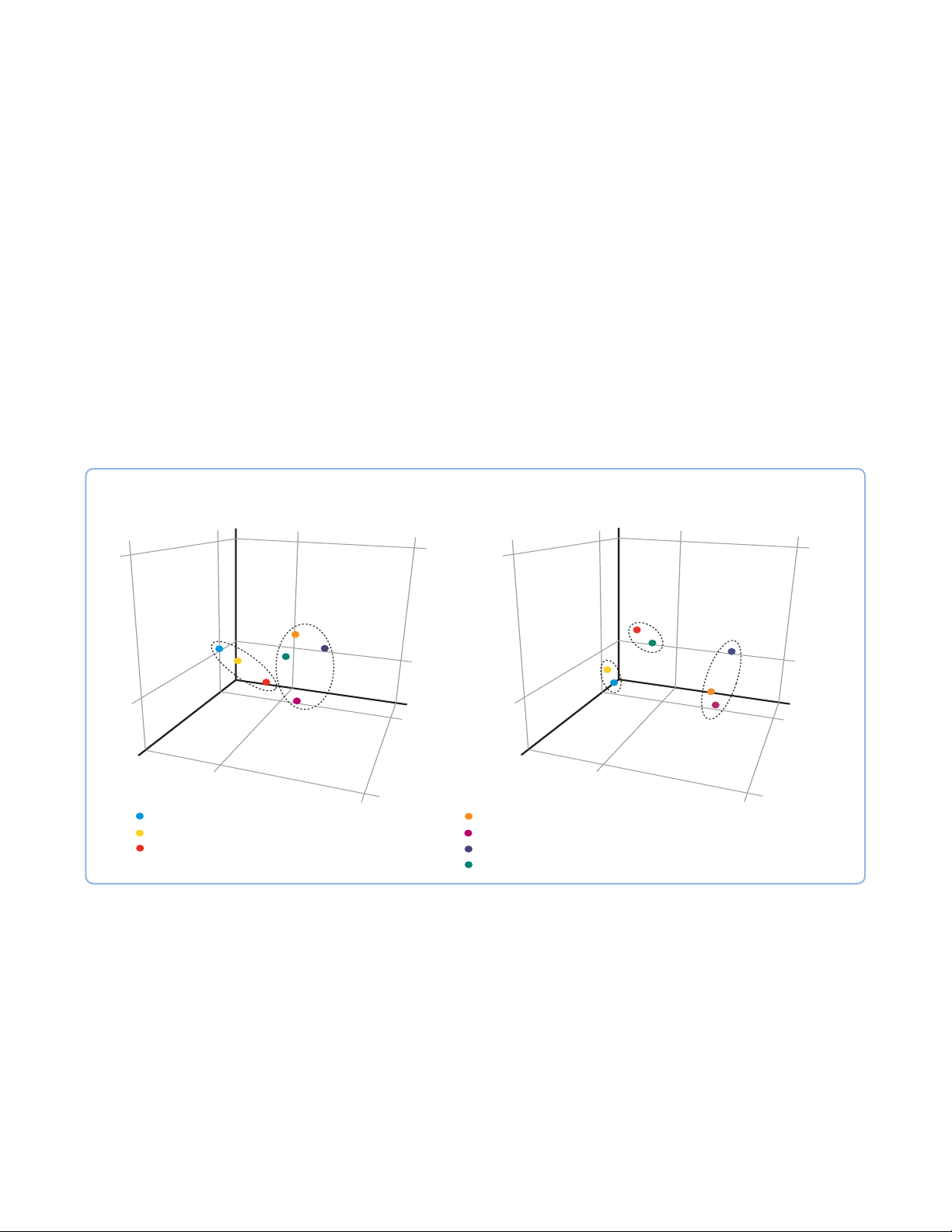
15
Aufgrund der Komplexität und Vielfalt von Metabolomanalysen ist es sehr wichtig, viele Optionen für die statistische Analyse und
die Visualisierung zu haben. In diesem komplexen Experiment mit mehreren Bedingungen zeigt nur die PCA Genotypunterschiede
auf. Wenn die PCA andererseits nach der ANOVA durchgeführt wird, lassen sich sowohl der Infektionsstatus als auch die
Genotypen nicht infizierter Proben unterscheiden.
Y
Z
X
0
0
0
1
1
1
Y
Z
0
0
0
1
1
1
X
TP309-Xa21-Linie
(transgen)
TP309-Linie
(wild)
Infiziert_wild
und transgen
Nicht infiziert
transgen
Nicht
infiziert_wild
TP309 Wildreis - keine Behandlung TP309-Xa21 transgener Reis - keine Behandlung
TP309 Wildreis - Pseudobelastung TP309-Xa21 transgener Reis - Pseudobelastung
TP309 Wildreis - Bakterientest TP309-Xa21 transgener Reis - Bakterientest
TP309-Xa21 transgener Reis - Knockout-Bakterientest
Nur PCA
1-Way-ANOVA und PCA
GeneSpring MS – eine übergreifende Plattform für die
Normalisierung und das
Vergleichen der Daten von
Massenspektren
Die Agilent GeneSpring MS-InformatikSoftware ist eine leistungsstarke Lösung
für das Erkennen von Metabolit-Biomarkern
mit Hilfe der Analyse von Massenspektrometerdaten. Daten von Massenspektren
aus großen Probenmengen und komplexen
Versuchsanordnungen lassen sich einfach
importieren, normalisieren, vergleichen
und anzeigen. Dazu gehören GC/MS- und
LC/MS- oder CE/MS-Daten von Agilent
TOF-, Q-TOF- und Triple-Quadrupol-Geräten.
Die GeneSpring MS-Software bietet
umfassende Werkzeuge für statistische
Analysen, Data-Mining und Visualisierungen:
• Varianzanalyse (ANOVA)
• Hauptkomponentenanalyse (PCA)
• t-Tests
• Vulkan-Diagramme
• Hierarchische Bäume (Dendrogramme)
• Selbstorganisierende Karten (SOMs)
• QT-Clusterbildung (Quality Threshold)
• Support-Vector-Machines (SVMs)
Die GeneSpring MS-Informatik-Software
lässt sich gut mit anderen StatistikSoftware-Paketen integrieren. Wenn
Sie ein bestimmtes, spezialisiertes
Analysenpaket bevorzugen, ist es recht
wahrscheinlich, dass die GeneSpring
MS-Software mit diesem kombiniert
werden kann.
Das Agilent Metabolomics Labor
www.agilent.com/chem/metabolomics
Page 16
Das Agilent Metabolomics Labor
Konzentration auf das
Wesentliche dank Agilent
Dienstleistungen
Die Agilent Dienstleistungsorganistation
genießt branchenweit eine hohe Anerkennung. Agilent bietet für Ihre
Metabolomics-Systeme relevante
Dienstleistungen. Damit können Ihre
Systeme in einwandfreiem Zustand gehalten und somit zuverlässig und mit
maximalem Durchsatz betrieben werden.
Vielfältige Agilent Dienstleistungen sind:
• Software-Updates zu einem festgelegten jährlichen Preis
• Vorbeugende Wartung vor Ort zur
Gewährleistung des zuverlässigen
Betriebs und zur Minimierung von
Ausfallzeiten
• Telefonunterstützung zur Diagnose
und Lösung von Software- und
Hardwareproblemen
• Remote-Updates, Diagnose und
Reparatur über sichere Internetverbindungen
• Reparaturarbeiten vor Ort
Agilent Dienstleistungen können bei
Bedarf erworben werden oder sind
gebündelt in kostengünstigen Dienstleistungsverträgen zu einem Festpreis
erhältlich. Sie bieten Schutz vor unerwarteten Reparaturkosten.
Agilent Dienstleistungsgarantie
Das Vertrauen in die Qualität der
Dienstleistungen und die Zuverlässigkeit
der Geräte ist so hoch, dass Agilent eine
eigene Dienstleistungsgarantie anbietet.
Wenn Sie eine Agilent Dienstleistungsvereinbarung abgeschlossen haben und
Unterstützung für Ihre Agilent Geräte
benötigen, sind die Reparatur oder der
kostenlosen Ersatz des Geräts durch die
Garantie abgedeckt.
Agilent bietet ein hohes Maß an
Unterstützung, damit der Betrieb Ihres
Labors mit maximaler Produktivität
aufrecht erhalten wird.
Agilent Technologies
Agilent Technologies ist führender Anbieter
von Forschungssystemen für Life Sciences,
mit deren Hilfe Wissenschaftler Einblicke in
komplexe biologische Prozesse gewinnen,
Krankheitsmechanismen verstehen lernen und
die Medikamentenentwicklung beschleunigen. Die auf Empfindlichkeit, Reproduzierbarkeit und Workflow-Produktivität ausgerichteten Agilent Life Sciences-Lösungen
umfassen Geräte, Mikrofluide, Software,
Microarrays, Verbrauchsmaterialien und
Dienstleistungen für die Genomics-,
Proteomics- und Metabolomics-Forschung.
Online-Shop:
www.agilent.com/chem/store
Agilent Kundendienstzentrum in
Ihrer Nähe:
www.agilent.com/chem/contactus
Europa
info_agilent@agilent.com
Nur für Forschungszwecke. Nicht zur Diagnose einsetzen.
Änderungen vorbehalten.
Alle Rechte vorbehalten.
© Agilent Technologies, Inc. 2007
Gedruckt in den Niederlanden, 31. Januar 2007
5989-5472DEE
 Loading...
Loading...