


Amplatzer Piccolo™ Occluder
Instructions for Use
Caution, consult accompanying documents
Not made with natural rubber latex
MR Conditional
Inner diameter
Outer diameter
Length
Usable length
Recommended delivery sheath dimensions
Do not use if package is damaged
Manufacturer
Catalog number
Lot number
Duct Occluder
Use-by date
Date of manufacture
Do not re-use
Sterilized using ethylene oxide
Unique device identification
Quantity
Consult instructions for use
Follow instructions for use on this website
Keep dry; keep away from rain.
Do not resterilize
Caution: Federal law restricts this device to sale by or on the order of a physician.
Amplatzer Piccolo™ Occluder
Device Description
The Amplatzer Piccolo™ Occluder is a self-expanding, nitinol mesh occlusion device for use
in a patent ductus arteriosus (PDA). The
retention discs. The central waist is designed to be positioned within the ductus. The
retention discs are deployed in the pulmonary and aortic ends of the ductus, or may be
deployed completely within the duct when treating small infants. The device may be delivered
via an anterograde (venous) or a retrograde (arterial) approach.
Radiopaque marker bands at each end of the occluder permit visibility during fluoroscopy.
Refer to F1 and F2 for more information about the device. Refer to T1 - T3 in the back for
occluder dimensions and sizing.
F1
F
device configuration is a central waist with two
G
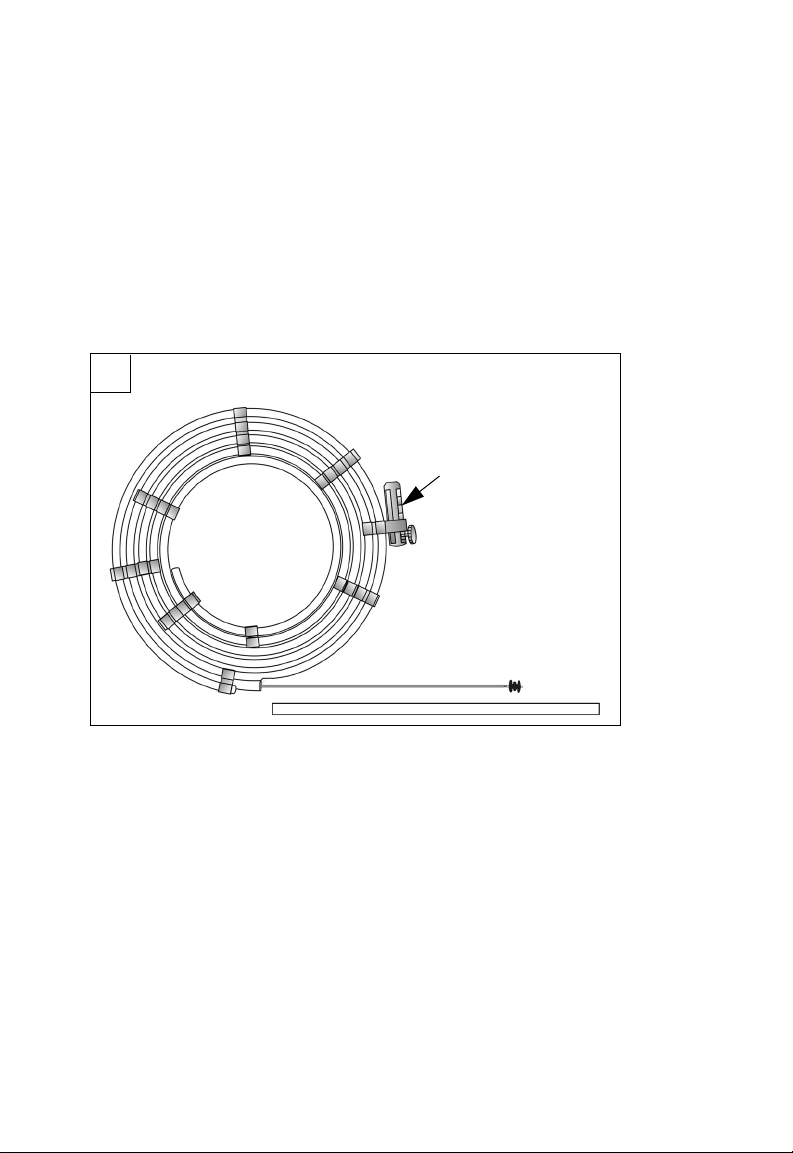
F1 Components
F. Hoop dispenser
G. Vise
H. Delivery wire
J. Occluder
K. Occluder protector tube
H
J
K
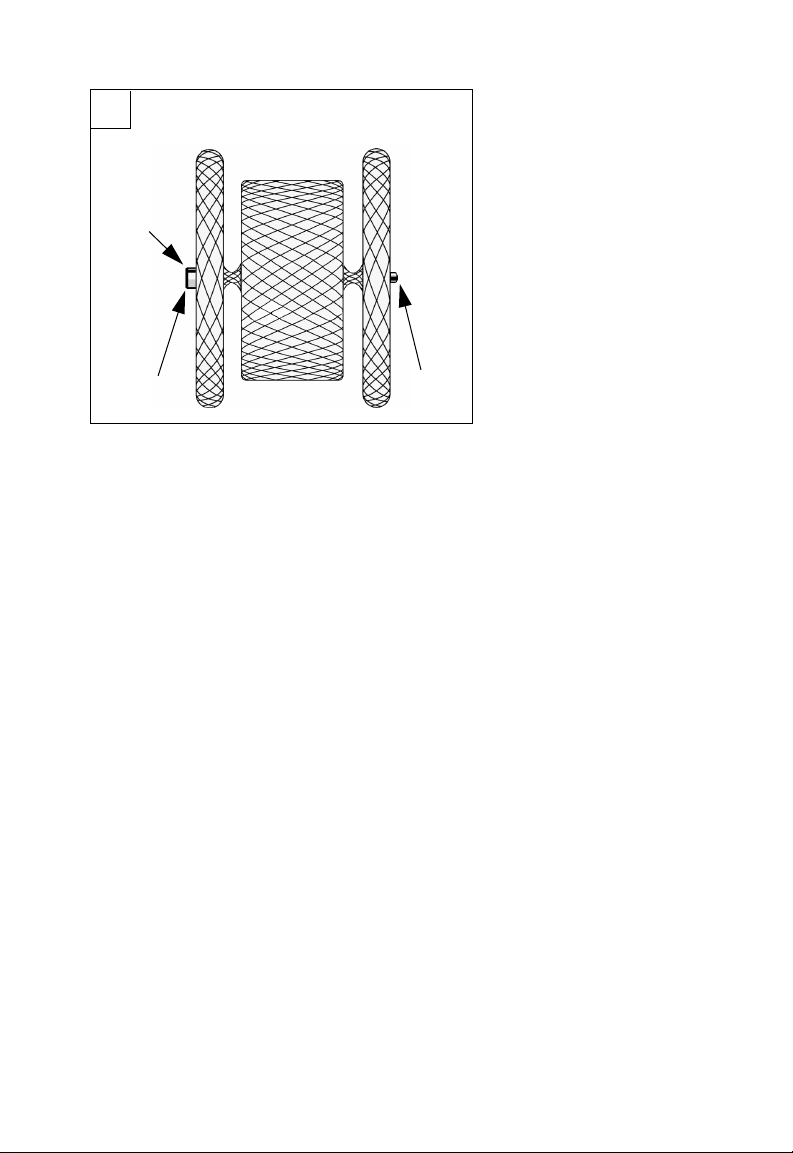
F2
L
M
F2 Components
L. Micro screw attachment
M. Radiopaque marker bands
M
Indications and Usage
The Amplatzer Piccolo™ Occluder is a percutaneous, transcatheter occlusion device
intended for the nonsurgical closure of a patent ductus arteriosus (PDA).
Contraindications
• Weight <700 grams at time of the procedure
• Age <3 days at time of procedure
• Coarctation of the aorta
• Left pulmonary artery stenosis
• Cardiac output that is dependent on right to left shunt through the PDA due to
pulmonary hypertension
• Intracardiac thrombus that may interfere with the implant procedure
• Active infection requiring treatment at the time of implant
• Patients with a PDA length smaller than 3 mm
• Patients with a PDA diameter that is greater than 4 mm at the narrowest portion
Warnings
• This device was sterilized with ethylene oxide and is for single use only. Do not reuse or
re-sterilize this device. Attempts to resterilize this device can cause a malfunction,
insufficient sterilization, or harm to the patient.
• Do not use the device if the sterile package is open or damaged.
• Use on or before the last day of the expiration month that is printed on the product
packaging label.
• Patients who are allergic to nickel can have an allergic reaction to this device.
• Prepare for situations that require the removal of this device. Preparation includes
access to a transcatheter snare kit and
• Accurate measurements of the ductus are crucial for correct occluder size selection.
an on-site surgeon.

• Do not release the occluder from the delivery wire if either a retention disc protrudes
into the pulmonary artery or aorta; or if the position of the occluder is not stable.
• Remove embolized devices. Do not remove an embolized occluder through intracardiac
structures unless the occluder is fully recaptured inside a catheter.
Precautions
• This device should be used only by physicians who are trained in standard transcatheter
techniques. Determine which patients are candidates for procedures that use this device.
• The physician should exercise clinical judgment in situations that involve the use of
anticoagulants and antiplatelet drugs before, during, and/or after the use of this device.
• Patients should have an activated clotting time (ACT) of greater than 200 sec prior to
device placement, unless the patient has a significant risk for bleeding and is unable to
be anti-coagulated.
• The device may be delivered via an anterograde (venous) or a retrograde (arterial)
approach. However, in small infants (≤2 kg), the device should be delivered using the
anterograde (venous) approach since small infants are at an increased risk for arterial
injury.
• In small infants (≤2kg) the occluder size is chosen so that the entire device with both
retention discs is placed within the duct (intraductal placement) to minimize the potential
for protrusion into the aorta or left pulmonary artery.
• In larger infants (>2kg), the occluder size is chosen so that the central waist spans the
entire length of the duct with the retention discs placed just outside the duct or within
the ampulla (extraductal disc placement) to achieve improved positional stability and
minimize the potential for device embolization.
• Prior to releasing the device from the delivery wire, it is important to rely on imaging to
ensure there is no obstruction of the aorta or left pulmonary artery.
• The Amplatzer Piccolo™ Occluder contains nickel-titanium alloy, which is generally
considered safe. However, in vitro testing has demonstrated that nickel is released from
this device for a minimum of 60 days following implant. Patients who are allergic to
nickel may have an allergic reaction to this device, especially those with a history of
metal allergies. Certain allergic reactions can be serious; patients should seek
immediate medical attention if there is suspicion of an allergic reaction. Symptoms may
include difficulty in breathing or swelling of the face or throat. While data are currently
limited, it is possible that some patients may develop an allergy to nickel if this device is
implanted.
• Use in specific populations
- Pregnancy – Minimize radiation exposure to the fetus and the mother.
- Nursing mothers – There has been no quantitative assessment for the presence
of leachables in breast milk.
• Store in a dry place.
• Do not use contrast power injection with delivery catheter.

MR Conditional
Non-clinical testing has demonstrated that the Amplatzer Piccolo™ Occluder is MR
Conditional. A patient with the Amplatzer Piccolo™ Occluder can be safely scanned in
an MR system under the following conditions:
- Static magnetic field of 1.5 Tesla (1.5T) or 3.0 Tesla (3.0T)
- Maximum spatial gradient field of 19T/m (1900 G/cm)
- Maximum MR system-reported, whole-body-averaged specific absorption rate
(SAR) of 2.0W/kg (normal operating mode)
Under the scan conditions defined above, the device is expected to produce a
maximum temperature rise of less than or equal to 3ºC after 15 minutes of continuous
scanning.
In non-clinical testing, the image artifact caused by the device extends approximately
9 mm from the Amplatzer Piccolo™ Occluder when imaged with a gradient echo pulse
sequence and a 3.0T MRI System.
Potential Adverse Events
Potential adverse events that may occur during or after a procedure using this device may
include, but are not limited to:
• Air embolus • Infection
• Allergic reaction • Myocardial infarction
• Anemia • Palpitations
• Anesthesia reactions • Partial obstruction of aorta
• Apnea • Partial obstruction of pulmonary artery
• Arrhythmia • Pericardial effusion
• Bleeding • Pericarditis
• Cardiac perforation • Peripheral embolism
• Cardiac tamponade • Pleural effusion
• Chest pain • Pulmonary embolism
• Device embolization • Re-intervention for device removal
• Device erosion • Respiratory distress
• Death • Stroke
• Endocarditis • Thrombus
• Fever • Transient ischemic attack
• Headache/migraine • Valvular regurgitation
• Hemolysis • Vascular access site injury
• Hematoma • Vascular occlusion
• Hypertension • Vessel perforation
• Hypotension

Clinical Summary
Design
The Amplatzer Piccolo™ Occluder (studied under the name Amplatzer™ Duct Occluder II
Additional Sizes—ADO II AS IDE Study) was a single arm, open-label, multi-center, study
designed to characterize the safety and effectiveness of the Amplatzer Piccolo™ Occluder
device. A total of 50 subjects were enrolled at eight centers in the United States from
June 5, 2017 to January 25, 2018. At the time of implant, 18 subjects were ≤2 kg and
32 subjects >2 kg in weight.
Subject follow-up occurred post-procedure, at 30 days and six months. The primary
effectiveness endpoint was the rate of effective closure of the ductus arteriosus (defined as a
grade 0 or grade 1 shunt) at the six-month follow-up visit. The primary safety endpoint was
the rate of major complications through 180 days after device implant. Major complications
were defined as device or procedure related adverse events resulting in death, lifethreatening adverse event, persistent or significant disability/incapacity, and/or a major open
surgical intervention performed by a surgeon under general anesthesia.
Patients Studied
Key inclusion criteria included a PDA that was ≤4mm in diameter and ≥3mm in length.
Patients were excluded if they weighed <700 grams or were younger than 3 days of age.
Other key exclusion criteria included:
- Coarctation of the aorta
- Left pulmonary artery stenosis
- Cardiac output that is dependent on right to left shunt through the PDA due to
pulmonary hypertension
- Intracardiac thrombus
- Active infection requiring treatment at the time of implant
Baseline characteristics are presented in Table 1 and PDA type is presented in Table 2.
Table 1. Baseline Characteristics
Age, months
Mean ± SD (n)
Range (Min, Max)
Sex, male 50.0% (9/18) 37.5% (12/32)
Weight (kg)
Mean ± SD (n)
Range (Min, Max)
Minimal PDA Diameter (mm)
Mean ± SD (n)
Range (Min, Max)
Maximal PDA Diameter (mm)
Mean ± SD (n)
Range (Min, Max)
PDA Length (mm)
Mean ± SD (n)
Range (Min, Max)
≤2 kg
(N=18)
1.23 ± 0.55 (18)
(0.49, 2.30)
1.34 ± 0.38 (18)
(0.76, 1.90)
2.72 ± 0.65 (18)
(1.4, 4.0)
3.75 ± 0.44 (18)
(3.0, 4.7)
8.81 ± 2.55 (18)
(4.6, 14.0)
>2 kg
(N=32)
24.88 ± 38.17 (32)
(0.66, 168.54)
10.29 ± 10.42 (32)
(2.03, 47.80)
2.64 ± 0.58 (32)
(1.5, 4.0)
4.37 ± 1.42 (32)
(2.0, 8.7)
7.98 ± 2.78 (32)
(3.1, 16.0)

Table 2. PDA Type
≤2 kg >2 kg
Type A: Conical 16.7% (3/18) 53.1% (17/32)
Type B: Window — 6.3% (2/32)
Type C: Tubular 38.9% (7/18) 6.3% (2/32)
Type D: Saccular — 6.3% (2/32)
Type E: Elongated 5.6% (1/18) 15.6% (5/32)
Type F: Fetal 38.9% (7/18) 12.5% (4/32)
Results
A total of 46 subjects were successfully implanted with the Amplatzer Piccolo™ Occluder
device, resulting in an implant success rate of 92% (18/18 ≤2 kg and 28/32 >2 kg). There
were two subjects >2 kg who had an intra-procedural device embolization, where the
embolized devices were successfully snared without complications. Both subjects were
subsequently implanted with a different commercially available device. There were two
additional subjects where the device was not released due to the inability to achieve a stable
position. One subject received a different commercially available device and one underwent
surgical ligation.
The primary effectiveness and safety endpoints are summarized in Table 3. Effective closure
was assessed with echocardiography and defined as having no or a trivial residual shunt at
six months. Echocardiography data were suitable for core lab assessment at 6 months for
44 subjects. Effective closure was achieved in all subjects (N=44) and no major
complications were encountered in any subjects (N=50). Serious adverse events that did not
meet the definition of the primary safety endpoint occurred in two subjects (4%)
(1 desaturation and 1 coarctation of the aorta).
Table 3. Primary Endpoints
≤2kg >2kg
Rate of effective closure (%) 100.0% (17/17) 100.0% (27/27)
Rate of major complications (%) 0.0% (0/18) 0.0% (0/32)
Supplemental Clinical Information
Design
Following completion of enrollment in the IDE study, additional subjects were enrolled under
a Continued Access Protocol (CAP). A total of 150 additional subjects were enrolled under
the CAP from 26 March 2018 to 1 February 2019. At the time of implant, 82 subjects were
≤2 kg and 68 subjects were >2 kg in weight. The CAP allowed for enrollment of up to
150 subjects using a similar protocol to the IDE study with equivalent inclusion/exclusion
criteria.
Patients Studied
Baseline characteristics are presented in Table 4, and PDA type is presented in Table 5.
Table 4. Baseline Characteristics — CAP
Age, months
Mean ± SD (n)
Range (Min, Max)
Sex, male 62.2% (51/82) 44.1% (30/68)
≤2 kg
(N=82)
1.26 ± 0.61 (82)
(0.30, 3.15)
>2 kg
(N=68)
27.38 ± 47.18 (68)
(0.49, 216.80)

Weight (kg)
Mean ± SD (n)
Range (Min, Max)
Minimal PDA Diameter (mm)
Mean ± SD (n)
Range (Min, Max)
Maximal PDA Diameter (mm)
Mean ± SD (n)
Range (Min, Max)
PDA Length (mm)
Mean ± SD (n)
Range (Min, Max)
≤2 kg
(N=82)
1.22 ± 0.34 (82)
(0.70, 2.00)
2.64 ± 0.63 (74)
(1.5, 4.0)
3.54 ± 0.80 (73)
(2.0, 5.2)
9.41 ± 2.76 (73)
(3.1, 18.0)
>2 kg
(N=68)
11.68 ± 14.80 (68)
(2.02, 68.50)
2.58 ± 0.70 (52)
(1.0, 4.0)
4.44 ± 1.46 (52)
(2.5, 10.0)
9.66 ± 3.28 (52)
(4.0, 16.0)
Table 5. PDA Type — CAP
≤2 kg >2 kg
Type A – Conical 3.7% (3/81) 40.0% (26/65)
Type B – Window 1.2% (1/81) —
Type C – Tubular 11.1% (9/81) 15.4% (10/65)
Type D – Saccular — 4.6% (3/65)
Type E – Elongated 4.9% (4/81) 12.3% (8/65)
Type F – Fetal 77.8% (63/81) 26.2% (17/65)
Results
A total of 145 subjects were successfully implanted with the Amplatzer Piccolo™ Occluder
device, resulting in an implant success rate of 96.7% (81/82 ≤2 kg and 64/68 >2 kg).
Unsuccessful implants occurred in one subject ≤2 kg and four subjects >2 kg. In all five
subjects, the device was not released due to inability to achieve a stable position. Two
subjects were implanted with a commercially available device, while two subjects were
scheduled to undergo surgical ligation of the PDA. PDA closure for one additional subject
was postponed to a later date. There were two subjects ≤2 kg and one subject >2 kg who
had an intra-procedural device embolization, where the embolized devices were successfully
snared without complications. All three subjects were subsequently implanted with a larger
Amplatzer Piccolo™ Occluder without complications.
The primary effectiveness and safety endpoints are summarized in Table 6. Effective closure
was assessed with echocardiography and defined as having no or a trivial residual shunt at
six months. Eleven subjects discontinued prior to the 6-month visit and seven subjects did
not complete the 6-month visit. Echocardiography data was suitable for core lab assessment
at six months for 129 subjects. Effective closure was achieved in 99.2% of subjects
(128/129).
The primary safety endpoint is the rate of major complications through 180 days after an
attempted Amplatzer Piccolo™ Occluder implant, as adjudicated by the CEC. Six subjects
with a successful implant withdrew before 180 days without an event and are excluded from
the analysis. Major complications occurred in four CAP subjects (2.8%). Two subjects
experienced procedural blood loss requiring transfusion ≤20 cc/kg. One subject with a history
of congenital thrombocytopenia experienced hemolysis and required transfusions totaling
≤20 cc/kg until the event resolved without sequelae. One subject experienced device-related
obstruction of the aorta six days post-procedure that was treated by stent implantation. The
subject died 14 days post-procedure secondary to severe respiratory failure, severe
pulmonary hypertension leading to cardiorespiratory arrest.

Table 6. Primary Endpoints — CAP
≤2 kg >2 kg
Rate of effective closure (%) 100.0% (72/72) 98.2% (56/57)
Rate of major complications (%) 5.1% (4/78) 0.0% (0/66)
Directions for Use
Materials recommended for use with this device
• AmplatzerTM TorqVueTM LP Catheter (9-TVLPC4F90/080)
• 0.035-inch (0.89-mm) Guidewire
Procedure
1. Prepare the patient for a standard transcatheter procedure. Once vascular access is
achieved administer anticoagulation to achieve an activated clotting time (ACT) of
greater than 200 sec prior to
risk for bleeding and is unable to be anticoagulated. With small infants, it is
recommended to deliver the device using an anterograde transvenous approach
and to avoid arterial access whenever possible.
CAUTION: Whenever possible, do not deliver the device in small infants
(≤2 kg) using the retrograde approach since small infants are at an increased
risk for arterial injury.
2. Do a right-heart catheterization or perform intra-operative echocardiography.
3. Take hemodynamic or echocardiographic measurements.
4. Use angiography or echocardiography to measure the PDA diameter at the
narrowest portion (D) and the length (E) of the PDA. Refer to F3 for an example of
measurement locations.
WARNING: Accurate measurements of the PDA is critical for correct occluder
selection.
device placement, unless the patient has a significant
F3
F3 Measurements
D.
Minimal ductus diameter
E. Ductus length

5. Use the PDA measurements to find the appropriate occluder size in T2 for patients
>2 kg.
NOTE: For small infants (≤2 kg), the length of the occluder may be shorter than the
length of the PDA to minimize the potential for protrusion into the aorta or left
pulmonary artery. For small infants, find the appropriate occluder size in T3. For
small infants, the occluder is chosen so that the entire device including the retention
discs is implanted within the duct (intraductal placement).
NOTE: If there is inconsistency between angiography and echocardiography
regarding the device size selection, then consider selecting the larger device size
unless the use of a larger device size would result in protrusion into the aorta or left
pulmonary artery. It is important to ensure that the most reliable imaging modality is
utilized for guiding device size selection. Inconsistencies between imaging
modalities may be due to multiple factors, such as variation in imaging angulation
and windows, amount of contrast injected, and/or ductal vascular tone.
6. Prepare the device for use.
- Inspect the sterile pouch.
CAUTION: Do not use the device if the sterile pouch is open or damaged.
- Open the sterile pouch. Inspect the device.
7. Prepare the catheter according to the manufacturer's instructions for use.
8. Prepare the occluder within the loader as follows:
- Insert the proximal end of the delivery wire forward through the distal end of the
loader and through the self-sealing hemostasis valve.
- Make sure the occluder is threaded tightly onto the delivery wire. Turn the
occluder counterclockwise 1/8 of a turn to make disconnection easier.
CAUTION: Do not overtighten the connection.
- Put the occluder and the loader assembly (loader + self-sealing hemostasis
valve) in sterile saline. Retract the occluder into the loader.
- Flush the loader and the occluder with sterile saline through the self-sealing
hemostasis valve.
9. Introduce the guidewire into the vasculature and advance through the PDA. Move
the catheter forward over the guidewire and through the ductus.
CAUTION: Do not advance the catheter over the guidewire without imaging
guidance if resistance is encountered, or if there is significant mismatch
between the catheter lumen and the guidewire diameter. Advancing the
catheter under such circumstances has the potential to result in
cardiovascular injury.
10. Utilize fluoroscopic and/or echocardiographic guidance to identify the catheter
position. If using angiographic guidance for the procedure, do a test injection with
contrast medium to see the position of the catheter. Placement of an esophageal
temperature probe pre-procedure may serve as a useful landmark of the aortic isthmus
in small infants (≤2 kg).
11. Remove the guidewire.
12. Permit blood backflow through the Tuohy-Borst hemostasis valve to remove air from
the system and flush the delivery catheter with heparinized saline.
13. Move the loader forward through the Tuohy-Borst hemostasis valve and into the
catheter until the loader stops.
14. Tighten the Tuohy-Borst hemostasis valve onto the loader. Remove any air that may
have entered the delivery catheter system by aspirating and flushing with heparinized
saline.
15. Hold the catheter, Tuohy-Borst hemostasis valve, and loader assembly as a single
unit. Move the occluder forward from the loader into the catheter.
NOTE: If it is difficult to transfer the occluder into the catheter, recapture the occluder
in the loader and adjust the position of the loader.
16. Move the occluder forward to the distal tip of the catheter.

CAUTION: Do not turn or twist the delivery wire.
17. Hold the delivery wire and retract the catheter to deploy the distal disc.
WARNING: Do not push the delivery wire to deploy the occluder.
CAUTION: Move the occluder carefully under imaging guidance to prevent
damage to the vessels or cardiac tissue.
18. Retract the catheter and delivery wire as one unit until the distal retention disc of the
occluder touches the vessel wall at the PDA.
NOTE: For small infants (≤2 kg), it may be necessary to deploy the distal disc within
the PDA to achieve an intraductal position.
19. Use angiography or echocardiography to make sure the distal retention disc of the
occluder is placed correctly against the vessel wall.
20. Stabilize the delivery wire and slowly retract the catheter to deploy the waist of the
occluder inside the PDA. The waist of the occluder must appose the ductus wall.
21. Stabilize the delivery wire and slowly retract the catheter to deploy the proximal
retention disc.
NOTE: For small infants (≤2 kg), it may be necessary to push the device forward
while retracting the catheter to fully pack the device within the duct and achieve an
intraductal position.
22. Do an aortic angiogram or evaluate device position with echocardiography.
- Make sure the occluder is in the correct position and orientation without
obstructing the aorta or left pulmonary artery.
- Make sure the occluder is stable.
- Make sure the occluder shape is correct.
- Measure occlusion of the PDA and assess whether there is a residual shunt.
NOTE: The discs should not protrude or bulge into the surrounding vessels. Refer to
F4 for an example of the correct shape for a deployed occluder when using an
intraductal device placement in small infants (≤2 kg). Refer to F5 for an example of
an incorrectly deployed occluder.
WARNING: Do not release the occluder from the delivery wire if a retention
disc extends into a vessel or if the occluder is not stable or if there is a
clinically relevant residual shunt. To recapture the occluder, move the catheter
forward over the occluder. Redeploy the occluder or replace it with a new
occluder.
CAUTION: Recapture and redeploy the occluder a maximum of two times. If
the position of the occluder is still unsatisfactory after the second
deployment, remove and replace the occluder and the catheter.

F4
F5
23. Connect the vise to the delivery wire. Turn the vise counterclockwise to disconnect
the occluder from the delivery wire.
WARNING: Do not push the delivery wire or catheter forward after the
occluder is released.
24. Remove the delivery wire and the catheter.
WARNING: Slowly remove the catheter from the patient to prevent the
introduction of air.
Post-procedure care
• Monitor the patient post-procedure. Do a transthoracic echocardiogram or X-ray to
make sure the occluder is in the correct position before the patient is released.
• Give endocarditis prophylaxis for six months. Prophylaxis beyond six months is at the
physician’s discretion.

Retrieval of embolized device
In the event a device embolizes, the following steps are recommended for transcatheter
device retrieval:
1. Heparinize the patient to achieve an activated clotting time (ACT) of greater than
200 sec.
2. Advance a catheter over a guidewire into the vessel containing the embolized
device.
WARNING: To avoid injury to intracardiac structures during device retrieval,
do not remove an embolized occluder through intracardiac structures unless
the occluder is fully recaptured inside a catheter.
3. Using a goose-neck snare (5 mm or 10 mm in size), grab the occluder tightly, based
on the instructions for use of the transcatheter snare kit, and recapture into the distal
end of the catheter.
4. Once the device is fully recaptured into the catheter, pull the device through the
catheter under fluoroscopic guidance and externalize.
In the event the device cannot be retrieved using a transcatheter approach, consult
with a surgeon for surgical retrieval of the device.
Post-procedure Instructions
• Instruct the patient on when to seek medical attention.
• Temporary patient ID card: A temporary patient ID card is included in the product
packaging. Complete this card and give it to the patient.
• Registration form: An implant registration form is located in each device box. Complete
the patient information section and send the form to Abbott Medical.
Disposal
• The carton and instructions for use are recyclable. Discard all packaging materials
appropriately.
• Devices can be returned to Abbott Medical for disposal. Contact an Abbott Medical
representative or returns@amplatzer.com for instructions.
• Use solid biohazard waste procedures to discard devices.
Warranty
Abbott Medical warrants to buyer that, for a period equal to the validated shelf life of the
product, this product shall meet the product specifications established by the manufacturer
when used in accordance with the manufacturer's instructions for use and shall be free from
defects in materials and workmanship. Abbott Medical's obligation under this warranty is
limited to replacing or repairing at its option, at its factory, this product if returned within the
warranty period to Abbott Medical and after confirmed to be defective by the manufacturer.
EXCEPT AS EXPRESSLY PROVIDED IN THIS WARRANTY, ABBOTT MEDICAL
DISCLAIMS ANY REPRESENTATION OR WARRANTY OF ANY KIND, EXPRESS OR
IMPLIED, INCLUDING ANY WARRANTY AS TO MERCHANTABILITY OR FITNESS FOR A
PARTICULAR PURPOSE.
See the Terms and Conditions of Sale for further information.
For U.S. --- California Only:
WARNING: This product can expose you to chemicals including ethylene oxide,
which is known to the State of California to cause cancer and birth defects or other
reproductive harm. For more information, go to www.P65Warnings.ca.gov.

Occluder dimensions
T1
B
A
C
ABC
mm (in) mm (in) mm (in)
9-PDAP-03-02-L 4.00 (0.157) 3.00 (0.118) 2.00 (0.079)
9-PDAP-03-04-L 4.00 (0.157) 3.00 (0.118) 4.00 (0.157)
9-PDAP-03-06-L 4.00 (0.157) 3.00 (0.118) 6.00 (0.236)
9-PDAP-04-02-L 5.25 (0.207) 4.00 (0.157) 2.00 (0.079)
9-PDAP-04-04-L 5.25 (0.207) 4.00 (0.157) 4.00 (0.157)
9-PDAP-04-06-L 5.25 (0.207) 4.00 (0.157) 6.00 (0.236)
9-PDAP-05-02-L 6.50 (0.256) 5.00 (0.197) 2.00 (0.079)
9-PDAP-05-04-L 6.50 (0.256) 5.00 (0.197) 4.00 (0.157)
9-PDAP-05-06-L 6.50 (0.256) 5.00 (0.197) 6.00 (0.236)
T1 Dimensions
A. Retention disc diameter
B. Waist diameter
C. Length between retention discs

Occluder sizing for patients >2kg using extraductal disc placement
T2
D
mm (in)
E
mm (in)
3–4 (0.118–0.157) 4.1–6 (0.161–0.236) 6.1–8 (0.240–0.315)
≤2 (≤0.079) 9-PDAP-03-02-L 9-PDAP-03-04-L 9-PDAP-03-06-L
2.1–3 (0.083–0.118) 9-PDAP-04-02-L 9-PDAP-04-04-L 9-PDAP-04-06-L
3.1–4 (0.122–0.157) 9-PDAP-05-02-L 9-PDAP-05-04-L 9-PDAP-05-06-L
T2 and T3 Sizing
D. Minimal ductus diameter
E. Ductus length
T3
Occluder sizing for patients ≤2kg using intraductal placement
D
mm (in)
3–12 (0.118–0.472) ≥12.1 (≥0.476)
≤1.7 (≤0.067) 9-PDAP-03-02-L 9-PDAP-03-04-L
1.8–3.2 (0.071–0.126) 9-PDAP-04-02-L 9-PDAP-04-04-L
3.3–4 (0.130–0.157) 9-PDAP-05-02-L 9-PDAP-05-04-L
E
mm (in)

‡ Indicates a third party trademark, which is property of its respective owner.
Abbott Medical
5050 Nathan Lane North
Plymouth, MN 55442 USA
+1 855 478 5833
+1 651 756 5833
™ Indicates a trademark of the Abbott group of companies.
Pat. http://www.abbott.com/patents
© 2020 Abbott. All Rights Reserved.
ARTEN600042307 B
2020-07
 Loading...
Loading...